Introduction
This Supplement marks 30 years of cooperation in sarcoma treatment in Scandinavia. The focus of sarcoma management has changed dramatically since the SSG was founded in 1979. Then referral of patients to a sarcoma center before surgery was the most important treatment associated factor that could be changed.
Today, approximately 90% of bone and soft tissue sarcoma patients in orthopedic sites are referred untouched to Scandinavian sarcoma centers. The improvement of referral practices of patients with intraabdominal and retroperitoneal sarcomas has been much slower. With the advent of more preoperative imaging in undiagnosed abdominal conditions and with the focus on GIST, the referral of these patients is improving dramatically. In gynecological sarcoma patients the need for centralized management is the same as for retroperitoneal tumors. The SSG Registry was primarily designed for orthopedic tumors and the recording of sarcomas in other sites has been haphazard at best. In the last edition of the SSG Registry (www.ssg-org.net) specific forms to register abdominal, retroperitoneal and uterine sarcomas have been designed. We expect that knowledge of diagnostic practices and treatment results for this large patient group, mostly treated outside of clinical trials, will increase as more patients are reported to the SSG Registry.
The population-based series of all chondrosarcomas in Sweden 1980–2002 of the chest wall shows that correct diagnostics at a sarcoma center are at least as important as the treatment. Almost half of the chondrosarcoma patients were treated outside of sarcoma centers and there was often a long doctor's delay. Interestingly, doctor's delay was often associated with a needle biopsy performed outside of a sarcoma center, providing incorrect assurance that the swelling of the chest wall was benign. This type of clinical series is important to show what happens when sarcoma care is not centralized and shows that the cost and inconvenience of traveling to a sarcoma center are far outweighed by the benefits.
The first clinical trial run by the SSG was the SSG I protocol of doxorubicin treatment in high-grade soft tissue sarcoma. 30-years later the benefits of adjuvant chemotherapy remains unclear. However, we know much more about prognostic factors in soft tissue sarcoma and can now select patients who are at high risk of metastatic disease. Many trials of adjuvant treatment for soft tissue sarcoma are flawed by the inclusion of patients with a good prognosis. By applying the clinical risk factors that have been proven relevant we can select a cohort of high-risk patients where it will be easier to assess the effect of adjuvant treatment.
In 1979, adjuvant and neo-adjuvant chemotherapy in osteosarcoma and Ewing sarcoma still remained to be explored and proven. Today we know the benefits but also the limitations of chemotherapy as still about 30% of the patients will eventually die of their sarcoma. The first SSG trial of osteosarcoma was initiated in 1982. We now realize that to improve survival we need randomized multi-group trials of new approaches to osteosarcoma treatment. Here EURAMOS constitutes a unique collaboration between sarcoma groups in Europe and the USA. Although we are moving towards more international collaboration, regional sarcoma groups such as the SSG will remain necessary to ensure compliance and quality of data.
One pillar of quality control within the SSG is the pathology board. This group now has a vast experience in reviewing common and uncommon sarcoma entities, and also in assessing chemotherapy response in different SSG protocols. All treatment protocols rest upon assurance that the diagnosis is correct, and that histological malignancy grade and risk factors are correctly assessed. When the Central Registry is used for in-depth studies of specific entities, such as liposarcoma in the present volume, uniform diagnostic criteria and grading are a prerequisite to assess treatment results. The SSG pathology group has always maintained close collaboration with cytogenetics primarily for diagnosis, for example in Ewing and synovial sarcoma, but the paper on genetic profiling shows that cytogenetics also has a role in prognostication.
We thank you for taking the time to read this volume dedicated to the SSG experience
Kirsten Sundby Hall
Thor Alvegård
Henrik Bauer
Chairpersons of the SSG
The Scandinavian Sarcoma Group
Summary of the first 30 years
Thor A Alvegård1, Henrik C F Bauer2, Paula Lindholm3, Anders Rydholm4, Svante Sigurdsson5, Kerstin Sundby Hall6
1Department of Cancer Epidemiology, Lund University Hospital, Lund, 2Orthopedic Oncology Service, Karolinska Hospital, Stockholm, Sweden, 3Department of Oncology, University Hospital, Turku, Finland, 4Department of Orthopedics, University Hospital, Lund, Sweden, 5Department of Oncology, Landspitalinn Hospital, Reykjavik, Iceland, 6Department of Oncology, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway
Correspondence Thor Alvegård: [email protected]
Musculoskeletal sarcomas call for multidisciplinary management by a “tumor team” of specialized orthopedic surgeons, radiologists, pathologists, tumor biologists (e.g. molecular and cytogenetics, DNA cytometry), cytologists, radiotherapists, and oncologists (). Only a few such teams existed in Scandinavia during the 1970s. With the inception of the Scandinavian Sarcoma Group (SSG) in 1979, several new teams were started, each with regional responsibility for centralized treatment of sarcoma patients. Together, Denmark, Finland, Iceland, Norway and Sweden have a population of 27 million. These countries have similar social structures, with modern medical services covering all inhabitants and an effective registration of all cancer patients. The similarity of the medical care systems in the Scandinavian countries makes multicenter studies easier to perform. The activities reported at the annual Scandinavian meetings (SSG) (Rydholm and Alvegård Citation1994a, [Citation1994b], [Citation1995], [Citation1996], [Citation1997], [Citation1998], 1999, 2000, 2001, 2002, [Citation2003]) stimulated Scandinavian sarcoma research, which is reflected in an increasing number of reports in the scientific literature.
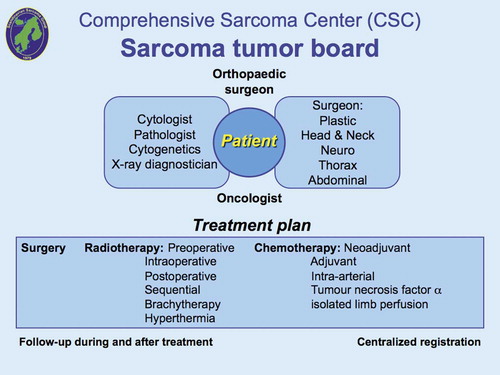
Figure 1. The sarcoma tumor board defines the diagnosis and determines the treatment and centralized registration. It is important that all sarcoma experts jointly to define diagnosis, treatment and follow-up.
Organization of the Scandinavian Sarcoma Group
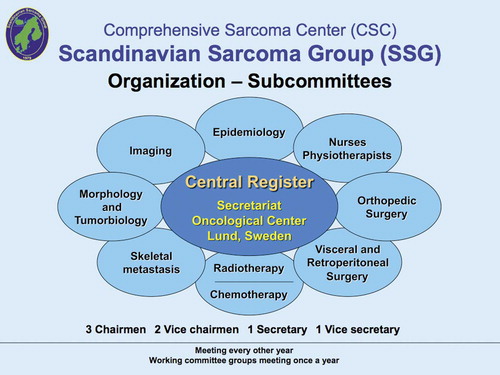
The Scandinavian Sarcoma Group (SSG) was constituted in 1979 and is composed of oncologists (pediatric and adult), surgeons, radiologists, pathologists, tumor biologists, nurses and physiotherapists from the Nordic countries (). The aim of the SSG is to uphold and improve the quality of diagnostics, treatment and care of sarcoma patients by sharing information and education, and stimulate and coordinate basic and clinical research. The SSG maintains two patient registers, i. e., the SSG Register of Bone and Soft Tissue Sarcoma Patients and the SSG Skeletal Metastasis Register, both financed by grants (Bauer et al. Citation2004, Hansen et al. Citation2004). The SSG is open to all specialists in the Nordic countries interested in sarcoma and has no membership fee. The Swedish Cancer Society, Nordic Cancer Union (NCU), several pharmaceutical companies and private donors have supported our Scandinavian research and development of treatment strategies for musculoskeletal tumors. The salary of the full-time secretary is paid by the Southern Swedish Oncologic Center, University Hospital of Lund, Sweden.
Figure 2. Organization of the Scandinavian Sarcoma Group. The morphology group meets 2–3 times a year for peer-review of all registrated sarcomas.
The SSG holds meetings yearly, Subcommittee Meetings in December yearly and the General Assembly in the spring every other year. Notes of the meetings are kept by the SSG secretary and chairmen of the subcommittees. The SSG Board consists of two Chairmen, two Vice-Chairmen, one Secretary, one Vice-Secretary and the respective chairmen of the 10 subcommittees (SSG Sarcoma Register, Epidemiology, Imaging, Morphology, Tumor Biology, Orthopedic Surgery, Visceral and Retroperitoneal Surgery, Oncology (pediatric and adult), SSG Metastasis Register and SSG Nurses and Physiotherapists). The Chairmen and Secretaries are elected by the General Assembly for 5 years. The Subcommittees elect their own chairmen.
The SSG office is located in Lund and is responsible for the preparation of meetings, keeping the Register of SSG members, and for the applications and details concerning grants. All subcommittees have a joint meeting once a year, to develop new strategies regarding research and treatments for musculoskeletal tumors. At our general meeting every other year (with about 130 active SSG members), new developments and strategies are submitted and discussed. Guest lectures are given by Scandinavian and international experts in various fields.
Goal
The main goal of the group is to improve the treatment of sarcoma patients in Scandinavian countries. Their outcome depends on a number of factors, some of which can be influenced. These include patient's and doctor's delay, referral to a highly specialized tumor center, the abilities of the diagnostic and therapeutic teams, the principles of treatment, the available equipment and details of the treatment schedules. Better treatment requires clinical and basic research. Our SSG register in connection with the national cancer registries and new biobank registry will in the future make translational sarcoma research easier to perform ().
Figure 3. SSG – Central register, cancer registries and biobank registries makes translational sarcoma research easier in the future.

Communication lines
The local groups are represented in the Scandinavian Sarcoma Group by one or several members. This permits direct contact between the SSG and the doctors treating the patients. The chairmen, the vice-chairmen, the secretary, the vice-secretary and most subcommittee chairmen are members of the European Musculoskeletal Oncology Society (EMSOS). Other members participate in and report about meetings of the Société Internationale d' Oncologie Pédiatrique (SIOP), European Organization for Research and Treatment of Cancer (EORTC), Connective Tissue Oncology Society (CTOS) and International Society of Limb Salvage (ISOLS). The SSG is thus part of the international sarcoma society network.
Centralization
Physicians outside the tumor treatment centers, who are the first to see the patient, must know when to suspect a sarcoma. This is a simple matter in most cases of skeletal sarcomas: pain and/or a palpable tumor lead to a conventional radiographic examination, which almost always arouses suspicion of a sarcoma. Therefore most patients with skeletal sarcomas were referred to tumor treatment centers before the Scandinavian Sarcoma Group was founded. However, at the time of inception of the SSG, many patients who had soft tissue sarcomas were treated after considerable delay in local hospitals and often with inadequate surgery. They therefore arrived at the tumor centers with advanced tumors, local recurrences or metastases. To improve the prognosis for these patients, the following recommendations were made:
All patients with soft tissue lesions suspected of malignancy should be referred to a tumor center, without prior biopsy.
Indications for referral to a tumor center before surgery:
– deep tumor of any size
– subcutaneous tumors larger than 5 cm and
– all other tumors, suspected of being malignant.
If a soft tissue sarcoma has been diagnosed by fine needle aspiration biopsy, incisional biopsy or excision, the patient should be referred to a tumor center, without further surgery.
This recommendation was signed by all active SSG members in Helsinki in 1982 from 4 countries representing 9 specialties and 21 tumor centers. The recommendation has been published in each country in the national medical journals, in books and has been presented at meetings. Copies have been sent to local hospitals and individual doctors. Since many years 9 of 10 patients with soft tissue sarcomas in southern Sweden, are referred to the regional tumor center. Among patients with deep sarcomas, 80% are referred before biopsy. During recent years all centers in the Scandinavian Sarcoma Group have achieved this favourable referral pattern (Rydholm Citation1997, Bauer et al. Citation2004).
Clinical investigations
The following studies have been started by the SSG since 1979:
SSG I: Soft tissue sarcoma. Malignancy grades III and IV. Wide ± adj. doxorubicin. Marginal surgery + radiotherapy ± adj. doxorubicin. A randomized study. Started 1981, ended Feb. 1986; 240 patients (Alvegård et al. Citation1989, Alho et al. Citation1989, Alvegård et al. Citation1989, Alvegård et al. Citation1989, Alvegård et al. Citation1990, Alvegård Citation1989, Wiklund et al. Citation1993). This was the second largest study included in the individual data meta-analysis reported by Tierney et al. 1997.
SSG II: Osteosarcoma. Combined primary treatment, ad modum Rosen T 10 protocol. Nonrandomized. Started 1982, ended 1989; 114 patients (Solheim et al. Citation1989, Saeter et al. Citation1991, Solheim et al. Citation1992).
SSG III: Soft tissue sarcoma. Planned in 1983 as a randomized study on the effects of various irradiation schedules on inoperable tumors. However, too few patients were included, and the study was discontinued.
SSG IV: Ewing's sarcoma. Combined modality treatment ad modum Rosen T 11 protocol. Nonrandomized. Started 1984, ended 1990; 52 patients (Alvegård et al. Citation1989, Nilbert et al. Citation1998).
SSG V: Treatment program for soft tissue sarcoma (all malignancy grades). Nonrandomized.
SSG VI: Osteosarcoma metastases. Combined modality. Nonrandomized. Started summer of 1987, ended 1989; 15 patients.
SSG VII: Centralized register of patients with sarcoma in Scandinavia. Started 1986, ongoing; >10 000 patients.
SSG VIII: Osteosarcoma. Combined primary treatment with high doses of methotrexate, cisplatinum and adriamycin preoperatively. Nonrandomized. Started 1990, ended December 1997; 113 patients (Saeter Citation1996a, Saeter Citation1996b, Smeland Citation2003).
SSG IX: Ewing's sarcoma. Combined modality treatment with cisplatinum, vincristin, adriamycin, ifosfamide, surgery ± hyperfractionated irradiation. Nonrandomized. Started 1990, ended April 1999; 133 patients (Elomaa et al. Citation1996, [Citation1999], [Citation2000]).
SSG X: Treatment of metastatic soft tissue sarcoma with ectoposide, ifosfamide and GCSF. Started 1991, ended 1995; 114 patients (Saeter et al. Citation1995, Saeter et al. 1996, Saeter et al. Citation1994).
SSG XI: Treatment of metastatic soft tissue sarcoma with trofosfamide. Started 1994, ended 1996; 40 patients.
SSG XII: Metastasectomy and chemotherapy for lung metastasis from soft tissue sarcoma. EORTC/ SSG randomized phase III study. Started July 1996, ended 1998; 15 patients.
SSG XIII: A Scandinavian Sarcoma Group treatment protocol for adult patients with high-risk soft tissue sarcoma of the extremities and trunk wall. Started 1998, ended 2007; 143 patients.
SSG XIV: A Scandinavian treatment research protocol for extremity localized high-grade osteosarcoma. Started 2001, ended 2005; 73 patients.
SSG XV: Phase III randomized, intergroup, international trial assessing the clinical activity of STI-571 at two levels in patients with unresectable or metastatic gastrointestinal stromal tumors (GIST) expressing the KIT receptor tyrosine (CD117). The Scandinavian Sarcoma Group was not accepted to participate in this trial by the EORTC because of patient health insurance problems.
SSG XVI: Registration of patients with surgically treated skeletal metastases. Started April 2000, ongoing; 1 000 patients.
SSG XVII: Recommendations for the diagnosis and treatment of abdominal, pelvic and retroperitoneal sarcomas. Started May 2002.
SSG XVIII: Short (12 months) versus long (36 months) duration of adjuvant treatment with the tyrosine kinase inhibitor imatinib mesylate of operable GIST with a high-risk for recurrence: A randomized phase II study. Started January 2004, ended 2008; 400 patients.
SSG XX: Phase II non-randomized treatment protocol for adult patients with non-metastatic high-risk soft tissue sarcoma of the extremities and trunk wall. Started 2007, ongoing; 25 patients.
ISG/SSG I: An Italian - Scandinavian treatment and research protocol for high-grade osteosarcoma of the extremities. Localized disease and metastatic relapse. Started March 1997, ended September 2000; 187 patients (Ferrari et al. Citation2005, CitationSerra et al. 2006).
ISG/SSG II: An Italian - Scandinavian treatment protocol for metastatic and pelvic osteosarcoma. Started March 1998, ended December 2003; 55 patients (Del Prever et al Citation2005).
ISG/SSG III: An Italian - Scandinavian treatment protocol for standard-risk Ewing's sarcoma. Started June 1999, ongoing; 106 patients (Ferrari et al. Citation2007).
ISG/SSG IV: An Italian - Scandinavian treatment protocol for high-risk Ewing's sarcoma. Started June 1999, ongoing; 70 patients.
Euroboss I: A European treatment protocol for bone sarcoma in patients older than 40 years. Started February 2003, ongoing; 220 patients.
Euramos I: A randomized trial of the European and American Osteosarcoma Study Group to optimize treatment strategies for resectable osteosarcoma based on histological response to preoperative chemotherapy. Started spring 2004, ongoing; 60 patients.
The Scandinavian Sarcoma Group Register
A register for data makes possible multicenter studies concerning treatment results and prognostic factors for local recurrence and survival of patients with soft tissue and bone sarcomas. Such studies are needed to determine more exactly how these patients should be treated. Our position is unique because of the close to 100% follow-up that is possible in Scandinavian countries. The SSG Register of soft tissue and bone tumors was started on March 1, 1986. The Register is now used for detailed studies on treatment and prognosis. It gives important information on how the treatment of patients with musculoskeletal tumors is evolving in Scandinavian countries. For example, important changes in referral patterns, preoperative diagnostic techniques and surgical margins have been found (Bauer et al. Citation2004).
Results and strategies
Soft tissue sarcoma
In our first randomized study (SSG I, 1981–1986), we reported that adjuvant chemotherapy with doxorubicin had no effect on metastasis-free and overall survival rates (Alvegård et al. Citation1989). SSG participated in a review and meta-analysis of the published results of all 15 randomized clinical trials (Tierney et al. Citation1997). In a multivariate analysis of the SSG I material, the following factors were identified as independent variables for predicting the development of distant metastases: malignancy grade IV, tumor size >10 cm, intratumoral vascular invasion and necrosis, as well as male sex. Recently, the Lund group devised a system based on three factors: tumor size, necrosis and vascular invasion. In a population-based study from the southern health region of Sweden, two prognostic groups were identified: 1) a good prognosis group with one or no factors present with a 5-year metastasis-free survival of 81% and 2) a poor prognosis group with two or three factors present and a metastasis-free survival of 32%. The good and poor prognosis groups included approximately 70% and 30% of the patients (Gustafson 1993, [Citation1994]). Adjuvant treatment strategies have been developed, based on preliminary good results of this analysis (SSG XIII). A phase II non-randomized treatment protocol for adult patients with non-metastatic high-risk soft tissue sarcoma of the extremities and trunk wall started in October 2007 (SSG XX).
Intraabdominal, retroperitoneal, pelvic and uterine sarcoma working group
All patients should at suspicion or diagnosis of a sarcoma be referred to a specialized centre for further evaluation and treatment. The management of intraabdominal retroperitoneal and pelvic soft tissue tumors is complex and prognosis of patients with such tumours can be affected from the earliest stages of work-up. All patients should therefore be treated by a multidisciplinary group with interest and experience in sarcoma. That includes all categories involved in the evaluation and treatment as surgeon, oncologist, cytologist, pathologist and radiologist. The only way to achieve this is by gather these rare patients to only a few units; centralization is the only way to be able to collect patients enough to get and maintain skill and experience, to develop and improve treatment, to collect patients and biological material enough for e.g. tissue bank and scientific studies and to be able to report outcome and follow-up.
Our SSG recommendations are based on proposals made by the Scandinavian Sarcoma Group (SSG) members, mainly from the recently established group responsible for intraabdominal, retroperitoneal, pelvic and uterine soft tissue sarcoma. The guidelines are aimed to give a general overview for the most important and initial decisions to be made and will provide recommendations that are based on the best available evidence. They will be updated periodically in accordance to the current knowledge of these disease entities.
Soft tissue sarcomas arising in the retroperitoneal space or in the intraabdominal cavity traditionally carry a poor prognosis. Many factors contribute to the fact that both the disease free and overall survival figures are poor among patients with sarcomas within these areas. However, the introduction of tyrosine kinase inhibitors has dramatically changed the treatment and course of GIST. Even in metastatic disease the maximum duration of response to Imatinib and other tyrosine kinase inhibitors is not yet known, and some patients may respond for longer than 5 years.
With the goal of increasing the survival of this group of sarcoma patients, the subsequent recommendations will focus on the:
– anatomical evaluation
– pathological diagnosis
– surgical management
– adjuvant and palliative therapy
– clinical trials
– follow-up
Osteosarcoma
In our first neo-adjuvant chemotherapy protocol for osteosarcoma (SSG II) we had a good tumor response in 19% using four treatment cycles with high doses of methotrexate. 5-year overall and metastasis-free survival rates were 62% and 58%, (Solheim et al. Citation1989, Saeter et al. Citation1991, Solheim et al. Citation1992). In our SSG VIII protocol the good tumor response rate is 60% after preoperative chemotherapy with high doses of methotrexate, cisplatinum and adriamycin (Smeland et al. Citation2003). Two new protocols (ISG/SSG I, II) have been started in collaboration with the Rizzoli Institute, Bologna. Increasing preoperative chemotherapy, including high doses of methotrexate, ifosfamide, cisplatinum and doxorubicin did not show increase of the metastasis-free or overall survival rates (Bacci et al. Citation2002). The SSG XIV protocol started in 2001 and ended in 2005 with 73 patients. A publication is under preparation. Since spring 2004 SSG is joining the European and American Osteosarcoma Study Group (EURAMOS I).
Ewing's sarcoma
The first report on our first study (SSG IV) was made by Nilbert et al. (Citation1998) with a long-time follow-up time by Smeland et al. (Citation2004). Our second study (SSG IX), using a combination of high doses of chemotherapy, surgery and accelerated fractionated radiation therapy, has so far resulted in a good tumor response following preoperative chemotherapy and preliminary results show a 5-year overall survival of approximately 70% (Elomaa et al. Citation1994, [Citation1996], [Citation1999]). Collaboration with the Rizzoli Institute has been started to develop a high dose treatment for poor responders, following preoperative chemotherapy (ISG/SSG III and IV) and started June 1999.
Surgical sarcoma network
In the Nordic countries surgical sarcoma treatment is centralized to larger university clinics. Many studies show that the most important prognostic factor for local control is centralization of untouched sarcomas to centres where treatment is supervised by formally organized multidisciplinary sarcoma teams. To ensure compliance with centralization guidelines for such rare tumours as sarcomas, the guidelines and procedures of centralization must be well known at local hospitals. This is best achieved when the geographical distance between local hospital and centre is not too large. The organisational guidelines was pioneered in the Southern Swedish Health region 30 years ago, and the University Hospital in Lund now has more than 90% of sarcomas referred as virgin tumours.The guidelines are implemented throughout Scandinavia and the organization has served as a model to other regions in Europe.
As the Nordic countries are a relatively sparsely populated large geographical area, the organizational arrangements imply that no sarcoma centre has more than 3–4 million inhabitants in their uptake area. This is less than many European centres and necessitates closer cooperation among surgeons in Scandinavia. To facilitate this, the SSG has since 2005 organised an internet based discussion forum called the surgical sarcoma network. It is e-mail based communication with attached radiological material, chosen by the surgeon initiating discussion, and distributed to all sarcoma surgeons in Scandinavia. This normally results in several suggestions of treatment within hours after a problem is posted. The suggestions are informal opinions “among equals”, much like the discussion among senior sarcoma surgeons at in-house meetings at the larger sarcoma centres in Europe and USA. The discussion has on occasions concluded that an unusual operation are best performed by a multi-institutional team of surgeons, and details have been arranged using the net-work facility. The legal responsibility for all actions based on the discussions remains with the treating surgeon.
The current treatment protocol for high grade soft tissue sarcomas has an optional arm for preoperative radiation treatment when there is “an obvious risk of intralesional surgery”. The surgical sarcoma network is used to establish a uniform interpretation of this inclusion criterion.
Annually about 20 new cases are presented from 13 institutions in Sweden, Finland, Denmark, Norway and Iceland, resulting in 115 e-mailed contributions to the discussion.
Centralized registration of patients with surgically treated skeletal metastases
Current surgical treatment for pathologic fractures is based on retrospective analyses of single institution experience. The reported series comprise heterogeneous patient populations regarding types of primary cancer, extent of the metastatic disease and location of the lesions. Areas of uncertainty include operative methods, indications for prophylactic surgical treatment and need for postoperative radiotherapy as radiation decreases the risk of local tumor progression but increases bone-healing complications.
The Scandinavian Sarcoma Group started the Skeletal Metastasis Registry in 1999 to improve the surgical treatment of skeletal metastases. Criteria for inclusion are patients surgically treated for either impending or complete non-spinal fractures due to skeletal metastase. 9 orthopedic oncology centres from Sweden, Denmark, Norway and Finland participate and data regarding more than 900 surgically treated patients have so far been registered. Additional aims of the registry are to provide a tool for quality assessment as measured in terms of reoperation date, operation morbidity and operation frequency for impending fractures.
SSG Nurses and physiotherapists group
At the annual SSG meeting 2007, May, 8–11, in Bergen, Norway, this group was officially established. Increased cooperation between nurses, physiotherapists and doctors is necessary for the outcome of the Scandinavian sarcoma patients. The main goal of this group is to: increase cooperation, knowledge and motivation. See also page 91 in this issue
SSG's publications
Since start 1986 more than 1000 articles have been published by members of the Scandinavian Sarcoma Centers i.e., 1979–1989 (Solheim et al. Citation1989), 1989–1993 (Alvegård and Rydholm 1994), 1993–1998 (Rydholm and Alvegård Citation1998), 1998–2003 (Alvegård and Rydholm Citation2004). For publications 2004–2008 see 92–104 in this issue. These publications represent research from the various Scandinavian Tumor Centers and the Scandinavian Sarcoma Group Research program. 31 members wrote their Ph.D. theses on issues relevant to sarcoma in this period (Table).
Table 1. Overview of Ph.D. theses by SSG members on issues relevant to sarcoma, 1979–2007
The Scandinavian Sarcoma Group Register 1986–2008
Henrik CF Bauer, Jan åhlén, Thor A Alvegård, Örjan Berlin, Gunnar Follerås, Anders Rydholm, Kirsten Sundby Hall, Clement S Trovik, Fredrik Vult von Steyern
The Scandinavian Sarcoma Group, University Hospital, SE-221 85 Lund, Sweden. Tel +46 (0)46 177560, Fax +46 (0)46 188143.
The Scandinavian Sarcoma Group Register (SSG Register) was initiated in 1986 with the aim of collecting prospective sarcoma data from all Scandinavian countries. There was a consensus that we needed population based data on all sarcoma patients, not only the few who qualified for clinical treatment trials. The SSG Register was based on the experience from the Southern Sweden Sarcoma Register founded in 1975 (Rydholm Citation1983). The SSG Register was designed to acquire data on referral, tumor characteristics, treatment and outcome with a minimum follow-up of 5 years. All centers in Sweden and Norway participated from the conception whereas no centers from Denmark and only Helsinki in Finland chose to participate. In the early 1990ies Helsinki advised that they did not have the resources to continue so the SSG Register has essentially become a Norwegian and Swedish affair. With a population base of approximately 14 million people and all centers participating we accomplished our goal of creating a population based sarcoma Register. Comparisons with patients entered in the National Cancer Registries show that more than 90% of sarcomas of extremities and the trunk wall are reported to the SSG Register.
9 052 sarcoma patients have been prospectively recorded in the SSG Register until September 2008. There are 4 583 soft tissue sarcomas of extremity and trunk wall and 1 852 soft tissue sarcomas of “non-orthopedic” sites, i.e. visceral, retroperitoneal, gynecological, and head-neck. There are 2 671 bone sarcomas including also benign giant cell tumors.
The SSG Register serves two purposes: to monitor referral pattern, treatment, and outcome over time, and to identify subsets of patients for in-depth studies. Regarding monitoring we have shown that referral of extremity sarcoma patients has continuously improved and since several years almost all these patients are referred to sarcoma centers, four fifths of them with primary tumors before biopsy or surgery (Bauer et al. Citation2001).
We have also shown that we have been too restrictive in our indications for adjuvant radiotherapy in soft tissue sarcoma. Wide surgical margins (including myectomy) for deep-seated high-grade sarcomas were associated with a 25% risk of local recurrence (Trovik et al. Citation2000). Hence, since 1998 we advise radiotherapy for all high-grade deep seated soft tissue sarcomas, irrespective of surgical margin and SSG Register data shows that the 5-year local recurrence rate has decreased to 15% (Jebsen et al. Citation2008).
In-depth studies based on SSG Register data are of importance to maintain interest in the Register. There have been 4 doctoral theses based on patients from the SSG Register and there are 3 ongoing projects (Table). Since these in-depth studies involves going back to the original patient charts they lead to control and improvement of data quality. They also lead to improved follow-up. Most importantly all patients included in these projects have had their diagnosis reviewed by the SSG Pathology Board. This ensures consistency in applying diagnostic criteria across Scandinavian Sarcoma Centers.
Thesis projects based on the SSG Register
The question arises whether collecting data to the SSG Register and assuring follow-up and data quality is worth the effort. There has been a slowing of reporting both new patients and of follow-up. This may be related to generation changes, the individuals who started registration at the different sarcoma centers in Norway and Sweden are being replaced by a younger generation. But steps are now taken to revitalize to local coordinators. We firmly believe that the type of quality control that is maybe the most important feature of the SSG Register is paramount for maintaining excellent sarcoma care. We do not need the Register to monitor osteosarcoma care in the young, but we need it as a means of defining and defending our treatment recommendations in the 90-year-old with a soft tissue sarcoma.
The SSG Register has been extensively revised during the last year (SSG VII: 4 www.ssg-org.net). Dr. Clement Trovik in Bergen ([email protected]) has taken over the Chairmanship of the SSG Register Subcommittee. He has worked closely with the SSG secretariat and data manager to revise the entry and follow-up forms and to insert checkpoints to identify incongruent or missing data. The forms for visceral and retroperitoneal sarcomas have been better adapted to treatment and classification applied in these regions. Trials of online registration are under way.
The viability of the SSG Register will be ensured if it is used for quality control and as a research tool. All SSG members are invited to submit plans for in-depth studies of Scandinavian sarcoma care.
Genetic profiling – implications for refined diagnosis and treatment of soft tissue sarcomas
Ana Carneiro1 and Mef Nilbert1,2
1Department of Oncology, Institute of Clinical Sciences, Lund University and Lund University Hospital, Lund, Sweden
2Clinical Research Centre, Hvidovre Hospital, Copenhagen University, Hvidovre, Denmark
Correspondence AC: [email protected]
Summary Soft tissue sarcomas (STS) are challenging as they represent a morphologically and genetically heterogeneous groups of tumors. A multitude of genetic changes, often in the form of fusion genes, were recognized during the 1980's and now constitute a diagnostic lexicon in several STS subtypes, whereas many of the more common subtypes are genetically complex without distinct alterations. Refined STS management requires improved diagnostic reproducibility, novel prognosticators, and introduction of targeted therapies. In recent years, a number of genetic profiling studies – analyzing copy-number alterations as well as gene expression changes – have deepened our understanding of STS development through demonstration of recurrently deregulated tumorigenic pathways. The challenge is now to bring the genetic profiles into clinical decision-making. This review, in conjunction with the Scandinavian Sarcoma Group's (SSG) 30 years jubilee, discusses how the information from genetic profiling studies may be translated into clinical practice for refined diagnostics, prognostics, and treatment of STS.
In the past 3 decades cytogenetics and molecular genetics have revealed specific chromosomal translocations and fusion genes in a large number of soft tissue sarcoma (STS) subtypes, but have also demonstrated extensive genetic complexity in other subtypes. Within the last decade, high-throughput techniques have enabled simultaneous analysis of multiple genes and thereby provided a deeper insight into the genomic aberrations and their interrelations with tumor associated pathways in STS ().
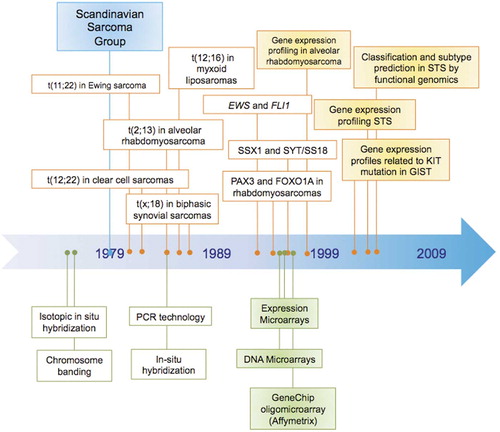
Figure 1. Timeline with the most important genetic and expression findings in soft tissue sarcomas and the techniques used for cytogenetic, genomic and expression studies in the past 30 years. Studies that first used a given approach are highlighted in yellow: Khan et al. published the first expression profiling study in sarcomas (alveolar rhabdomyosarcoma) in 1998, while the first study profiling different STS histotypes was published in 2002 by Nielsen et al.; Segal et al. (2003) first applied genomic profiling for classification and subtype prediction in STS; correlation of genomic and expression profiles with given tumor characteristics was first performed in GIST with KIT mutation by Allander et al. (2001).
Simple and complex karyotypes
The identification of chromosomal abnormalities in STS has added a new dimension to diagnostics by complementing traditional microscopic examination. Detection of tumor specific chromosomal aberrations by conventional cytogenetics is nowadays especially useful in confirming the diagnosis of poorly differentiated sarcomas, in particular those arising in unusual locations or age groups, or tumors exhibiting atypical histopathologic features. From a genetic perspective, sarcomas fall into two major categories; one characterized by relatively simple, near-diploid karyotypes and another characterized by highly complex, severely deranged chromosomes and aneuploidy. Sarcomas with relatively simple karyotypes harbor reciprocal chromosome translocations resulting in the formation of fusion genes. Although these translocations are probably crucial in tumorigenesis, they are typically associated with additional genetic alterations, the roles of which remain to be defined (CitationDeneen et al. 2001).
The genetically complex subtypes continue to puzzle clinicians and pathologists due to extensive heterogeneity, frequent pleomorphism, and lack of discriminative genetic alterations. STS with complex karyotypes can be exemplified by leiomyosarcoma, pleomorphic liposarcoma, and undifferentiated pleomorphic sarcoma (UPS, formally referred to as malignant fibrous histiocytoma – MFH). Cell-cycle gene disturbances are commonly present with a high prevalence of p53 checkpoint alterations, including inactivating mutations of TP53 and homozygous deletion of CDKN2A (CitationStratton et al. 1990, CitationLatres et al. 1994, CitationDei Tos et al. 1996, CitationOrlow et al. 1999, Creager et al. Citation2001). Frequent amplification of CDK4 and MDM2 have also been reported in sarcomas with complex karyotypes, although the exact roles and interactions of these two genes in sarcoma development and progression remains unknown. Amplification of CDK4 and MDM2 occur both in well differentiated lesions and in pleomorphic tumors, suggesting that these events may occur early in tumorigenesis. Genetically complex STS frequently show dramatically lengthened and heterogeneous telomeres (CitationMontgomery et al. 2004). The large brightly stained telomeric regions are reminiscent of those observed in immortalized cells that maintain telomeres in a telomerase-independent manner, consistent with deregulation of the alternative lengthening of telomeres (ALT) pathway. The discovery of heterogeneous telomere lengths and ALT pathway involvement in most sarcomas with complex karyotypes supports the existence of a telomere maintenance pathway that is incapable of karyotypic stabilization. In addition, both ALT activation and telomerase expression have been correlated to poor prognosis (CitationAvigad et al. 2007, CitationCairney et al. 2008).
Methodological considerations in genetic profiling
In STS, most diagnostically applied alterations are apparent using low-resolution techniques such as cytogenetics, but detailed mapping of the resultant fusion genes and high-throughput genetic profiling provides detailed insights into the various signalling pathways involved. Moreover, high-resolution mapping of copy-number alterations and expression changes represent powerful techniques to identify novel markers for diagnostic, prognostic, and predictive applications. High-density gene arrays simultaneously allow characterization of the expression of more than 30.000 genes (or more in oligonucleotide arrays) in a single experiment.
Pre-processing and gene filtering are essential steps before data analysis, the latter aiming at eliminating genes that are poorly measured or that lack variation across the samples. By eliminating genes from further analysis, this step irreversibly influences the number of informative genes and thereby the final results. Depending on the aims of the study, data can be analyzed for class discovery, class prediction, and class comparison. Class discovery refers to analyses aimed at discovering clusters within a sample set, and is generally achieved by unsupervised clustering algorithms. Such analyses are performed without use of information about the expected samples class. Supervised data analyses are generally applied for class prediction studies (in which the objective is to predict a given feature, e.g. treatment response) and class comparison studies (in which the objective is to identify biological differences between groups of samples). The latter analyses use data in which samples show a given asset e.g. development of metastasis, and herein aim to identify genes that are differentially expressed. In order to not introduce bias, the findings should be validated in an independent sample set. In this type of analysis, the number of false discoveries is crucial. The conventional level of significance, p=0.05, is not applicable since it would imply 500 false positive genes per 10.000 genes analyzed and frequently re-sampling methods are therefore preferred for the identification of consistently deregulated genes linked to the variable studied. Limited sample size represents a weakness of many microarray studies. The required number of samples depends on the fold-difference in mean expression as well as on the variation in expression within each class, but formulas for calculation of appropriate sample sizes are available. Data management and statistical methods differ between studies and the many steps in the analysis, subject to variable handling, influence the top-ranked gene lists in different studies. However, if interacting genes and pathways, rather than single genes, are considered, the currently available genetic profiling data in STS demonstrate considerable reproducibility with deregulated pathways recurrently demonstrated.
Genetic profiling for refined STS diagnosis
Gene expression profiling has suggested novel classification patterns in several malignancies, including STS, breast cancer, lymphoma, malignant melanoma, and leukemia (CitationGolub et al. 1999, CitationAlizadeh et al. 2000, CitationBittner et al. 2000, CitationPerou et al. 2000, CitationSorlie et al. 2001). In STS, array-based copy-number alterations and gene expression profiling have broadly supported the division into the genetically simple and complex subtypes, but have also provided promising insights into sarcoma biology for application in refined diagnostics.
CitationNielsen et al. (2002) analyzed 41 STS, including GIST, synovial sarcomas, liposarcomas, leiomyosarcomas, MFH and malignant peripheral nerve sheath tumors (MPNST), by cDNA microarray (CitationNielsen, Lancet 2002). Hierarchical clustering yielded 5 major clusters with synovial sarcomas, GIST, and MPNST forming distinct groups. Among the leiomyosarcomas, 6/11 samples formed a separate cluster, distinguished by e.g. calponin expression, whereas the remaining leiomyosarcomas clustered with MFH/UPS and liposarcomas. Class prediction for GIST involved 125 genes, including KIT. Synovial sarcomas were recognized by a 104-gene cluster containing e.g. SSX, EGFR, and retinoic acid receptor pathway genes, leiomyosarcomas were associated with muscle-related genes, and MPNST showed expression of nerve sheath-related genes.
CitationSegal et al. (2003) used Affymetrix GeneChip arrays to analyze 51 STS of 9 subtypes. Distinct clustering was observed among STS with specific translocations, but not among subtypes with genetic complexity and pleomorphism. The discriminating genes within this study included SCF and KIT for GIST, WNT5A, and FZD1 in synovial sarcoma, melanocytic genes in clear-cell sarcomas, and CDK4 and MDM2 in dedifferentiated liposarcoma.
CitationBaird et al. (2005) used oligonucletides-arrays to obtain gene expression profiles of 181 tumors representing 16 histotypes of STS and osteosarcomas. 2766 genes showed differential expression among the sarcoma subgroups and separated the tumors into two distinct clusters: one harboring cytogenetically simple STS and the other composed STS with cytogenetic complexity. Up-regulation of tyrosine kinases and tyrosine kinase receptors was identified in half of the samples. Altered expression of genes in the WNT signaling pathway, e.g. WNT5A and FZD1, was observed in all synovial sarcomas, whereas homeobox genes were overexpressed in several sarcoma subtypes.
Francis et al. (Citation2007) applied cDNA expression profiling to 177 STS. Unsupervised analysis resulted in two major clusters – one mainly containing STS characterized by type-specific genetic alterations and the other predominantly genetically complex and pleomorphic STS. Synovial sarcomas, myxoid/round-cell liposarcomas, and GIST clustered tightly within the former cluster and discriminatory signatures for these were characterized by developmental genes from the EGFR, FGFR, WNT, Notch, Hedgehog, RAR, and KIT signaling pathways. The more pleomorphic STS subtypes, e.g. leiomyosarcoma, MFH/UPS, and dedifferentiated/pleomorphic liposarcoma, were part of the latter cluster and were characterized by relatively heterogeneous profiles, although subclusters herein were identified.
CitationHeidenblad et al. (2006) applied tiling resolution microarrays to sarcomas with ring chromosomes. The DNA copy number profiles revealed multiple amplification targets, with a large number of small amplicons. More than 40% of all amplicons, in STS as well as bone tumors, mapped to chromosome 12 with recurrent amplifications in 12q13.3-14.1 and 12q15.1, encompassing SAS and CDK4, and MDM2, respectively. This study also showed amplification of genes involved in the c-JUN pathway in most of the tumors with 12q amplification.
CitationMeza-Zepeda et al. (2006) used aCGH to create a detailed map of DNA copy number changes in GIST and leiomyosarcomas and herein demonstrated multiple gains and losses in both tumor types. Leiomyosarcomas showed more frequent losses of 10q21.3 and 13q14.2-q14.3 and recurrent high-level amplification of the 17p13.1-p11.2 region. Hierarchical clustering analysis separated GIST from leiomyosarcomas and herein identified 6 discriminating regions, suggesting that aCGH could provide independent and distinct information in histologically similar tumors.
CitationPrice et al. (2007) used whole-genome gene expression to study 68 GIST and leiomyosarcomas in order to identify distinct genetic classifiers. Obscurin and Prune2 were found to accurately distinguish most GIST from leiomyosarcoma, and this classifier was validated in 19 independent samples.
CitationSegal et al. (2003) studied 21 cell lines and 60 primary STS, melanoma, and clear-cell sarcoma. Unsupervised cluster analysis of the gene expression profiles clearly distinguished between STS and melanoma and identified the clear-cell sarcoma cluster as a distinct group with genes involved in melanocyte differentiation as discriminators.
CitationFritz et al. (2002) performed expression profiling and aCGH in pleomorphic, dedifferentiated liposarcomas. The highest amplification peaks contained the genes MDM2, GLI and CDK4, which served as class predictors for this tumor type. Involvement of 12q13-15 indicated a close relationship between dedifferentiated and well-differentiated liposarcomas. Clustering based on the expression levels of 1600 genes allowed most of the tumors to be separated into pleomorphic or dedifferentiated liposarcomas, with the heat shock protein HSP90 and the adaptor protein gene SCAP showing high expression in pleomorphic liposarcomas.
CitationFernebro et al. (2006) applied 27k spotted cDNA microarray slides to assess gene expression profiles in synovial sarcomas with various fusion genes e.g. SS18-SSX1/SSX2 types and with correlations to clinicopathological data. Oncogenes (TCF7 and NOTCH), G-protein coupled receptors, histones, and metallothioneins were more frequently upregulated in tumors with the SSX1 fusion, suggesting different downstream effects for synovial sarcomas with the SSX1 and SSX2 gene fusion types.
Carneiro et al. (Citation2009) applied aCGH and gene expression profiling to 18 leiomyosarcomas and 31 UPS with the aim to identify molecular subtype signatures. Both the gains/losses and the gene expression signatures revealed striking similarities between these STS subtypes. Leiomyosarcomas and UPS were indistinguishable using unsupervised hierarchical cluster analysis and significance analysis for microarrays, which suggests a shared lineage.
Genetic profiling of for refined STS prognostics
Genetic signatures have been recognized to serve not only as diagnostic adjuncts, but also as prognostic markers. Francis et al. (Citation2007) reported a potential metastatic signature in high grade pleomorphic sarcomas characterized by expression of hypoxia-related genes, e.g. HIF-1 alpha (HIF-1a), which independently predicted risk of metastasis. Lee et al. (CitationLee et al. 2004) identified a gene signature related to cell-cycle signal transduction genes that predicted metastasis in leiomyosarcomas. CitationFernebro et al. (2006) reported a set of 30 genes (among them TOP2A) related to metastatic potential in synovial sarcomas, albeit in a small sample set. Carneiro et al. (Citation2009) used aCGH and identified loss of 4q31 and loss of 18q22 as markers of metastasis.
Despite the limited data, these studies suggest that the use of microarrays may provide novel information, which may be particularly relevant in high-grade, pleomorphic, and genetically complex STS, 50% of which metastasize and in which conventional prognostic markers have reached their limit. Moreover, these studies point to specific pathways/aberrations that could be further pursued to unravel mechanisms of metastasis.
Towards targeted therapies in STS
Surgery remains the mainstay treatment for STS. Treatment options in metastatic STS are limited since highly effective chemotherapy regimens are lacking. The two most commonly used drugs are doxorubicin, which yields a response rate of about 20%, and ifosfamide, the added value of which is debated. Subtype-specific treatment and most likely the introduction of targeted therapies are therefore needed in STS. Most clinical trials have considered STS as a single pathological entity, although more than 50 different subtypes exist, many of which develop through specific tumorigenic mechanisms as outlined. Not surprisingly, these subtypes differ in drug sensitivity and a major undertaking in STS is to translate better knowledge of sarcoma biology into application of targeted therapies. GIST with c-kit mutations and the successful introduction of imatinib represent the prime example of a targeted therapy. Although several ongoing trials evaluate targeted therapies in STS, these therapeutic options have not yet reached clinical application.
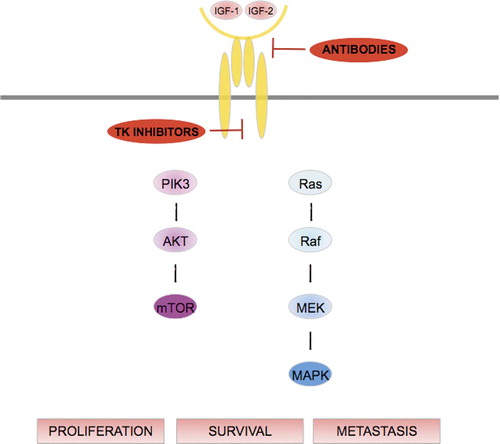
For STS containing fusion genes, strategies specifically inhibiting the fusion genes or their products are attractive as reviewed by CitationTomescu et al. (2001). Such approaches could include antisense therapies, direct targeting of deregulated transcription factors or their pathways, and applications related to immune response mechanisms. For STS with complex karyotypes identification of deregulated pathways through genomic profiling studies constitutes a valuable source to identify biologically relevant tumor subsets and pathways that can be therapeutically targeted. Deregulation of multiple targets within the mammalian target of rapamycin (mTOR) pathway and of factors involved in hypoxia and angiogenesis have repeatedly been observed in STS. Among the more specific targets are e.g. the insulin growth factor receptor (IGFR), members of the WNT pathway in synovial sarcoma, and MDM2 and CDK4 in pleomorphic liposarcoma.
AKT and Mammalian target of rapamycin (mTOR)
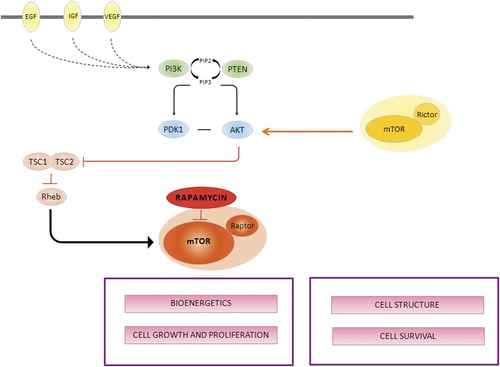
Using high-density microarrays, CitationHernando et al. (2007) identified upstream modulators or intrinsic components of the PI3K-AKT pathway overexpressed at the mRNA level in different STS types. The functional relevance of AKT upregulation was confirmed by the concomitant activation of upstream regulators, suggesting a potential involvement of deregulated AKT-mTOR activity, which was particularly common in MFH/UPS and leiomyosarcomas. mTOR is a key part of AKT activation and therefore a potential drug target for tumors with deregulated AKT. Unlike other kinases, mTOR seems to be a stable target and has not been shown to be mutated in human cancers. mTOR acts as a master modulator of cellular catabolism and anabolism, thereby compromising cell grow and proliferation. Rapamycin and derivatives that specifically block mTOR have recently been developed. Rapamycin, also known as sirolimus, is a lipophilic macrolide demonstrated to have anti-proliferative properties attributed to the modulation of the synthesis of proteins required for ribosome biosynthesis, protein translation, and cell cycle progression, resulting in G1 cell cycle arrest through mTOR inhibition (). Three rapamycin analogs are currently being evaluated i.e. the cell-cycle inhibitor-779 (CCI-779, temsirolimus), RAD-001 (everolimus), and AP23573 (deforolimus). No other proteins have been identified as rapamycin targets, thus making mTOR–rapamycin interaction specific. Results from phase I studies with mTOR inhibitors have shown favorable tolerability and have suggested antitumor activity in several tumor types, including sarcomas. Several phase II trials have been performed with promising clinical efficacy of AP23573, which is currently being evaluated in a phase III trial (ClinicalTrials.gov identifier: NCT00538239).
Figure 2. mTOR is a large protein kinase that nucleates at least two distinct multi-protein complexes: the rapamycin-sensitive complex, also called mTOR complex 1 (mTORC1) and the rapamycin-insensitive complex (also called mTOR complex 2, mTORC2), which is defined by its interaction with RICTOR (rapamycin-insensitive companion of mTOR). In response to growth factors and nutrients mTORC1 regulates cell growth by modulating translation of eukaryotic translation ignition factor 4E binding protein 1 (4EBP1) and ribosomal S6 kinase 1 (S6K1). mTORC2 is considered part of the PI3K–AKT pathway as it directly phosphorylates AKT in response to growth-factor signaling mediated by tyrosine kinases receptors (EGF, VEGF, IGF). After being recruited to the membrane, PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) and thereby converts PIP2 to PIP3. Subsequently, PIP3 recruits AKT and 3-phosphoinositide-dependent protein kinase 1 (PDK1) to the plasma membrane resulting in partial activation of AKT. Phosphatase and tensin homologue deleted on chromosome ten (PTEN) dephosphorylates PIP3 back to PIP2 and thus shuts off PI3K signaling. Activated AKT leads to the inhibition of the tumor suppressors tuberous sclerosis complex (TSC1 and TSC2), thus promoting the activation of mTOR through Rheb.
Insulin growth factor (IGF)
Elevated expression of IGF2 has been demonstrated in several STS histotypes as well as in osteosarcomas. In synovial sarcomas, IGF2 overexpression is induced by the SYT/SSX oncoprotein (Citationde Bruijn et al. 2006, CitationSun et al. 2006). High levels of IGF2 have also been reported in rhabdomyosarcomas (CitationZhan et al. 1994, CitationZhan et al. 1998). In desmoplastic small-cell tumors, high expression of IGF-1 receptor (IGF-1R) is related to an effect from the fusion protein EWS-WT1 on the IGF-1R promoter. These findings have also been confirmed in gene expression studies. CitationBaird et al. (2005) identified IGF2 among the top expressed genes in synovial sarcomas, which was confirmed by immunostaining. Binding of the IGF1 and IGF2 ligands to IGF-R activates its intrinsic tyrosine kinase activity resulting in signaling through cellular pathways that modulate proliferation and inhibit apoptosis. The key downstream signaling pathways include PI3K-AKT-mTOR and RAF-MEK-ERK (). Antibodies as well as tyrosine kinase inhibitors are clinically evaluated as IGF-1R targets. Several monoclonal antibodies that block ligand binding have been developed and are now in phase I-II clinical trials. One of these, R1507, has shown activity against Ewing sarcoma, but gene expression studies suggest that such agents may be of value also in other sarcoma types, including synovial sarcoma and rhabdomyosarcoma. Another monoclonal antibody against IGF-1R, IMC-A12 (cixutumumab), is being evaluated in a phase I/II trial in STS (ClinicalTrials gov identifier: NCT00720174). Tyrosine kinase inhibitors have also been designed against IGF1R, but because the insulin receptor and the IGF-1R are 95% homologous at the tyrosine kinase ATP-binding site, small molecules will to some degree also inhibit the insulin receptor (CitationRodon et al. 2008). Currently, one tyrosine kinase inhibitor (OSI906) is being evaluated in phase I trials (ClinicalTrials.gov identifier: NCT00514306 and NCT00514007).
Figure 3. Binding of IGF-1 to its receptor triggers the autophosphorylation of the latter and leads to activation of different signaling cascades through the phosphatidylinositol 3-kinase/AKT and ras/raf/mitogen-activated protein/extracellular signal-regulated kinase pathways. The IGF pathway thus modulates cell proliferation, survival and metastasis.
Anti-angiogenesis and hypoxia
Angiogenesis is a key factor in cancer progression (CitationFolkman et al. 1989). Most tumors develop their own blood vessels already at 1-2 mm in size. Using gene expression profiling several angiogenic genes have been found to be upregulated in STS, including the PDGF receptor (PDGFR), MMP-2, and Notch-1 and Notch-4 (CitationYoon et al. 2006). Overexpression of PDGF-B has been associated with increased cell growth and high-grade tumors (CitationWang et al. 1994). Anti-angiogenic agents have proven effective in several cancer types and various types of angiogenesis inhibitors, e.g. monoclonal antibodies and tyrosine kinase inhibitors of VEGF and other angiogenic factors, thalidomide, and retinoic acids are being investigated, also in metastatic sarcoma (CitationGasparini et al. 2005). Initial investigation of the monocloncal anti-VEGF antibody bevacizumab in combination with doxorubicin in metastatic STS demonstrated stable disease, but no objective response (CitationD'Adamo et al. 2005). Small-molecule inhibitors of VEGF signaling, e.g. sorafenib, sunitinib, and pazopanib are being examined in phase II studies in patients with metastatic STS and recently, pazopanib showed activity against STS in a phase II multicentre EORTC study. A phase III study is currently recruiting (ClinicalTrials.gov identifier:NCT00794521).
The extent of hypoxia within the tumor micro-environment has been linked to metastatic spread and radiotherapy resistance. CitationDetwiller et al. (2005) demonstrated upregulation of hypoxia-related genes. Francis et al. (Citation2007) identified a gene expression signature in metastazing tumors and among the top ranked genes were HIF-1a and its targets, which suggests that the HIF-1a related pathway may represent a potential target. Preclinical data indicate that local control can be improved using tirapazamine before surgery and radiotherapy in STS (CitationLunt et al. 2005).
CDK4 and MDM2 as potential therapeutic targets
Well-differentiated and dedifferentiated liposarcoma harbour a characteristic 12q13-15 amplicon encompassing both CDK4 and MDM2 (CitationWunder et al. 1999). CitationHeidenblad et al. (2006) and Carneiro et al. (Citation2009) have reported recurrent amplification of CDK4 (and MDM2) in a subset of pleomorphic STS. On the basis of CDK4 amplification and overexpression, cyclin-dependent kinase inhibitors (CDKI) are a compelling group of agents to pursue as new therapeutics in STS (CitationSchwartz et al. 2005). Flavopiridol is the best studied of these agents and phase I studies are currently under way. Nutlins are a family of MDM2-specific agents with proven activity against sarcoma cell lines (CitationAmbrosini et al. 2007) and also represent promising targets for clinical evaluation.
Frizzled pathway/WNT
The WNT pathway has demonstrated consistent upregulation in synovial sarcoma. Nagayama et al. first reported to have identified 26 genes upregulated in synovial sarcomas (CitationNagayama et al. 2002), including the Frizzled homologue 10 (FZD10), whose product belongs to the Frizzled family of seven-pass transmembrane receptors in the WNT signaling pathway. Pre-clinical studies in mice with synovial sarcomas, have shown that treatment with monoclonal antibody against native FZD10, increases time to tumor progression. Despite being a promising therapeutic target, no clinical application directed to FZD10 is yet available.
Conclusions
STS elegantly demonstrate how conventional histopathology and molecular genetics can complement each other (CitationHelman et al. 2003, CitationOliveira et al. 2004, CitationAntonescu 2006, CitationAntonescu 2008, CitationBridge 2008). Molecular pathology is increasingly applied as a diagnostic adjunct to morphology and molecular characterization has in several STS types identified recurrent derangement of central signaling pathways. Examples include KIT in GIST, EGFR, WNT, frizzled, and retinoic acid receptor pathways in synovial sarcoma, CDK4 and MDM2 in dedifferentiated liposarcoma, muscle-related genes in leiomyosarcoma, and nerve-sheath related genes in MPNST. A major undertaking in STS is to translate this knowledge into targeted therapies with inhibitors of mTOR, IGF-1, angiogenesis, CDK4, and MDM2 as potential targets. The detailed mapping of fusion genes, amplified regions, altered expression, and deranged signaling pathways leads to an increasingly diversified diagnosis and treatment of STS, and the integration of genetic profiles into the successes and failures of targeted therapies is likely to improve outcome for future STS patients.
Acknowledgments
Ana Carneiro holds a fellowship from the Portuguese Foundation for Science and Technology: SFRH / BD / 27561 / 2006. Financial support was granted by the Swedish Research Council, the Swedish Cancer Fund, the Swedish Children´s Cancer Fund, and the Lund University Hospital.
Clinical and molecular studies of liposarcoma
Katarina EngstrÖm
Department of Oncology, The Sahlgrenska Academy, Gothenburg, Sweden
Correspondence: [email protected]
Liposaroma in the Scandinavian Sarcoma Group Register
Liposarcoma (LS) represents a uniform nomenclature for several subtypes with different morphological and cytogenetic characteristics and clinical behavior. In 2002, the World Health Organization (WHO) published the third edition of its classification of soft tissue tumors. The 5 LS subtypes are well differentiated LS (WDLS), dedifferentiated LS (DDLS), pleomorphic LS (PLS), myxoid/round cell LS (MLS/RCLS) and mixed-type LS. The term WDLS should be used for tumors in deep locations, difficult to surgically access with risk for local recurrence (e.g. the retroperitoneum and mediastinum) and the preferred term for tumors in the extremities and trunk wall is atypical lipomatous tumor (Citation[1]).
Liposarcoma comprises 10–16% of all soft tissue sarcomas (Citation[2]). In the last 20 years, several large studies dealing with LS have been published by different sarcoma centers (Citation[3-7]). The Scandinavian Sarcoma Group (SSG) Register started in March 1986. In Norway and Sweden, approximately 90% of all soft tissue sarcomas of extremities and chest wall are reported to the SSG Register, and so the Register is considered population-based for these two countries (Citation[8]). The SSG Pathology Board has previously reviewed liposarcomas and malignant fibrous histiocytomas in the Register (Citation[9]). We recently performed an analysis of 237 patients diagnosed with LS of extremities and chest wall between 1986 and 1998 (Citation[10]), with a focus on the clinicopathological characteristics, treatment, and outcome. Since only 2 patients in this material received adjuvant chemotherapy, the results, that is, the rate of first local recurrence or distant metastasis, represent the outcome of surgery and radiotherapy. The analysis revealed a high proportion of low-grade LS (histopathological grade I and II on a IV-grade scale), in particular WDLS, which accounted for 53% of low-grade LS and 36% of all subtypes. In the subcutaneous location, there was a 1:1 ratio between the low-grade and high-grade (histopathological grade III and IV) group, but the corresponding ratio in the deep location was 2.6:1.
Well-differentiated liposarcomas may recur locally, but they do not metastasize unless they undergo dedifferentiation, which occurs in only a few percent on extremities and chest wall (Citation[2], Citation[11]). The local recurrence rate of WDLS on extremities and trunk is reported in the literature as ranging from 8% to 52% (Citation[12-18]). In our study, the local recurrence rate was 13% with surgically treatable recurrences. Well-differentiated liposarcomas with non-wide surgery and without postoperative radiotherapy (45 patients) showed no local recurrence in 82% of cases, and so the routine use of radiotherapy cannot be recommended. No patient developed metastases. Our results support the use of the term “atypical lipomatous tumor” for these lesions when they arise in the extremities or trunk wall.
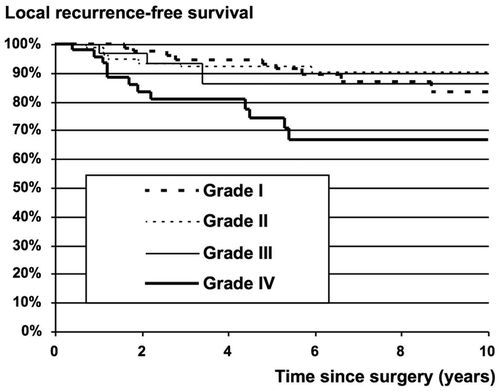
During the investigation period, SSG recommended that soft tissue sarcomas operated on with intralesional and marginal surgical margins should be treated with postoperative radiotherapy. In the low-grade group (WDLS and MLS), radiotherapy was applied in 17% of cases; the local recurrence rate was 13% with radiotherapy and 18% without. In the high-grade group (MLS/RCLS, RCLS, DDLS, and PLS), radiotherapy was applied in 58% of tumors with non-wide surgery; the local recurrence rate was 19% with radiotherapy and 47% without. Univariate analysis of radiotherapy as a prognostic factor for local recurrence revealed no statistical significance in the low-grade group, the high-grade group, or the total group. However, this factor became significant in the multivariate analysis, when radiotherapy was analyzed together with final surgical margins and grade, a result which was interpreted as an effect of radiotherapy for the same grade and margin. This result is supported by Jebsen et al., who, in a Scandinavian Sarcoma Group study, showed that adjuvant radiation therapy effectively prevents local recurrence in soft tissue sarcoma of the extremities and trunk wall irrespective of the tumor depth, malignancy grade, and surgical margin status (Citation[19]). In multivariate analysis, surgery outside a sarcoma centre and the subtype DDLS were adverse prognostic factors for local recurrence, while old age, large tumor size, high grade, and histological type RCLS were prognostic for metastatic disease. The estimated 10-year metastasis-free survival rate was 95% for low-grade LS and 61% for high-grade LS. Local recurrence-free survival rates were 87% for low-grade LS and 75% for high-grade LS ( and ).
Figure 1. Metastasis-free survival for liposarcoma grade I–IV.
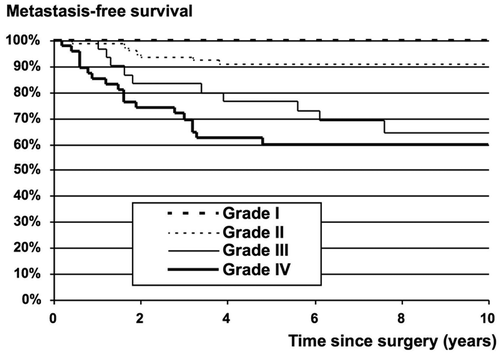
Figure 2. Local recurrence-free survival for grade I–IV.
The study revealed high rates of metastases in the different high-grade subtypes. Dedifferentiated liposarcoma patients developed metastases in 57% of cases, followed by MLS/RCLS (46%), PLS (36%), and RCLS (33%). There are several reports of sensitivity in myxoid liposarcoma to cytotoxic drugs such as doxorubicine, ifosfamide, and trabectedin (Yondelis®) (Citation[20-22]). The ongoing Scandinavian Sarcoma Group study, SSG XX, on adjuvant chemotherapy in high-grade soft tissue sarcomas used as inclusion criteria either vascular invasion or 2 of the following 3 criteria: tumor size > 8 cm, necrosis, and infiltrative growth pattern. Our study showed no vascular invasion in MLS/RCLS, and necrosis and infiltrative growth pattern was seldom present; applying the SSG XX protocol on this tumor group would result in exclusion of 50% of patients that later metastasize. Conversely, the prognostic factors of tumor size > 10 cm and a diagnosis of MLR/RCLS or RCLS identified 90% of the high-grade MLS/RCLS that later developed metastases. Considering the reported chemosensitivity in this subtype of LS, it is important that future prognostic studies are able to better identify high-risk MLS/RCLS patients.
Well-differentiated liposarcomas show the presence of extra ring and/or giant marker chromosomes derived from the chromosome 12q(Citation[13-15]). Amplification of mouse double minute (MDM) 2 and cyclin-dependent kinase (CDK) 4 genes from this region is frequently seen in WDLS, but TP53 is very rarely mutated (Citation[23-25]). Dedifferentiated liposarcomas display the same chromosomal abnormality associated with WDLS, but the amplification levels of MDM2 and CDK4 are frequently higher (Citation[25]). The MDM2 gene is a proto-oncogene, and the phosphoprotein MDM2 negatively regulates P53. MDM2 binds to the transactivation domain of P53, inhibits the P53-mediated transcriptional activity, and promotes P53 degradation through ubiquitination. It even degrades pRB, and thereby supports the transcription activity of the E2F family and cell cycle progress (Citation[26-29]). Nutlin-3a, a recently-developed small-molecule antagonist of MDM2 (Citation[30-32]), has been shown to work as a P53 inducer in DDLS-derived cell lines (Citation[33]) and WDLS-derived cell lines (Citation[34]), leading to apoptosis. This is a promising therapeutic agent, especially needed for treatment of WDLS in abdominal and thoracic cavity and for DDLS.
Clinical and molecular studies of myxoid/round cell liposarcoma
The myxoid/round cell liposarcoma subgroup constitutes approximately 40% of all LS (Citation[2], Citation[35]). Round cell liposarcoma is a morphologic continuum of MLS which carries the same unique cytogenetic features as MLS. Presence of round cell differentiation > 5%, presence of necrosis, and over-expression of P53 are associated with poorer disease-specific survival (Citation[36]).
More than 95% of MLS cases carry the translocation t(12; 16)(q13; p11), which results in the FUS-DDIT3 (also known as ¨TLS-CHOP¨) fusion oncogene. A few percent carry a variant translocation, t(Citation[12]; Citation[22]), and an EWS-DDIT3 fusion oncogene (Citation[37], Citation[38]).
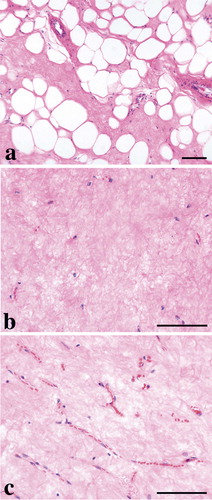
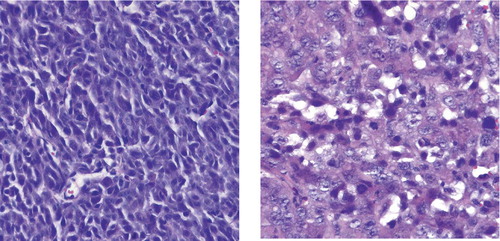
Until 1990, a few reports on MLS showed post-radiotherapy reduction in tumor size with radiation doses as low as 30 and even 10 Gy (Citation[39], Citation[40]). At the Department of Oncology, Sahlgrenska University Hospital, Gothenburg, 15 patients with 33 MLS tumors were treated with radiotherapy between 1994 and 2004 (Citation[41]). 30 tumors were evaluated with radiology before and after radiation therapy; 17 responded with more than 30% tumor volume reduction (4 with complete response and 8 with more than 50% response). This response was achieved with a radiation dose of 40-46 Gy. In recent years, several larger studies of radiation response in MLS have shown both excellent reduction of tumor size (Citation[42]) and high local control rate (Citation[43], Citation[44]). The SSG study also demonstrated excellent local control in MLS, MLS/RCLS, or RCLS treated with radiation therapy. No patient developed local recurrence after postoperative radiation therapy (24 out of 99 patients), but 15% of the non-irradiated patients had local recurrences. In the Gothenburg study (Citation[41]), all tumors surgically removed after preoperative radiation (27 tumors) showed a dramatic morphologic response, with paucicellularity, hyalinization, and in most lesions a lipomatous appearance with mostly univacuolated adipocyte-like cells that varied in size (). Minimal microscopic foci of characteristic MLS were also occasionally seen. The presence of round cells did not correlate to either radiological response or the post-irradiation morphology.
Figure 3. Irradiated MLS/RCLS with lipogenic maturation with mostly univacuolated adipocyte-like cells, (10x) (a), hyalinisation and pausicelluarity, (20x) (b) and preserved delicate capillary network, (20x) (c). Bar denote 100 micrometres.
The Scandinavian Sarcoma Group does not recommend preoperative radiation therapy, but the tumor volume reduction and the elimination of tumor cells seen in this study support the use of preoperative radiotherapy whenever a non-wide surgery is overhanging.
The mechanism behind the morphologic response to radiation therapy is unclear. The immature spindle cells were to a large extent eradicated in our tumors. According to Kuroda et al. (Citation[45]), FUS-deficient mice exhibited increased radiosensitivity, and it may be that it is the translocated FUS gene that contributes to the high radio-responsiveness in MLS. The lipoma-like appearance after radiotherapy could be due to the more differentiated lipoblasts having been left unaffected, or it could be due to radiation-induced cell cycle arrest leading to maturation of the lipoblasts. In the literature, it is suggested that a mixed MLS with WDLS is rare (Citation[46], Citation[47]), but in our material 21 out of 27 lesions had areas with lipoma-like appearance.
Demetri et al. reported in 1999 that treatment with troglitazone, a PPAR-γ synthetic ligand, induced adipocytic differentiation in 2 patients with MLS and MLS/RCLS (Citation[48]). In addition, in 2007, Grosso et al. reported paucicellularity, hyalinization, and adipocyte-like maturation in patients treated with preoperative trabectedin (Yondelis®) (Citation[22]). The possible mechanism behind the lipoma-like appearance after radiation therapy could be an induction of P53, which is seldom mutated in MLS (Citation[49-52]). Wild-type P53 activates transcription of P21, and the P21 protein in turn inactivates the cyclin E/CDKs – pRB pathway and arrests the cell at the G1-S checkpoint. Unpublished data from our group showed an induction of P21 expression after irradiation of cultured MLS cells.
Further support for our hypothesis can be found in the work of Ventura et al. (Citation[53]), who showed that mice carrying a reactivable TP53 knockout allele developed lymphomas and sarcomas, and that the lymphomas underwent apoptosis when the TP53 gene was reactivated. However, regression of sarcomas was delayed, and no extensive apoptosis was evident. Instead, restoration of P53 expression suppressed proliferation and induced cell cycle arrest, and the cells changed morphology and expressed senescence-associated β-galactosidase, P15 and P16. This report supports our hypothesis that radiation-induced expression of P53 may lead to cell cycle arrest in MLS and terminal differentiation of lipoblasts.
The question remains of whether the gene fusion in MLS arises in normal mesenchymal stem cells or whether it occurs at a later differentiation stage; and it may also be asked whether the fusion gene is sufficient for neoplastic transformation. It has been shown that the fusion gene can block terminal differentiation of pre-adipocytes in vivo and in vitro. Also, transgenic mice that expressed FUS-DDIT3 in all tissues developed MLS/RCLS-like tumors arising in adipose tissue. These reports led to the hypothesis that MLS/RCLS develops from pre-adipocytes carrying FUS-DDIT3 which are incapable of terminal differentiation. However, since myxoid liposarcomas in humans preferentially develop in or between the large muscles of the thigh, and rarely in adipose tissue, this hypothesis has been called into question. At the Lundberg Laboratory for Cancer Research in Gothenburg, we observed that the pFUS-DDIT3-EGFP and pDDIT3-EGFP-transfected, xenografted HT1080 cell line induced a switch from a poorly differentiated sarcoma to an MLS/RCLS-like morphology () (Citation[54]). Microarray expression profiling supported the switch in morphology by showing a changed expression pattern of transfected HT1080 cells toward an MLS/RCLS-like profile. These findings support the idea that FUS-DDIT3 can drive a primitive mesenchymal tumor cell toward an MLS/RCLS phenotype. Further support comes from Riggi et al., who reported that mesenchymal progenitor cells isolated from mouse bone marrow transformed and grew as MLS-like tumors in SCID mice when transfected with the FUS-DDIT3 fusion gene (Citation[55]). Pérez-Losada et al. showed that transgenic mice expressing high levels of DDIT3 did not develop liposarcoma tumors (Citation[56]). In our experiments, DDIT3, when expressed in a primitive fibrosarcoma cell, induced a switch in morphology and expression pattern toward an MLS/RCLS type. This supports the hypothesis that it is the DDIT3 gene that determines the tumor entity when fused with the FUS gene.
Figure 4. Microphotographs of section from SCID mouse tumors of HT1080 cells (left column) and the pFUS-DDIT3EGFP-transfected variant (right column).
The above results show that forced expression of FUS-DDIT3 and DDIT3 induces a switch in HT1080 cells toward MLS/RCLS-like morphology and gene expression. The results also indicate that the transcription factor partner DDIT3 of FUS-DDIT3 is the tumor type-determining part of this EWS-group fusion oncogene.
SSG pathology review experiences and histological grading of malignancy in sarcomas
Bodil Bjerkehagen 1, Johan Wejde 2, Magnus Hansson 3, Henryk Domanski 4, Tom BÖhling 5
1 Division of Pathology, Radiumhospitalet, Rikshospitalet University Hospital, Oslo, Norway,
2 Department of pathology, Karolinska University Hospital, Stockholm, Sweden,
3 Department of clinical cytology and pathology, Sahlgrenska University Hospital, Gothenburg, Sweden,
4 Department of Pathology and Cytology, Lund University Hospital, Lund, Sweden,
5 Department of Pathology, HUSLAB and University of Helsinki, Helsinki Finland
Correspondence: [email protected]
The rarity of sarcomas, the histological variation with often complex diagnostic work-up and overlap with benign mesenchymal tumors emphasize the importance to have dedicated pathologists working in the field. Sarcoma pathology is in continuous development with new histological entities and changes of diagnostic criteria. One example of refinement in diagnostics in mesenchymal tumors was the identification of the gastrointestinal stromal tumors (GISTs) in the late 1990ths with the use of the antibody CD117 and identification of activating mutations in KIT or PDFGRA (Joensuu and Kindblom Citation2004). Previously most of these tumors were diagnosed as benign or malignant epithelioid leiomyomatous tumors or other spindle cell tumors. This means that projects including abdominal sarcomas diagnosed before 2000 have to be reviewed.
The Scandinavian Group (SSG) was established in 1979 and from the beginning pathologists took part in the organization. In the first years the morphology group was lead by Lennart Angervall. The aim for the pathology group has been to standardize and improve the quality of diagnostics in sarcoma. They have contributed to treatment protocols and recommendations for diagnosis and research. For study cases the pathologists have reviewed the diagnosis, malignancy grade and evaluated the histological response after preoperative chemotherapy in bone sarcomas.
Lars-Gunnar Kindblom established in 1995 a panel of pathologists from Scandinavia in a peer-review committee with the aim to review all cases in the SSG Central database. Since 1995 the panel has reviewed 2304 cases.
Review process
The morphology group in SSG consists of trained sarcoma pathologists from tumor centers in the Nordic countries (). The SSG Central Register contains data on 9384 sarcoma cases (October 2008) treated at sarcoma centres in Scandinavia from 1986. (http://www.ssg-org.net/). Histopathological data of the tumors is included in the database. The pathology group has met 2-4 times annually reviewing histopathological diagnosis of sarcomas from different institutions in SSG. Many of the cases are from patients included in clinical trials, but cases in specific research projects are also included.
Table 1. Participants of the review group 1995–2008
The review is usually performed with a multiheaded microscope (). The SSG secretary participates in the meeting. From the pathology report the following parameters are extracted: sex, age at diagnosis, site, localization, depth of infiltration, size, and the contributor's diagnosis. If ancillary methods like immunohistochemistry, ultrastructural pathology, FISH, RT-PCR or cytogenetic studies were used, it is noted. From the review process the histopathological diagnosis, diagnostic alternatives, grade of malignancy, tumor necrosis, vascular invasion, mitotic rate, and pushing or diffuse infiltrative growth pattern are evaluated. The status of the surgical margins has not been re-evaluated; information about the status of the margins is based on the original reports. In special cases new immunohistochemical analyses as well as genetic analyses were performed to aid classification. Most cases diagnosed with only cytology were excluded. Furthermore sarcomas pretreated with radiotherapy and/or chemotherapy were difficult to review because the treatment can induce necrosis and morphological changes. All cases are classified according to the WHO recommendations (Fletcher et al. Citation2002). A special protocol was used for registration of data ().
Figure 1. The multiheaded microscope.

Figure 2. The SSG review protocol

Histological grading of malignancy
The grading of soft tissue and bone sarcomas is important because of the prognostic impact. The clinical course of sarcomas varies within a particular histopathological entity. There is no generally accepted grading system for sarcomas (Deyrup and Weiss, Citation2006) and there is an ongoing discussion which grading system to prefer. The French Federation of Cancer Centers system has been tested in several studies and is probably the best documented system, see Table 2 (Guillou et al. Citation1997, Coindre et al. Citation1996, Miettinen Citation2006). Because there are so many types of sarcomas and subgroups therein it is difficult to validate grading systems for sarcomas. The different morphologic variables are of varying importance for the different types of sarcomas (Angervall and Kindblom Citation1993). Ideally there should be one specific grading system for every subtype of sarcoma, but that is not clinically meaningful.
In the SSG, the histological grading of sarcomas has been based on a four tiered grading system primarily based on Broders grading model (Broders Citation1920, CitationBroders et al. 1939). Generally only untreated primary sarcomas using good quality slides and representative tissue should be used for grading. Grade 1 and 2 means low grade malignancy and grade 3 and 4 tumors are regarded as high-grade malignant sarcomas. This system considers the degree of cellularity, cell and nuclear polymorphism, differentiation, the presence and amount of necrosis, hemorrhage, mitotic activity and growth pattern, but does not score the different parameters (Angervall and Kindblom Citation1993).
For many soft tissue sarcomas the grade is given by the diagnosis. For instance atypical fibroxanthoma of skin, dermatofibrosarcoma protuberans and well differentiated abdominal or retroperitoneal liposarcoma are all grade 1 sarcomas. Typical examples of grade 2 sarcomas include myxoid liposarcoma and many subcutaneous myxofibrosarcomas. For high grade sarcomas (grades 3 and 4) the grade is partly based upon histogenetic diagnosis and partly upon the morphologic features. Examples of grade 4 sarcomas include round cell liposarcoma and pleomorphic liposarcoma. Immunostains for evaluating proliferative activity with Ki-67 (MIB1) is also helpful (Hasegawa 2007, Engellau et al. 2004).
Grading specific for myxofibrosarcoma has been described by Angervall et al. (Citation1977). A good association between grade as defined by this system and outcome (metastatic disease) has been reported in many studies by the SSG, for review see Gustafson (Citation1994) and recently in liposarcoma (Engstrom et al. Citation2008, see also pp 24–30 in this issue) and in nonvisceral soft tissue leiomyosarcoma (Svarvar et al. Citation2007).
A widely used system is the French grading system (FNCLCC). This is based on tumor differentiation, mitotic count and the amount of tumor necrosis (Guillou et al.Citation1997, Deyrup and Weiss Citation2006). The total score of each of these parameters gives the grade. Grade 2 and grade 3 are considered high-grade malignant tumors.
In SSG the so called SIN system also has been applied for prognostication of soft tissue sarcoma. Here one use information about the size of the tumor, necrosis, and tumoral vascular invasion instead or as a complement to the histological grade of the tumor (Gustafson et al. Citation2003). More recently also the peripheral tumor growth pattern, defined as infiltrative or pushing border, has been included in the prognostic work-up (Engellau et al. Citation2005, [Citation2007], see also pp 44–50 in this issue).
Reviews of soft tissue saroma
The pathology group started to review malignant fibrous histiocytoma/pleomorphic and spindle cell sarcoma, followed by synovial sarcoma and liposarcoma. Several doctoral theses and projects based on the sarcoma database has included the revised pathology data (Engellau et al. Citation2004, Svarvar et al. Citation2007, Engstrom et al. Citation2008,). For details about the reviews and methods for the first 1000 soft tissue sarcoma see Meis-Kindblom et al. (Citation1999).
The SSG XIII protocol includes patients with high risk soft tissue sarcoma and required pathology review not only to confirm the diagnosis and the malignancy grade but also to evaluate defined risk factors according to the SIN system. Likewise pathology required in the SSG XX protocol opened in 2007 which replace SSG XIII and adds growth pattern to the risk factors. SSG XVIII is a closed protocol for adjuvant treatment of patients with a high risk GIST. According to the protocol all cases had to be reviewed by a reference pathologist.
Currently an ongoing project initiated by the pathologists in SSG includes the review of angiosarcomas and solitary fibrous tumors.
Reviews of bone tumors
Within SSG the pathologists have been involved in 7 osteosarcoma and 4 Ewing sarcoma treatment protocols. The diagnosis has to verified by review in each case. Another important role for the pathologist is the evaluation of the histological response to the preoperative chemotherapy. The response grade is an important prognostic marker, and in most protocols decides the postoperative treatment. The response has to be evaluated shortly after surgery, and cannot wait until the pathologists have a review meeting. The response is therefore usually evaluated by the local pathologist, but in some protocols by a specifically named single pathologist within the SSG. However, both the diagnosis and treatment response have later been evaluated by the review group for several protocols (Bohling et al. Citation2004).
Osteosarcoma
In osteosarcoma SSG has been involved in 5 closed protocols; SSG II, SSG VIII, ISG/SSG I, ISG/SSG II and SSG XIV. In these protocols three different ways of evaluating the response grade has been used. In SSG II and SSG VIII the criteria defined by Huvos was used, which uses a four-grade scale, where grades III and IV were considered as good response (Huvos Citation1991). In the collaborative protocols with the Italian sarcoma group ISG/SSG I and II a new evaluation system was developed in collaboration with the Italian pathologists. In these protocols the criteria for good response was rather tight, and therefore less than 20% of the cases were classified as such (Ferrari et al. Citation2005). In the SSG XIV protocol (Smeland et al. Citation2009) the criteria for good response were not as tight as in the ISG/SSG protocols, and the response was defined as a good in tumors where < 10% of examined tumor area revealed unquestionable viable tumor and no single area of unaffected viable tumor exceeded 2,5 mm in largest diameter.
At the moment SSG participates in a multinational osteosarcoma treatment protocol together with many European and American centres, called EURAMOS-1 (http://www.ctu.mrc.ac.uk/euramos/). This protocol requires that only named reference pathologists are allowed to give the final response grade. In the Nordic countries all cases are sent to Helsinki for evaluation. So far approximately 60 cases have been evaluated by the reference pathologist (TB). The response is considered good if less than 10% of the tumor is viable.
Ewing sarcoma
SSG has used two closed treatment protocols, SSG IV and SSG IX, in Ewing sarcoma. In these, the effect of the induction chemotherapy was evaluated using a modified Huvos grading system (Bohling et al. Citation2004). However, in 1999 SSG entered together with the Italian sarcoma group two protocols, one for localized and one for metastatic tumors. In these protocols the diagnosis requires molecular pathology, either a wide range of immunohistochemistry, but preferably genetic analysis to demonstrate the typical 11;22 translocation. The chemotherapy response is evaluated by the Picci-method (Picci et al. Citation1997), where the response is graded into 3 categories. This system has shown to be very accurate, and the results are reliable also in review settings.
Chondrosarcoma and other bone tumors
So far, there is only one SSG project on chondrosarcomas. This is an ongoing project and includes primary chondrosarcomas of the thoracic wall. The main aspect for the review group has been to verify the diagnosis and to grade the chondrosarcomas.
Conclusion
The pathology group contributes on several aspects in SSG. Histopathology data in the central SSG register is updated. The work in the peer-review committee standardizes diagnostic practice among SSG centres. The group gives mandatory support to research projects and clinical trials. In addition the review process offers an optimal opportunity to train young pathologists in sarcoma diagnostics.
Acknowledgements
The authors would like to thank Evy Nilsson and Eva-Mari Olofsson for secretarial assistance.
Adjuvant systemic therapy of gastrointestinal stromal tumors
The Scandinavian Sarcoma Group perspective
Heikki Joensuu1, and Mikael Eriksson2
1Department of Oncology, Helsinki University Central Hospital, Helsinki, Finland;
2Department of Oncology, Lund University Hospital, Lund, Sweden
Correspondence: [email protected]
ABSTRACT – Gastrointestinal stromal tumors (GISTs) have a malignant potential varying from virtually no risk of recurrence (microscopic GISTs) to a high risk. Acquired secondary mutations that interfere with imatinib binding have been identified as a common mechanism for drug resistance and tumor progression in advanced GIST. Patients with advanced GIST but with a small tumor mass have the longest time to disease progression suggesting that the rate of secondary mutations that confer drug resistance may be low when the number of cancer cells is small. Thus, early administration of imatinib in the adjuvant setting might result in a low rate of secondary mutations and might improve survival. The results of the ACOSOG Z9001 trial demonstrated a significantly longer recurrence-free survival among patients treated with adjuvant imatinib as compared to placebo. Although data on influence of adjuvant imatinib on overall survival is lacking, its administration may be warranted to patients who have a high risk of recurrence and death from GIST that likely exceeds the risks related to adjuvant administration of imatinib. Many aspects of adjuvant treatment remain unknown including the overall benefits and harms of adjuvant treatment, optimal selection of patients for treatment, and the duration of adjuvant treatment, which is currently being investigated in the ongoing Scandinavian Sarcoma Group/AIO trial (SSG/AIO XVIII).
Gastrointestinal stromal tumor (GIST), the most common sarcoma of the gastrointestinal tract, has an annual incidence of 10 to 15 cases per million in the Western populations (Nilsson et al. Citation2005, Tryggvason et al. Citation2005, Steigen et al. Citation2006). Small GISTs, a few mm in diameter, occur in a large proportion of the normal population and have no to minimal malignant potential (Kawanowa et al. Citation2006, Agaimy et al. Citation2007, Abraham et al. Citation2007). The larger GISTs are often highly malignant and commonly give rise to intraabdominal implants and liver metastases. GISTs generally do not respond to conventional chemotherapy agents, but they are excellent targets for small molecule tyrosine kinase inhibitors that selectively inhibit the KIT and PDGFRA receptor tyrosine kinases, which have an important role in the molecular pathogenesis of GIST. KIT and PDGFRA are frequently mutated in GISTs leading to genesis of constitutively active mutant proteins.
Imatinib mesylate was the first effective systemic treatment for advanced GIST, and it has a generally favorable adverse effect profile (Joensuu et al. Citation2001, Demetri et al. Citation2002). A few other tyrosine kinase inhibitors, such as sunitinib, nilotinib, sorafenib and vatalanib (PTK787/ZK222584) have also been found to be active in the treatment of advanced GIST. Most (up to 85%) patients achieve a durable response or a long-lasting disease stabilization with imatinib therapy, and the median survival time of patients diagnosed with inoperable/metastatic GIST has now increased to approximately 5 years as calculated from the date of treatment initiation (Blanke et al. Citation2008).
These results represent a giant leap forward as compared to the era that preceded the times of targeted therapies. Unfortunately, secondary drug resistance was soon found to develop in most patients, although a few patients diagnosed with advanced GIST have now responded to imatinib for up to 8 years, and it is currently not known whether secondary drug resistance will develop in all GIST patients with advanced disease. The median time to GIST progression was approximately 2 years in the B2222 trial that was the first randomized trial to address efficacy of imatinib in the treatment of inoperable/advanced GIST (Blanke et al. Citation2008).
Delay or prevention of development of secondary resistance to tyrosine kinase inhibitors and other targeted agents is a key for a major improvement in the outcome of GIST patients who present with advanced disease, since responses to subsequently administered agents tend to be short-lived once resistance to the first-line tyrosine kinase inhibitor has emerged. Administration of a targeted agent early in the course of the disease in the adjuvant setting is potentially one of the most promising strategies to avoid early emergence of secondary drug resistance. Here we discuss the rational for adjuvant systemic therapy for GIST and the ongoing clinical trials.
Tumor mutation rate and secondary resistance to tyrosine kinase inhibitors
Although responses to targeted agents are usually durable, most responses remain partial. When metastatic deposits are removed at surgery while the patient is still responding to imatinib or sunitinib, almost all responding tumors contain at least some cells that immunostain for KIT and that are likely persisting GIST cells (Rutkowski et al. Citation2006, CitationRuka et al. 2008). GIST cells thus have a variable responsiveness to tyrosine kinase inhibition, which often allows a few GIST cells to survive despite persistent treatment.
Only one mutation in KIT or PDGFRA has invariably been found in mutation analyses of GISTs that have never been exposed to tyrosine kinase inhibitors, whereas secondary KIT mutations occur in most GISTs that grow under tyrosine kinase treatment. Interestingly, the secondary mutations are commonly found in KIT exons 13, 14 or 17, which exons are rarely mutated in primary GISTs (Heinrich et al. Citation2006, CitationWardelmann et al. 2006). These mutations are often located adjacent to the binding site of imatinib likely interfering with its binding, are often associated with a decreased responsiveness to imatinib (Heinrich et al. Citation2006), and confer a growth advantage to the mutated cell population. Apparently, GIST cells that survive under imatinib administration continue to mutate at multiple sites of the genome. When a mutation that interferes with imatinib binding is generated, the cell harboring the mutation will gain a substantial growth advantage relative to other surviving GIST cells, and will eventually progress to a clinically detectable drug-resistant tumor.
Recent evidence suggests that several KIT mutations may coexist in a GIST lesion that grows under imatinib treatment, and that the number of mutations detected in a mutation analysis of such tumors may depend on the number of biopsies analyzed (CitationWardelmann et al. 2006). It is not known why multiple coexistent mutations cannot be found in GISTs that have not been treated with imatinib or other tyrosine kinase inhibitors. Hypothetically, such cell clones fail in competition with cells with only one mutation in the absence of imatinib, and escape detection in a conventional mutation analysis.
These findings lend support to presence of clinically significant mutational activity in most GISTs. In one study, nuclear and mitochondrial microsatellite instability were detected in 3 and 10 of the 62 GISTs examined, respectively, suggesting that mitochondrial microsatellite instability may play a role in the development of some GISTs, whereas nuclear microsatellite instability may have little importance (Kose et al. Citation2006). Yet, aberrant methylation of CpG islands of DNA repair genes may be common in GISTs. One recent study found aberrant methylation of human mutL homolog 1 (hMLH1) in 60% of the GISTs examined (Saito et al. Citation2008).
Clinical consequences of mutational activity
Large GISTs are likely to generate mutations that lead to drug-resistance earlier than small GISTs. This hypothesis is supported by the data from the B2222 trial, where GIST patients diagnosed with overtly metastatic disease were treated with imatinib administered either 400 mg or 600 mg per day. In this study the time to GIST progression was the longest (median 6 years) among the quartile of patients who had the smallest tumor volume at the time of imatinib initiation and the shortest (median 1.5 years) among the quartile of patients who had the largest tumor volume (Blanke et al. Citation2008, supplement ). However, the more favorable outcome of GISTs with a small tumor volume may also be due to either lead bias (tumors with a small volume are at an earlier stage of the course of the disease) or length bias (the tumors with a small volume may be biologically less aggressive). There was no apparent plateau in the proportions of tumors that eventually progressed within the first 5 years of imatinib treatment in this study.
The potential propensity of large GISTs to develop drug-resistant mutations sooner than smaller GISTs may be an argument for administering targeted therapy early during the course of the disease when the number of cancer cells is still small. If the rate of mutations remains the same irrespective of cancer volume, emergence of imatinib resistance may take a considerably longer time when imatinib is administered in the adjuvant setting than in advanced disease, because the number of GIST cells that may potentially develop a mutation leading to drug resistance is much smaller. Little is known about the mutation rates of GISTs during the natural course of the disease vs. during tyrosine kinase administration, but cancers in general tend to become less differentiated with time due to tumor progression, which might lead to a greater genomic instability and higher spontaneous mutation rates as the tumor bulk increases with time.
Other factors associated with drug resistance
Besides the inherent mutation rate and the overall number of GIST cells, many other cancer biological and pharmacokinetics related factors may influence emergence of drug resistance. In one cohort study, GIST patients who had imatinib plasma trough concentration below 1110 ng/mL (the lowest quartile of plasma concentrations) had a significantly shorter median time to disease progression as compared to those who had a higher imatinib plasma trough concentration (11 months vs. over 30 months, respectively), and they also had a lower response rate to imatinib (44% vs. 67%) (von Mehren et al. Citation2008). Low imatinib plasma concentrations might allow more GIST cells to survive and to maintain a relatively high cancer cell proliferation rate, which might favor emergence of drug-resistant mutations. Administration of adequate drug doses and good compliance to treatment may be important strategies to delay emergence of drug resistance.
Adjuvant trials in GIST
Two prospective, single-arm, multicenter trials have evaluated imatinib administered 400 mg/day for 12 months as adjuvant systemic treatment of GIST patients considered to have a high risk for disease recurrence (DeMatteo et al. Citation2005, Zhan et al. Citation2007). Both trials found imatinib to be well tolerated by most patients and GIST recurrence to be infrequent (<10%) during imatinib administration.
Patients with KIT-positive GIST, no prior adjuvant therapy, and with a tumor 3 cm or larger in diameter were randomly allocated after macroscopically complete surgery to receive either adjuvant imatinib 400 mg/day or placebo in the American College of Surgeons Oncology Group (ACOSOG) Z9001 trial. The primary endpoint was recurrence-free survival in this Phase III trail with planned cross-over between the study arms, and secondary endpoints included overall survival and safety. The trial was interrupted early when 732 patients had entered the study due to markedly improved recurrence-free survival in the imatinib arm as compared with placebo (97% vs 83% at 1-year of follow-up; P < 0.001) (DeMatteo et al. 2008).
Two other large randomized trials that address imatinib as adjuvant treatment of operable GIST have recently completed accrual. EORTC 62024 (NCT00103168) compares 2 years administration of adjuvant imatinib (400 mg/day) to observation following macroscopically complete surgery. The study population consists of patients diagnosed with a GIST of either intermediate or high risk of recurrence as defined by the NIH Consensus Classification criteria. 900 patients have entered this prospective Phase III trial that has overall survival as the primary endpoint.
The Scandinavian Sarcoma Group/AIO trial (SSGXVIII/AIO) (NCT00116935) compares 12 to 36 months of adjuvant imatinib administered 400 mg/day following macroscopically complete surgery. The study population consists of GIST patients considered to have a high risk of recurrence based on the National Institute of Health (NIH) of the United States Consensus Classification, but includes also patients with tumor rupture (either spontaneously or during surgery). The primary endpoint in this prospective Phase III trial is recurrence-free survival. The accrual of the trial (400 patients) was completed in September 2008, and the first efficacy results are anticipated in 2010.
The main focus of the SSG/AIO trial is whether a prolonged adjuvant treatment, as compared to a potential standard of 12 months as used in the ACOSOG Z9001, is beneficial or harmful to patients with a high risk of GIST recurrence. The recurrence rate during the first year off treatment will be compared as one of the secondary endpoints between the two arms. A smaller relapse rate during the first year after stopping adjuvant imatinib in the 3-year arm might reflect curative potential of adjuvant treatment. The efficacy of imatinib rechallenge among patients who relapse after adjuvant treatment will also be studied within the frame of the SSG/AIO trial, a topic that is discussed further below.
Response to imatinib rechallenge after adjuvant imatinib
The ACOSOG Z9001 trial found that adjuvant imatinib prolongs the time to GIST recurrence (DeMatteo et al. 2008). There has been a concern of whether patients treated with adjuvant imatinib will respond to imatinib in cases where GIST recurs during the follow-up. The prospective, randomized BRF-14 trial carried out in advanced GIST found that interruption of imatinib treatment among the subset of patients who continued to respond to imatinib usually resulted in rapid disease progression after imatinib withdrawal, but the most patients responded again after imatinib rechallenge (Blay et al. Citation2007). This scenario resembles the one where adjuvant imatinib administration is discontinued as planned without detectable macroscopic GIST, and is radically different from the one where GIST progresses during imatinib treatment.
No data have been published regarding sensitivity of GISTs that recur after completion of adjuvant therapy to imatinib. One of the authors (H.J.) treated 7 consecutive GIST patients who were estimated to have a high risk of recurrence with adjuvant imatinib for 6 to 12 months between May 2002 and April 2004, and whose GIST subsequently recurred during the follow-up. 5 of these 7 patients responded to imatinib rechallenge, and one further patient had stabilized disease. One patient with a high risk GIST treated by the other author (M.E.) had GIST recurrence 18 months after a 2-year adjuvant treatment period, and rechallenge with imatinib resulted in a partial remission that has now continued for longer than 2.5 years. Although based on small numbers of patients treated, these findings suggests that most patients who have been treated with adjuvant imatinib will respond to imatinib rechallenge when the disease recurs (unpublished data).
Effect of adjuvant treatment on survival
Due to the short follow-up time at the time of the ACOSOG Z9001 trial interruption, only a few deaths had occurred, which prevented carrying out a meaningful analysis of overall survival. It is currently not known whether adjuvant imatinib influences overall survival.
It has been speculated that administration of adjuvant imatinib might decrease frequency or the duration of response to imatinib if GIST recurs after adjuvant therapy. There are currently no data to support this hypothesis. The overall rate of a secondary drug-resistant mutation is likely the slower the fewer GIST cells are exposed to imatinib. This may be reflected as a longer time to emergence of a drug-resistant cell clone when imatinib is administered in the adjuvant setting when the tumor burden is small as compared to the overtly metastatic stage of the disease. Importantly, most tumors that recur after adjuvant imatinib will respond to imatinib rechallenge, although the median duration of the response is still unknown. Thus, even if adjuvant imatinib might not cure any patient, which is not known, early administration of imatinib and other targeted agents in the adjuvant setting could be beneficial in terms of overall survival provided that systemic adjuvant treatments do not shorten survival by toxicity, especially of those patients who might be cured by surgery alone.
The target group for adjuvant therapy
Patients who are likely to be cured by surgery alone should preferably not be exposed to adjuvant therapies. To address the question of patient selection for adjuvant therapy, many prognostic factors have been studied in GIST. The generally accepted most important adverse prognostic features are a large tumor size, a high mitotic count, and tumor origin in the intestine. The NIH Consensus Classification uses 2 of these 3 features, the tumor size and the mitotic count (Fletcher et al. Citation2002). The Consensus Classification has now been shown to predict survival of GIST patients in several large population-based series (reviewed in Joensuu 2008). Of note, patients with GIST with a low or intermediate risk of recurrence by the Consensus Classification have in most studies favorable survival not markedly inferior of that of the general population suggesting that many patients with either a low or an intermediate risk GIST may not be ideal candidates for adjuvant therapies.
The NIH Consensus Classification does not take into account the tumor site. Evidence from 2 large series from the Armed Forces Institute of Pathology (AFIP) of the United States suggests that fairly small GISTs of intestinal origin are frequently associated with metastases and death (Miettinen et al. Citation2005, [Citation2006], Miettinen and Lasota Citation2006) and, therefore, the Consensus Classification probably needs to be modified accordingly. The AFIP risk classification, in turn, does not include ruptured GISTs, which have a high risk of recurrence (Takahashi et al. Citation2007). A suggested modification of risk classification to define the group with a high risk of GIST recurrence after macroscopically complete surgery is presented in . Limiting of systemic adjuvant treatment to the modified high risk group would result in administration of adjuvant treatment to approximately 50% of all patients diagnosed with GIST (Joensuu Citation2008).
Table 1. Proposed modification of consensus classification for selecting GIST patients for adjuvant therapy (with permission from Joensuu H. Hum Pathol 2008)
Adjuvant systemic therapy: current practise
Adjuvant administration of imatinib to GIST patients who present with localized disease has been considered investigational (Casali et al. Citation2008). Yet, imatinib has now been found to increase considerably progression-free survival in a large randomized study (DeMatteo et al. 2008), and the time to appearance of a mutation leading to drug resistance is likely often relatively long when imatinib is administered in the adjuvant setting to patients who have a minimal tumor burden. These factors make administration of adjuvant imatinib appealing especially to patients who have a high (>50%) risk for GIST recurrence and thus a high risk of death from GIST, since risk of fatal toxicity related to imatinib appears to be rare.
At present, there are different opinions regarding adjuvant administration of imatinib. The views of experts range from abstaining from treatment even in cases with close to 100% risk of recurrence (such as patients with a spontaneously ruptured tumor) to administration of adjuvant imatinib to GIST patients diagnosed with a relatively small single tumor (3 cm or larger in diameter). Use of the 3 cm cut-off as the sole criterion for patient selection results in administration of adjuvant imatinib to some patients with an intermediate or even a low risk of recurrence and who often have a favorable outcome with surgery alone, whereas some patients with GIST with a high mitotic count and thus with a relative unfavorable prognosis might be deprived from adjuvant therapy. A more rational choice may be to use the high risk group of the modified Consensus Classification in selection of patients for adjuvant treatment.
The optimal duration of adjuvant therapy is currently not known. If adjuvant imatinib therapy is chosen to be administered, a feasible choice is imatinib administration at the dose of 400 mg/day for a total of 12 months. Patients with the imatinib-resistant PDGFRA mutation D842V may not benefit from adjuvant imatinib therapy, and adjuvant imatinib might not be effective for neurofibromatosis-1 associated GIST (Mussi et al. Citation2008). It is not known whether patients with KIT exon 9 mutation or with no mutation (wild type GIST) will benefit less from adjuvant imatinib than those with GIST with KIT exon 11 mutation, which could be anticipated from data generated in the metastatic setting (Heinrich et al. Citation2003).
Future directions
The effect of adjuvant systemic administration of tyrosine kinase inhibitors on overall survival will not be known for several years, although survival data are being collected in the ongoing adjuvant trials. Prevention of secondary drug resistance will remain an important research target, as well as methods to identify patients who are at a clinically significant risk for GIST recurrence.
Combinations of drugs and sequential treatments of novel targeted agents deserve to be investigated not only in advanced GIST but in the adjuvant setting as well. These approaches might lead to improved eradication of GIST cells, and to a reduced risk of drug resistance. Studies addressing individualized dosing of targeted agents based on patient metabolic properties and tumor biological features are also warranted.
Prognostic systems for soft tissue sarcoma of the extremities and trunk wall in adults
A review and the Scandinavian Sarcoma Group experience
Jacob Engellau
Department of Oncology, Lund University Hospital, Lund, Sweden.
In soft tissue sarcoma (STS) of the extremities and trunk wall population-based patient series have demonstrated that more than one-third of patients will develop metastases, most commonly to the lungs, and most of these patients will die from their sarcoma (Gustafson Citation1994, MÖller-Nielsen Citation2001). For a long time the value of chemotherapy for patients with STS was unclear. The scarcity of the tumors and the many histological subtypes, sometimes difficult to delineate even for experienced pathologists, have hampered adequate recruitment and analysis of studies investigating the effects of chemotherapy. Meta-analyses of several randomized controlled trials (RCT) have, however, demonstrated a marginal efficacy of chemotherapy in STS (Sarcoma Meta-analysis Collaboration Citation1997, Meta-analysis Group Citation2000, Pervaiz et al. Citation2008). Despite this, the use of adjuvant chemotherapy for STS patients has become more and more common. Most STS patients are elderly, the median age at diagnosis for the commonest histotypes is 60-70 years. The chemotherapy regimens used in neoadjuvant and adjuvant settings usually contain doxorubicin and ifosfamide, sometimes together with other cytotoxic drugs. Thus, these regimens portend considerable toxicity. Therefore, identification of true high-risk patients for developing metastases is of obvious clinical importance in order to avoid over-treatment in actual low-risk patients as the modest improvement in outcome may well be outweighed by treatment induced toxicity and mortality. For this purpose, there has been a strong interest in designing clinically applicable prognostic systems for STS patients. For practical clinical use, an ideal prognostic system should clearly identify high-risk and low-risk patients for metastasis, be unambiguous in applicability and based on strong and reproducible prognostic factors for metastasis. Due to the pattern of dissemination in STS, with uncommon involvement of regional lymph nodes, the TNM-system is not applicable in prognosticating STS without overt metastasis at diagnosis. Local recurrence as a marker of an aggressive phenotype, or as a causative risk factor in itself for metastasis, has been the subject for extensive debate in STS. A review of this, still ongoing, issue is beyond the scope of this review, but suffice to say is that the development of a local recurrence after surgery with negative margins is associated with a high risk for metastatic disease in high-grade STS. This risk is particularly high if the local recurrence develops a short time (< 2 years) after the primary surgery.
Malignancy grading
Determining histotype does for most STS not provide adequate information on which to base treatment decisions of neoadjuvant or adjuvant chemotherapy. Histologic classification is also subject to periodic variations due to changes in diagnostic methods and concepts of classification. Therefore, combinations of histopathologically determined specific factors have been combined into malignancy grading systems to improve prognostication. Over the years there have been many such systems in use, most based on assessment of histological subtype, degree of differentiation, cellularity, cellular pleomorphism, mitotic count, and presence of tumor necrosis (Kilpatrick Citation1999). Subsequently, two systems have come to prevail internationally. These are the III-tiered malignancy grading systems devised by the Fédération Nationale des Centres de Lutte Contre le Cancer (FNCLCC) (Coindre Citation2006) and the National Cancer Institute (NCI) system (Costa Citation1990). However, in the Scandinavian countries Sweden, Norway and Finland a IV-tiered system is used (Markhede et al. Citation1982, Angervall et al. Citation1986, Meis-Kindblom et al. Citation1999), and Denmark uses a separate III-tiered system (Myhre-Jensen et al. Citation1983). This lack of agreement on a uniform malignancy grading system is, of course, a problem when comparing different tumor series. There is also no consensus as to the weights that should be attributed to the various components included in the grading systems. Furthermore, none of the common systems for malignancy grading includes presence of vascular invasion, which repeatedly has been shown to be one of the strongest prognostic factors for metastasis in STS (Trojani et al. Citation1984, Gustafson Citation1994, Mandard et al. Citation1989, Coindre et al. Citation2001, Gustafson et al. Citation2003, Engellau et al. Citation2005, [Citation2007]). The different systems of malignancy grading all identify about 10% of the tumors as of low-grade with low risk of metastasis (< 10%), and a large proportion of high-grade tumors which metastasize in about 60%, but almost half of the patients are classified in an intermediate group with a risk of metastasis of about 30%. The low-grade tumors are virtually never considered for systemic adjuvant therapy whereas the histologic intermediate-grade and high-grade tumors, comprising the large majority of STS, need to be considered for chemotherapy. For clinical purposes, these patients need to be classified further in categories of low-risk and high-risk tumors to avoid both over-treatment and omitting treatment of patients at true high risk of metastasis.
Although determining malignancy grade clearly improves prognostication compared to histologic classification, malignancy grade does not provide enough prognostic information for clinical treatment decisions.
Current common prognostic systems in STS of the extremities and trunk wall
The nomogram
A model for addressing the problem of clinical prognostication in STS has been developed at Memorial Sloan Kettering Cancer Center (MSKCC). Based on patients treated at their institution, a series of 2 327 patients for which clinicopathological factors, treatment related factors and follow-up had been prospectively entered into a database were used to construct a nomogram (Kattan et al. Citation2002). The nomogram included all sites of a STS, also head and neck, intra-thoracic, visceral, and retroperitoneal localizations and has henceforth been in use at MSKCC. It provides an estimate of projected risk of sarcoma-specific death, combining both the risk of death as a result of metastasis and the risk of treatment-related mortality. The nomogram can be used for patient counseling and determining whether adjuvant chemotherapy should be advocated, or argued against. The original nomogram has subsequently been validated and adapted for extremity STS in an independent series of 642 patients treated at the Instituto Nazionale per lo Studio e la Cura dei Tumori in Milano, Italy (Mariani et al. Citation2005, ). In this validation and adaptation, the III-tiered FNCLCC system for malignancy grading was used, instead of the II-tiered system proposed by Hajdu which was used in the original nomogram. Although providing a useful tool for assessment of sarcoma-related survival, the nomogram includes prognostic factors that could be questioned, and lacks other, strong prognostic factors. In population-based series age has not been found to be a prognostic factor (Gustafson Citation1994). Also tumor depth may be questioned as there is a strong correlation between tumor size and depth, and when tumor tumor size and malignancy grade are accounted for, tumor depth loses its prognostic value (Rydholm and Gustafson Citation2003). One of the strongest prognostic factors, the abovementioned factor vascular invasion is not included in the set of parameters on which the nomogram is based. The nomogram does not categorize patients, and may therefore paradoxically be difficult to employ clinically. At what level of risk of metastasis is cytotoxic treatment warranted? A large proportion of patients with localized STS will be attributed an intermediate risk of metastasis, and given the modest gain of the chemotherapy used, patient counseling may thus still be difficult using the nomogram.
Figure 1. Modified MSKCC Nomogram for postoperative prediction of sarcoma-specific survival in adult (>16 years) patients with localized soft tissue sarcoma of the extremities. From Mariani et al. 2005 (Reprint with permission from Wiley-Blackwell).

The American AJCC/UICC system
The system, originally presented in 1977, classifies tumors based on the TNM concept. In soft tissue sarcoma, the classification takes into account histopathological malignancy grade dichotomized as low-grade or high-grade. If a III-tiered grading system is used grade 1 is low-grade and, grades 2–3 are denoted as high-grade, whereas in a IV-tiered grading system grades 1–2 are considered low-grade, and 3–4 high grade. The staging system also incorporates tumor size, tumor depth, and presence of positive regional lymph node(-s) or distant metastasis. In the current 6th edition staging manual, this translates into 4 stages () (Greene et al. Citation2002).
Table 1. Stage grouping in the AJCC/UICC staging system (6th ed.) for soft tissue sarcomas
The patients that are commonly considered for adjuvant treatment are patients with stage II and stage III tumors. These comprise almost 88% in the series of 1097 patients from MSKCC on which the basis for staging of STS was based (Greene et al. Citation2002). In subsequently reported series the patients with the highest risk of metastasis (excluding stage lV tumors with overt metastases and dismal prognosis), stage III with histological high-grade, large (> 5 cm), and deep-seated tumors constitute almost 50% (Wunder et al. Citation2000). However, almost 50% of tumors are classified as stage II, with a risk of metastasis of about 30% and whether these patients should be candidates for adjuvant chemotherapy is unclear. Although in widespread use, there are obvious shortcomings in the AJCC system for staging STS due to the fact that several known strong prognostic factors for a highly malignant phenotype are not incorporated into the system. The inclusion of nodal involvement has also been questioned as it is a vary rare manifestation in most STS, reported to occur in less than 5% of patients during their course of disease (Behranwala et al. Citation2004, Fong et al. Citation1993). Although nodal involvement at diagnosis carries a considerable risk of also developing distant metastasis, the prognosis is better than overt distant metastasis at the time of diagnosis (Behranwala et al. Citation2004).
An assessment of possible improvements to the current 6th edition of the AJCC staging system has recently been demonstrated in a large series from University of Texas MD Anderson Cancer Center. In this series of 1091 patients, clear improvements to the AJCC staging system could be demonstrated if further known prognostic factors were incorporated. These included age, primary site, histologic subtype, and surgical margin status (Lahat et al. Citation2008). It remains to be seen to what extent the AJCC staging system will be modified in the forthcoming 7th edition expected to be introduced during 2009.
Prognostic systems—the Scandinavian experience
In the realm of the Scandinavian Sarcoma Group (SSG), composed of centers where most of Scandinavian sarcoma patients are treated, there have been a longstanding interest in prognostic factors for metastasis in STS. Much of the understanding thus acquired has come from collaborative studies on population-based tumor series. Already in 1983 the benefits of a centralized management of soft-tissue tumors and soft tissue sarcomas was demonstrated (Rydholm Citation1983). In this work, the importance of adequate surgical margins was also shown and an algorithm for management of soft-tissue lesions suggested. This has henceforth been adopted by virtually all sarcoma centers in the SSG. Also prediction of survival was assessed; in a series of STS of extremity and trunk wall the following risk factors and their weights (coefficient of the survival function) were identified: grade IV (weight 1.78), grade III (1.0), tumor pain (0.94), male sex (0.86), age > 50 years (0.86), size < 6 cm (0.84), marginal surgery (0.77) extracompartmental site (0.66). The sum of the weigths for a specific patient determined the survival probability which could be easily found in a diagram: a sum > 6 implied almost 100% mortality, 3 or lower was associated with 80% survival. This system for prognostication is similar to the Nomogram used by the MSKCC.
In 1987 Rööser et al. explored the prognostic factors for metastasis in a population-based series of 237 patients with STS of the extremities and trunk wall. For high-grade tumors the strongest independent prognostic factors were found to be malignancy grade IV, tumor size > 10 cm, presence of tumor necrosis, and vascular invasion. Vascular invasion was defined as intratumoral invasion of vascular elements with tumor cells adherent to the vessel wall or associated with fibrin, red or white blood cells. This definition has henceforth been employed. A prognostic system based on presence of these factors was shown to identify almost 50% of high-grade tumors with 3–4 factors present and a long-term tumor-related survival of < 0.25. Tumors with 0–1 factors present had a survival comparable to histological low-grade tumors (Rööser Citation1987).
In 1989 it was again demonstrated that metastasis correlated with a high malignancy grade, presence of vascular invasion, large tumor size (> 10 cm), and male sex. The results were based on one of the first RCT's of adjuvant doxorubicin in STS, the SSG I trial which recruited 240 patients from 1981–1986 (Alvegård et al. Citation1989).
These findings were further explored in a population-based series of 508 histologically mixed soft tissue sarcomas of the extremities and trunk wall. In this series the prognostic factors vascular invasion, presence of tumor necrosis, and tumor size > 10 cm were identified as the strongest factors predicting metastasis (Gustafson Citation1994). Malignancy grade lost its prognostic value in multivariate analysis when presence of vascular invasion and tumor necrosis were adjusted for, which accounts for the considerable importance these factors have been attributed in subsequent prognostic systems implemented by the SSG. Based on the results in the latter series a prognostic system was suggested that included tumor Size, vascular Invasion, and presence of tumor Necrosis, the SIN-system (Gustafson Citation1994). In this system patients were categorized as low-risk if 0–1 of the abovementioned factors were present, and high-risk with 2–3 factors. In the original series, low-risk tumors constituted two-thirds of the patients and had a 5-year metastasis-free survival rate of 0.8. The one-third of the patients which were classified as high-risk tumors had a corresponding 5-year metastasis-free survival of 0.3. The system has been prospectively implemented in a prospective non-randomized adjuvant treatment protocol, the SSG XIII protocol, in which in adjunction to local treatment recommendations high-risk patients were treated with doxorubicin and ifosfamide. The results in the SSG XIII compare favorably with previous trials which support the selection of prognostic factors on which patient accrual was based.
The reproducibility of the SIN-system was demonstrated in a bi-institutional study of 200 STS analyzed in Lund and Institut Bergonié. In this study, the strong importance of vascular invasion, size, and necrosis was again shown, and also that the extent of necrosis had no bearing on the impact of necrosis as a prognostic factor. The SIN-system also favorably compared with the AJCC/UICC 5th edition staging system (Gustafson et al. Citation2003).
Clinically applied there were, however, drawbacks with the SIN-system. Vascular invasion is not included into any of the systems of malignancy grading. This, and the fact that vascular invasion may be difficult to detect if not specifically sought for, probably explains the low rate of detection of vascular invasion which in turn led to a pivotal importance of tumor size when applying the SIN-system. Based on pathology peer-review data in the SSG tumor registry, a retrospective analysis of the probability of being classified as high-risk and thus be a candidate for adjuvant chemotherapy was calculated. This demonstrated that for tumors < 8 cm, the risk, or opportunity, to be classified as high-risk was 1%, whereas for tumors > 8 cm the corresponding likelihood for adjuvant treatment was 46% (unpublished data). It should be added, though, that vascular invasion was not a principal aim of the pathology peer-review, which focused on histological classification and attribution of malignancy grade.
In a study of 140 histologically mixed STS of the extremities or trunk wall on which whole-tumor sections had been established clinically useful prognostic factors were explored (Engellau et al. Citation2005). The whole-tumor section facilitates detection of necrosis and vascular invasion, and the peripheral tumor growth pattern (pushing or infiltrative) is also readily discernable. This series clearly supported the importance of the established prognostic factors tumor size and necrosis, and also vascular invasion which was demonstrated in one-third of the cases, of which more than two-thirds metastasized. The tumor growth pattern was also an independent strong prognostic factor for both local recurrence and metastasis. These findings was further explored in a retrospective study including two independent series of 434 patients with high-grade STS of the extremities or trunk wall treated in Sweden and Norway, and 175 patients treated at the sarcoma center in Lund and at the Norwegian Radium Hospital in Oslo, Norway (Engellau et al. Citation2007). In the larger series, the importance of the prognostic factors tumor size > 8 cm, presence of tumor necrosis, vascular invasion, and infiltrative growth pattern was again demonstrated, and formed the basis for a novel prognostic system. This system of recursive partitioning, or a decision tree, was based on a step-wise attribution of risk where the strongest prognostic factor, presence of vascular invasion provided the primary fork ( and ). The prognostic system was independently validated in the series of 175 tumors, and its prognostic usefulness confirmed. A comparison was also made with the SIN-system and the American AJCC/UICC staging system 6th edition. Compared to the SIN-system, half of the tumors were discordantly classified and all of these were classified as low-risk in the SIN-system and high-risk in the decision tree and one-third of these patients developed metastasis. In comparison with the AJCC/UICC staging system, there were one-third discordantly classified tumors. 11 tumors classified as high-risk in the AJCC/UICC system were attributed low-risk in the decision tree, and 1 metastasized, and 43 tumors were discordantly classified as low-risk in the AJCC/UICC system, and 23 metastasized. Of particular interest was the capacity of the decision tree to identify patients with high-grade malignant STS, but at low-risk for metastasis. These patients comprised almost half of the series, also including large and deep-seated tumors, and only 15% metastasized making them phenotypically comparable to histological low-grade STS (Engellau et al. Citation2007).
Figure 2. Cumulative incidence of metastasis in a series of 434 histological high-grade STS: (A) tumors with vascular invasion (n=36, dashed line) versus tumors without vascular invasion (n=398, solid line). (B) Cumulative incidence of metastasis in tumors without vascular invasion (n=398) with presence of 0–3 risk factors (size >8 cm, tumor necrosis, infiltrative peripheral tumor growth pattern), 3 factors (n=61), 2 risk factors (n=130), 1 risk factor (n=169) and 0 risk factors (n=38). (C) Cumulative incidence of metastasis in the 227 high-risk tumors with vascular invasion (n=36) or presence of 2–3 risk factors (n=191) versus the 207 low-risk tumors without vascular invasion and presence of 0–1 risk factors.
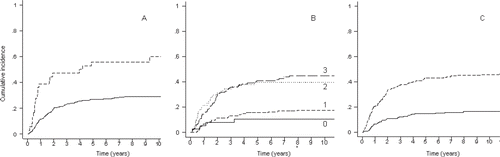
Figure 3. The SSG decision tree for classification of risk for metastasis in high-grade STS of the extremities or trunk wall based on the factors vascular invasion, tumor size, tumor necrosis and, peripheral tumor growth pattern.
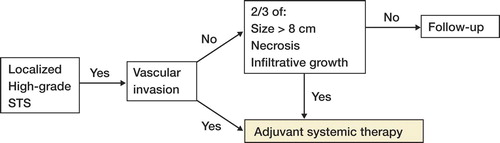
The decision tree thus validated is now implemented in Scandinavia in the SSG XX trial.
In summary, there has been a continuous interest of the SSG to improve patient selection for adjuvant chemotherapy. For this purpose the population-based series made possible by the high degree of centralization of diagnosis and treatment of STS in Scandinavia provide excellent conditions. In repeated series the importance of tumor vascular invasion, presence of tumor necrosis, and tumor size has been demonstrated and recently also tumor growth pattern, forming the basis of prognostic systems for patients with STS of the extremities and trunk wall. Future work in this setting will undoubtedly enable histotype-specific prognostication with incorporation of important prognostic factors identified by molecular genetics of STS.
Treatment of soft tissue sarcoma of the extremity and trunk wall
The Scandinavian Sarcoma Group perspective
Kirsten Sundby Hall 1, Mikael Eriksson 2, Øyvind S Bruland 1,3, Jacob Engellau 2 and Clement S Trovik 4
1Department of Oncology, The Norwegian Radium Hospital, Rikshospitalet University Hospital, Oslo, Norway,
2Department of Oncology, University hospital, Lund, Sweden, 3University of Oslo, Norway,
4Department of Orthopaedics, Haukeland University Hospital, Bergen, Norway
Correspondence: [email protected]
Since the founding of the Scandinavian Sarcoma Group (SSG) 30 years ago, development of an optimal management of soft tissue sarcomas (STS) has been an important task based on clinical studies, review based treatment recommendations and biological research. Surgery remains the mainstay of STS treatment and has been used as the sole treatment in most patients in Scandinavia. However, the use of adjuvant radiotherapy and chemotherapy has steadily increased. In this overview, we briefly describe our experiences with no ambitions of reviewing the entire field as such.
Epidemiology and centralization
Soft tissue sarcomas account for less than 1% of all malignant tumors (Jemal et al. Citation2004, Olsson Citation2004). Physicians outside tumor treatment centers, who most often are first to see the patients, must know when to suspect a sarcoma and refer the patient to specialized centers with multidisciplinary sarcoma teams (Rydholm Citation1998). Evidence points to significant improvements in outcome for patients treated in specialist centers (Bauer et al. Citation2001)
A recent paper from SSG demonstrated that the referral pattern has shown a gradual increase of virgin STS referred to and treated at sarcoma centers; from 62% in the late 1980s to 77% in the last time period of 1998–2005 (Jebsen et al. Citation2008).
Surgical treatment
During the early years of SSG, compartmental excisions according to Enneking et al. (1980) were attempted. However, improved referral patterns with more patients referred to tumor centers before surgery (biopsy or excision), now most often makes it possible to avoid the loss of function seen with compartmental excisions. Also, the routine use of CT/MRI in the preoperative planning facilitates safe resection margins with less loss of normal tissue. At present, the surgical goal is to obtain a wide margin; i.e. a cuff of healthy tissue surrounding the tumor. For strictly intramuscular tumors such a margin is often obtained by a myectomy.
The quality of the surgical margin obtained is still classified according to Enneking (1980) with some modifications. It is done in cooperation between the surgeon and the pathologist. An intralesional margin is recorded when the plane of excision, in any part of the tumor, passes through the tumor, leaving microscopic or macroscopic tumor tissue behind. The intralesional margin is categorized into two types depending on whether macroscopic tumor tissue remains or not. A marginal margin is recorded when the plane of excision passes outside the tumor, but in any part too close to the tumor to merit for a wide margin. With another terminology an intralesional margin (both types) reflects a positive margin, whereas a marginal margin corresponds to a negative margin.
A wide margin is recorded when the excised tumor is surrounded all around by a cuff of healthy tissue or uninvolved fascia. The necessary thickness of this cuff to merit for a wide margin has been discussed during the years. In the latest soft tissue sarcoma protocol (SSG XX – activated 2007, see www.ssg-org.net) a cuff of fatty or muscular or loose areolar tissue must be minimum 10 mm in a formalin fixed specimen to qualify for a wide margin. Unengaged fascia, even if close to the tumor, is also sufficient for a wide margin. The margin obtained by myectomy is regarded as a subtype of the wide margin and has been applied for strictly intramuscular lesions (not subjected to open biopsy) when the involved muscle, from origin to insertion, is completely removed (Rydholm & Rööser 1987).
To examine whether classification of the surgical margin obtained at different institutions adhered to SSG guidelines, a random sample, comprising one quarter of patients included in a recent study of radiotherapy effects, were evaluated (Jebsen et al. Citation2008). A panel of Scandinavian sarcoma surgeons independently scrutinized the surgical and pathology reports. The few discrepancies in the individual classifications of surgeons in this panel were solved by consensus. The panel disagreed with the original margin assessment in 6% of cases. In 4% of all cases this reclassification changed from wide to marginal /intralesional or vice versa. Considering the element of judgment inherent in all margin assessment, we find this validity and reliability acceptable for using the Scandinavian Sarcoma Group Register for studies of local tumor control.
Compiled since 1986, this Register contains data on more than 9000 sarcoma patients who had their definitive treatment of the primary tumor at a sarcoma center in Scandinavia. The surgical margins were wide in 76% of subcutaneous lesions, and wide in 58% of deep-seated lesions. The amputation rate was 7% and has been declining. The rate of wide margins obtained is lower than sometimes reported in international sarcoma series, but the risk of local recurrence () is, however, similar (Jebsen et al. Citation2008). This may imply that our definition/classification of surgical margins could be somewhat more rigorous than that applied by other sarcoma centers.
Table 1. Local recurrence rates, wide surgical margin and use of radiotherapy in three treatment time periods
When SSG was established in 1979, surgery with a wide margin was considered sufficient treatment of STS, and radiotherapy was used only for tumors which had been resected with an intralesional or marginal margin. In a series of STS of the extremities and trunk wall from the SSG register it was found that a wide surgical margin in high-grade deep STS was associated with a 25% risk of local relapse (Trovik et al. Citation2001a). This has influenced the radiotherapy practice in SSG, as described below.
Radiotherapy
A high malignancy grade and the quality of the surgical margins are the two most important risk factors for a local recurrence (Gustafson et al. Citation1994, Eilber et al. Citation2003, Zagars et al. Citation2003, Pisters et al. Citation2007). The impact of radiotherapy on improved local control of STS in conjunction with surgery has been demonstrated in several prospective randomized trials (Harrison et al. Citation1993, Pisters et al. Citation1994, [Citation1996], Yang et al. Citation1998), as well as in retrospective reviews (Lindberg et al. Citation1981, Suit and SpiroCitation1994, Wilson et al. Citation1994, Trovik et al. Citation2001a). This effect is mainly demonstrated after intralesional or marginal surgery, but improvement in local control after wide margin surgery has also been reported (Stotter et al. Citation1990, Pisters et al. Citation1996, Trovik et al. Citation2001b).
After wide and marginal surgery for deep-sited tumors a dose of 50–60 Gy in conventional 2 Gy daily fractions is generally recommended as well as following marginal surgery of superficial tumors. If macroscopic tumor tissue is left behind, doses above 60 Gy should be attempted (Zagars et al. Citation2003a, Pisters et al. Citation2007). The benefit of radiotherapy on low grade malignant sarcomas is still controversial (Pisters et al. Citation1996, Mollabashy et al. Citation2002, Strander et al. Citation2003, Jebsen et al. Citation2008).
The timing of radiotherapy varies between studies. In a prospective trial involving 94 patients randomized to preoperative radiotherapy with 50 Gy in 25 fractions versus 96 patients postoperatively given 66 Gy in 33 fractions a slightly better overall survival was demonstrated in the preoperative arm (O'Sullivan et al. Citation2002). A higher rate of wound complications was demonstrated in the preoperative arm after a median follow up of 3.3 years. However, no significant difference was demonstrated at 5 years follow up with recurrence free survival rates of 58 % and 59 % respectively, albeit with a slightly higher incidence of late radiation related complications in the latter arm (Davis et al. Citation2005).
With increasing use of adjuvant radiotherapy within SSG, the local 5-year local recurrence rates have decreased, despite similar distributions of surgical margins among patients receiving primary treatment at a sarcoma center (Jebsen et al. Citation2008). 1093 adult patients with extremity and trunk wall STS with a median follow up of 5 years, treated at 4 Scandinavian sarcoma centers during 1986–2005, were stratified according to the treatment period. The use of radiotherapy, with 77% of the patients given postoperative radiotherapy, increased from 28% to 53%. The 5-year local recurrence rate decreased from 27% to 15%, without any obvious change in the rate of wide surgical margins (). The positive impact of radiotherapy on local recurrence was also significant after a surgery with wide margin and also in low grade STS (Jebsen et al. Citation2008).
At present, SSG recommends that postoperative radiotherapy starts as soon as the wound has healed. To our knowledge, only one study has specifically studied this aspect and demonstrated an improved local recurrence-free survival rate in patients starting less than 4 months after surgery compared to those who started radiotherapy later (Schwartz et al. Citation2002). In the SSG study mentioned above (Jebsen et al. Citation2008) the median interval from surgery to start of radiotherapy was 7 weeks with as many as 98 % starting within 4 months. However, no difference between the two groups in local recurrence rate was demonstrated.
Both experimental and clinical evidence in several tumor types indicate that accelerated tumor cell proliferation occurs during prolonged radiotherapy regimens, or in treatment schedules containing breaks/split course regimes (Bese et al. Citation2007). There is clinically no clear evidence for this in sarcomas. However, the high expression of proliferation markers such as mitotic index and MIB-1 expression in STS indicate that prolonged radiotherapy treatment time, low doses per fraction or split-course treatment is less effective for local control also in STS (MØller Nilsen 2001). Within the two latest adjuvant studies in STS performed by SSG; SSG XIII (completed, but unpublished) and the ongoing SSG XX, hyperfractionated/accelerated radiotherapy (1.8 Gy twice 2 daily, 5 days a week) is given in between chemotherapy cycles in order to shorten the overall treatment period. The interval between the 2 daily fractions should be at least 6 hours to allow for repair of sub-lethal radiation damage in normal tissues. The preliminary survival data from SSG XIII are promising and with a low, acceptable rate of late radiation tissue sequelae. This has encouraged us to explore a modification of this multimodal treatment algorithm, within the framework of the current SSG XX. Both acute and late radiation toxicity will be prospectively studied based on the RTOG/EORTC scoring schemes. In this protocol, most patients will be treated postoperatively (, group A). The protocol also includes a treatment group with preoperative chemo- and radiotherapy, aimed for patients in whom resection of the tumor carries an obvious risk for an intralesional surgical margin (, group B). The radiotherapy schedule and doses are similar to SSG XIII. For further details see: www.ssg-org.net where the SSG XX protocol may be downloaded.
Figure 1. The SSG XX protocol. Group A: Adjuvant therapy arm for high-risk STS in extremities and trunk wall with primary surgery.
Group B: Preoperative and adjuvant treatment for STS when primary resection carries an obvious risk of an intralesional margin.
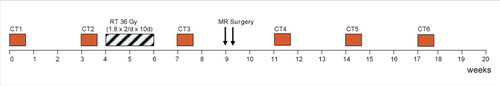
Chemotherapy
Adjuvant chemotherapy
A considerable fraction of all STS patients will, however, develop metastatic disease, most commonly in the lungs, and with the highest risk within the first 2 years after surgery. STSs are in general considered to be moderately responsive to chemotherapy (Clark et al. Citation2005). The role of adjuvant chemotherapy has been an area of great interest for SSG since it's inception and has been tested prospectively in two earlier adjuvant protocols, and with a third one on-going (SSG XX), activated in October 2007.
The first Scandinavian adjuvant chemotherapy study, carried out 1981–1986 (SSG I), was a randomized trial (Alvegård Citation1989). The study could not demonstrate any effect of adjuvant doxorubicin on metastasis-free- or overall survival in patients with high-grade malignant soft tissue sarcomas of the extremities and trunk wall. This study was included in a meta-analysis of results from 14 published randomized clinical trials in STS. Here, however, the use of adjuvant chemotherapy improved metastasis-free survival and local tumor control, with a trend toward better overall survival (Tierney et al. Citation1995). A recent update of this meta-analysis, which included the addition of 4 new eligible trials, showed a statistical significance for overall survival ascribed to the use of adjuvant chemotherapy (Pervaiz et al. Citation2008). This new finding may be attributed to either the larger sample size resulting in narrower confidence interval, or to the evolution in the chemotherapeutic regimens used, involving dose intensification and the addition of ifosfamide to doxorubicin-based protocols. The EORTC-STBSG 62931 adjuvant trial, which was recently closed, was not included in this meta-analysis. Results from this trial failed to demonstrate a benefit in overall or relapse-free survival among 351 STS patients included (Woll et al. Citation2007).
The second adjuvant STS protocol by SSG (SSG XIII) was a phase II non-randomized trial for a subgroup of patients operated for soft tissue sarcoma of the extremity and trunk wall with high risk to develop metastases. It was opened in 1998, and the prognostic SIN-system was used for selection of high risk patients (Gustafson Citation1994, Gustafson et al. Citation2003). The study included patients operated for STS grade III and IV, in a four-graded scale, and in addition displaying at least two of the following factors: tumor size > 8 cm, microscopic and/or macroscopic necrosis, and vascular invasion. Patients above 70 years of age were not included. Chemotherapy consisted of 6 cycles of doxorubicin and ifosfamide with hyperfractionated/accelerated radiotherapy as described above interpolated between chemotherapy cycles 2 and 3. The preliminary survival data of SSG XIII is promising and also toxicity seems moderate. The protocol had recruited 114 eligible patients when it was replaced by the present SSG XX trial October 1th., 2007. The results of SSG XIII will be published when all analyses are finalized.
Based on the experience from SSG XIII, and improved knowledge of prognostic factors in STS, the SSG decided to initiate a new protocol for high-risk STS (SSG XX) with a modification of the current system of prognostication. During the period 1998-2005 1074 primary, high-grade STS of the extremities and the trunk wall have been registered in the SSG Central Register. In SSG XIII only 114 patients have been included since the protocol was launched in 1998. The identification of high risk patients eligible for adjuvant treatment may have been hampered by the difficulties of identifying vascular invasion in the tumor, one of the adverse prognostic factors in the SIN-system. Consistent with other investigators, SSG found in retrospective reviews a considerable variation in the frequency of which this prognostic factor was identified. When present it is a strong risk factor for metastases, but a non-finding may not be informative. Data from Engellau et al. (Citation2007) supported the prognostic importance of the peripheral tumor growth pattern, infiltrating worse than pushing, which provided independent prognostic information in addition to tumor size, necrosis, and vascular invasion (Engellau et al. Citation2005). Based on the relative importance of the prognostic factors vascular invasion, size > 8 cm, necrosis, and infiltrative growth pattern, a novel prognostic system for histologically high-grade malignant tumor was designed (Engellau et al. Citation2007). Tumors with vascular invasion were thus shown to entail a high risk. Following this algoritm, a further selection into the high risk-group is based on the presence of two of the 3 factors tumor size > 8 cm, necrosis, and infiltrative tumor growth pattern ().
Figure 2. Decision algoritm for adjuvant treatment.
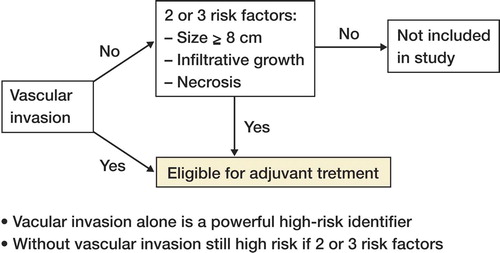
This new prognostic algorithm is the basis for selection of eligible patients in the on-going adjuvant SSG XX protocol (). There are biological research studies linked to the protocol with focus on genetic profiling, and also pharmacogenetic studies attempting to study the relationship between genetic variants and side-effects of treatment.
Treatment of metastatic disease
Most patients (80–90%) with STS have clinically localized disease at diagnosis. Nevertheless, STS has a propensity for early distant metastases despite local treatment with a curative intent (Clark et al. Citation2005). A subset of patients with operable distant metastases and chemosensitive disease may have a long disease-free interval, or might even be cured. The role of chemotherapy combined with metastasectomy has been uncertain in metastatic STS. In 1996 EORTC and SSG started a prospective trial (SSG XII) randomizing operable patients to excisions of lung metastases with or without aggressive chemotherapy, but this study was closed in 2001 due to low inclusion rate. However, promising results have been reported when effective chemotherapy was combined with complete surgical removal of all metastatic lesions. Good histopathological response to chemotherapy was associated with good prognosis and more relevant for outcome than a good radiological response (Sæter et al. 1995, Wiklund et al. Citation1997).
SSG X was a prospective phase II study designed to evaluate response and toxicity to ifosfamide in combination with continuous infusions of etoposide (VIG). In 26 patients treated with VIG + complete surgery of metastases the relapse free and overall survival at 2 years postsurgery were 39% and 74%, respectively (Sæter et al.1997).
In 2004, SSG presented treatment recommendations that may guide therapy for metastatic STS patients (SSG XIX, see www.ssg-org.net). Because there are relatively few studies of sufficient standard (small studies, retrospective studies, limited randomized data, diversity of histological diagnosis etc) on treatment for these patients, it is difficult to draw conclusions from the literature on the role of various treatment regimens. Therefore definitions like “Levels of evidence” and “Grade of Recommendations” were used in order to show the degree of uncertainty of the current recommendations.
Proposals for decision of chemotherapy should be based on the consideration of curative or palliative treatment intent. In principle, curative intention implies that surgery of the metastases is possible. Further, choice of treatment should be planned individually according to age, performance status, symptoms, and co-morbidity that may influence tolerability. Tumor-related factors like size, histological type and grade, location of metastases, rate of tumor growth, and current status at the primary tumor site also have to be taken into account.
So far, doxorubicin and ifosfamide are the most effective combination regimen against STS with regard to response rates, but no clear advantage has been demonstrated in survival. For the anthracyclines and ifosfamide, a dose response has been documented in phase II studies (Reichardt et al. Citation1998, Patel et al. Citation2000). Since there are no randomized trials showing that dose-intensified chemotherapy regimens will result in a survival benefit, standard dose chemotherapy should be the preferred treatment in metastatic or unresectable STS. The combination of doxorubicin and ifosfamide (doxorubicin: 50 mg/m2 + ifosfamide: 5 g/m2) with or without dacarbazine (250 mg/m2) are two options.
Isolated limb perfusion
Hyperthermic isolated limb perfusion (ILP) with tumor necrosis factor alpha (TNF-alpha) and melphalan has been adopted at 3 Scandinavian centers, Sahlgrenska sjukhuset (Gothenburg), Rigshospitalet (Copenhagen) and Radiumhospitalet (Oslo). TNF-alpha-ILP is an established treatment in large, bulky tumors in the limbs to avoid amputations (Eggermont et al. Citation2003). Further indications for this treatment modality in extremity located STS may be: attainment of primary tumor control in metastatic disease, recurrent tumor in an irradiated field, multiple tumors in the extremity, aggressive fibromatosis, and elderly patients where surgery is not feasible (Verhoef et al. Citation2007).
Regional hyperthermia
Regional hyperthermia in combination with chemotherapy and radiotherapy (Issels Citation2008) has shown improved clinical outcome (both regarding local control and disease-free survival) in high-risk soft tissue sarcomas in an EORTC study (Issels et al. Citation2007). Haukeland University Hospital in Bergen contributed to this study and is so far the only center in Scandinavia with facilities and experience with this treatment modality in bone and soft tissue sarcoma. A phase II protocol is currently running.
Future prospects
The new ESMO recommendations for diagnosis, treatment and follow-up of STS to which SSG has participated, define the current best standard of care (Casali et al Citation2008).
With the improvements of imaging, surgical techniques and increased use of adjuvant radiation therapy most patients with localized disease can be cured. However, a proportion of patients will develop metastatic disease, and there is need for new therapies to improve the long-term prognosis for such patients.
Trabectedin is a novel chemotherapeutic agent which has been shown to slow the growth and stabilize the tumor, especially in lipo- and leiomyosarcoma, as recently reviewed by Cassier (Citation2008). It has been registered in Europe for use as second line treatment in metastatic soft tissue sarcomas. The combination of gemcitabine and docetaxel is also effective as second-line treatment in some patients (Maki et al. Citation2007). Taxanes seems to be an option in angiosarcoma, at least as second line treatment. Presently, some SSG centers are participating in clinical trials of other drugs for STS such as tyrosine kinase inhibitors and monoclonal antibodies targeting the insulin-like growth factor-1 receptor (IGF-1R).
There seems to be a shift in the strategy from broad-spectrum cytotoxic chemotherapy to more molecular molecular-targeted therapies. Microarrays have been used to profile gene expression in several subtypes (Nilbert et al. Citation2004). This information might be used to develop new targeted therapies that later may be integrated into standard treatment such as has been the case for gastrointestinal stromal cell sarcoma. Moreover, there is need for broad international collaboration in accruing sufficient numbers of patients into clinical studies given the rarity of sarcoma. SSG has on several occasions shown a positive attitude to join relevant intergroup studies.
Scandinavian experience in classical osteosarcoma
Results of the SSG XIV protocol
Sigbjørn Smeland1, Øyvind S Bruland1,2, Lars Hjorth3, Otte Brosjö4, Bodil Bjerkehagen5, Gustaf Österlundh6, åke Jakobson7, Kirsten Sundby Hall1, Odd R Monge8, Olle BjÖrk7 and Thor A Alvegaard9
Correspondence: [email protected]
1Division of Cancer Medicine and Radiotherapy, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway
2University of Oslo, Norway
3Department of Pediatric Oncology, Lund University Hospital, Lund, Sweden
4Department of Orthopaedics, Karolinska Hospital, Stockholm, Sweden
5Division of Pathology, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway
6Department of Pediatric Hematology and Oncology, The Queen Silvia Children's Hospital, Sahlgrenska University Hospital, Gothenburg,
7Pediatric Oncology Unit, Astrid Lindgren Children's Hospital, Stockholm, Sweden
8Department of Oncology, Haukeland University Hospital, Bergen, Norway
9Department of Cancer Epidemiology, Lund University Hospital, Lund, Sweden
Background and purpose The Scandinavian Sarcoma Group (SSG) XIV protocol was based upon the organisations experience from 3 previous osteosarcoma trials and was considered best standard of care for patients with extremity localised, non-metastatic osteosarcoma. We report the outcome of this protocol.
Patients and methods From March 2001 to April 2005, 63 patients recruited from 10 centres in Finland, Sweden and Norway were included in this analysis. Patients received pre-operative chemotherapy consisting of 2 cycles of paired methotrexate (12 g/m2), cisplatin (90 mg/m2) and doxorubicin (75 mg/m2). Good histological responders continued with 3 cycles postoperatively whilst poor responders were salvaged with the addition of 3 cycles of ifosfamide (10–12 g/m2). Outcome data was compared to previous SSG osteosarcoma trials.
Results With a median follow-up of 64 months for survivors, the projected metastasis-free and sarcoma-related survivals at 5 years were 69% and 77%, respectively. 84% of the patients were treated with limb salvage surgery (49 patients) or rotationplasty (4 patients). 3 toxic deaths (5%) were recorded, all related to acute chemotherapy toxicity. The 5-year metastasis-free survival of patients receiving salvage therapy was 47% compared to 89% for good histological responders that only received the 3 drug combination postoperatively.
Interpretation Outcome in the SSG XIV protocol compares favourably to previous SSG osteosarcoma trials and other published trials. The addition of ifosfamide to poor responders as an add on treatment did not improve outcome for poor responders to a similar level as for good responders. In a multi-institutional setting limb salvage surgery can safely be used in more than 80% of the patients.
Current management of high-grade osteosarcoma comprises pre- and postoperative chemotherapy in combination with complete surgical removal of all tumour sites (Link et al. Citation1986, Fuchs et al. Citation1998, Bacci et al. Citation2000). With this strategy, long-term overall survival rates of 70% are reported for patients with non-metastatic, extremity localized (classical) osteosarcoma and more than 80% of the patients are managed by limb-salvage surgery (Bielack et al. Citation2002, Smeland et al. Citation2003, Ferrari et al. Citation2005). Preoperative chemotherapy offers an opportunity to modify postoperative chemotherapy according to histological response. Although never proven effective in a randomized trial, this principle has been used in several osteosarcoma trials (Rosen et al. Citation1979, Rosen et al. Citation1982, Winkler et al. Citation1988).
The active drugs in osteosarcoma are doxorubicin, methotrexate, cisplatin, and ifosfamide, but there is no general agreement on their optimal combination (Fuchs et al. Citation1998, Ferrari et al. Citation2005, Meyers et al. Citation2005, Lewis et al. Citation2007). In the SSG VIII study all patients received a preoperative combination of methotrexate, doxorubicin, and cisplatin (Smeland et al. Citation2003). Poor responders were salvaged with an exchange to a combination of ifosfamide/etoposide postoperatively. The overall results were good with 5-year metastasis-free and sarcoma-related survival of 63% and 74%, respectively. However, the ifosfamide/etoposide replacement combination resulted in a poor outcome of poor responders. In the recent Italian/Scandinavian ISG/SSG 1 protocol dose intensification by addition of high-dose ifosfamide up-front to all patients failed to improve outcome (Ferrari et al. Citation2005). In a previous trial from the Rizzoli institute addition of ifosfamide (9 g/m2) to poor responders resulted in a similar outcome for poor as for good responders and, this together with the COSS-86 study also using all 4 active drugs, represent the best survival data published (Bacci et al. Citation1993, Fuchs et al. Citation1998). Thus, based on SSG's own experience and that of other intergroups, the SSG XIV protocol was considered best standard therapy in classical osteosarcoma. All patients received a 3-drug combination of methotrexate, doxorubicin and cisplatin both pre- and postoperatively and poor histological responders were salvaged with the addition of high-dose ifosfamide.
Patients and methods
Patients
From March 2001 to April 2005, 71 patients with high-grade extremity localized osteosarcoma from 6 centres in Sweden, 3 centres in Norway and 2 centres in Finland, were treated according to the SSG XIV protocol. Eligibility criteria were age < 40 years and no evident metastases by mandatory use of chest CT and whole body bone scan at presentation. The primary tumor was assessed by plain radiographs, technetium 99-MDP bone scan, CT scan and MRI of the entire bone involved. 8 patients were excluded due to metastatic disease (n=6), a revised diagnosis of clear cell sarcoma (n=1) or malignant fibrous histocytoma (n=1) rendering 63 patients eligible for this analysis. The diagnosis of osteosarcoma was confirmed by open biopsy in all cases. The SSG pathology panel reviewed all slides and agreed on diagnosis, subtype, and malignancy grade. The median age at diagnosis was 15 (8-39) years. 40 (64%) patients were male. Tumor localizations were femur (n=34), tibia (n=15), humerus (n=6), fibula (n=4), radius (n=2) and other (n=2).
Chemotherapy
Chemotherapy consisted of 2 cycles of paired methotrexate (MTX), 12 g/m2, doxorubicin (ADM), 75 mg/m2 and cisplatin (CDP), 90 mg/m2 pre-operatively and 3 cycles post-operatively (). Poor histological responders continued to receive 3 courses of ifosfamide (IFO), 10 g/m2 as an add on treatment.
Figure 1. Treatment outline in SSG XIV protocol.
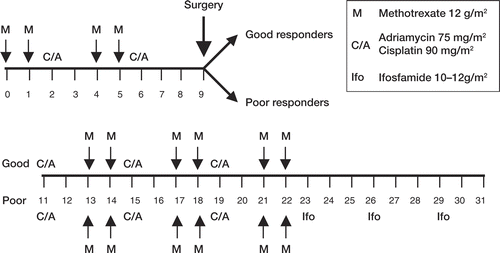
With good bone marrow tolerance the protocol allowed escalation of the IFO dose with 20% for the next course. MTX was administered in a 4-hour infusion with 11 doses of leucovorin (folinic acid) as rescue (8 mg/m2) every 6th hour, beginning 24 hours after starting the MTX infusion (Ferrari et al. Citation2005). CDP was delivered as a 48-hour continuous infusion intravenously and was followed by ADM given as a 4-hour continuous infusion. IFO, in combination with an equal amount of mesna, was delivered as continuous infusion at a dose of 2 g/m2/day for 5 consecutive days. Postoperative chemotherapy was scheduled to begin 7 days after surgery. All drugs were given as single agents per course.
Complete blood counts and renal and liver function were monitored before each chemotherapy administration. No dose reduction was allowed, and if the absolute granulocyte count was equal to or less than 1000/µL (500 for MTX cycles), and/or the platelet count was equal to or less than 100.000/µL (60.000 for MTX cycles), chemotherapy was delayed until recovery. After each cycle, the blood count was monitored twice weekly starting on day 7 from the beginning of the chemotherapy infusion. G-CSF support was given according to the ASCO guidelines (1994).
Surgery and histological response assessment
The type of surgery was chosen depending on the size and the location of the tumor, neurovascular involvement and skeletal maturity. For limb salvage surgery, it was mandatory that the preoperative staging showed the possibility of achieving adequate surgical margins. In resected patients, the type of reconstruction was chosen according to tumor location and extension, patient age and preferences. After surgery, the surgical margins were assessed according to Enneking et al. (Citation1980) as radical, wide, marginal or intralesional. The grading of histological response analysis was done with a two-grade scale. Good response was defined as < 10% of the examined tumor area revealing unquestionably viable tumor and no single area of unaffected viable tumor exceeding 2.5 mm in largest diameter. Poor response was defined as any of the two above criteria unfulfilled. Unaffected was defined as a morphologic appearance closely resembling that of the pretreatment biopsy and unquestionably viable tumor as various degrees of response, including decreased cellularity, and signs of maturation with bone and cartilage matrix production, but with remaining clearly viable tumor cells. The initial pathologic evaluation at each institution determined the postoperative chemotherapy. Sections from each patient were reviewed for histological response by the SSG reference pathology panel.
Response criteria and statistical analyses
Projected metastasis-free survival was calculated from the date of diagnosis until the date of distant metastases or last follow-up. Event-free survival was calculated from the date of diagnosis until the date of distant metastases, local recurrence, treatment-related death or last follow-up. Sarcoma-related survival was calculated from the date of diagnosis until death from osteosarcoma, treatment-related causes or last follow-up. For statistical analyses SPSS for Windows (Release 15.0, SPSS Inc., Chicago, IL, USA) was used. The Kaplan Meier method was used for survival analysis and curves were compared by the log-rank test.
Results
Compliance
2 patients did not receive neoadjuvant chemotherapy. 1 was in need of prompt surgical treatment and the other had a preoperative diagnosis of chondrosarcoma, later revised to high-grade osteosarcoma. 1 patient was postoperatively treated according to a different chemotherapy protocol as a decision of the treating physician. 3 treatment-related deaths were recorded, 1 shortly after completion of preoperative chemotherapy, 1 during postoperative therapy and 1 during completion of chemotherapy. The median time from start of chemotherapy to surgery was 80 days which represents a median delay of 17 days to protocol. The median treatment duration was 219 days in good responders and 275 days in poor responders, representing median delays of 65 days for good responders and 67 days for poor responders. Data on chemotherapy compliance was available for 57 patients. 88% of the patients received all 5 courses of CDP/ADM of whom 75% were given with no dose reduction. 2 patients received only 2 courses, one due to early death and the other due to change in postoperative therapy. 5 patients received only 4 courses due to toxicity. 47 of the 63 patients received all 10 courses of MTX. No dose reduction was observed for MTX. Regarding IFO courses, 23 of the 26 patients received all 3 courses with a dose reduction for 12% of the patients. Except the 3 treatment-related deaths and 1 case of change in postoperative chemotherapy, 91% of the patients received 4 or more courses of CDP/ADM and 8 or more courses of MTX.
Toxicity
Detailed toxicity data was available for 48 (75%) of the patients. 36% of CDP/ADM courses and 26% of the IFO courses were followed by grade IV leucopenia. 21% of the patients did not experience any episode of grad IV leucopenia. Regarding platelet toxicity, 28% of the CDP/ADM courses were followed by grade IV thrombocytopenia and 31% by platelet transfusion. For the IFO courses, only 1 of 81 recorded courses were followed by grade IV toxicity and no platelet transfusion was given. 9 patients (14%) experienced a mild to moderate renal impairment mainly after MTX administration. This caused changes in planned schedule for 1 patient. None of these patients required dialysis. We recorded 3 treatment related deaths all related to acute chemotherapy toxicity. 1 patient (boy, 8 years) developed neutropenic fever and septic shock after completion of preoperative therapy. 1 patient (good responder) developed typhlitis and septicemia after the first postoperative cycle of chemotherapy and 1 patient (poor responder) developed septic shock in relation to the last cycle of postoperative chemotherapy. In addition, 2 cases of life-threatening toxicity were observed. A male patient (age 21) stayed 10 days at the intensity care unit after completion of preoperative chemotherapy due to development of severe enterocolitis in combination with grade IV neutropenia and thrombocytopenia. The second case was a girl (age 15) that developed grade IV cardiotoxicity before the third course of IFO and was in need of prompt medical treatment. The cardiac function is not fully normalized more than 3 years from end of therapy and the patient is in need of permanent medical treatment.
Surgery and local control
53 (84%) of the patients were treated with limb salvage surgery (n=49) or rotationplasty (n=4). 8 patients (13%) were amputated and 2 patients were not operated upon due to early progression or toxic death. 63% of the operated patients obtained wide or radical margins. 2 patients, both poor responders, developed local recurrence at 18 and 29 months from diagnosis. One had a primary wide and the other a marginal margin at surgery. 1 of these patients later developed distant metastases and died 30 months from diagnosis. The other is alive in second complete remission 64 months from diagnosis and 35 months from local recurrence. The 5-year projected local-recurrence free survival is 96% (95% CI, 91–100%).
Histological response and postoperative chemotherapy
Histological response was documented in 59 patients. 2 patients were treated up-front with amputation. 1 patient died a toxic death before surgery and 1 patient was never operated upon due to early dissemination of disease. According to the assessment by the review pathologists, 27 (46%) obtained a good response. The review process changed the response assessment in 4 patients, in 3 from good to poor response and in 1 from poor to good response. Regarding postoperative chemotherapy for the 59 patients that received preoperative chemotherapy, information on postoperative chemotherapy is lost for1 patient (poor responder). For the remaining 58, 1 good responder received postoperative chemotherapy according to another protocol as decided by the treating physician, and in 1 patient with poor response alpha-interferon was exchanged for IFO as salvage therapy. Thus, 4 patients with a definite evaluation of poor response did not receive IFO. 30 patients received unchanged MTX-CDP-DOX postoperatively and 26 patients received salvage therapy with addition of IFO according to protocol. In 5 of 26 patients the IFO dose was escalated 20% for the second and/or third cycle. No patient had a second escalation of the IFO dose.
Survival and postrelapse outcome
With a median follow-up of 64 (16–85) months for survivors, 49 patients are alive and 46 patients are in complete remission (). The projected sarcoma-related survival at 5 years is 77% (95% CI 65–89%). 40 patients (64%) are alive in first complete remission. The projected metastasis-free survival at 5 years is 69% (95% CI 57–81%). The projected event-free survival at 5 years is 65% (95% CI 53–77%), including 1 local recurrence and 3 treatment related deaths in addition to 18 metastatic relapses (). 5-year metastasis-free survival by response (n=59) was 89% for good responders (n=27) and 53% (n=32) for poor responders (Log Rank p= 0.004). 5-year metastasis-free survival by postoperative chemotherapy (n= 56) was 89% (n=30) for patients receiving unchanged MTX-CDP-DOX and 47% (n=26) for patients receiving addition of IFO (Log Rank p=0.001). Metastasis-free survival by sex revealed no differences with 5-year survival of 68% for men and 73% for women (Log Rank p=0.7). Of the 18 patients that developed distant metastases, 14 had lung metastases, 2 bone metastases and no information is available for 2 patients. Relapse treatment was not defined by protocol and varied between centres according to previous therapy; 14 patients were treated by second line surgery and surgery only was offered to 7 patients. Second line chemotherapy was given to 7 patients and 1 patient received interferon adjuvant to surgery. 13 patients obtained a second complete remission and 7 of those are alive, 12–65 months from relapse. All 5 patients that did not obtain a second complete remission are dead of osteosarcoma.
Figure 2. SSG XIV: Sarcoma-related (—) and metastasis-free (….) survival (n = 63).
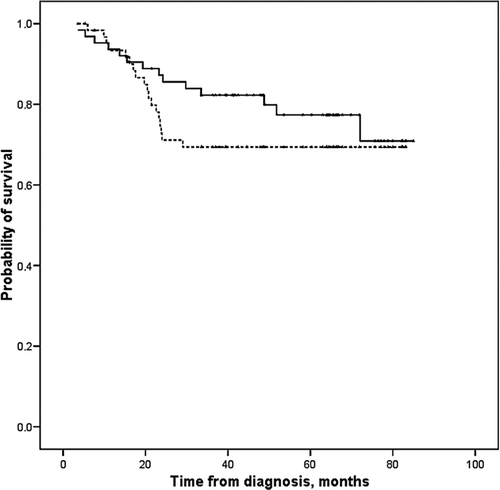
Table 1. Outcome in SSG osteosarcoma protocols
Discussion
The survival data in the present analysis compares favorably with previous SSG studies and other osteosarcoma trials reported (Saeter et al. 1991, Fuchs et al. 1998, Smeland et al. 2003, Ferrari et al. 2005, Meyers et al. 2005). The 5-year sarcoma-related survival was 77% and metastasis-free survival 69% compared to 64% and 55% in the SSG II study, 74% and 63% in the SSG VIII study and 72% and 60% in the ISG/SSG 1 study, respectively (). Metastasis-free survival probably best reflects the efficacy of the chemotherapy regimen. Interestingly the metastasis-free survival is higher in SSG XIV using a 3-drug regimen for all patients with addition of IFO only for poor responders, compared to the recent ISG/SSG 1 trial using all 4 drugs up-front for all patients. This may reflect that not only cumulative doses but also dose-intensity of individual drugs are important for the effect (Bruland and Pihl Citation1997).
The percentage of patients that received limb salvage surgery or rotationplasty was at the same level as for the ISG/SSG 1 trial (). Thus, it seems appropriate to conclude that in a multi-institutional setting more than 80% of the patients can safely receive limb-salvage surgery or rotationplasty without compromising the risk of local recurrence when compared to previous trials in which amputation rate were much higher, as in SSG II (). The low number of local recurrences in SSG XIV may also have contributed to the favorable sarcoma-related survival rate since local recurrences are often followed by distant metastases and death from disease (Bacci et al. Citation2007). In SSG XIV, 2/63 patients had a local recurrence in contrast to 8/113 in SSG VIII. Another issue that may influence overall results is stage migration due to more consistent use of high-resolution CT chest scans. Patients with metastases at presentation have a worse prognosis and for the 6 patients not included in this analysis due to metastatic disease, but treated according to the protocol, 4 are dead of disease. Postrelapse therapy was not defined by the protocol, but the data reveals a substantial practise of metastasectomy. 13 of 18 relapsed patients obtained a second complete remission and of these 7 are alive. This is comparable to published results from other intergroups and SSG's own experience (Saeter et al. Citation1995, Ferrari et al. Citation2003, Kempf-Bielack et al. Citation2005). In SSG VIII, three quarters of the relapsed patients received second line surgery and the overall survival at 5 years from relapse was 21%, all in the group that obtained a second complete remission (Smeland et al. Citation2003)
Table 2. Surgery and local recurrence (LR) rate in SSG osteosarcoma protocols
The preoperative chemotherapy in this protocol was similar to SSG VIII. However, the percentage of good responders differs from 46% in SSG XIV to 58% in SSG VIII. This probably reflects the modification of the assessment criteria that was defined for this protocol. The median delay to surgery was similar in SSG XIV and SSG VIII with 17 and 20 days, respectively. Hence a difference in preoperative drug intensity cannot explain the difference in histological response. The difference in outcome between good and poor histological responders in the current report was 42%. Thus, the strategy to add IFO failed to improve prognosis for poor responders to the level of good responders. This emphasizes the importance to address the salvage question in a randomized trial, such as in the ongoing EURAMOS-1. The result from SSG XIV may reflect an underlying chemoresistance, including to IFO, but may also be influenced by the late introduction of IFO in this protocol. The reason for the chosen design was not to interfere with the dose intensity of the other drugs.
SSG has previously reported a sex dependent outcome in both the SSG II and the SSG VIII trials. In SSG XIV, however, there was no such difference in outcome between men and women. This is in accordance with the data in ISG/SSG 1 and published results from other groups. It probably reflects that data from the previous protocols are not due to a major underlying difference in tumor aggressiveness and/or chemosensitivity by sex (Saeter et al. Citation1991, Bielack et al. Citation2002, Smeland et al. Citation2003, Ferrari et al. Citation2005).
3 treatment related deaths occurred. In general, the reported percentage of treatment related death in SSG osteosarcoma trials is 1–3%; 3/113 in SSG VIII and 1/57 in ISG/SSG. The relatively high number of treatment related deaths in this trial, and all due to acute chemotherapy induced toxicity, emphasizes the importance of organizing a safe osteosarcoma care. One may speculate that the more frequent use of growth factor support does not reduce but rather shift the pattern of serious life-threatening toxicity. However, by Aug 2008, SSG has recruited 57 patients to the EURAMOS-1 study with a slightly more intensive chemotherapy regimen than SSG XIV and a similar use of growth factor support. So far no toxic death has been reported among SSG patients.
In conclusion, the outcome in the SSG XIV protocol compares favorably with both previous SSG and other reported osteosarcoma trials. However, the use of high-dose IFO as an add on treatment did not improve the outcome for poor responders to a similar level as for good responders. The salvage question has to be addressed in a randomized trial in order to demonstrate whether the addition of IFO to the 3-drug regimen of MTX, ADM, and CDP improves the prognosis or not. Current data shows that more than 80% of the patients in a multi-institutional setting can expect to be operated upon with limb salvage surgery with no increased risk of local recurrence.
Acknowledgement
The authors thank Eva-Mari Olofsson at the SSG study centre secretariat for her active participation in the project and collegues at all recruiting centers. This work was supported by the Swedish Cancer Foundation and the Nordic Cancer Union.
The European postmarketing adult Osteosarcoma Surveillance Study: characteristics of patients
A preliminary report
Birgitta von Schéele1, Rebecca D. Martin2, Alicia W. Gilsenan2, Jeanette Ceberg3, Elizabeth B. Andrews2, Daniel Masica4, Thor Alvegård3
1 RTI Health Solutions, Williams House, Manchester Science Park, the United Kingdom
2 RTI Health Solutions, Research Triangle Park, NC, United States
3 Scandinavian Sarcoma Group, Department of Cancer Epidemiology, University Lund, Sweden
4 Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN, United States
Correspondence BvS: [email protected]
SUMMARY The Scandinavian Sarcoma Group (SSG) registry participates in a multinational postmarketing drug surveillance study evaluating potential medication exposures (including teriparatide) in a population-based series of adult osteosarcoma cases. We present preliminary data from this study.
The SSG registry systematically identifies eligible cases in collaboration with regional and national cancer registries in Sweden, Norway, Denmark, Finland, and Iceland. All cases aged ≥40 years initially diagnosed in January 2004 or later with histologically confirmed osteosarcoma or 5 other prespecified types of bone sarcomas are eligible. Data were collected from the medical records.
This review includes all information abstracted to date from patient records of 49 of 85 cases diagnosed between January 2004 and September 2008 (estimated to be all reported adult cases). All patients were Caucasian, mean age 59 (41-88 years), the majority were men. The most prevalent morphology subtypes were osteosarcoma NOS and chondroblastic osteosarcoma. Leg bones were the most frequent tumor site. Potential risk factors for osteosarcoma included prior history of cancer (27%), radiation treatment (24%), or prior injury or infection at the site of the tumor (14%). Site of prior radiation treatment and osteosarcoma tumor matched for 7/9 cases. One prior history of Paget's disease was reported. Treatment with teriparatide before diagnosis had not been reported.
Data collected in this study present population based demographic and risk-factor data and are consistent with prior research reporting a link between radiation site and tumor site, and a possible association between osteosarcoma and prior history of cancer, and prior injury or infection at the site of the tumor.
As an element of the post-approval surveillance program for teriparatide (Forsteo™), the European Agency for the Evaluation of Medicinal Products (EMEA) requested a 10-year safety surveillance study to monitor for a trend signaling a possible association between teriparatide and adult osteosarcoma. Teriparatide is a biosynthetic human parathyroid hormone (PTH [1-34]) used for treatment of osteoporosis. In clinical studies, osteoporosis treated for up to 2 years with teriparatide demonstrated significantly increased bone mineral density with a decreased incidence of fractures, compared with the placebo group in clinical studies (Neer et al. Citation2001). As a part of the routine preclinical testing program, teriparatide was administered to rats in varying doses. In rats, teriparatide caused increases in bone mass and a dose-dependent increase in the incidence of osteosarcoma (Vahle et al. Citation2002). Subsequent rat studies targeted for the detection of osteosarcoma documented a “no-effect” dose (Vahle et al. Citation2004). In a long-term study in cynomolgus monkeys (spanning 18 months of treatment plus 3 years of follow up observation), no bone tumors were detected by radiographic or histological evaluation in any monkey in the study (Vahle et al. Citation2008). Studies have shown that the rat skeleton is more sensitive to the pharmacological effects of PTH in formation of new bone and osteosarcoma than monkey or human skeletons (Miller Citation2008).
Due to the rare incidence of adult osteosarcoma and of the drug exposure (daily injections of teriparatide), traditional case control or cohort study designs were not feasible to address the potential association between osteosarcoma in humans and teriparatide exposure. A surveillance study design was chosen to monitor cases; if exposure rates exceeded the expected exposure level, a traditional study design could be implemented to measure statistical association between the rare exposure and the rare outcome.
Bone sarcomas are rare (0.2% of all tumors), and the United States (US) Surveillance, Epidemiology and End Results (SEER) program reports that osteosarcoma accounts for one third of all sarcomas of the bone (Dorfman and Czerniak Citation1995). The incidence of osteosarcoma in patients aged 40 years and older is estimated to be approximately 3 per million per year in Sweden (CitationSocialstyrelsen 2007, Stark et al. Citation1990). The incidence of osteosarcoma has a bimodal distribution,with the first peak between ages 10 and 25 years and the second peak starts at 50 and rises up to 80 years of age (Dorfman and Czerniak Citation1995, CitationSocialstyrelsen 2007, Stark et al. Citation1990, Unni and Dahlin Citation1996). Most data reveal a higher incidence of osteosarcoma among males, with a male-to-female ratio ranging from 1.3:1 to 1.6:1 (Stark et al. Citation1990, Dorfman and Czerniak Citation1995, Unni and Dahlin Citation1996).
Little is known about the etiology of osteosarcoma among adults (Unni and Dahlin Citation1996, International Agency for Research on Cancer [IARC] Citation2002). Potential risk factors for osteosarcoma include family history of osteosarcoma or retinoblastoma, Li-Fraumeni syndrome, or a prior history of radiation treatment, Paget's disease, injury or infection at the tumor site or metallic implant at tumor site (IARC Citation2002, Unni and Dahlin Citation1996). Among adults, Paget's disease and prior radiation treatment are more common among those aged over 40 years than among younger patients (Unni and Dahlin Citation1996). The male-to-female ratio changes to a predominance of females among those with prior exposure to radiation. Radiation therapy accounts for 5 % of all osteosarcomas (Unni and Dahlin Citation1996) and 8% of the osteosarcoma cases aged of 40 years and above (Grimer et al. Citation2003). The postradiation interval to osteosarcoma diagnosis among the Mayo cohort reported on by Unni and Dahlin (Citation1996) ranged from 1 to 55 years with a median interval of 5 to 9 years. Nearly half of osteosarcomas occur in the knee region (Unni and Dahlin Citation1996). In the Mayo cohort (Unni and Dahlin Citation1996), among those aged 40 years and older, half of the cases occurred in long bones. Atypical sites are more common among older patients and include pelvis, sternum, clavicle, scapula, and spine, in descending order of frequency. Although nonclassical osteosarcoma represents 40% of the high-grade osteosarcoma population (Smeland et al. Citation2004), few of the osteosarcomas are extraskeletal osteosarcomas (Unni and Dahlin Citation1996). Stark and colleagues (Citation1990) reported that 6% of their Swedish cohort were diagnosed with soft tissue osteosarcomas. The Mayo cohort also reported that atypical sites were more common among those exposed to radiation therapy, sites such as clavicle, scapula, ribs, innominate bone, and sacrum being more common (Unni and Dahlin Citation1996).
This 10-year case-series surveillance study is a multinational study (comprised of the US and a European component) that includes data from 5 Nordic countries. The European Osteosarcoma Surveillance study identifies cases through the Scandinavian Sarcoma Group (SSG) registry—primarily reported to them from the population-based regional and national cancer registries in Sweden, Norway, Denmark, Finland, and Iceland—and collects data through review of medical records. Primarily, the study aims to identify newly diagnosed cases of osteosarcoma in men and women aged 40 years and older and to identify incident cases of adult osteosarcoma with a history of treatment with teriparatide, if any occur. The study also aims to systematically collect, for descriptive epidemiologic purposes, additional patient information including demographics, other drug exposures, relevant risk factors, and comorbid conditions in this large series of adults with osteosarcoma.
Nordic countries were selected for participation in the European Osteosarcoma Surveillance study because of their high incidence of osteoporosis and, therefore, the potential for higher teriparatide exposure rates than other European countries. These countries also have high quality, population-based, national cancer registries with mandatory reporting of new tumor cases. In addition, the well-established SSG registry has the capability to coordinate the identification and data collection of cases.
The Osteosarcoma Surveillance Study is being conducted by RTI International (RTI), acting as the Coordinating Epidemiology Unit, and is sponsored by Eli Lilly and Company (a pharmaceutical company) with advice from the Osteosarcoma Surveillance Study Advisory Board, composed of members independent of RTI and Lilly.
Objective
This paper provides a descriptive review of data from the ongoing surveillance study to characterize the environmental and treatment exposures in adult osteosarcoma patients according to tumor type. This review also examines demographic information, morphologic distribution of tumors by anatomical site, and the distribution of potential risk factors among adult osteosarcoma patients from the ongoing European Osteosarcoma Surveillance Study in the 5 Nordic countries.
Methods
Design
This case-series surveillance study collects information on demographics, cancer descriptors, brief medical history, and environmental and drug exposures in adult patients diagnosed with osteosarcoma and other prespecified tumors where site equals bone (i.e “other bone sarcomas”).
The definition of a case is histologically confirmed disease based on International Classification of Diseases for Oncology, Third Edition, (ICD-O-3) codes. All types of osteosarcoma are eligible (ICD-O-3 codes 9180, 9181, 9182, 9183, 9184, 9185, 9186, 9187, 9192, 9193, 9194, 9195). To ensure a broad-based review and to capture potentially misclassified cases of true osteosarcoma, data are also collected from patients diagnosed with five other prespecified tumors—sarcoma NOS (ICD-O-3 code 8800), spindle cell sarcoma (8801), fibrosarcoma NOS (8810), malignant fibrous histiocytoma (8830), and dedifferentiated chondrosarcoma (9243)—where the primary site is bone. Descriptive analyses characterize the demographic profile, tumor cell morphology, and topography among all reported cases and quantify the prevalence of potential risk factors among cases.
Setting
The SSG registry acts as the European coordinator to identify and collect data from population-based regional and national cancer registries in Sweden, Norway, Denmark, Finland, and Iceland. These 5 Nordic countries comprise a total population of approximately 25 million. Cancer has been a reportable disease for 50 years in Scandinavia, and the national cancer registries have a long history of producing high quality cancer surveillance data.
Data collection
Study cases are identified by the SSG registry. Once an incident case of osteosarcoma is diagnosed in the Nordic countries, the treating physician routinely reports it directly by case report to the regional or national cancer registry and also to the SSG registry. Upon notification of an eligible case, the SSG registry contacts the local investigator to obtain consent from the treating physician to contact the patient and then to obtain patient consent (when applicable). The local investigator abstracts data from the patient's medical record and returns a completed data collection form to the SSG. Patient identifiers are removed, and a limited data form without any patient identifiers is sent to RTI for data entry and analysis. A summary of the study progress in Europe and the US is regularly reported to the EMEA and the US Food and Drug Administration.
In parallel with the US study, the European study collects data on the following dimensions: personal cancer information; demographics including race, age, country of residence and vital status; a brief medical history including cancer, osteoporosis, Paget's disease, bone fracture, infection at tumor site, and history of medication use and treatments such as use of osteoporosis medications, and radiation and chemotherapy treatment; family medical history of osteosarcoma, selected cancers, and Paget's disease; lifestyle habits such as smoking and alcohol use; and occupational and environmental exposures.
Ethics committee
The Osteosarcoma Surveillance Study has been approved by the RTI Institutional Review Board (IRB) under a Federalwide Assurance in the US and applicable ethics committees' approvals in each Nordic country. Data collection was initiated on cases diagnosed January 1, 2004, and is currently planned to include incident cases diagnosed until December 31, 2013.
Analysis
Descriptive analyses were conducted to summarize the main outcomes including demographic profile (age and age range, gender, race, and ethnicity), tumor topography and morphology distribution, prevalence of potential risk factors (lifestyle exposures, treatment, injury, and infection exposures, environmental exposures, and personal and family health history), and matching of prior radiation treatment site and tumor site.
Results
Between January 1, 2004, and September 30, 2008, 85 cases (61 osteosarcoma and 24 other bone sarcomas) were identified by the SSG. Sweden reported most of the cases, as expected, due to larger population size and the use of rapid case reporting by the Swedish cancer registries. Iceland has reported no cases to date, well within statistical projections for Iceland due to the small population size of that country (Figure). At present, out of 85 identified cases, 49 abstraction forms have been completed and returned, 6 cases are pending abstraction, and 25 cases are pending patient consent. Only 5 patients or physicians (6% of reported cases) have denied consent to access the medical records. summarizes the current status of the data collection process.
Figure. Geographic distribution of identified cases of osteosarcoma and other bone sarcoma cases by country as of September 2008.
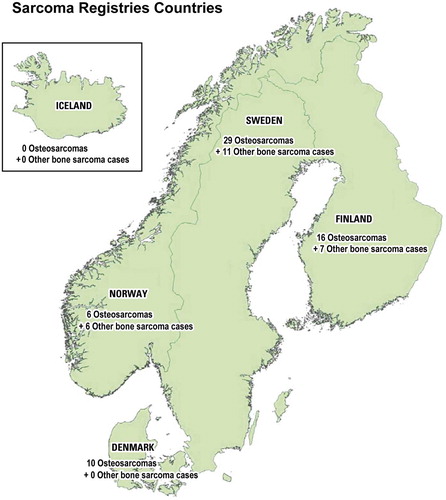
Table 1. Case status summary for osteosarcoma and other bone sarcomas by patient consent and records abstracted
Demographic profile
All cases with abstracted medical records were Caucasian, and the majority were men (59% of the osteosarcomas and 75% of the other bone sarcomas). The mean age at diagnosis of osteosarcoma was 59 (41–88) years. The other bone sarcoma cases had a mean age of 60 (42–86) years. Nearly half of the cases were deceased (21/49) when reported to the SSG registry.
Tumor morphology and topography
Of the 49 abstracted cases, 31 patients were diagnosed with osteosarcoma NOS, 5 patients with chondroblastic osteosarcoma, and 1 with parosteal osteosarcoma (). In addition, data were collected on cases with the other bone sarcomas (sarcoma NOS, spindle cell sarcoma, fibrosarcoma NOS, malignant fibrous histiocytoma and dedifferentiated chondrosarcoma). Nearly one quarter (12/49) of all the abstracted cases were other bone sarcomas, with spindle cell sarcoma and malignant fibrous histiocytoma being most common.
Table 2. Tumor morphology by ICD-O-3 Code and tumor topography
The primary tumor site demonstrated great variability between cases, but was most prevalent in the lower part of the body, with more than half of the cases occurring in the legs and pelvic region.
Personal and family medical history
One fourth of the cases (10/37) had a history of any other cancer prior to the osteosarcoma diagnosis. Prior to the diagnosis, 5 cases had a history of some kind of injury or infection at the site of the osteosarcoma (). Among the osteosarcoma patients, 1 case had a family history of breast cancer and 1 case had a family history of leukemia. Among cases of the other bone sarcomas, 1 case had a family history of breast cancer. None of the cases had any family history of osteosarcoma, retinoblastoma, brain cancer, or Paget's disease.
Table 3. Selected medical history among osteosarcoma cases and cases with other bone sarcomas
Drug exposure
History of osteoporosis and medication use was recorded, primarily to detect use of teriparatide. There were no reports of teriparatide use in any of the cases abstracted. Overall, the cases had a history of no or little use of medications related to osteoporosis before diagnosis of the tumor. Although 3 cases reported a history of osteoporosis, only one of these cases was actively treated with calcium and alendronate. Four out of a total of 18 women were treated with estrogens. Four of 49 cases have been continuously treated for at least 1 month with corticosteroids.
Treatment exposure
9 of the 37 osteosarcoma cases and 1 case of the 12 other bone sarcomas had a history of radiation treatment before the osteosarcoma diagnosis. Six of the osteosarcoma cases and none of the other bone sarcomas were treated with chemotherapy. None of the cases received any radioactive iodine treatment.
displays the individual characteristics of the cases that had been exposed to radiation. The site of prior radiation treatment and site of tumor matched for 7/9 osteosarcoma cases. Among the site-matched cases, the mean interval between radiation treatment and tumor diagnosis was 20 (7–36) years.
Table 4. Patients with history of radiation treatment (n=10), by tumor type
Almost half of the cases were current or former smokers and/or consumed alcohol. Eight osteosarcoma cases (n=37) were current cigarette smoker and 11 additional cases had stopped smoking. 2 osteosarcoma cases (n=37) and 1 case with other bone sarcoma (n=12) had been exposed to petrochemicals in their occupation. 1 case had been exposed to pesticides, and no case had been exposed to nuclear power or nuclear waste.
Discussion and conclusion
Data collected in this ongoing surveillance study from a population-based case series provides information regarding characteristics of adult patients with osteosarcoma and other tumors where the primary site is bone. In addition to demographic information, the study examines morphology distribution by site and the distribution of potential risk factors among adult osteosarcoma patients. While the numbers are expected to be small, they do present a population-based view of these rare tumors in adults in the Nordic countries.
The average age of this cohort is younger than others in the literature, especially among the osteosarcoma patients, where 43% were aged 50 to 59 years. The majority of the cases were reported in long bones, but approximately a third were reported in the pelvic and craniofacial bones. Among the osteosarcoma cases, a positive history was reported for bone fractures, Paget's disease of bone, joint replacement, and infection or trauma at the site of the tumor prior to diagnosis, as well as a history of cancer resulting in prior radiation therapy. The results are consistent with those of Unni and Dahlin (Citation1996) and Grimer and colleagues (Citation2003), who reported an association between radiation site and tumor site, and an possible association between osteosarcoma and prior history of cancer, Paget's disease, and possibly of infection or injury at the site. The study has yet not identified any case with a history (or family history of) retinoblastoma or Li-Fraumeni syndrome, which are often referred to as risk factors for osteosarcoma.
Participating investigators
Very little medication exposure was reported among this cohort. There were no reports of teriparatide exposure. This 10-year population-based surveillance study of adult osteosarcoma patients will continue to add to our body of knowledge concerning the epidemiology of adult osteosarcoma.
Treatment of localized Ewing sarcoma family of tumors
The Scandinavian Sarcoma Group experience
Sigbjørn Smeland1, Thomas Wiebe2, Odd R Monge3, Otte Brosjö4, Tom BÖhling5, Lars Hjorth2, Kirsten Sundby Hall1, åke Jakobson6 and Thor A Alvegaard7
Correspondence: [email protected]
1 Division of Cancer Medicine and Radiotherapy, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway
2 Department of Pediatric Oncology, Lund University Hospital, Lund, Sweden
3Department of Oncology, Haukeland University Hospital, Bergen, Norway
4 Department of Orthopaedics, Karolinska Hospital, Stockholm, Sweden
5 Department of Pathology, HUSLAB and University of Helsinki, Helsinki, Finland
6 Pediatric Oncology Unit, Astrid Lindgren Children's Hospital, Stockholm, Sweden
7 Department of Cancer Epidemiology, Lund University Hospital, Lund, Sweden
The Ewing sarcoma family of tumors (ESFT) consists of Ewing sarcoma, peripheral primitive neuroectodermal tumor (PNET) and malignant small cell tumor of the thoracopulmonary region (Askin tumor) (Khoury, Citation2008). SSG has conducted two trials in ESFT, SSG IV, SSG IX and in cooperation with the Italian Sarcoma Group ISG/SSG III (localized disease) and IV (metastatic disease).
SSG IV
The SSG IV protocol for Ewing's sarcoma of bone, eligible for patients with localized and metastatic disease, recruited 52 patients in the period 1984 to 1990. Patients received five blocks of 12 week cycles of vincristine, methotrexate, doxorubicin, cyclofosfamide, bleomycin and dactinomycin. After two induction cycles, local treatment was performed (week 24), surgery and/or radiotherapy as daily fractions of 2 Gy to a total dose of 40 Gy or 60 Gy. Sixty Gy was delivered to patients receiving radiotherapy alone or after incomplete surgery (Alvegård et al. Citation1989, Nilbert et al. Citation1998). Local recurrence developed in 10 patients. Of the 47 patients with localized disease at presentation, 27 later developed metastases. With 10 year median follow-up time, the metastasis-free and sarcoma-related survival at 5 years was 43% and 46%, respectively (Nilbert et al. Citation1998).
SSG IX
The high local recurrence rate in SSG IV (19%) compared to other studies (Jürgens et al. Citation1988, Burgert et al. Citation1990) lead to a changed strategy for local treatment in the following SSG IX protocol. That included earlier timing (week 9) and introduction of hyperfractionated and accelerated radiotherapy, 1.5 Gy twice daily. Based on the CESS 86 study, cyclofosfamide was replaced by ifosfamide (Jürgens et al. Citation1988) and cisplatin was introduced based on reported effects (Castello et al. Citation1988, Tursz et al. Citation1989). Thus, the chemotherapy regimen in SSG IX consisted of four cycles of a VAI (vincristine, adriamycin, ifosfamide) and PAI (cisplatin, adriamycin, ifosfamide) in combination. The treatment duration was scheduled to 35 weeks. SSG IX was open for all patients with Ewing's sarcoma, also extraosseous tumors. The aims of the study were to improve the sarcoma-related survival and local control rate at 5 years to 70% and 90%, respectively. In the period 1990–1999, 88 patients were recruited. The sarcoma-related and metastasis-free survival rates at 5 years for patients with localized disease were 70% and 58%, respectively. 9 patients (10%) developed local recurrence. Thus, the aims of the study were achieved (, Figure) (Elomaa et al. Citation2000). The improved local control probably reflects a combined effect of more and better surgery, earlier timing of local treatment and the use of hyper fractionated/accelerated radiotherapy. Multivariate analyses of prognostic factors for outcome revealed weight loss, presence of metastasis at presentation, inadequate surgical margins and poor histologic response to chemotherapy as independent adverse factors.
Figure. SSG's EFST trials. Sarcoma-related survival in localized disease.
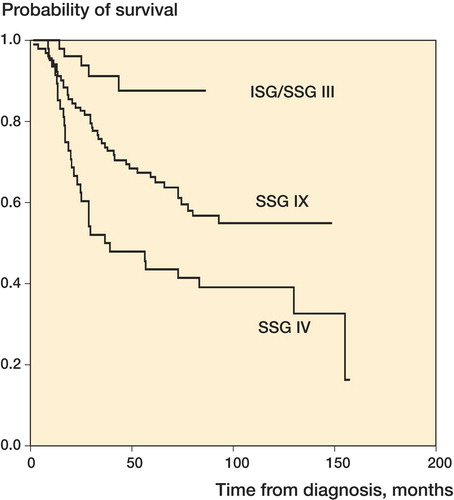
Table 1. Outcome in SSG's ESFT trials for localized disease
The Italian/Scandinavian ISG/SSG III protocol
The chemotherapy regimen in the ISG/SSG III protocol for non-metastatic disease was based upon the Rizzoli Institute REN-1 protocol utilizing a 4-drug regimen containing vincristine, doxorubicin, dactinomycin and cyclofosfamide (Bacci et al. Citation1998) and a later study demonstrating a benefit of adding etoposide, ifosfamide and dactinomycin to the VACA regimen (Bacci et al. Citation2002). Response to induction chemotherapy is a strong prognostic factor in ESFT (Picci et al. Citation1993, Picci et al. Citation1997, Elomaa et al. Citation2000, Paulussen et al. Citation2001). Accordingly, in the protocol, patients with poor response (histological or radiological) were salvaged by high-dose chemotherapy (HDCT), busulfan 4mg/kg x 4 days and melfalan 140 mg/m2, with autologous stem cell support in an attempt to improve their prognosis.
The ISG/SSG III study was closed by Dec 2006 and 296 Scandinavian and Italian patients were enrolled. Preliminary data was reported at the 2007 ASCO meeting (Ferrari et al. Citation2007). 52% of the patients were poor responders and 86% of those received HDCT. With a median follow-up of 37 months the 5-year overall and event-free survival were 74% and 66%, respectively and importantly the event-free survival for poor responders who received HDCT (68%) and for the good responders (71%) were in the same range. No toxic deaths were recorded. The key conclusion was that HDCT was feasible and in a large non-randomized study seems to improve outcome for poor responders. An updated analysis of the Scandinavian patients (n=56), at a median follow-up time of 40 months, revealed a 5 year sarcoma-related survival of 88%, event-free survival of 87% and metastasis-free survival of 91% (). The sarcoma-related survival for good responders and poor responders receiving HDCT was 86% and 88%, respectively. Only one patient experienced local recurrence (after radiotherapy alone as local therapy). 23 patients (44%) received surgery, 20 (39%) combined therapy and 9 patients (17%) radiotherapy alone (). Two treatment-related deaths were observed among the Scandinavian patients, one patient died of septic emboli 17 months from diagnosis and one patient died of secondary leukemia 29 months from diagnosis.
Table 2. Local treatment and local control rate in SSG's ESFT trials for localized disease
The survival data for the Scandinavian patients in ISG/SSG III represents a substantial improvement compared to SSG IV and SSG IX () and also compares favorably with published results from other intergroups (Oberlin et al. Citation2001, Paulussen et al. Citation2001, Grier et al. Citation2003). The outcome for poor responders was much better than reported from previous Scandinavian and Italian studies (Elomaa et al. Citation2000, Bacci et al. Citation2002). Thus, the data suggest a substantial benefit of adding HDCT to the group of poor responders in a setting in which about 50% of the patients were assessed as poor responders. Another factor that may have contributed to the good result in ISG/SSG III was the improved local therapy with a more combined approach of surgery and radiotherapy (). Only one patient has experienced local recurrence in ISG/SSG III in contrast to 10% in SSG IX and 19% in SSG IV. Regarding long-term effects after treatment for Ewing's sarcoma cautions should be addressed to the risk of developing secondary malignancies due to the high doses of alkylating agents and topoisomerase inhibitors, radiotherapy and in ISG/SSG III also high-dose therapy (Paulussen et al. Citation2001). Among SSG's study patients (n=176) four patients have so far developed secondary malignancies, acute myelogenous leukemia or radiation-induced osteosarcoma (one patient in ISSGSSG III, two patients in SSG IX and one in SSG IV).
Future aspects and concluding remarks
No new drugs have been included in first line treatment for ESFT the last decades and currently none are tested out in randomized trials. In the ongoing studies, strategic questions are addressed including intensity of induction therapy and HDCT or not to poor responders or patients with metastatic disease. Recently, an improved effect of more condensed (every 2-weeks) chemotherapy compared to standard every 3-week has been reported (Womer et al. Citation2008). Currently the most promising new agent is the insulin-like growth factor antibody, anti-IGFR. IGFR signaling is important in the oncogene pathway in ESFT and mechanistic data is supported by clinical results from a phase I study (Le Roith et al. 2004, Olmos et al. Citation2008). The drug is currently tested out in several phase II studies including relapsed and/or refractory ESFT and a randomized trial for ESFT patients in first relapse is underway.
To conclude, conventional poly-agent chemotherapy including the six active drugs (doxorubicin, ifosfamide, cyclofosfamide, etoposide, dactinomycin and vincristine) in combination with surgery and/or radiotherapy is highly successful in standard-risk patients and long-term survival rates of more than 70 % are reported. The results from ISG/SSG III offering salvage therapy with HDCT suggests a benefit in poor responders but is not proven in a randomized trial or in a context with more intensified induction therapy as in EuroE.W.I.N.G.99. Novel drugs targeting the oncogene pathway are currently tested out with promising early results. In addition to optimize therapy with new drugs and compare treatment strategies the international community should aim to have a common language in ESFT as in osteosarcoma established by the EURAMOS-1 protocol. This will optimize the care and better standardize relapse treatment, but also facilitate ancillary biology projects.
Giant cell tumor of bone
The Scandinavian Sarcoma Group experience
Henrik CF Bauer1, Gunnar Follerås2, Arne Kivioja3, Anders Rydholm4, Fredrik Vult von Steyern4
1Department of Orthopedics, Karolinska Hospital, Stockholm Sweden, 2the Norwegian Radium Hospital, Oslo, Norway, 3Departments of Orthopedics and Traumatology, Helsinki, Finland, 4Department of Orthopedics, University Hospital, Lund, Sweden
Correspondence: [email protected]
The method of filling bone defects with cement after curettage of giant cell tumor (GCT) was introduced simultaneously in Sweden and in the Netherlands (Persson and Wouters Citation1976) ().
Figure 1. 17-year-old girl with a large destruction of the tibial condyle. Cytology proved GCT.
Although curettage and cementation of GCTs has become widely accepted there remain several controversies as regards indications and long-term complications. Does the cement – especially the heat generated when setting – kill remaining tumor cells leading to lower recurrence rates? Should a fracture through the tumor lead to resection and endoprosthetic replacement instead of curettage and cementation? What are the results for treatment of recurrences after curettage and cementation? GCTs are almost always located juxtaarticularly in the epiphyseal bone causing destruction of the subchondral bone. Does the cement induce osteoarthritis due to heat or disturbed nutrition to the cartilage?
These issues have been addressed in a number of studies based on all GCT patients reported to the SSG Registry (Vult von Steyern et al. Citation2006, Kivioja et al. Citation2008) or on those treated in Lund and Stockholm (DreinhÖfer Citation1995, Vult von Steyern et al. Citation2007).
294 GCT patients from 13 Scandinavian hospitals were prospectively collected in the SSG Registry 1986–2003 (Kivioja et al. Citation2008). The overall local recurrence rate was 0.22. A resection with marginal or wide margin was performed in 92 patients leading to a recurrence rate of 0.12. 200 patients underwent intralesional curettage and these had a recurrence rate of 0.27. In this group, filling with cement was associated with a 0.20 recurrence, compared to 0.56 after filling with bone graft or no filling at all.
We concluded that our recurrence rate was not as low as the best reported, i. e. around 10% from single institutions, but acceptable taking into account that 13 hospitals were involved and patients were accrued since 1986. We did not use high-speed burring during most of the time period that the operations were performed. The large difference in recurrence rates between cementation of the defect after curettage compared to bone grafting implies that the cement does indeed kill remaining tumor cells. This remains controversial since in other series even better results have been obtained without cement. We don't consider leaving the cavity without filling since we don't recognize any adverse effects of the cement. In addition, cement with contrast enables early detection of a local recurrence, as pointed out already by Persson and Wouters (Citation1976).
In our series better local control rates were obtained by marginal or wide excision as opposed to curettage. Nevertheless we consider that excision is only indicated for expendable bones like the head of the fibula. For femur, tibia and humerus, resection and endoprosthetic replacement will indeed give low recurrence rates but involves the risk of considerably higher morbidity through implant wear and loosening. Such procedures should be reserved for recurrent cases and when the joint is destroyed ().
Figure 2. There are cases where curettage and cementation is not an option:
In a further study based on the same patients, we analyzed 19 patients treated for local recurrence with a follow-up time of 4 years after the last recurrence (Vult von Steyern et al. Citation2006). 6 patients had resection but in 13 further curettage and cementation was performed, in 2 patients twice as they had another local recurrence. Hence the local recurrence rate was 0.15 after the repeat curettage. This means that if the recurrence rate is 0.15 after both primary and secondary curettage. Hence, 98% of GCT patients will have local control with 2 operations involving curettage and cementation.
Fracture at presentation may be considered an indication to opt for resection rather than curettage in GCT (). We reviewed 15 patients with a pathological fracture at presentation of the primary GCT (DreinhÖfer et al. Citation1995). In 10 cases curettage and cementation was performed leading to good or excellent function and only 2 local recurrences were seen. We concluded that fracture does not preempt curettage and cementation but sometimes the reconstruction must be reinforced by plates or pins.
Finally we reviewed 9 patients 6 to 16 years after curettage and cementation for GCT around the knee (Vult von Steyern et al. Citation2007). To assess the condition of the cartilage plain radiography, gadolinium-enhanced MRI and measurement of the cartilage oligomeric matrix protein was performed. Only 1 patient had signs of degenerative changes to the knee joint and had both intraarticular fracture and local recurrence in the medical history. All were physically active and the Lysholm knee score was excellent to good in all patients. We conclude that there is no evidence that the cement placed directly under the cartilage in GCT is harmful to the joint. There is no reason to replace cement for newer bone-filling agents, which are more expensive and not evaluated for long-term outcome and do not produce the heat which may prevent local recurrence.
Chest wall chondrosarcoma
BjÖrn Widhe and Henrik C F Bauer
Department of Molecular Medicine and Surgery, Section of Orthopedics, Karolinska Institute, Stockholm Sweden
Correspondence: [email protected]
Chondrosarcoma is the second most common primary bone malignancy after osteosarcoma. Chondrosarcoma has been reported in almost every bone in the body but the more common sites are pelvis, proximal and distal femur, proximal humerus and ribs. Approximately 10-15 % of the chondrosarcoma are located in the chest wall, which makes chondrosarcoma the most common primary malignant tumor of the chest wall (Burt et al. Citation1992).
There are limited number of studies of chondrosarcoma of the chest wall and most of them are case reports or single institutional experiences with small number of patients included (McAfee et al. Citation1985, Burt et al. Citation1992, Agrawal et al. Citation1995, Lioulias et al. Citation2003, Fong et al. Citation2004)
We have conducted a population based study on chest wall chondrosarcoma (Widhe and Bauer Citation2009. All 106 patients with chondrosarcoma of the chest wall in Sweden (1980-2002) were identified from the Scandinavian Sarcoma Group Register and the Swedish Cancer Register. The diagnoses were confirmed by a blinded review by the SSG pathology board. That study forms the basis for this presentation of chest wall chondrosarcoma in general.
Symptoms and clinical picture
The most prominent symptom of chest wall chondrosarcoma is a bony-hard, palpable mass. Previous reports have shown that two thirds to three quarters of the patients had a palpable mass, often for a long period of time before diagnosis (McAfee et al. Citation1985, Burt et al. Citation1992). The mass slightly increased in size but remained often painless. In our patient series the duration of symptoms varied from a couple of days to several years and occasionally even decades. 14% of the patients were diagnosed accidentally from a regular chest radiograph prescribed for other reasons. A minority of the patients, approximately 15%, related the onset of symptoms to a minor trauma.
Diagnosis
Plain chest wall radiographs may detect a chest wall chondrosarcoma but other radiological examinations are more accurate. CT and/or MRI are now routine procedures before treatment of chest wall chondrosarcoma. With these two radiological modalities the tumor extension within the bone and soft tissues can be visualized for preoperative planning.
Several SSG reports have demonstrated the importance and accuracy of fine-needle aspiration cytology in the diagnosis of primary bone tumors (Kreicbergs et al. Citation1996, Willen Citation1997). Fine-needle analysis and/or core biopsies can be used to confirm the chondrosarcoma diagnosis before treatment. However, the pathological diagnosis is difficult and requires specialized pathologists with knowledge and experience of sarcoma. A false benign diagnosis by the pathologist might result in a long doctor's delay, in our series more than 1 year in several patients. A benign or inconclusive cytological analysis not only might prolong doctor's delay but also result in poor surgical margins; many patients at non-specialty centers were operated with shelling out for what was believed to be a benign lesion. This might be one reason why non-specialty centers obtained poor surgical margins (Figure).
Figure. Surgical margins.
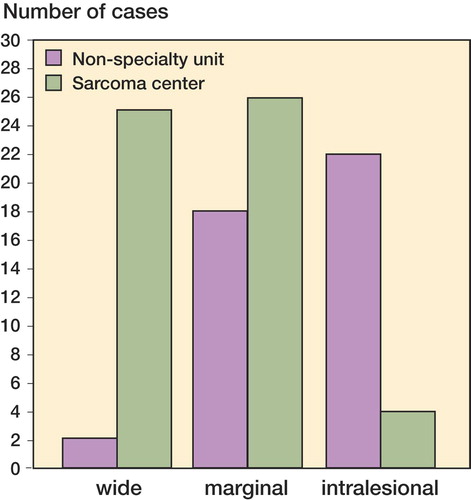
Treatment
Radiotherapy and chemotherapy are of little help in chondrosarcomas (Bjornsso et al. 1998, Bruns et al. Citation2001). Surgery, with wide margins, still remains the only effective treatment. A wide excision not only reduces the risk of local recurrences but also results in better overall survival (Burt et al. Citation1992, Fong et al. Citation2004, Widhe and Bauer Citation2009). Postoperative radiotherapy have been used after narrow surgical margins, the benefit, however, remains unclear.
In Sweden there are specialized sarcoma centers diagnosing and treating sarcoma patients. This is strictly adhered to for patients with sarcomas of long bones. However, chest wall chondrosarcoma have also been treated by thoracic surgeons and general surgeons at non-specialty centers. In our study of chondrosarcoma of the chest wall, only half of the patients were operated at a sarcoma center.
In chest wall chondrosarcoma, a wide surgical margin requires intact pleura internally, and a transverse rib resection > 2 cm from the tumor at both sides. If the tumor penetrates into the subcutaneous tissue also the overlying subcutaneous tissue and often skin has to be removed. In Sweden, better surgical margins were achieved at SSG centers (Widhe and Bauer Citation2009) (Figure).
Most series show low or no perioperative mortality. A few patients in our series were not treated with a curative intent due to high age and poor general condition. They experienced severe social distress as the tumor expanded to an enormous mass. A decision not to operate must be carefully considered given the poor outcome with nonoperative treatment.
Local recurrence
Local recurrence after chondrosarcomas may develop after a long period of time - even more than 10 years after the primary operation. Surgical margin and histological grade are two independent factors related to development of local recurrence. In our study, the local recurrence rate after a wide surgical margin was 0.04 compared to 0.76 after an intralesional operatio. The difference in surgical margin achieved between sarcoma centers and non-specialty centers explained why local recurrence rate was 57% (24/42) at non-specialty centers and 16% (9/55) at sarcoma centers. Previous studies of chest wall chondrosarcomas reported local recurrence rates of 0.3–0.5 (McAfee et al. Citation1985, Burt et al. Citation1992; Fong et al. Citation2004). Fiorenza et al. (Citation2002) found that local recurrence in chondrosarcoma in general (all locations) does not influence survival. However, in our series of chest wall chondrosarcoma local recurrence per se was a risk factor for death even without metastasis. The progression of a local recurrence in the chest wall can result in an enormous mass leading to respiratory failure and death.
Metastases
The metastasis rate in chest wall chondrosarcoma is reported to be 0.2–0.3 (McAfee et al. Citation1985, Burt et al. Citation1992). This is slightly less than in chondrosarcoma of the pelvis but more than in chondrosarcoma of the extremities. In our series, tumor size was one independent factor related to development of metastasis, others were histological grade and local recurrence. None of the patients with malignant grade 1 tumors developed metastasis, whereas 7/8 of grade 4 metastasized. Patients who developed metastases had a dismal prognosis. In selected cases surgical resections of metastases might be of benefit as a palliative treatment.
Outcome
In a previous Scandinavian study by Soderstrom et al, the reported 10 year survival rate was 0.49 for patients with chest wall chondrosarcoma (Soderstrom et al. Citation2003). In our study, surgical margin was an independent factor in predicting outcome, where patients treated with a wide surgical margin had a 10-year survival rate of 0.92, with a marginal surgical margin 0.66 and with an intralesional margin 0.47. Patients treated at a sarcoma center had a 10-year survival rate of 0.75 compared to 0.59 for patients treated at non-specialty centers. The difference in outcome between sarcoma centers and non-specialty centers can be explained by the surgical margins obtained. At sarcoma centers 4/55 operations were intralesional compared to 22/42 at non-specialty centers.
In the previous Scandinavian study of chondrosarcoma no improvement in outcome over time was seen. In fact, the survival rates were higher in the first half of the study (1970–1980) compared to the last (1980–1990) (Soderstrom et al. Citation2003). Neither could we find any improvement of survival over time.
Patients with chest wall chondrosarcoma should be referred to sarcoma centers for diagnosis and treatment. Better referral practices would improve overall survival rates in chondrosarcoma.
The Scandinavian Sarcoma Group Skeletal Metastasis Registry
Functional outcome and pain after surgery for bone metastases in the pelvis and extremities
Bjarne H Hansen1, Johnny Keller1, Minna Laitinen2, Peter Berg3, Sigmund Skjeldal4, Clement Trovik5, Johan Nilsson6, Anders Walloe7, Anders Kalén8, Rikard Wedin9
1Dept of Orthopedic Surgery, University Hospital, Aarhus, Denmark, 2Dept of Orthopedic Surgery, University Hospital, Tampere, Finland, 3Dept of Orthopedic Surgery, Sahlgren University Hospital, Gothenburg, Sweden,4Dept of Orthopedic Surgery, The Norwegian Radium Hospital, Oslo, Norway, 5Dept of Orthopedic Surgery, Haukeland University Hospital, Bergen, Norway, 6Dept of Orthopedic Surgery, Lund University Hospital, Lund, Sweden, 7Dept of Orthopedic Surgery, Ulleval Hospital, Oslo, Norway, 8Dept of Orthopedic Surgery, University Hospital, LinkÖping Sweden, 9Dept of Orthopedic Surgery, Karolinska University Hospital, Stockholm, Sweden.
Correspondence: Bjarne Hauge Hansen: [email protected]
Background Few authors have investigated function and pain after surgical treatment of patients with bone metastases. In 1999 the Scandinavian Sarcoma Group (SSG) initiated the Skeletal Metastasis Registry as a multi-centric, prospective study to provide a scientific basis for recommendations of treatment.
Patients and methods We have analyzed function and pain in 530 patients ( mean age 65 yr) operated on ( 599 operations) for non-spinal skeletal metastases at 9 SSG centres. 7 % were operated for more than 1 metastasis. Carcinoma of the breast, prostate, kidney, and lung were the dominating sites for primary tumors.
Results 25% of the patients died within 6 weeks after operation. 11 % of the patients had complications. 6 % had reoperation. In patients surviving more than 1 year the reoperation rate was 12 %. 92 % of the patients had no, light or moderate pain from metastasis at 6 weeks ( first control) and 6 months follow-up. Patients using opioids were reduced from 40 % preoperative to 30 % at 6 months after surgery. In patients with metastases in pelvis or lower extremity 79 % were walking with or without crutches, 6 weeks and 88 %, 6 months after surgery. More patients with metastases in proximal femur were mobile at 6 weeks and 6 months when treated with prosthetic replacement compared to internal fixation.
Interpretation Palliative surgery for bone metastases improves function and reduce pain. Mobility is improved by surgery in patients with metastases in the pelvis or lower extremity. Prosthetic replacement seems to do better than internal fixation for metastases in the proximal femur. We need to analyze function and pain earlier than 6 weeks postoperative to investigate the benefit of surgery in patients with short time survival.
Skeletal metastasis is a severe complication in malignant disease and often dramatically decrease quality of life by causing pain, pathologic fracture, hypercalcemia, anemia, and paraparesis. During recent years, more attention has been directed towards improved palliative care for cancer patients with skeletal metastases. Radiotherapy remains the mainstay for painful skeletal metastases (Sze et al. Citation2003), but hormone therapy, chemotherapy, and biphosphonates (Diel et al. Citation2004, Pavlakis et al. Citation2005) are also important treatment modalities. The aim of surgical treatment is to restore function while alleviating pain. Immediate improvement in pain and functional status is particular important in patients with short life expectancy. Outcome after surgery for bone metastases have been reported (Yazawa et al. Citation1990, Allan et al. Citation1995, Vena et al. Citation1999, Kunisada et al. Citation2000, Wedin et al. Citation2001, Ward et al. Citation2003) but only few authors have reported prospective data (Clohisy et al. Citation2000, Marco et al. Citation2000, Cheng Citation2003, Talbot et al. 2005). Most of the reported series have a small number of patients. SSG has since 1986 maintained a sarcoma registry and in 1999 the Skeletal Metastasis Registry was started with prospective registration of patients operated for skeletal metastases in the long bones and pelvis. In this paper we analyze function and pain in patients surgically treated in 9 SSG centres.
Patients and methods
530 patients with bone metastases, myeloma or lymphoma in the extremities or pelvis were included (). They underwent a total of 599 operations and were followed prospectively. 7% of the patients were operated for more than 1 metastasis. There were 283 (53%) women and 247 (47%) men with a median age of 65 (19–96) years at the first operation. Carcinomas of the breast, prostate, kidney and lung were the most common sites of primary tumor . 71% of the patients had more than 1 known bone metastasis at the time of the first surgical procedure and 34% had known visceral metastases. The indication for surgery was a complete pathological fracture in 75%, impending fracture in 21%, and pain in 4%. Operations concerned femur in 66%, humerus in 19%, pelvis in 8%, tibia in 3%, and other locations in 4%. 226 (43%) patients were treated with prosthetic procedures and 274 (51%) with internal fixation. The rest were treated with other surgical procedures. 129 (24%) patients had received preoperative radiation of the operated bone metastasis.
Table 1. Clinical features of the 530 patients operated for bone metastases
The patients were registrated at operation and followed at least 6 months or until death. Pain, use of analgesics, functional ability assessed with the Karnofsky score as good, moderate, or poor performance and mobility were registrated preoperatively, at 6 weeks and at 6 months. In patients treated for more than 1 metastasis, survival was assessed from the time of the first operation. No outcome data concerning function and pain are available in patients who died before the 6 weeks follow-up. In case of a fracture the preoperative data were just before the fracture. Postoperative complications and reoperations were registrated.
Results
25% of the patients died within the first 6 weeks after operation and 55% within the first 6 months. The 1-year survival rate, estimated with the Kaplan-Meier method, was 0.4.
Of the 411 patients treated for bone metastases in the pelvis or lower extremity, 61% were walking, with or without crutches, before surgery compared to 79% of the survivng patients at 6 weeks and 87% at 6 months after the operation (). 9% of the patients were confined to bed at 6 weeks and 5% at 6 months. In patients with a fracture, 75% were walking at 6 weeks and 84% at 6 months. In patients with metastases in the proximal femur 65% walked preoperatively. After treatment with internal fixation (46/189) 68% of the surviving patients walked at 6 weeks compared to 83% having prosthetic treatment (142/189). 1 patient undervent a Girdlestone procedure and had no walking ability after operation. At 6 months 80% of the patients with internal fixation compared to 90% with prosthesis were walking with or without crutches ().
Table 2. Mobility in 411 patients operated for bone metastases in pelvis or lower extremity and the surviving patients at 6 weeks and 6 months
Table 3. Mobility in 189 patients operated for bone metastases in proximal femur with prosthesis or internal fixation
25% of the patients had pain from the metastasis for less than 1 month before the operation. 10% had pain for more than 6 months. Only 13% of the patients had no pain from the metastasis before operation. 92% of the surviving patients had no, light, or moderate pain from the metastasis at 6 weeks and 6 months after operation compared to 45% preoperatively. 36% and 38% had no pain at 6 weeks and 6 months. 28% and 35% of the patients used no analgetics at 6 weeks and 6 months after operation. 40% used opioids preoperatively compared to 39% at 6 weeks and 30% at 6 months ( ).
Table 4. Pain and use of analgesics in the 530 operated patients preoperative and in the surviving patients at 6 weeks and 6 months after operation
84 patients were treated with internal fixation for metastases in humerus and 14 had prosthetic treatment. In the proximal humerus 17 patients had internal fixation and 14/31 had a prosthesis. All of these patients had no, light or moderate pain 6 weeks and 6 months after operation with no difference in treatment. Half of the patients treated with internal fixation used no or only light analgesics compared to two thirds of the patient treated with prosthesis at 6 weeks follow-up. All patients treated with prosthesis used no or light analgesics at 6 months compared to three quarters treated with internal fixation.
Preoperative 81% of the operated patients had a good or moderate Karnofsky score. At 6 weeks postoperative 77% had the same score and 85% at 6 months after operation ().
Table 5. Karnofsky performance score in the 530 operated patients, and the surviving patients at 6 weeks and 6 months
56 of the 530 operated patients (11%) had complications, mainly infections (40%) and prosthetic dislocations (36%). 33/530 (6%) had reoperation related to the first operated metastasis. In patients surviving less than 6 weeks 6% had complications and 3% were reoperated. In patients surviving more than 1 year the complication rate was 13 % and the reoperation rate 12%.
Discussion
As shown in other studies (Talbot et al. 2005) evaluation of the impact of surgical treatment on quality of life in patients with skeletal metastases is complicated. There is always loss of follow-up data making it difficult to make conclusions.
Talbot et al. (2005) used SF36 in a series of 65 patients with non-spinal bone metastases and showed no improvement in quality of life postoperatively, but showed substantial improvements in functional scores (MSTS and TESS). In another prospective study on non-spinal bone metastases by Clohisy et al. (2001) pain was the only improvement among the 8 dimensions of SF36 at 6 weeks and 12 weeks postoperatively. Bauer (Citation2005) stated that assessment of outcome after surgery for long bone or pelvic metastases should focus on failures instead of functional outcome; the surgeon can do no more than try to stabilize impending or manifest fractures. Reoperation because of failure is a severe complication for these often gravely ill patients. Mobility as well as pain treatment is important for quality of life, and operative treatment for a pathological fracture in the femur may give immediate reduction in pain, facilitate patient nursing and re-establish walking function. Functional ability in this series measured by Karnofsky performance score showed a small decrease in the group of good and moderate 6 weeks after operation, 77% compared to 81% preoperative and a small increase at 6 months to 85%. This is indicating that functional ability is maintained and not improved. This should be compared to the improvement in functional scores found by Talbot et al. (2005) at both 6 and 12 weeks by measuring MSTS and TESS.
Mobility is particularly affected when the patients have bone metastases in the pelvis or lower extremity. Most of the operations in this series involved these localizations (77%). In patients with impending or manifest fracture we found that 79 % were walking with or without crutches at 6 weeks and 87% at 6 months (). This strongly indicates that surgery improves mobility in patients with bone metastases in pelvis or lower extremity. In patients with metastases in the proximal femur 75 % where treated with prosthetic replacement and we found a tendency showing that more patients were mobile after 6 weeks and 6 months when treated by prosthesis compared to internal fixation (). CitationWedin et al. (2005 ) found that the failure rate leading to reoperation 2 years after operation with internal fixation was 0.35 compared to 0.18 after endoprosthetic reconstruction in patients with fracture in the proximal femur. Our and Wedins findings indicate that prosthetic replacement should be the choice of treatment in these patients.
Function was not specifically analyzed in patients having metastases in the proximal humerus. We found a tendency indicating that prosthetic treatment in these patients reduced the use of analgesics compared to internal fixation. This supports the findings of Frassica et al. (Citation2003). In treatment of metastases in humerus there is no absolute indications for operative treatment because most patients can be treated effectively with analgesics and irradiation. Function is not affected in the same degree as in patients having metastases in pelvis and lower extremity.
92% of the patients had no, light or moderate pain from the operated bone metastasis at 6 weeks and 6 months postoperative (). At the same time 6% and 7% had strong or severe pain compared to at least 37% of the patients that had strong or severe pain preoperatively. This support the studies of Talbot et al. (2005) and Barwood et al. (Citation2000 finding that surgery can improve pain control. We found that 39% of the patients were using opioids at 6 weeks compared to at least 40% preoperatively. Patients using opioids were reduced to 30% at 6 months, suggesting that surgery might reduce the use of strong analgesics at late follow-up (). In this study we do not know the exact number of patients treated with postoperative radiation as pain treatment. Talbot et al. (2005) was not able to show a decrease in the number of patients receiving pain medication, maybe because he stopped the follow-up 12 weeks postoperatively. The complication rate of 11% in our series is lower than reported in other series (Yazawa Citation1990, Allan et al. Citation1995, Kunisada et al. Citation2000, Wedin et al. Citation2001, Talbot et al. 2005). Despite our intention to have all reoperations reported we cannot excude missing reports. Wedin et al. (Citation1999) have shown, that the greatest risk factor for failure leading to reoperation after operative treated bone metastases is long survival. This is supported by a tendency in our study with an increase in reoperation rate in patients surviving more than 1 year after operation.
In patients with very limited life expectancy improvement from surgery must occur within short time. We have no good tools for prognostication of expected survival although different prognostic systems have been suggested (Tokuhashi et al.Citation1991, Bauer et al. Citation1995, Hansen et al. Citation2004, Nathan et al. Citation2005). In most studies of metastatic bone disease, including the present, the prognosis has been poor. In our series the 1-year survival was 0.4, which is similar to other series. (Bono et al. Citation1991, Bauer et al. Citation1995, Dürr et al. Citation1998, Bohm et al. Citation2002). One quarter of the patients died within the first 6 weeks postoperatively as also found in other studies (Clohisy at al. 2001, Talbot et al. 2005). We have no follow-up data on these patients, so we do not know if they had any benefit from surgery. Obviously we need to analyze pain and function even earlier than 6 weeks after operation to assess short-time benefit from surgery. In our series patients who died before 6 weeks had a higher frequency of fracture and higher percentage of known visceral and multiple bone metastases. They also more often had lung cancer and a higher frequency of poor Karnofsky performance score indicating that we might be able to identify this group of patients.
Our study shows that surgery for bone metastases in the pelvis and extremities can improve function and pain control in palliative care to help improving quality of life in these often gravely ill patients, which support the findings of others (Talbot et al. 1995, Barwood et al. Citation2000, Kelly et al. Citation2003).
The Scandinavian Sarcoma Group for Nurses and Physiotherapists
Charlotta Våde
Department of Oncology, The Norwegian Radium Hospital, Rikshospitalet University Hospital, Oslo, Norway
Correspondence: [email protected]
In August 2006 Reg. Nurses and Physiotherapists at The Norwegian Radiumhospital in Oslo invited health care professionals working with sarcoma patients to a presymposium to assess the need for cooperation within the Scandinavian countries. There were 46 participants attending this meeting. A working group was established to work towards SSG's next meeting. Denmark, Sweden, and Norway were all represented in this group. Hard work ended in our first official seminar in Bergen in May 2007.
The SSG for Nurses and Physiotherapists was formed at the annual SSG meeting in Bergen in May 2007 by health personnel from Scandinavia who primarily work with sarcoma patients.
A board consisting of nurses and physiotherapists from Denmark, Sweden, and Norway was elected for a period of 2 years. Our Chairman, Lotta Våde, is at The Norwegian Radiumhospital in Norway and the rest of the members are geographically divided between Sweden, Denmark, and Norway
SSG for Nurses and Physiotherapists participates in the SSGs meetings and has so far primarily worked with practical issues on how to establish the group. In the process there has been a lot of work and learning. The group is now established with it's own board, web-site, separate seminars/workshops and an independent economy, see www.ssg-nurses-physiotherapists.org.
In May 2008 the group arranged a Scandinavian day during EMSOS meeting in Warszawa with 25 participants.
In addition, each country has been working with different issues. Swedish nurses and physiotherapists have arranged national meetings once a year for health care professionals working with sarcoma patients This has been a great success. and health care professionals from several different professions have attended.
Our main goal for the future is to improve the cooperation between Scandinavian health care professionals. We think we can achieve this by:
– establishing research projects within the Scandinavian countries, with a multi disciplinary project involving nurses, physiotherapist, oncologists, orthopedists and other health care professionals;
– introducing mutual guidelines within the Scandinavian countries e.g. routines for administration of MTX and isolation routines;
– introducing mutual guidelines for physical therapy during all phases of the treatment;
– participating and arranging conferences/seminars/workshops.
It is also an important goal for us to give other professions working with sarcoma knowledge about sarcoma and the existence of our group. We look forward to continue our cooperation and develope our relationship with the SSG in the years to come.
Notes
Related Research Data
References
- Alho A, Alvegård TA, Berlin Ö, , for the Scandinavian Sarcoma Group, et al. Surgical margin in soft tissue sarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand 1989; 60(6)687–92
- Alvegård TA, Solheim Ø, Elomaa I. Ewing's sarcoma - a preliminary report from the Scandinavian Sarcoma Group. Acta Oncol 1989; 28(Suppl 2)59–64
- Alvegård TA, Solheim Ø, Åkerman M, Berg NO. Ewing's sarcoma. A preliminary report from the Scandinavian Sarcoma Group. Acta Orthop Scand 1989; 28(Suppl 2)59–64
- Alvegård TA. Management and prognosis of patients with high-grade soft tissue sarcomas. An evaluation of a Scandinavian joint care program. University of Lund, Lund 1989, (thesis)
- Alvegård TA, Sigurdsson H, Mouridsen H, et al. Adjuvant chemotherapy with Doxorubicin in high-grade soft tissue sarcoma: A randomized trial of the Scandinavian Sarcoma Group. J Clin Oncol 1989; 7: 1504–13
- Alvegård TA, Berg NO. Histopathology peer review of high-grade soft tissue sarcoma. The Scandinavian Sarcoma Group experience. J Clin Oncol 1989; 7: 1845–51
- Alvegård TA, Berg NO, Ranstam J, et al. Prognosis in high-grade soft tissue sarcomas. The Scandinavian Sarcoma Group experience in a randomized adjuvant chemotherapy trial. Acta Orthop Scand 1989; 60(5)517–21
- Alvegård TA, Berg NO, Baldetorp B, et al. Cellular DNA content and prognosis of high grade soft tissue sarcoma. The Scandinavian Sarcoma Group experience. J Clin Oncol 1990; 8: 538–47
- Alvegård TA. List of publications from the Scandinavian Sarcoma Centers. Scandinavian Sarcoma secretariat. 1994; 125: 1989–93
- Alvegård TA, Rydholm A. List of publications from the Scandinavian Sarcoma Centers. Acta Orthop Scand 2004; 75(Suppl 311)99–114
- Bacci G, Ferrari S, Longhi A, Picci P, Mercuri M, Alvegard TA, Saeter G, Conati D, Manfrini M, Lari S, Briccoli A, Forni C. High Dose Ifosfamide in Combination with High Dose Methotrexate, Adriamycin and Cisplatin in the Neoadjuvant Treatment of Extremity Osteosarcoma: Preliminary Results of an Italian Sarcoma Group/Scandinavian Sarcoma Pilot Study. J Chemoth 2002; 14(2)198–206
- Bauer HCF, Alvegård TA, Berlin Ö, Erlanson M, Kalén A, Lindholm P, Gustafson P, Smeland S, Trovik CS. The Scandinavian sarcoma register 1986-2002. Acta Orthop Scand 2004; 75(Suppl 31)8–15
- Bauer HCF, MÖller T, Alvegård TA, Rydholm A, Gustafson P, Solheim Ö, Erlanson M, Karlsson M, Monge O, Trovik C, Blomqvist C, Wiklund T, Berlin Ö, Turesson I. Monitoring referral and treatment in soft tissue sarcoma: Study based on 1851 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand 2001; 72: 150–9
- Del Prever AB, Smeland S, Tienghi A, et al. High-risk osteosarcoma (OS): Preliminary results of the ISG-SSG II protocol. ASCO meeting abstract 2005; 23(26)9002
- Elomaa I, Blomqvist C, Saeter G, et al. Three–year results of the SSG IX protocol in Ewing's sarcoma. Acta Orthop Scand 1994; 65(Suppl 262)77–8
- Elomaa I, Blomqvist C, Saeter G, et al. For the Scandinavian Sarcoma Group. Five-year results of the SSG IX protocol in Ewing's sarcoma. Med Ped Oncol 1996; 27(4)225
- Elomaa I, Blomqvist C, Saeter G, et al. Chemotherapy in Ewing's sarcoma-the Scandinavian Sarcoma Group experience. Acta Orthop Scand 1999; 285(70)69–73
- Elomaa I, Blomqvist CP, Saeter G, et al. Five-year results in Ewing's sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Eur J Cancer, 2000; 36: 875–80
- Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand 1994; 65(Suppl 259)131
- Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 2005; 34: 8845–52
- Ferrari S, Alvegard T, et al. Non-metastatic Ewing´s family tumors: High dose chemotherapy with stem cell rescue in poor responder patients. Preliminary results of the Italian/Scandinavian ISG/SSG III protocol. J Clin Oncol 2007, In press. ASCO Proceedings 2007; 25: 548 (abstract 10014)
- Gustafson P., Åkerman M., Alvegård TA., Coindre JM., Fletcher CDM., Rydholm A., Willén H. Prognostic information in soft tissue sarcoma using tumour size, vascular invasion, and microscopic tumour necrosis - the SIN-system. Eur J Cancer 2003; 39: 1568–76
- Hansen BH, Wedin R, Keller J, Laitinen M, Berg P, Skjeldal S, Trovik C, Nilsson J, WallØe A, Kalén A. The Scandinavian sarcoma group skeletal metastasis register. Survival after surgery for bone metastases in the pelvis and extremities. Acta Ortop Scand 2004; 75(Suppl. 31)11–15
- Nilbert M, Saeter G, Elomaa I, Monge O, Wiebe T, Alvegård T. Ewing's sarcoma treatment in Scandinavia 1984-1990. Acta Oncol 1998; 37(4)375–78
- Rydholm A. Management of patients with soft-tissue tumors. Strategy developed at a regional oncology center. Acta Orthop Scand 1983, Suppl 203: 54
- Rydholm A. Centralization of soft tissue sarcoma. The southern Sweden experience. Acta Orthop Scand 1997; 68(Suppl 273)4–8
- Rydholm A, Alvegård T. Scandinavian Sarcoma Group, GÖteborg, Sweden, May 5-7, 1993. Acta Orthop Scand A 1994a; 65(Suppl 265)55–72
- Rydholm A, Alvegård TA. Scandinavian Sarcoma Group, Helsinki, April 10-22, 1994. Acta Orthop Scand 1994b; 65(Suppl 262)63–80
- Rydholm A, Alvegård TA. Scandinavian Sarcoma Group, Stockholm, April 26-28, 1995. Acta Orthop Scand 1995; 66(Suppl 265)89–100
- Rydholm A, Alvegård TA. Scandinavian Sarcoma Group, Reykjavik, May 7-9, 1996. Acta Orthop Scand 1996; 67(Suppl 272)41–59
- Rydholm A, Alvegård TA. Scandinavian Sarcoma Group, Trondheim, April 16-18, 1997. Acta Orthop Scand 1997; 68(Suppl 274)65–77
- Rydholm A, Alvegård TA. Scandinavian Sarcoma Group, Snekkersten, April 22-24, 1998. Acta Orthop Scand 1998; 69(Suppl 282)25–43
- Smeland S, Müller C, Alvegård TA, Wiklund T, Wiebe T, BjÖrk O, Stenwig AE, Willén H, HolmstrÖm T, Follerås G, Brosjö O, Kivioja A, Jonsson K, Monge O, Saeter G. Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur J of Cancer 2003; 39: 488–94
- Smeland S, Wiebe T, Brosjö O, BÖhling T, Alvegård TA. Chemotherapy in Ewing's sarcoma - The Scandinavian Sarcoma Group experience. Acta Orthop Scand 2004; 75(Suppl. 311)87–91
- Solheim Ø, Alvegård TA, Elomaa I. Adjuvant chemotherapy for osteosarcoma a preliminary report from the Scandinavian Sarcoma Group. Acta Oncol 1989; 28(Suppl 2)53–7
- Solheim Ø, Alvegård TA. The Scandinavian Sarcoma Group. 10 Years' experience. Acta Oncol 1989; 28(Suppl 2)175
- Solheim Ø, Saeter G, Elomaa I, Alvegård TA. The treatment of osteosarcoma: Present trends. The Scandinavian Sarcoma Group experience. Annals of Oncology 1992; 3(Suppl 2)711
- Saeter G, Alvegård TA, Elomaa I, et al. Treatment of osteosarcoma of the extremities with the T10 protocol, with emphasis on the effects of preoperative chemotherapy with single agent high–dose methotrexate. A Scandinavian Sarcoma Group study. J Clin Oncol 1991; 9: 1766–75
- Saeter G, Strander H, Monge OR, Alvegård TA. Dose escalating etoposide (E), ifosfamide (I) and granulocyte colony stimulating factor (GCSF) (VIGregimen) in advanced adult soft tissue sarcoma. A Scandinavian Sarcoma Group study. Ann Oncol 1994; 8(5 suppl)171
- Saeter G, Talle K, Solheim Ø. Treatment of advanced, high–grade soft–tissue sarcoma with ifosfamide and continuos infusion etoposide. Cancer Chemotherapy and Pharmacology 1995; 36: 172–5
- Saeter G., for the Scandinavian Sarcoma Group. Treatment of osteosarcoma with high-dose methotrexate containing neoadjuvant chemotherapy. Scandinavian Sarcoma Group data. Med Ped Oncol 1996a; 27(4)263
- Saeter G., for the Scandinavian Sarcoma Group. Treatment strategies and outcome in metastatic (relapsed) osteogenic sarcoma. The Scandinavian Sarcoma Group (SSG) experience. Med Ped Oncol 1996b; 27(4)264
- Serra M, Pasello M, Manara MC, et al. May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int J Oncol Dec 29, 2006, 6: 1459–68
- Tierney JF, Mosseri V, Stewart L, et al. Adjuvant chemotherapy for soft-tissue sarcoma: review and meta-analysis of the published results of randomized clinical trials. Br J Cancer 1995; 72: 469–75
- Tierney JF. Adjuvant chemotherapy for localized resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma meta-analysis collaboration. Lancet 1997; 350(9092)1647–54
- Wiklund TA, Alvegård TA, Mouridsen HT, et al. Marginal surgery and postoperative radiotherapy in soft tissue sarcomas. Eur J Cancer 1993; 29A: 30–69
- Bauer HCF, Möller T, Alvegård TA, Rydholm A, Gustafson P, Solheim ö, Erlanson M, Karlsson M, Monge O, Trovik C, Blomqvist C, Wiklund T, Berlin ö, Turesson I. Monitoring referral and treatment in soft tissue sarcoma: Study based on 1851 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand 2001; 72: 150–9
- Jebsen NL, Trovik CS, Bauer HCF, Rydholm A, Monge O, Hall KS, Alvegård T, Bruland Ø. Radiotherapy to improve local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma: a Scandinavian Sarcoma Group study. Int J Radiation Oncology Biol Phys 2008; 71(4)1196–203
- Rydholm A. Management of patients with soft-tissue sarcoma. Strategy developed at a regional oncology center. Acta Orthop (Suppl 203) 1983; 54
- Trovik CS, Bauer HCF. Local recurrence of soft tissue sarcoma a risk factor for late metastases: 379 patients followed for 0.5-20 years. Acta Orthop Scand 2000; 65: 553–8
- Trovik CS, Bauer HCF, Alvegård TA, Anderson H, Blomqvist C, Berlin ö, Gustafson P, Sæter G, Wallöe A. Surgical margin, Local recurrence and Survival in Soft Tissue Sarcoma. Study based on 620 patients from the Scandinavian Sarcoma Group Register. Europ J Cancer 2000; 36: 710–6
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. Feb 3, 2000; 403(6769)503–11
- Ambrosini G, Sambol EB, Carvajal D, Vassilev LT, Singer S, Schwartz GK. Mouse double minute antagonist Nutlin-3a enhances chemotherapy-induced apoptosis in cancer cells with mutant p53 by activating E2F1. Oncogene. May 24, 2007; 26(24)3473–81
- Antonescu CR. The role of genetic testing in soft tissue sarcoma. Histopathology. Jan, 2006; 48(1)13–21
- Antonescu CR. Targeted therapy of cancer: new roles for pathologists in identifying GISTs and other sarcomas. Mod Pathol. May, 2008; 21(Suppl 2)S31–6
- Avigad S, Naumov I, Ohali A, Jeison M, Berco GH, Mardoukh J, et al. Short telomeres: a novel potential predictor of relapse in Ewing sarcoma. Clin Cancer Res. Oct 1, 2007; 13(19)5777–83
- Baird K, Davis S, Antonescu CR, Harper UL, Walker RL, Chen Y, et al. Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer Res. Oct 15, 2005; 65(20)9226–35
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. Aug 3, 2000; 406(6795)536–40
- Bridge JA. Contribution of cytogenetics to the management of poorly differentiated sarcomas. Ultrastruct Pathol. Mar-Apr, 2008; 32(2)63–71
- Cairney CJ, Hoare SF, Daidone MG, Zaffaroni N, Keith WN. High level of telomerase RNA gene expression is associated with chromatin modification, the ALT phenotype and poor prognosis in liposarcoma. Br J Cancer. Apr 22, 2008; 98(8)1467–74
- Carneiro A, Francis P, Bendahl P-O, Fernebro J, Åkerman M, Fletcher C, et al. Indistinguishable genomic profiles and shared prognostic markers in undifferentiated pleomorphic sarcoma and leiomyosarcoma: different sides of a single coin?. Laboratory Investigation. 2009, in press
- Creager AJ, Cohen JA, Geradts J. Aberrant expression of cell-cycle regulatory proteins in human mesenchymal neoplasia. Cancer Detect Prev. 2001; 25(2)123–31
- D'Adamo DR, Anderson SE, Albritton K, Yamada J, Riedel E, Scheu K, et al. Phase II study of doxorubicin and bevacizumab for patients with metastatic soft-tissue sarcomas. J Clin Oncol. Oct 1, 2005; 23(28)7135–42
- de Bruijn DR, Allander SV, van Dijk AH, Willemse MP, Thijssen J, van Groningen JJ, et al. The synovial-sarcoma-associated SS18-SSX2 fusion protein induces epigenetic gene (de)regulation. Cancer Res. Oct 1, 2006; 66(19)9474–82
- Dei Tos AP, Maestro R, Doglioni C, Piccinin S, Libera DD, Boiocchi M, et al. Tumor suppressor genes and related molecules in leiomyosarcoma. Am J Pathol. Apr, 1996; 148(4)1037–45
- Deneen B, Denny CT. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. Oct 11, 2001; 20(46)6731–41
- Detwiller KY, Fernando NT, Segal NH, Ryeom SW, D'Amore PA, Yoon SS. Analysis of hypoxia-related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res. Jul 1, 2005; 65(13)5881–9
- Fernebro J, Francis P, Eden P, Borg A, Panagopoulos I, Mertens F, et al. Gene expression profiles relate to SS18/SSX fusion type in synovial sarcoma. Int J Cancer. Mar 1, 2006; 118(5)1165–72
- Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. May 4, 1989; 339(6219)58–61
- Francis P, Namlos HM, Muller C, Eden P, Fernebro J, Berner JM, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics. 2007; 8 73
- Fritz B, Schubert F, Wrobel G, Schwaenen C, Wessendorf S, Nessling M, et al. Microarray-based copy number and expression profiling in dedifferentiated and pleomorphic liposarcoma. Cancer Res. Jun 1, 2002; 62(11)2993–8
- Gasparini G, Longo R, Fanelli M, Teicher BA. Combination of antiangiogenic therapy with other anticancer therapies: results, challenges, and open questions. J Clin Oncol. Feb 20, 2005; 23(6)1295–311
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. Oct 15, 1999; 286(5439)531–7
- Heidenblad M, Hallor KH, Staaf J, Jonsson G, Borg A, Hoglund M, et al. Genomic profiling of bone and soft tissue tumors with supernumerary ring chromosomes using tiling resolution bacterial artificial chromosome microarrays. Oncogene. Nov 9, 2006; 25(53)7106–16
- Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer. Sep, 2003; 3(9)685–94
- Hernando E, Charytonowicz E, Dudas ME, Menendez S, Matushansky I, Mills J, et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat Med. Jun, 2007; 13(6)748–53
- Latres E, Drobnjak M, Pollack D, Oliva MR, Ramos M, Karpeh M, et al. Chromosome 17 abnormalities and TP53 mutations in adult soft tissue sarcomas. Am J Pathol. Aug, 1994; 145(2)345–55
- Lee YF, John M, Falconer A, Edwards S, Clark J, Flohr P, et al. A gene expression signature associated with metastatic outcome in human leiomyosarcomas. Cancer Res. Oct 15, 2004; 64(20)7201–4
- Lunt SJ, Telfer BA, Fitzmaurice RJ, Stratford IJ, Williams KJ. Tirapazamine administered as a neoadjuvant to radiotherapy reduces metastatic dissemination. Clin Cancer Res. Jun 1, 2005; 11(11)4212–6
- Meza-Zepeda LA, Kresse SH, Barragan-Polania AH, Bjerkehagen B, Ohnstad HO, Namlos HM, et al. Array comparative genomic hybridization reveals distinct DNA copy number differences between gastrointestinal stromal tumors and leiomyosarcomas. Cancer Res. Sep 15, 2006; 66(18)8984–93
- Montgomery E, Argani P, Hicks JL, DeMarzo AM, Meeker AK. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am J Pathol. May, 2004; 164(5)1523–9
- Nagayama S, Katagiri T, Tsunoda T, Hosaka T, Nakashima Y, Araki N, et al. Genome-wide analysis of gene expression in synovial sarcomas using a cDNA microarray. Cancer Res. Oct 15, 2002; 62(20)5859–66
- Nielsen TO, West RB, Linn SC, Alter O, Knowling MA, O'Connell JX, et al. Molecular characterisation of soft tissue tumours: a gene expression study. Lancet. Apr 13, 2002; 359(9314)1301–7
- Oliveira AM, Fletcher CD. Molecular prognostication for soft tissue sarcomas: are we ready yet?. J Clin Oncol. Oct 15, 2004; 22(20) 4031–4
- Orlow I, Drobnjak M, Zhang ZF, Lewis J, Woodruff JM, Brennan MF, et al. Alterations of INK4A and INK4B genes in adult soft tissue sarcomas: effect on survival. J Natl Cancer Inst. Jan 6, 1999; 91(1)73–9
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. Aug 17, 2000; 406(6797)747–52
- Price ND, Trent J, El-Naggar AK, Cogdell D, Taylor E, Hunt KK, et al. Highly accurate two-gene classifier for differentiating gastrointestinal stromal tumors and leiomyosarcomas. Proc Natl Acad Sci U S A. Feb 27, 2007; 104(9) 3414–9
- Rodon J, DeSantos V, Ferry RJ, Jr., Kurzrock R. Early drug development of inhibitors of the insulin-like growth factor-I receptor pathway: lessons from the first clinical trials. Mol Cancer Ther. Sep, 2008; 7(9)2575–88
- Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. Dec 20, 2005; 23(36)9408–21
- Segal NH, Pavlidis P, Antonescu CR, Maki RG, Noble WS, DeSantis D, et al. Classification and subtype prediction of adult soft tissue sarcoma by functional genomics. Am J Pathol. Aug, 2003; 163(2)691–700
- Segal NH, Pavlidis P, Noble WS, Antonescu CR, Viale A, Wesley UV, et al. Classification of clear-cell sarcoma as a subtype of melanoma by genomic profiling. J Clin Oncol. May 1, 2003; 21(9)1775–81
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. Sep 11, 2001; 98(19)10869–74
- Stratton MR, Moss S, Warren W, Patterson H, Clark J, Fisher C, et al. Mutation of the p53 gene in human soft tissue sarcomas: association with abnormalities of the RB1 gene. Oncogene. Sep, 1990; 5(9)1297–301
- Sun Y, Gao D, Liu Y, Huang J, Lessnick S, Tanaka S. IGF2 is critical for tumorigenesis by synovial sarcoma oncoprotein SYT-SSX1. Oncogene. Feb 16, 2006; 25(7)1042–52
- Tomescu O, Barr FG. Chromosomal translocations in sarcomas: prospects for therapy. Trends Mol Med. Dec, 2001; 7(12) 554–9
- Wang J, Coltrera MD, Gown AM. Cell proliferation in human soft tissue tumors correlates with platelet-derived growth factor B chain expression: an immunohistochemical and in situ hybridization study. Cancer Res. Jan 15, 1994; 54(2)560–4
- Wunder JS, Eppert K, Burrow SR, Gokgoz N, Bell RS, Andrulis IL. Co-amplification and overexpression of CDK4, SAS and MDM2 occurs frequently in human parosteal osteosarcomas. Oncogene. Jan 21, 1999; 18(3)783–8
- Yoon SS, Segal NH, Park PJ, Detwiller KY, Fernando NT, Ryeom SW, et al. Angiogenic profile of soft tissue sarcomas based on analysis of circulating factors and microarray gene expression. J Surg Res. Oct, 2006; 135(2)282–90
- Zhan S, Helman LJ. Glimpsing the cause of rhabdomyosarcoma. Nat Med. May, 1998; 4(5)559–60
- Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest. Jul, 1994; 94(1)445–8
- Fletcher CD. The evolving classification of soft tissue tumours: an update based on the new WHO classification. Histopathology 2006; 48(1)3–12
- Weiss SWGJ. Enzinger and Weiss's soft tissue tumors, 5th ed. CV Mosby Co, St. Louis (Mo) 2008
- Orson GG, Sim FH, Reiman HM, Taylor WF. Liposarcoma of the musculoskeletal system. Cancer 1987; 60(6)1362–70
- Zagars GK, Goswitz MS, Pollack A. Liposarcoma: outcome and prognostic factors following conservation surgery and radiation therapy. Int J Radiat Oncol Biol Phys 1996; 36(2)311–9
- Pearlstone DB, Pisters PW, Bold RJ, Feig BW, Hunt KK, Yasko AW, et al. Patterns of recurrence in extremity liposarcoma: implications for staging and follow-up. Cancer 1999; 85(1)85–92
- Linehan DC, Lewis JJ, Leung D, Brennan MF. Influence of biologic factors and anatomic site in completely resected liposarcoma. J Clin Oncol 2000; 18(8)1637–43
- Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg 2006; 244(3)381–91
- Bauer HC, Trovik CS, Alvegard TA, Berlin O, Erlanson M, Gustafson P, et al. Monitoring referral and treatment in soft tissue sarcoma: study based on 1,851 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand 2001; 72(2)150–9
- Meis-Kindblom JM, Bjerkehage B, Bohling T, Domanski H, Halvorsen TB, Larsson O, et al. Morphologic review of 1000 soft tissue sarcomas from the Scandinavian Sarcoma Group (SSG) Register. The peer-review committee experience. Acta Orthop Scand Suppl 1999; 285: 18–26
- Engstrom K, Bergh P, Gustafson P, Hultborn R, Johansson H, Lofvenberg R, et al. Liposarcoma: outcome based on the Scandinavian Sarcoma Group register. Cancer 2008; 113(7)1649–56
- Fletcher CDM, Unni KK, Mertens F. Pathology and Genetics of Tumours of Soft Tissue and Bone. WHO Classification of Tumours. IARC Press, Lyon 2002; 35–46
- Evans HL. Liposarcomas and atypical lipomatous tumors: a study of 66 cases followed for a minimum of 10 years. Surg Pathol 1988; 1(1)41–54
- Evans HL. Atypical Lipomatous Tumor, its Variants, and its Combined Forms: A Study of 61 Cases, With a Minimum Follow-up of 10 Years. Am J Surg Pathol 2007; 31(1)1–14
- Rozental TD, Khoury LD, Donthineni-Rao R, Lackman RD. Atypical lipomatous masses of the extremities: outcome of surgical treatment. Clin Orthop Relat Res 2002, 398: 203–11
- Sommerville SM, Patton JT, Luscombe JC, Mangham DC, Grimer RJ. Clinical outcomes of deep atypical lipomas (well-differentiated lipoma-like liposarcomas) of the extremities. ANZ J Surg 2005; 75(9)803–6
- Weiss SW, Rao VK. Well-differentiated liposarcoma (atypical lipoma) of deep soft tissue of the extremities, retroperitoneum, and miscellaneous sites. A follow-up study of 92 cases with analysis of the incidence of “dedifferentiation”. Am J Surg Pathol 1992; 16(11)1051–8
- Bassett MD, Schuetze SM, Disteche C, Norwood TH, Swisshelm K, Chen X, et al. Deep-seated, well differentiated lipomatous tumors of the chest wall and extremities: the role of cytogenetics in classification and prognostication. Cancer 2005; 103(2)409–16
- Billing V, Mertens F, Domanski HA, Rydholm A. Deep-seated ordinary and atypical lipomas: histopathology, cytogenetics, clinical features, and outcome in 215 tumours of the extremity and trunk wall. J Bone Joint Surg Br 2008; 90(7)929–33
- Jebsen NL, Trovik CS, Bauer HC, Rydholm A, Monge OR, Hall KS, et al. Radiotherapy to Improve Local Control Regardless of Surgical Margin and Malignancy Grade in Extremity and Trunk Wall Soft Tissue Sarcoma: A Scandinavian Sarcoma Group Study. Int J Radiat Oncol Biol Phys 2008
- Eilber FC, Eilber FR, Eckardt J, Rosen G, Riedel E, Maki RG, et al. The impact of chemotherapy on the survival of patients with high-grade primary extremity liposarcoma. Ann Surg 2004; 240(4)686–95, discussion 695-7
- Jones RL, Fisher C, Al-Muderis O, Judson IR. Differential sensitivity of liposarcoma subtypes to chemotherapy. Eur J Cancer 2005; 41(18)2853–60
- Grosso F, Jones RL, Demetri GD, Judson IR, Blay JY, Le Cesne A, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007; 8(7)595–602
- Dei Tos AP, Doglioni C, Piccinin S, Maestro R, Mentzel T, Barbareschi M, et al. Molecular abnormalities of the p53 pathway in dedifferentiated liposarcoma. J Pathol 1997; 181(1)8–13
- Pilotti S, Della Torre G, Lavarino C, Di Palma S, Sozzi G, Minoletti F, et al. Distinct mdm2/p53 expression patterns in liposarcoma subgroups: implications for different pathogenetic mechanisms. J Pathol 1997; 181(1)14–24
- Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: liposarcoma. Cancer Genet Cytogenet 2004; 155(1)1–24
- Levav-Cohen Y, Haupt S, Haupt Y. Mdm2 in growth signaling and cancer. Growth Factors 2005; 23(3)183–92
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 2003; 13(1)49–58
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69(7)1237–45
- Xiao ZX, Chen J, Levine AJ, Modjtahedi N, Xing J, Sellers WR, et al. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 1995; 375(6533)694–8
- Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle 2004; 3(4)419–21
- Klein C, Vassilev LT. Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer 2004; 91(8)1415–9
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303(5659)844–8
- Singer S, Socci ND, Ambrosini G, Sambol E, Decarolis P, Wu Y, et al. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res 2007; 67(14)6626–36
- Muller CR, Paulsen EB, Noordhuis P, Pedeutour F, Saeter G, Myklebost O. Potential for treatment of liposarcomas with the MDM2 antagonist Nutlin-3A. Int J Cancer 2007; 121(1)199–205
- Dei Tos AP. Liposarcoma: new entities and evolving concepts. Ann Diagn Pathol 2000; 4(4)252–66
- Antonescu CR, Tschernyavsky SJ, Decuseara R, Leung DH, Woodruff JM, Brennan MF, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res 2001; 7(12)3977–87
- Panagopoulos I, Mandahl N, Ron D, Hoglund M, Nilbert M, Mertens F, et al. Characterization of the CHOP breakpoints and fusion transcripts in myxoid liposarcomas with the 12; 16 translocation. Cancer Res 1994; 54(24)6500–3
- Panagopoulos I, Hoglund M, Mertens F, Mandahl N, Mitelman F, Aman P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 1996; 12(3)489–94
- Friedman M, Egan JW. Irradiation of liposarcoma. Acta Radiol 1960; 54: 225–39
- Perry H, Chu FC. Radiation therapy in palliative management of soft tissue sarcomas. Cancer 1962; 15: 179–83
- Engstrom K, Bergh P, Cederlund CG, Hultborn R, Willen H, Aman P, et al. Irradiation of myxoid/round cell liposarcoma induces volume reduction and lipoma-like morphology. Acta Oncol 2007; 46(6)838–45
- Pitson G, Robinson P, Wilke D, Kandel RA, White L, Griffin AM, et al. Radiation response: an additional unique signature of myxoid liposarcoma. Int J Radiat Oncol Biol Phys 2004; 60(2)522–6
- Guadagnolo BA, Zagars GK, Ballo MT, Patel SR, Lewis VO, Benjamin RS, et al. Excellent local control rates and distinctive patterns of failure in myxoid liposarcoma treated with conservation surgery and radiotherapy. Int J Radiat Oncol Biol Phys 2008; 70(3)760–5
- Hatano H, Ogose A, Hotta T, Kawashima H, Sugita T, Sasamoto R, et al. Treatment of myxoid liposarcoma by marginal or intralesional resection combined with radiotherapy. Anticancer Res 2003; 23(3C)3045–9
- Kuroda M, Sok J, Webb L, Baechtold H, Urano F, Yin Y, et al. Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. Embo J 2000; 19(3)453–62
- Mentzel T, Fletcher CD. Dedifferentiated myxoid liposarcoma: a clinicopathological study suggesting a closer relationship between myxoid and well-differentiated liposarcoma. Histopathology 1997; 30(5)457–63
- Mentzel T, Palmedo G, Hantschke M, Woziwodzki J, Beck C. Mixed-type liposarcoma: clinicopathological, immunohistochemical, and molecular analysis of a case arising in deep soft tissues of the lower extremity. Virchows Arch 2008; 453(2)197–201
- Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, et al. Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci U S A 1999; 96(7)3951–6
- Dei Tos AP, Piccinin S, Doglioni C, Vukosavljevic T, Mentzel T, Boiocchi M, et al. Molecular aberrations of the G1-S checkpoint in myxoid and round cell liposarcoma. Am J Pathol 1997; 151(6)1531–9
- Pilotti S, Lavarino C, Mezzelani A, Della Torre C, Minoletti F, Sozzi G, et al. Limited role of TP53 and TP53-related genes in myxoid liposarcoma. Tumori 1998; 84(5)571–7
- Schneider-Stock R, Walter H, Rys J, Radig K, Hoang-Vu C, Roessner A. No correlation of c-myc overexpression and p53 mutations in liposarcomas. Virchows Arch 1998; 433(4)315–21
- Oda Y, Yamamoto H, Takahira T, Kobayashi C, Kawaguchi K, Tateishi N, et al. Frequent alteration of p16(INK4a)/p14(ARF) and p53 pathways in the round cell component of myxoid/round cell liposarcoma: p53 gene alterations and reduced p14(ARF) expression both correlate with poor prognosis. J Pathol 2005; 207(4)410–21
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445(7128)661–5
- Engstrom K, Willen H, Kabjorn-Gustafsson C, Andersson C, Olsson M, Goransson M, et al. The myxoid/round cell liposarcoma fusion oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma phenotype in transfected human fibrosarcoma cells. Am J Pathol 2006; 168(5)1642–53
- Riggi N, Cironi L, Provero P, Suva ML, Stehle JC, Baumer K, et al. Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res 2006; 66(14)7016–23
- Perez-Losada J, Sanchez-Martin M, Rodriguez-Garcia MA, Perez-Mancera PA, Pintado B, Flores T, et al. Liposarcoma initiated by FUS/TLS-CHOP: the FUS/TLS domain plays a critical role in the pathogenesis of liposarcoma. Oncogene 2000; 19(52)6015–22
- Angervall L, Kindblom LG. Principles for pathologic-anatomic diagnosis and classification of soft-tissue sarcomas. Clin Orthop 1993, 289: 9–18
- Angervall L, Kindblom LG, Merck C. Myxofibrosarcoma. A study of 30 cases. Acta Pathol Microbiol Scand [A] 1977, 85A: 127–140
- Bohling T, Bacchini P, Bertoni F, Bjerkehagen B, Domanski H, Kindblom LG, Meis-kindblom J, Wejde J. Diagnosis and tumor response in osteosarcoma and Ewing's sarcoma. Acta Orthop Scand Suppl 2004; 75(Suppl 311)72–76
- Broders AC. Squamous-cell epithelioma of the lip. A study of five hundred and thirty-seven cases. JAMA 1920; 74: 656–64
- Broders AC, Hargrave R., Meyerding HW. Pathological features of soft tissue fibrosarcoma with special reference to the grading of its malignancy. Surgery gynecolology and obstetrics 1939; 69: 267–80
- Coindre JM, Terrier P, Bui NB, et al. Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol 1996; 14: 869–77
- Deyrup AT, Weiss SW. Grading of soft tissue sarcomas: the challenge of providing precise information in an imprecise world. Histopathology 2006; 48: 42–50
- Engellau J, Bendahl PO, Persson A, et al. Improved prognostication in soft tissue sarcoma: independent information from vascular invasion, necrosis, growth pattern, and immunostaining using whole-tumor sections and tissue microarrays. Hum Pathol 2005; 36: 994–1002
- Engellau J, Persson A, Bendahl PO, et al. Expression profiling using tissue microarray in 211 malignant fibrous histiocytomas confirms the prognostic value of Ki-67. Virchows Arch 2004; 445: 224–30
- Engellau J, Samuelsson V, Anderson H, et al. Identification of low-risk tumours in histological high-grade soft tissue sarcomas. Eur J Cancer 2007; 43: 1927–34
- Engstrom K, Bergh P, Gustafson P, et al. Liposarcoma: outcome based on the Scandinavian Sarcoma Group register. Cancer 2008; 113: 1649–56
- Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 2005, 23: 8845–52
- Fletcher CD, Unni KK, Mertens FE. World Health Organization Classification of tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press, Lyon 2002
- Guillou L, Coindre JM, Bonichon F, et al. Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 1997; 15: 350–62
- Gustafson P. Soft tissue sarcoma Epidemiology and prognosis in 508 patients. Acta Orthop Scand 1994; 65: 259, Thesis
- Gustafson P, Akerman M, Alvegard TA, et al. Prognostic information in soft tissue sarcoma using tumour size, vascular invasion and microscopic tumour necrosis-the SIN-system. Eur J Cancer 2003; 39: 1568–76
- Hasegawa T. Histological grading and MIB-1 labeling index of soft-tissue sarcomas. Pathol Int 2007; 57: 121–5
- Huvos AG. Bone Tumors: Diagnosis, treatment and prognosis. Pathologic assessment of preoperative (neoadjuvant) chemotherapy. W.B.Saunders, Philadelphia 1991
- Joensuu H, Kindblom LG. Gastrointestinal stromal tumors–a review. Acta Orthop Scand Suppl 2004; 75(Suppl 311)62–71
- Meis-Kindblom JM, Bjerkehage B, Bohling T, et al. Morphologic review of 1000 soft tissue sarcomas from the Scandinavian Sarcoma Group (SSG) Register. The peer-review committee experience. Acta Orthop Scand Suppl 1999, Suppl 285: 18–26
- Miettinen M. Soft tissue tumors - from grading to specific mutations. Arch Pathol Lab Med 2006; 130: 1446–7
- Picci P, Bohling T, Bacci G, et al. Chemotherapy-induced tumor necrosis as a prognostic factor in localized Ewing's sarcoma of the extremities. J Clin Oncol 1997, 15: 1553–9
- Smeland S, Bruland OS, Hjorth L, et al. Scandinavian experience in classical osteosarcoma, results of the SSGXIV protocol. Acta Orthop Scand Suppl 2009; 80(Suppl 334)60–66
- Svarvar C, Bohling T, Berlin O, et al. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian Sarcoma Group. Cancer 2007; 109: 282–91
- Abraham SC, Krasinskas AM, Hofstetter WL, Swisher SG, Wu TT. “Seedling” mesenchymal tumors (gastrointestinal stromal tumors and leiomyomas) are common incidental tumors of the esophagogastric junction. Am J Surg Pathol 2007; 31(11)1629–35
- Agaimy A, Wunsch PH, Hofstaedter F, Blaszyk H, Rümmele P, Gaumann A, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol 2007; 31(1)113–20
- Blanke CD, Demetri GD, von Mehren M, Heinrich M, Eisenberg B, Fletcher J, et al. Long-term results from a randomized phase II trial of standard versus higher dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008; 26(4)620–5
- Blay J-Y, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 2007; 25(9)1107–13
- Casali PG, Jost L, Reichardt P, Schlemmer M, Blay J-Y. Gastrointestinal stromal tumors: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 2008, Suppl 2: ii35–ii38
- DeMatteo RP, Antonescu CR, Chadaram V, You YN, McCall L, Maki R, et al. Adjuvant imatinib in patients with primary high risk gastrointestinal stromal tumor (GIST) following complete resection: Safety results from the U.S. Intergrup Phase II trial ACOSOG Z9000. J Clin Oncol 2005; 23, 818s (abstract #9009)
- DeMatteo RP, Owzar K, Maki R. Adjuvant imatinib mesylate increases recurrence free survival (RFS) in patients with completely resected localized primary gastrointestinal stromal tumor (GIST): North American Intergroup Phase III trial ACOSOG Z9001 [ASCO abstract 10079]. Presented at: 43rd Annual Meeting of the American Society of Clinical Oncology; Chicago, IL; June 1-5, 2007
- Demetri GD, von Mehren M, Blanke CD, van den Abbeele A, Eisenberg B, Roberts PJ, et al. Antitumor effects of an oral selective tyrosine kinase inhibitor, imatinib mesylate, in patients with advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347(7)472–80
- Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley B, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002; 33(5)459–65
- Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003; 21(23)4342–9
- Heinrich MC, Corless CL, Demetri G, Joensuu H, Eisenberg B, von Mehren M, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006; 24(29)4764–74
- Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 2008; 39(10)1411–19
- Joensuu H, Roberts P, Sarlomo-Rikala M, Andersson L, Tervahartiala P, Tuveson D, et al. Clinical response induced by the tyrosine kinase inhibitor STI571 in metastatic gastrointestinal stromal tumor expressing a mutant c-kit proto-oncogene. N Engl J Med 2001; 344(14)1052–6
- Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol 2006; 37(12)1527–35
- Kose K, Hiyama T, Tanaka S, Yoshihara M, Yasui W, Chayama K. Nuclear and mitochondrial DNA microsatellite instability in gastrointestinal stromal tumors. Pathobiology 2006; 73(2)93–7
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006; 23(2)70–83
- Miettinen M, Makhlouf H, Sobin LH, et al. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006; 30(4)477–89
- Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005; 29(1)52–68
- Mussi C, Schildhaus HU, Gronchi A, Wardelmann E, Hohenberger P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res 2008; 14(14)4550–5
- Nilsson B, Bumming P, Meis-Kindblom JM, Oden A, Dortok A, gustavsson B, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era–a population-based study in western Sweden. Cancer 2005; 103(4)821–9
- Ruka W, Rutkowski P, Szawłowski A, Nowecki Z, Debiec-Rychter M, Grzesiakowska U, et al. Surgical resection of residual disease in initially inoperable imatinib-resistant/intolerant gastrointestinal stromal tumor treated with sunitinib. Eur J Surg Oncol Feb 18, 2008, [Epub ahead of print]
- Rutkowski P, Nowecki Z, Nyckowski P, Dziewirski W, Grzesiakowska U, Nasierowska-Guttmejer A, et al. Surgical treatment of patients with initially inoperable and/or metastatic gastrointestinal stromal tumors (GIST) during therapy with imatinib mesylate. J Surg Oncol 2006; 93(4)304–11
- Saito K, Sakurai S, Sano T, Sakamoto K, Asao T, Hosoya Y, et al. Aberrant methylation status of known methylation-sensitive CpG islands in gastrointestinal stromal tumors without any correlation to the state of c-kit and PDGFRA gene mutations and their malignancy. Cancer Sci 2008; 99(2)253–9
- Steigen SE, Eide TJ. Trends in incidence and survival of mesenchymal neoplasm of the digestive tract within a defined population of northern Norway. APMIS 2006; 114(3)192–200
- Takahashi T, Nakajima K, Nishitani A, Souma Y, Hirota S, Sawa Y, et al. An enhanced risk-group stratification system for more practical prognostication of clinically malignant gastrointestinal stromal tumors. Int J Clin Oncol 2007; 12(5)369–74
- Tryggvason G, Gislason HG, Magnusson MK, Jonasson JG. Gastrointestinal stromal tumors in Iceland, 1990-2003: the icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer 2005; 117(2)289–93
- Von Mehren M, Wang Y, Joensuu H, Blanke CD, Wehrle E, Demetri GD. Imatinib pharmacokinetics and its correlation with clinical response in patients with unresectable/metastatic gastrointestinal stromal tumor. J Clin Oncol 2008; 26: 218s, (abstract #4523)
- Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res Mar 15, 2006; 12(6)1743–9
- Zhan WH. Efficacy and safety of adjuvant post-surgical therapy with imatinib in patients with high-risk of relapsing GIST. J Clin Oncol 2007; 25: 556s, (abstract #10045)
- Alvegård TA, Berg NO, Ranstam J, Rydholm A, Rööser B. Prognosis in high-grade soft tissue sarcomas. The Scandinavian Sarcoma Group experience in a randomized adjuvant chemotherapy trial. Acta Orthop Scand 1989; 60: 517–21
- Angervall L, Kindblom LG, Rydholm A, Stener B. The diagnosis and prognosis of soft tissue tumors. Semin Diagn Pathol 1986; 3: 240–58
- Behranwala KA, A'Hern R, Omar AM, Thomas JM. Prognosis of lymph node metastasis in soft tissue sarcoma. Ann Surg Oncol 2004; 11: 714–9
- Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med 2006; 130: 1448–53
- Coindre JM, Terrier P, Guillou L, Le Doussal V, Collin F, Ranchere D, Sastre X, Vilain MO, Bonichon F, N'Guyen BB. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001; 91: 1914–26
- Costa J. The grading and staging of soft tissue sarcomas. Pathobiology of Soft Tissue Tumors., C. Fletcher, P. McKee. Churchill Livingstone, EdinburghU.K. 1990
- Engellau J, Bendahl PO, Persson A, Domanski HA, åkerman M, Gustafson P, Alvegård TA, Nilbert M, Rydholm A. Improved prognostication in soft tissue sarcoma: independent information from vascular invasion, necrosis, growth pattern, and immunostaining using whole-tumor sections and tissue microarrays. Hum Pathol 2005; 36: 994–1002
- Engellau J, Samuelsson V, Anderson H, Bjerkehagen B, Rissler P, Sundby-Hall K, Rydholm A. Identification of low-risk tumours in histological high-grade soft tissue sarcomas. Eur J Cancer 2007; 43: 1927–34
- Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg 1993; 217: 72–7
- Greene FL, Page DL, Fleming ID, Fritz AG, Balch CM, Haller DG, Morrow M. AJCC cancer staging handbook / American Joint Committee on Cancer (6:th ed). Science+Business Media, Springer 2002
- Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand Suppl 1994, 259: 1–31
- Gustafson P, åkerman M, Alvegård TA, Coindre JM, Fletcher CD, Rydholm A, Willen H. Prognostic information in soft tissue sarcoma using tumour size, vascular invasion and microscopic tumour necrosis-the SIN-system. Eur J Cancer 2003; 39: 1568–76
- Kattan MW, Leung DH, Brennan MF. Postoperative nomogram for 12-year sarcoma-specific death. J Clin Oncol 2002; 20: 791–6
- Kilpatrick SE. Histologic prognostication in soft tissue sarcomas: grading versus subtyping or both? A comprehensive review of the literature with proposed practical guidelines. Ann Diagn Pathol 1999; 3: 48–61
- Lahat G, Tuvin D, Wei C, Anaya DA, Bekele BN, Lazar AJ, Pisters PW, Lev D, Pollock RE. New perspectives for staging and prognosis in soft tissue sarcoma. Ann Surg Oncol 2008; 15: 2739–48
- Mandard AM, Petiot JF, Marnay J, Mandard JC, Chasle J, De Ranieri E, Dupin P, Herlin P, De Ranieri J, Tanguy A, et al. Prognostic factors in soft tissue sarcomas. A multivariate analysis of 109 cases. Cancer 1989; 63: 1437–51
- Mariani L, Miceli R, Kattan MW, Brennan MF, Colecchia M, Fiore M, Casali PG, Gronchi A. Validation and adaptation of a nomogram for predicting the survival of patients with extremity soft tissue sarcoma using a three-grade system. Cancer 2005; 103: 402–8
- Markhede G, Angervall L, Stener B. A multivariate analysis of the prognosis after surgical treatment of malignant soft-tissue tumors. Cancer 1982; 49: 1721–33
- Meis-Kindblom JM, Bjerkehage B, Böhling T, Domanski H, Halvorsen TB, Larsson O, Lilleng P, Myhre-Jensen O, Stenwig E, Virolainen M, Willen H, åkerman M, Kindblom LG. Morphologic review of 1000 soft tissue sarcomas from the Scandinavian Sarcoma Group (SSG) Register. The peer-review committee experience. Acta Orthop Scand Suppl 1999, 285: 18–26
- Meta-analysis Group, MRC Clinical Trials Unit. Adjuvant chemotherapy for loalized resectable soft tissue sarcoma in adults. Cochrane Database Systematic Reviews 2000; 4: 1
- Myhre-Jensen O, Kaae S, Madsen EH, Sneppen O. Histopathological grading in soft-tissue tumours. Relation to survival in 261 surgically treated patients. Acta Pathol Microbiol Immunol Scand [A] 1983; 91: 145–50
- Möller-Nielsen M. Biological characterisation of soft tissue sarcomas. Odense University, OdenseDenmark 2001, PhD thesis: 207
- Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008; 113: 573–81
- Rydholm A. Management of patients with soft-tissue tumors. Strategy developed at a regional oncology center. Acta Orthop Scand Suppl 1983, 203: 13–77
- Rydholm A, Gustafson P. Should tumor depth be included in prognostication of soft tissue sarcoma?. BMC Cancer 2003; 3: 17
- Rööser B. Prognosis in soft tissue sarcoma. Acta Orthop Scand Suppl 1987, 225: 1–54
- Sarcoma Meta-analysis Collaboration. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Lancet 1997; 350: 1647–54
- Trojani M, Contesso G, Coindre JM, Rouesse J, Bui NB, de Mascarel A, Goussot JF, David M, Bonichon F, Lagarde C. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer 1984; 33: 37–42
- Wunder JS, Healey JH, Davis AM, Brennan MF. A comparison of staging systems for localized extremity soft tissue sarcoma. Cancer 2000; 88: 2721–30
- Alvegård T, Sigurdsson H, Mouridsen H, Solheim Ø, Unsgaard B, Ringborg U, Dahl O, Nordentoft AM, Blomqvist C, Rydholm A, Stener B, Ranstam J. Adjuvant chemotherapy with doxorubicin in high-grade soft tissue sarcoma: a randomized trial of the Scandinavian Sarcoma Group. J Clin Oncol 1989; 7(10)1504–13
- Bauer HC, Trovik CS, Alvegard TA, et al. Monitoring referral and treatment in soft tissue sarcoma: study based on 1,851 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand 2001; 72: 150–9
- Bese NS, Hendry J, Jeremic B. Effects of prolongation of overall treatment time due to unplanned interruptions during radiotherapy of different tumor sites and practical methods for compensation. Int J Radiat Oncol Biol Phys. 2007; 68(3)654–61
- Casali PG, Jost L, Sleijfer S, Verweij J, Blay JY. On behalf of the ESMO Guidelines Working Group. Soft tissue sarcomas: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Ann Oncol 2008; 19(supplement 2)ii89–ii93
- Cassier PA, Dufresne A, Blay JY, Fayette J. Trabectedin and its potensial in the treatment of soft tissue sarcoma. Therapeutics and Clinical Risk management 2008; 4(1)109–16
- Clark MA, Fisher CS, Judson I, Thomas JM. Soft-tissue sarcomas in adults. N Engl J Med 2005; 353(7)701–11
- Davis AM, O'Sullivan BO, Turcotte R, et al. Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiotherapy and Oncology 2005; 75: 48–53
- Eggermont AMM, de Wilt JHW, ten Hagen TLM. Current uses of isolated limb perfusion in the clinic and a model system for new strategies. Lancet Oncol 2003; 4: 429–37
- Eilber FC, Rosen G, Nelson SD, et al. High-grade extremity soft tissue sarcomas: factors predictive of local recurrence and its effect on morbidity and mortality. Ann Surg 2003; 237: 218–26
- Engellau J, Bendahl PO, Persson A, et al. Improved prognostication in soft tissue sarcoma: independent information from vascular invasion, necrosis, growth pattern, and immunostaining using whole-tumor sections and tissue microarrays. Hum Pathol 2005; 36: 994–1002
- Engellau J, Samuelsson V, Anderson H, Bjerkehagen B, Rissler P, Hall KS, Rydholm A. Identification of low-risk tumors in histological high-grade soft tissue sarcomas. Eur J Cancer 2007; 43: 1927–34
- Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop 1980, 153: 106–20
- Gustafson P. Soft tissue sarcoma. Epidemiology and prognosis in 508 patients. Acta Orthop Scand Suppl 1994; 259: 1–31
- Gustafson P. Soft tissue sarcoma Epidemiology and prognosis in 508 patients. Acta Orthop Scand Suppl 1994; 65: 259, Thesis
- Gustafson P, åkerman M, Alvegård TA, et al. Prognostic information in soft tissue sarcoma using tumour size, vascular invasion and microscopic tumour necrosis–the SIN-system. Eur J Cancer 2003; 39: 1568–76
- Harrison LB, Franzese F, Gaynor JJ, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in the management of completely resected soft tissue sarcomas of the extremity and superficial trunk. Int J Radiat Oncol Biol Phys 1993; 27: 259–65
- Issels RD, Lindner LH, Wust P, et al. Regional hyperthermia (RHT) improves response and survival when combined with systemic chemotherapy in the management of locally advanced, high grade soft tissue sarcomas (STS) of the extremities, the body wall and the abdomen: A phase III randomized pros. ASCO Ann Meet Proc Proc Part I. J Clin Oncol 2007; 25(18S)10009
- Issels RD. Regional hyperthermia in high-risk soft tissue sarcomas. Curr Opin Oncol 2008; 20(4)438–43
- Jebsen NL, Trovik CS, Bauer HCF, Rydholm A, Monge O, Hall KS, Alvegård T, Bruland Ø. Radiotherapy to improve local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma: a Scandinvian Sarcoma Group study. Int J Radiat Oncol Biol Phys 2008; 71(4)1196–203, Epub 2008 Jan 22
- Jemal A, Tiwari RC, Murray T, et al. Cancer statistics 2004. CA Cancer J Clin 2004; 54: 8–29
- Lindberg RD, Martin RG, Romsdahl MM, et al. Conservative surgery and postoperative radiotherapy in 300 adults with soft-tissue sarcomas. Cancer 1981; 47: 2391–97
- Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, Fanucchi M, Harmon DC, Schuetze SM, Reinke D, Thall PF, Benjamon RS, Baker LH, Hensley ML. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcoma: results of Sarcoma Alliance for Research through Collaboration Study 002. J Clin Oncol 2007; 25(19)2755–63
- Mollabashy A, Virkus WW, Zlotecki RA, et al. Radiation therapy for low-grade soft tissue sarcoma. Clin Orthop 2002, 397: 190–95
- MØller Nielsen M. Biological characterization of soft-tissue sarcomas – STS. Faculty of Health Sciences, Odense University, OdenseDenmark 2001; 207, PhD thesis
- Nilbert M, Meza-Zepeda LA, Francis P, Berner JM, NamlØs HM, Fernebro J, Myklebost O. Lessons from genetic profiling in soft tissue sarcomas. Acta Orthop Scand 2004; 75(Suppl 311)35–50
- Olsson H. An updated review of the epidemiology of soft tissue sarcoma. Acta Orthop Scand 2004; 75(Suppl 311)16–20
- O'Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 2002; 359: 2235–41
- Patel SR. Dose-intensive chemotherapy for soft-tissue sarcomas. Proc Am Soc Clin Oncol 2000; 453–7
- Pervaiz N, Colterjohn N, Farrokhyar F, Tozer R, Figueredo A, Ghert M. A systemic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 2008; 113(3)573–81
- Pisters PW, Harrison LB, Woodruff JM, et al. A prospective randomized trial of adjuvant brachytherapy in the management of low-grade soft tissue sarcomas of the extremity and superficial trunk. J Clin Oncol 1994; 12: 1150–5
- Pisters PW, Harrison LB, Leung DH, et al. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 1996; 14: 859–68
- Pisters PW, O'Sullivan B, Maki RG. Evidence-based recommendations for local therapy for soft tissue sarcomas. J Clin Oncol 2007; 25: 1003–8
- Reichardt P, Tilgner J, Hohenberger P, Dorken B. Dose-intensified chemotherapy with ifosfamide, epirubicin, and filgrastim for adult patients with metastatic or locally advanced soft tissue sarcoma: A phase II study. J Clin Oncol 1998; 16(4)1438–43
- Rydholm A, Rooser B. Surgical margins for soft-tissue sarcoma. J Bone Joint Surg (Am) 1987; 69(7)1074–8
- Rydholm A. Improving the management of soft tissue sarcoma: diagnosis and treatment should be given in specialist center. BMJ 1998; 317: 93–4
- Schwartz DL, Einck J, Hunt K, et al. The effect of delayed postoperative irradiation on local control of soft tissue sarcomas of the extremity and torso. Int J Radiat Oncol Biol Phys 2002; 52: 1352–59
- Strander H, Turesson I, Cavallin-Stahl E. A systematic overview of radiation therapy effects in soft tissue sarcomas. Acta Oncol 2003; 42: 516–31
- Stotter AT, A'Hern RP, Fisher C, Mott AF, Fallowfield ME, Westbury G. The influence of local recurrence of extremity soft tissue sarcoma on metastasis and survival. Cancer 1990; 65: 1119–29
- Suit HD, Spiro IJ. The role of radiation in patients with soft tissue sarcomas. Cancer Control 1994; 1(6)592–8
- Saeter G, Alvegård TA, Monge OR, et al. VIG chemotherapy in advanced soft tissue sarcoma results of the Scandinavian Sarcoma Group SSG X trial. Acta Orthop Scand 1995; 66(suppl 265)93–4
- Saeter G, Alvegård TA, Monge OR, Strander H, Turesson I, Klepp R, Söderberg M, Wist E, Raabe N, Erlanson M, Solheim OP, Hannisdal E. Ifosfamide and continuous infusion etoposide in advanced adult soft tissue sarcoma. A Scandinavian Sarcoma group phase II study. Eur J Cancer 1997; 33: 1551–8
- Tierney JF, Mosseri V, Stewart L, et al. Adjuvant chemotherapy for soft-tissue sarcoma: Review and meta-analysis of the published results of randomized clinical trials. Br J Cancer 1995; 72: 469–75
- Trovik CS, Bauer HC, Berlin O, et al. Local recurrence of deep seated, high-grade, soft tissue sarcoma: 459 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand 2001; 72: 160–6, (a)
- Trovik CS. Local recurrence of soft tissue sarcoma. A Scandinavian Sarcoma Group Project. Acta Orthop Scand 2001; 72(suppl 300)1–31, (b)
- Verhoef C, de Wilt JHW, Grünhagen DJ, van Geel AN, ten Hagen TLM, Eggermont AMM. Isolated limb perfusion with Melphalan and TNF-α in the treatment of extremity sarcoma. Current treatment Options in Oncology 2007; 8: 417–27
- Wiklund T, Saeter G, Strander H, Alvegård T, Blomqvist C. The outcome of advanced soft tissue sarcoma patients with complete tumour regression after either chemotherapy alone or chemotherapy plus surgery. The Scandinavian Sarcoma Group experience. Eur J Cancer 1997; 33: 357–61
- Wilson AN, Davis A, Bell RS, et al. Local control of soft tissue sarcoma of the extremity: the experience of a multidisciplinary sarcoma group with definitive surgery and radiotherapy. Eur J Cancer 1994; 30A: 746–51
- Woll PJ, van Glabekke M, Hohenberger P, et al. EORTC Soft Tissue & Bone Sarcoma Group. Adjuvant chemotherapy (CT) with doxorubicin and ifosfamide in resected soft tissues sarcoma (STS): interim analysis of a randomised phase III trial. J Clin Oncol ASCO Annual Meeting Proc 2007; 25(18S), abstract 10008
- Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 1998; 16: 197–203
- Zagars GK, Ballo MT. Significance of dose in postoperative radiotherapy for soft tissue sarcoma. Int J Radiat Oncol Biol Phys 2003; 56: 473–81, (a)
- Zagars GK, Ballo MT, Pisters PW, et al. Prognostic factors for patients with localized soft-tissue sarcoma treated with conservation surgery and radiation therapy: an analysis of 225 patients. Cancer 2003; 97: 2530–43
- American Society of Clinical Oncology. Recommendations for the use of hematopoietic colony-stimulating factors: evidence-based clinical practice guidelines. J Clin Oncol 1994; 12: 2471–508
- Bacci G, Ferrari S, Bertoni F, et al. Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the istituto ortopedico Rizzoli according to the istituto orotpedico Rizzoli/osteosarcoma-2 protocol: an update report. J Clin Oncol 2000; 18: 4016–27
- Bacci G, Forni C, Longhi A, et al. Local recurrence and local control of non-metastatic osteosarcoma of the extremities: a-27-year experience in a single institution. J Surg Oncol 2007; 96: 118–23
- Bacci G, Picci P, Ferrari S, et al. Primary chemotherapy and delayed surgery for nonmetastatic osteosarcoma of the extremities. Cancer 1993; 72: 3227–8
- Bielack S, Kempf-Bielack B, Delling G, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: An analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 2002; 20: 776–90
- Bruland OS, Pihl A. On the current management of osteosarcoma. A critical evaluation and a proposal for a modified treatment strategy. Eur J Cancer 1997; 33: 1725–31
- Enneking WF, Spanier S, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop 1980, 153: 106–20
- Ferrari S, Briccoli A, Mercuri M, et al. Postrelapse survival in osteosarcoma of the extremities: prognostic factors for long-term survival. J Clin Oncol 2003; 21: 710–5
- Ferrari S, Smeland S., Mercuri M, et al. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 2005; 23: 8845–52
- Fuchs N, Bielack SS, Epler D, et al. Long-term results of the co-operative German-Austrian-Swiss osteosarcoma study group's protocol COSS-86 of intensive multidrug chemotherapy and surgery for osteosarcoma of the limbs. Ann Oncol 1998; 9: 893–9
- Huvos AG, Rosen G, Marcove C. Primary osteogenic sarcoma. Pathologic aspects in 20 patients after treatment with chemotherapy, en bloc resection and prosthetic bone replacement. Arch Pathol Lab Med 1977; 101: 14–8
- Kempf-Bielack B, Bielack SS, Jürgens H, et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 2005; 23: 559–68
- Lewis IJ, Nooij MA, Whelan J, et al. Improvement in histologic response but not survival in osteosarcoma patietns treated with intensified chemotherapy:a randomized phase III trial of the European Ostesarcoma Intergroup. J Natl Cancer Inst 2007; 99: 112–18
- Link MP, Goorin AM, Miser AW, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 1986; 314: 1600–6
- Meyers P, Schwartz CL, Krail M, et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamdie and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 2005; 23: 2004–11
- Rosen G, Caparros B, Huvos AG, et al. Preoperative chemotherapy for osteogenic sarcoma: Selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer 1982; 49: 1221–30
- Rosen G, Murphy ML, Huvos AG, et al. Primary osteogenic sarcoma. The rationale for preoperative chemotherapy and delayed surgery. Cancer 1979; 43: 2163–77
- Smeland S, Müller C, Alvegaard TA, et al. Scandinavian Sarcoma Group Osteosarcoma study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur J Cancer 2003; 39: 488–94
- Saeter G, Alvegård TA, Elomaa I, et al. Treatment of osteosarcoma of the extremities with the T-10 protocol, with emphasis on the effects of preoperative chemotherapy with single-agent high-dose methotrexate: A Scandinavian Sarcoma Group study. J Clin Oncol 1991; 9: 1766–75
- Saeter G, Høie J, Stenwig AE, et al. Systemic relapse of patients with osteogenic sarcoma: prognostic factors for long term survival. Cancer 1995; 75: 1084–93
- Winkler K, Beron G, Delling G, et al. Neoadjuvant chemotherapy of osteosarcoma: results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. J Clin Oncol 1988; 6: 329–37
- Dorfman HD, Czerniak B. Bone cancers. Cancer 1995; 75(1 Suppl)203–10
- International Agency for Research on Cancer (IARC). Pathology and Genetics of Tumours of Soft Tissue and Bone. World Health Organization Classification of Tumours., CDM Fletcher, K Unni, F Mertens, 2002; 227–8, Available from http://www.iarc.fr/en/Publications/PDFs-online/Cancer-Pathology-and-Genetics/World-Health-Organization-Classification-of-Tumours2
- Grimer RJ, Cannon SR, Taminiau AM, Bielack S, Kempf-Bielack B, Windhager R, Dominkus M, Saeter G, Bauer H, Meller I, Szendroi M, Folleras G, San-Julian M, van der Eijken J. Osteosarcoma over the age of forty. Eur J Cancer 2003; 39(2)157–63
- Miller PD. Safety of parathyroid hormone for the treatment of osteoporosis. Curr Osteoporos Rep 2008; 6(1)12–6
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344(19)1434–41
- Ries LAG, Wingo PA, Miller DS, Howe HA, Weir HK, Rosenberg HM, Vernon SW, Cronin K, Edwards BK. The annual report to the nation on the status of cancer, 1973–1997, with a special section on colonrectal cancer. Cancer 2000; 88: 2398–424
- Smeland S, Wiebe T, Böhling T, Brosjö O, Jonsson K, Alvegård TA. Chemotherapy in osteosarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand Suppl 2004; 75(S 311)92–8
- Socialstyrelsen. Cancer incidence in Sweden 2006. Statistics: health and diseases 2007:16 series. 2007 Dec 19. Available from http://www.socialstyrelsen.se/NR/rdonlyres/AAD7CA00-CCE3-4E3E-B998-1450BD7FB4A1/9428/20074216.pdf
- Stark A, Kreicbergs A, Nilsonne U, Silfverswärd C. The age of osteosarcoma patients is increasing. An epidemiological study of osteosarcoma in Sweden 1971 to 1984. J Bone Joint Surg Br 1990; 72(1)89–93
- Unni KK, Dahlin DC. Dahlin's bone tumor: general aspects and data on 11,087 cases. Lippincott-Raven, Philadelphia 1996; 5: 67–9; 143–4
- Vahle JL, Long GG, Sandusky G, Westmore M, Ma YL, Sato M. Bone neoplasms in F344 rats given teriparatide [rhPTH(1-34)] are dependent on duration of treatment and dose. Toxicol Pathol 2004; 32(4)426–38
- Vahle JL, Sato M, Long GG, Young JK, Francis PC, Engelhardt JA, Westmore M, Ma L, Nold JB. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone(1-34) for 2 years and relevance to human safety. Toxicol Pathol 2002; 30: 312–21
- Vahle JL, Zuehlke U, Schmidt A, Westmore M, Chen P, Sato M. Lack of bone neoplasms and persistence of bone efficacy in cynomolgus macaques following long-term treatment with teriparatide {rhPTH(1-34)}. J Bone Miner Res 2008, Published online August 6, 2008: 10.1359/jbmr.080807
- Alvegård TA, Solheim Ø, åkerman M, et al. Ewing's sarcoma. A preliminary report from the Scandinavian Sarcoma Group. Acta Oncol 1989; 28(Suppl 2)59–64
- Bacci G, Picci P, Avella M, et al. Neoadjuvant chemotherapy for localized Ewing's sarcoma of bone: experience at the Istituto Ortopedica Rizzoli. Cancer 1998; 82: 1174–83
- Bacci G, Ferrari S, Bertoni F, et al. Prognostic factors in Ewing's sarcoma of bone treated with adjuvant chemotherapy: analysis of 359 patients at the Istituto Ortopedico Rizzoli. J Clin Oncol 2000; 18: 4–11
- Bacci G, Mercuri M, Longhi A, et al. Neoadjuvant chemotherapy for Ewing's tumour of bone:recent experience at the Rizzoli Orthopaedic Institute. Eur J Can 2002; 38: 2243–51
- Burgert EOJ, Nesbit ME, Garnsey LA, et al. Multimodal therapy for the management of nonpelvic, localized Ewing's sarcoma of bone: Intergroup study IESS-II. J Clin Oncol 1990; 8: 1514–24
- Castello MA, Dominici C, Clerico A. A pilot study of 5-day continuous infusion of high-dose cisplatin and pulsed etoposide in childhood solid tumors. Am J Pediatr Hematol Oncol 1988; 10: 103–8
- Elomaa I, Blomqvist CP, Saeter G, et al. Five-year results in Ewing's sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Eur J Cancer 2000; 36: 875–80
- Ferrari S, Alvegard T, et al. Non-metastatic Ewing's family tumors: High-dose chemotherapy with stem cell rescue in poor responder patients. Preliminary results of the Italian/Scandinavian ISG/SSG III protocol. ASCO Meeting Abstracts 2007; 25: 10014
- Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003; 348: 694–701
- Jürgens H, Exner U, Gardner H, et al. Multidsciplinary treatment of primary Ewing's sarcoma of bone. A 6-year experience of a European cooperative trial. Cancer 1988; 61: 23–32
- Khoury JD. Ewing sarcoma family of tumors: a model for the new era of integrated laboratory diagnostics. Expert Rev Mol Diagn 2008; 8: 97–104
- LeRoith D, Helman L. The new kid on the block(ade) of the IGF-1 receptor. Cancer Cell 2004; 5: 201–2
- Nilbert M, Saeter G, Elomaa I, et al. Ewing's sarcoma treatment in Scandinavia 1984–1990. Ten-year results of the Scnadinavian Sarcoma group protocol SSG IV. Acta Oncol 1998; 37: 375–8
- Oberlin O, Delaey MC, Bui BN, et al. Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 2001; 30: 1646–54
- Olmos D, Okuno S, Schuetze SM, et al. Safety, pharmacokinetics and preliminary activity of the anti-IGF-IR antibody CP-751,871 in patients with sarcoma. ASCO Meeting Abstracts 2008; 26: 10501
- Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumour of bone: final results of the cooperative Ewing's sarcoma study CESS 86. J Clin Oncol 2001; 19: 1818–29
- Paulussen M, Ahrens S, Lehnert M, et al. Second malignancies after Ewing tumor treatement in 690 patietns from a cooperative German/Austrian/Dutch study. Ann Oncol 2001; 12: 1619–30
- Picci P, Rougraff BT, Bacci G, et al. Prognostic significance of histopathologic response to chemotherapy in nonmetastatic Ewing's sarcoma of the extremities. J Clin Oncol 1993; 11: 1763–9
- Picci P, Böhling T, Bacci G, et al. C Chemotherapy induced tumor necrosis as a prognostic factor in localized Ewing's sarcoma of the extremities. J Clin Oncol 1997; 15: 1553–9
- Tursz T, Guillot T, Le Chevalier T, et al. A pilot study of common combination chemotherapy (PAVI) for neuroectodermal tumors in young adults: Ewing's sarcoma, malignant neuroepithelioma, Askin's tumor. ASCO 1989; 8: A1231
- Womer RB, West DC, Krailo MD, et al. Randomized comparison of every-two-week vs. every-three-week chemotherapy in Ewing sarcoma family tumors (ESFT). ASCO Meeting Abstracts 2008; 26: 10504
- Dreinhöfer K, Rydholm A, Bauer HCF, Kreicbergs A. Giant cell tumors with fracture at diagnosis. Curettage and acrylic cementation in 10 of 15 cases. J Bone Joint Surg (Br) 1995; 77-B: 189–93
- Kivioja AH, Blomqvist C, Hietaniemi K, Trovik C, Walloe A, Bauer HC, Jorgensen PH, Bergh P, Follerås G. Cement is recommended in intralesional surgery of giant cell tumors: A Scandinavian Sarcoma Group study of 294 patients followed for a median time of 5 years. Acta Orthop 2008; 79(1)86–93
- Persson BM, Wouters HW. Curettage and acrylic cementation in surgery of giant cell tumors of bone. Clin Orthop 1976, 120: 125–33
- Vult von Steyern F, Bauer HCF, Trovik C, Kivioja A, Bergh P, Holmberg Jörgensen P, Follerås G, Rydholm A. Scandinavian Sarcoma Group. Treatment of local recurrences of giant cell tumour in long bones after curettage and cementing. J Bone Joint Surg (Br) 2006; 88-B: 531–5
- Vult von Steyern F, Kristiansson I, Jonsson K, Mannfolk P, Heinegård D, Rydholm A. Giant-cell tumour of the knee. The condition of the cartilage after treatment by curettage and cementing. J Bone Joint Surg (Br) 2007; 89-B(3)361–365
- Agrawal K, Subbarao K, et al. An innovative method of reconstruction of large skeletal chest wall defects. 30th Annual National Conference of Plastic-Surgeons-of-India. ThiruvananthapuramIndia 1995
- Bjornsson J, McLeod R, et al. Primary chondrosarcoma of long bones and limb girdles. Cancer 1998; 83(10)2105–19
- Bruns J, Elbracht M, et al. Chondrosarcoma of bone: An oncological and functional follow-up study. Annals of Oncology 2001; 12(6)859–64
- Burt M, Fulton M, et al. Primary bony and cartilaginous sarcomas of chest wall: results of therapy. Ann Thorac Surg 1992; 54(2)226–32
- Fiorenza F, Abudu A, et al. Risk factors for survival and local control in chondrosarcoma of bone. J Bone Joint Surg Br 2002; 84(1)93–9
- Fong YC, Pairolero PC, et al. Chondrosarcoma of the chest wall: a retrospective clinical analysis. Clin Orthop Relat Res 2004, 427: 184–9
- Kreicbergs A, Bauer HCF, et al. Cytological diagnosis of bone tumours. J Bone Joint Surg Br 1996; 78B(2)258–63
- Lioulias AG, Kokotsakis JN, et al. Radical resection of a giant chondrosarcoma of the anterior chest wall. Ann Thorac Surg 2003; 75(1)296
- McAfee MK, Pairolero PC, et al. Chondrosarcoma of the chest wall: factors affecting survival. Ann Thorac Surg 1985; 40(6)535–41
- Soderstrom M., Ekfors T. O., et al. No improvement in the overall survival of 194 patients with chondrosarcoma in Finland in 1971-1990. Acta Orthop Scand 2003; 74(3)344–50
- Widhe B, Bauer HCF. J Thorac Cardiovasc Surg. 2009, in press
- Willen H. Fine needle aspiration in the diagnosis of bone tumors. Acta Orthop Scand 1997; 68: 47–53
- Allan DG, Bell RS, Davis A, et al. Complex acetabular reconstruction for metastatic tumor. J Arthroplasty 1995; 10: 310–6
- Barwood SA, Wilson JL, Molnar RR. The incidence of acute cardiorespiratory and vascular dysfunction following intramedullary nail fixation of femoral metastasis. Acta Orthop Scand 2000; 71: 147–52
- Bauer HC. Controversies in the surgical management of skeletal metastases. J Bone Joint Surg Br 2005; 87(5)608–17
- Bauer HFC, Wedin R. Survival after surgery for spinal and extremity metastases. Prognostication in 241 patients. Acta Orthop Scand 1995; 66: 143–6
- Bohm P, Huber J. The surgical treatment of bony metastases of the spine and limbs. J Bone Joint Surg Br 2002; 84(4)521–9
- Bono B, Cazzaniga P, Zurrida SM, et al. Palliative surgery of metastatic bone disease: a review of 83 cases. Eur J Cancer 1991; 27(5)556–8
- Cheng EY. Prospective Quality of Life Research in bony metastatic disease. Clin Orthop 2003, 415: 289–97
- Clohisy DR, Le CT, Dykes DC, et al. Evaluation of the feasibility of and results of measuring health-status in patients undergoing surgical treatment for skeletal metastases. J Orthop Res 2000; 18: 1–9
- Frassica FJ, Frassica DA. Evaluation and treatment of metastases to the humerus. Clin Orthop 2003, 415: 212–18
- Diel IJ, Boby JJ, Lichinitser MR, et al. Improved quality of life after log-term treatment with the biphosphonate ibanronate in patients with metastatic bone disease due to breast cancer. Eur J Cancer 2004; 40(11)1704–12
- Dürr HR, Refior HJ. Prognosis of skeletal metastases. Orthopade 1998; 27(5)294–00
- Hansen BH, Keller J, Laitinen M, et al. The Scandinavian Sarcoma Group Skeletal Metastases Register. Survival after surgery for bone metastases in the pelvis and extremities. Acta Orthop Scand 2004; 75(Suppl 311)11–15
- Kelly CM, Wilkins RM, Eckardt JJ, et al. Treatment of metastatic disease of the tibia. Clin Orthop 2003, 415 Suppl: S219–29
- Kunisada T, Choong PF. Major reconstruction for periacetabular metastasis: early complications and outcome following surgical treatment in 40 hips. Acta Orthop Scand 2000; 71: 585–90
- Marco RAW, Sheth DS, Boland PJ, et al. Functional and oncological outcome of acetabular reconstruction for the treatment of metastatic disease. J Bone Joint Surg Am 2000; 82: 642–51
- Nathan SA, Healey JH, Mellano D. Survival in patients operated on for pathological fracture: Implications for end-of-life orthopaedic care. J Clin Oncol 2005; 23: 6072–82
- Pavlakis N, Schmidt R, Stockler M. Biphosphonates for breast cancer. Cochrane Database Syst Rev. 2005; 3: CD003474
- Tokuhashi Y, Kawano H, Toriyama S. Score assessment of metastatic bone tumour for operative treatment. Limb salvage- major reconstructions in oncologic and nontumoral conditions., F Langlais, B Tomeno. Springer Verlag, Berlin Heidelberg 1991; 725–32
- Ward WG, Holsenbeck SBA, Dorey FJ, et al. Metastatic Disease of the Femur: Surgical Treatment. Clin Orthop 2003, 415: 230–44
- Vena VE, James BS, Rosier RN, et al. Pelvic reconstruction for severe periacetabular metastatic disease. Clin Orthop 1999, 362: 171–80
- Wedin R, Bauer HC, Ruthquist LE. Surgical treatment for skeletal breast cancer metastases: A population-based study of 641 patients. Cancer 2001; 92: 257–62
- Wedin R., Bauer HC. Surgical treatment of skeletal metastatic lesions of the proximal femur?: endoprosthesis or reconstruction nail. J Bone Joint Surg Br 2005; 87(12)1653–7
- Wedin R, Bauer HC, Wersäll P. Failures after operation for skeletal metastatic lesions of long bones. Clin Orthop 1999, 358: 128–39
- Yazawa Y, Frassica FJ, Chao EY, et al. Metastatic bone diasease: A study of the surgical treatment of 166 patients with humeral and femoral fractures. Clin Orthop 1990, 251: 213–9
- Sze WM, Shelly MD, Held I, et al. Palliation of metastatic bone pain: single fraction versus multifraction radiotherapy – a systematic rewiew of randomized trials. Clin Oncol (R Coll Radiol) 2003; 15: 342–4
Denmark
- Le Cesne A, Blay JY, Judson I, van Oosterom A, Verweij J, Radford J, Lorigan P, Rodenhuis S, Ray-Coquard I, Bonvalot S, Collin F, Jimino J, Di Paola E, van Glabbeke M, Nielsen OS. Phase 2 study of ET-743 in advanced soft tissue sarcomas: An EORTC soft tissue and bone sarcoma trial. J Clin Oncol 2005; 23: 576–584
2006
- Hansen JK, Sørensen FB, Christensen MF. A five-week-old girl with inspiratory stridor due to infantile hemangiopericytoma. Eur Arch Otorhinolaryngol 2006; 8: 1–4
- Nielsen OS, Reichardt P, Pink D, Christensen TB, Daugaard S, Hermans C, Marreaud S, van Glabbeke M, Judson I. Phase 1 EORTC study determining safety of pegulated liposomal doxorubicin (Caelyx) in combination with ifosfamide in previously untreated adult patients with advanced or metastatic soft tissues. Eur J Cancer 2006; 42: 2303–09
2007
- Reichardt P, Nielsen OS, Bauer S, Hartmann JT, Schöffski P, Christensen TB, Pink D, Daugaard S, Marreaud S, van Glabbeke M, Blay JY. Exetecan in pretreated adult patients with advanced soft tissue sarcoma: Results of a phase II-study of the EORTC soft tissue and bone and sarcoma group. Eur J Cancer 2007; 43: 1017–22
Finland
- Airla N, Luomala M, Elovaara I, Kettunen E, Knuutila S, Lehtimäki T. Suppression of im-mune system genes by methylprednisolone in exacerbations of multiple sclerosis. Preliminary results. J Neurol 2004; 251: 1215–1219
- Alvegård TA, Bauer H, Blomqvist C, Rydholm A, Smeland S. The Scandinavian Sarcoma Group–background, organization and the SSG Register–the first 25 years. Acta Orthop Scand Suppl 2004; 75: 1–7
- Cerisano V, Aalto Y, Perdichizzi S, Bernard G, Manara MC, Benini S, Cenacchi G, Preda P, Lattanzi G, Nagy B, Knuutila S, Colombo MP, Bernard A, Picci P, Scotlandi K. Molecular mechanisms of CD99-induced caspase-independent cell death and cell-cell adhesion in Ewing's sarcoma cells: actin and zyxin as key intracellular mediators. Oncogene 2004; 23: 5664–5674
- Hattinger CM, Tarkkanen M, Benini S, Pasello M, Stoico G, Bacchini P, Knuutila S, Scotlandi K, Picci P, Serra M. Genetic analysis of fibrosarcoma of bone, a rare tumour entity closely related to osteosarcoma and malignant fibrous histiocytoma of bone. Eur J Cell Biol 2004; 83: 483–491
- Knuutila S. Cytogenetics and molecular pathology in cancer diagnostics. Review. Ann Med 2004; 36: 162–171
- Kresse SH, Berner JM, Meza-Zepeda LA, Gregory SG, Knuutila S, Myklebost O, Forus A. Mapping and characterisation of a novel amplicon at 1q23 in human sarcomas and desmoid tumors. AACR 95th Annual Meeting, Orlando, Florida, March, 27-312004. 45: 393, The Proceedings of the AACR 2004, Abstract
- Larramendy ML, Koljonen V, Böhling T, Tukiainen E, Knuutila S. Recurrent DNA copy number changes revealed by comparative genomic hybridization in primary Merkel cell carcinomas. Mod Pathol 2004; 17: 561–567
- Mandahl N, Mertens F, Panagopoulos I, Knuutila S. Genetic characterization of bone and tissue tumors. Acta Orthop Scand 2004; 75(Suppl 311)21–28
- Nissi R, Böhling T, Autio-Harmainen H. Immunofluorescence localization of propyl 4-hydroxylase isoenzymes in bone tumours. Type I enzyme predominates in osteo- and chon-drosarcomas, but type II enzyme is more clearly expressed in the benign counterparts. Acta Histochemica 2004; 18: 111–121
- Popov P, Tukiainen E, Asko-Seljavaara S, Huuhtanen R, Virolainen M, Virkkunen P, Blomqvist C. Soft-tissue sarcomas of the upper extremity - surgical treatment and outcome. Plastic and Reconstructive Surgery 2004; 113: 222–30
- Sirén V, Antinheimo J, Jääskeläinen J, Böhling T, Carpén O, Vaheri A. Plasminogen acti-vation in neurofibromatosis 2-associated and sporadic schwannomas. Acta Neurochir (Wien) 2004; 146: 111–118
2005
- Atiye J, Wolf M, Kaur S, Monni O, Böhling T, Kivioja A, Tas E, Serra M, Tarkkanen M, Knuutila S. Gene amplifications in osteosarcoma - CGH microarray analysis. Genes Chrom Cancer 2005; 42(2)158–63
- Ferrari S, Smeland S, Mercuri M, Bertoni F, Longhi A, Ruggieri P, Alvegard T, Picci P, Capanna R, Bernini G, Muller C, Tienghi A, Wiebe T, Comandone A, Böhling T, Brach Del Prever A, Brosjö O, Bacci G, Saeter G. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin and doxorubicin for patients with localized osteosarcoma of the extremity. A joint study by the Italian and Scandi-navian sarcoma groups. J Clin Oncol 2005; 23: 8845–8852
- Lassus P, Ristimäki A, Huuhtanen R, Tukiainen E, Asko-Seljavaara S, Andersson LC, Miettinen M, Blomqvist C, Haglund C, Böhling T. Cyclooxygenase-2 expression in human soft-tissue sarcomas is related to epithelial differentiation. Anticancer Res 2005; 25: 2669–2674
- Myllykangas S, Nagy B, Saharinen J, Knuutila S. Association of DNA copy number amplification and fragile sites in human neoplasias. Fragilome – Chromosomal Insta-bility, Fragile Sites, and Cancer, Heidelberg, German Cancer Research Center, DKFZ. February 17-19, 2005; 3, Abstract
- Sebenik M, Ricci A, Dipasquale B, Mody K, Pytel P, Jee KJ, Knuutila S, Scholes J. Undifferentiated intimal sarcoma of large systemic blood vessels: Report of thirteen cases with immunohistochemical profile and review of the literature. Am J Surg Pathol 2005; 29: 1184–1193
- Sihto H, Puputti M, Pulli L, Tynninen O, Koskinen W, Aaltonen L-M, Tanner M, Böhling T, Visakorpi T, Bützow R, Knuutila A, Nupponen N, Joensuu H. Epidermal growth factor receptor domain II, IV and kinase domain mutations in human solid tumors. J Mol Med 2005; 83: 976–983
2006
- Kaur S, Vauhkonen A, Böhling T, Mertens F, Mandahl N, Knuutila S. Gene copy number changes in dermatofibrosarcoma protuberans – a fine-resolution study using array comparative genomic hybridization. Cytogenet Genome Res 2006; 115: 283–288
- Kaur S, Larramendy M, Gentile M, Svarvar C, Koivisto-Korander R, Vauhkonen H, Scheinin I, Leminen A, Butzow R, Böhling T, Knuutila S. New insights into the cellular pathways affected in primary uterine leiomyosarcoma. Cancer Genomics Pro-teomics 2006; 3: 347–354
- Knuutila S. Kromosomi- ja geenimuutosten kliininen merkitys sarkoomissa. Review. Focus Oncologiae. Proceedings of the Annual Cancer Symposium of the Foundation for the Finnish Cancer Institute 2006; 7: 14–22
- Larramendy ML, Kaur S, Svarvar C, Böhling T, Knuutila S. Gene copy number profiling of soft-tissue leiomyosarcomas by array-comparative genomic hybridization. Cancer Gen Cytogen 2006; 169: 94–101
- Myllykangas S, Himberg J, Böhling T, Nagy B, Hollmén J, Knuutila S. DNA copy number amplification profiling of human neoplasms. Oncogene 2006; 25: 7324–7332
- Myllykangas S, Knuutila S. Manifestation, mechanisms and mysteries of gene am-plifications. Review. Cancer Lett 2006; 232: 79–89
- Svarvar C, Larramendy M, Blomqvist C, Gentile M, Koivisto-Korander R, Leminen A, Butzow R, Böhling T, Knuutila S. Do DNA copy number changes differentiate uterine from non-uterine leiomyosarcomas and predict metastasis?. Mod Pathology 2006; 19: 1068–82
- Tarkkanen M, Larramendy M, Böhling T, Serra M, Hattinger CM, Kivioja A, Elomaa I, Picci P, Knuutila S. Malignant fibrous histiocytoma of bone: Analysis of genomic imbalances by comparative genomic hybridization and C-MYC expression by immu-nohistochemistry. Eur J Cancer 2006; 42: 1172–1180
- Tyybäkinoja A, Knuutila S. Molekyylikaryotyypitys – raja sytogenetiikan ja mole-kyylibiologian väliltä häviämässä. Review. Duodecim 2006; 122: 2018–22
- Vauhkonen H, Savola S, Kaur S, Larramendy ML, Knuutila S. Molecular karyoty-ping in sarcoma diagnostics and research. Review. Adv Exp Med Biol 2006; 587: 53–63
2007
- Kaur S, Larramendy M, Vauhkonen S, Böhling T, Knuutila S. Loss of TP53 in sarcomas with 17p11∼p12 gain. A fine-resolution oligonucleotide array comparative genomic hybridization study. Cytogenet Genome Res 2007; 116: 153–157
- Kivinen K, Peterson H, Hiltunen L, Laivuori H, Heino S, Tiala I, Knuutila S, Rasi V, Kere J. Evaluation of STOX1 as a preeclampsia candidate gene in a population-wide sample. Eur J Hum Genet 2007; 15: 494–497
- Knuutila S. Molekyylipatologia – uudet diagnostiset syto- ja molekyyligenetiikan me-netelmät syöpätaudeissa. Moodi 2007; 6: 209–212
- Knuutila S. Geenit kromosomeissa; sytogenetiikan perusteet. Perinnöllisyyslääketie-de, 3rd edition, P Aula, H Kääriäinen, A Palotie. Kustannus Oy Duodecim. 2007; 31–47
- Myllykangas S, Böhling T, Knuutila S. Specificity, selection and significance of gene amplifications in cancer. Review. Semin Cancer Biol 2007; 17: 42–55
- Popov P, Böhling T, Asko-Seljavaara S, Tukiainen E. Microscopic margins and result in the surgery for dermatofibrosarcoma protuberans. Plast Reconstr Surg 2007; 119: 1779–1784
- Savola S, Nardi F, Scotlandi K, Picci P, Knuutila S. Microdeletions in 9p21.3 induce false negative results in CDKN2A FISH analysis of Ewing sarcoma. Cytogenet Geno-me Res 2007; 119: 21–26, (Epub 14 Dec 2007)
- Svarvar C, Böhling T, Berlin ö, Gustafson P, Follerås G, Bjerkehagen B, Domanski H, Tukiainen E. Blomqvist C and the SSG leiomyosarcoma working group. Clinical course of non-visceral soft tissue leiomyosarcoma in 225 patients from the Scandina-vian Sarcoma Group register. Cancer 2007; 15 109(2)282–291
- Vauhkonen H, Heino S, Myllykangas S, Lindholm PM, Savola S, Knuutila S. Etiology of specific molecular alterations in human malignancies. Review. Cytogenet Ge-nome Res 2007; 118: 277–283
2008
- Kivioja A, Blomqvist C, Hietaniemi K, Trovik C, Walloe A, Bauer H, Holmberg Jørgensen P, Bergh P, Follerås G. Cement is recommended in intralesional surgery of giant cell tumors. A study of 294 patients by the Scandinavian Sarcoma Group. Acta Orthop 2008; 79: 86–93
- Knuutila S. Molecular karyotyping in cancer diagnosis. Abstracts of the 34th European Congress of Cytopathology, Rovaniemi, 15-18 June 2008. Cytopathology 2008; 19(s1)30, Abstract
- Myllykangas S, Tikka J, Böhling T, Knuutila S, Hollmén J. Classification of human cancers based on DNA copy number amplification modeling. BMC Medical Genomics 2008; 1: 15
- Larramendy M, Gentile M, Soloneski S, Knuutila S, Böhling T. Does comparative genomic hybridization reveal distinct differences in DNA copy number sequence patterns between leiomyosarcoma and malignant fibrous histiocytoma?. Cancer Gen Cytogen 2008; 187: 1–11
- Scheinin I, Ferreira JA, Knuutila S, Meijer GA, van de Wiel MA, Ylstra B. Sample size calculations for array CGH experiments. Poster abstract. MC-GARD Workshop “Genome bioinformatic techniques”, Life and Health Science Research Institute (ICVS), School of Health Sciences (ECS), University of Minho. Braga September 8-13, 2008
- Scheinin I, Myllykangas S, Borze I, Knuutila S, Saharinen J. CanGEM: mining gene copy number changes in cancer. Nucleic Acids Res 2008; 36(Database issue)D830–835, (Epub 11 Oct 2007)
Norway
- Bauer HC, Alvegard TA, Berlin O, Erlanson M, Kalen A, Lindholm P, Gustafson P, Smeland S, Trovik CS. The Scandinavian Sarcoma Group Register 1986-2001. Acta Orthop Scand Suppl 2004; 75(311)8–10
- Hansen BH, Keller J, Laitinen M, Berg P, Skjeldal S, Trovik C, Nilsson J, Walloe A, Kalen A, Wedin R. The Scandinavian Sarcoma Group Skeletal Metastasis Register. Survival after surgery for bone metastases in the pelvis and extremities. Acta Orthop Scand Suppl 2004; 75(311)11–15
2005
- Haugland HK, Jebsen NL, Mannelqvist M, Skar R, Eide J, øvrebø K, Horn A, Jensen DK, Monge OR, Lilleng PK. Behandling av pasienter med. Treatment of patients with gastrointestinal stromal tumor (GIST) medtumour with imatinib mesylate. Tidsskr. Nor Lægeforen, 125, 868-872, 2005. Laegeforen 2005; 125(7)868–72
2006
- Helgestad J, Monge OR, Ulvestad E, Svendsen F, Eide J, Rosendahl K, Houge G, Trovik CS. Cancer in children–good results can be even better. Tidsskr Nor Laegeforen. 2006; 126(7)926–9
- Vult von Steyern F, Bauer HC, Trovik C, Kivioja A, Bergh P, Holmberg Jorgensen P, Folleras G, Rydholm A. Scandinavian Sarcoma Group. Treatment of local recurrences of giant cell tumour in long bones after curettage and cementing. A Scandinavian Sarcoma Group study. J Bone Joint Surg Br 2006; 88(4)531–5
2007
- Aksnes LH, Hall KS, Jebsen N, Fosså SD, Dahl AA. Young survivors of malignant bone tumours in the extremities: a comparative study of quality of life, fatigue and mental distress. Support Care Cancer 2007; 15(9)1087–96, Epub 2007 Mar 9
- Svarvar CD, Böhling T, Berlin ö, Gustafson P, Follerås G, Bjerkehagen B, Domanski HA, Sundby Hall K, Tukiainen E, Blomqvist C, the Scandinavian Sarcoma Group Leiomyosarcoma Working Group. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian Sarcoma Group. Cancer 2007; 109(2)282–291
2008
- Engström K, Bergh P, Gustafson P, Hultborn R, Johansson H, Löfvenberg R, Sundby Hall K, Trovik CS, Wahlstöm O, Bauer HCF. Liposarcoma - Outcome based on 237 patients from the Scandinavian Sarcoma Group Register. Accepted. Cancer Aug 20, 2008, (Epub ahead of print)
- Jebsen N, Trovik C, Bauer B, Rydholm A, Monge O, Sundby Hall K, Alvegård, Bruland ØS. Radiotherapy improves local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma. A study of 1093 patients from the Scandinavian Sarcoma Group. Int J Radiat Oncol Biol Phys 2008; 71(4)1196–203, Epub 2008 Jan 22
- Kivioja AH, Blomqvist C, Hietaniemi K, Trovik C, Walloe A, Bauer HCF, Holmberg Jorgensen P, Bergh P, Follerås G. Cement is recommended in intralesional surgery of giant cell tumors. A study of 294 patients followed for a median of 5.2 years. A Scandinavian Sarcoma Group study. Acta Orthop 2008; 79(1)86–93
Oslo
- Alvegard TA, Bauer H, Blomqvist C, Rydholm A, Smeland S. The Scandinavian Sarcoma Group–background, organization and the SSG Register–the first 25 years. Acta Orthop Scand Suppl 2004; 75(311)1–7
- Bauer HC, Alvegard TA, Berlin O, Erlanson M, Kalen A, Lindholm P, Gustafson P, Smeland S, Trovik CS. The Scandinavian Sarcoma Group Register 1986-2001. Acta Orthop Scand Suppl 2004; 75(311)8–10
- Brandal P, Bjerkehagen B, Heim S. Molecular cytogenetic characterization of tenosynovial giant cell tumors. Neoplasia 2004; 6(5)578–83
- Bruheim S, Bruland ØS, Breistøl K, Mælandsmo GM, Fodstad Ø. Human osteosarcoma xenografts and their sensitivity to chemotherapy. Pathol Oncol Res 2004; 10(3)133–41
- Böhling T, Bacchini P, Bertoni F, Bjerkehagen B, Domanski H, Kindblom L-G, Meis-Kindblom J, Weide J. Diagnosis and tumor response in osteosarcoma and Ewing's sarcoma. Acta Orthop Scand Suppl 2004; 75(311)72–6
- Cekaite L, Haug O, Myklebost O, Aldrin M, østenstad B, Holden M, Frigessi A, Hovig E, Sioud M. Analysis of the humoral immune response to immunoselected phagedisplayed peptides by a microarray-based method. Proteomics 2004; 4(9)2572–82
- Davies CLD, Lundstrøm LM, Frengen J, Eikenes L, Bruland ØS, Kaalhus O, Brekken C. Radiation improves the distribution and uptake of liposomal doxorubicin (Calyx TM) in human osteosarcoma xenografts. Cancer Res 2004; 64: 547–53
- Eikenes L, Bruland ØS, Brekken C, Davies Cde L. Collagenase increases the transcapillary pressure gradient and improves the uptake and distribution of monoclonal antibodies in human osteosarcoma xenografts. Cancer Res 2004; 64: 1555–60
- Engellau J, Anderson H, Bauer HCF, Hall KS, Gustafson P, Akerman M, Meis-Kindblom J, Alvegård TA, Nilbert M. Time-dependence of prognostic factors for patients with soft tissue sarcomas: A Scandinavian Sarcoma Group Study of 388 Malignant Fibrous Histiocytomas. Cancer 2004; 100(10)2233–9
- Engellau J, Persson A, Bendal P-O, åkerman M, Domanski HA, Bjerkehagen B, Lilleng P, Weide J, Rydholm A, Alvegård TA, Nilbert M. Expression profiling in 211 malignant fibrous histiocytomas confirms the prognostic value of Ki-67. Virchows Arch 2004; 445: 224–30
- Fernberg JO, Hall KS. Chemotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 2004; 75(311)77–86
- Henriksen G, Bruland ØS, Larsen R. Thorium- and actinium polyphosphonate compounds as bone-seeking alpha particle emitting agents. Anticancer Res 2004; 24: 101–6
- Henriksen G, Schoultz BW, Michaelsen TE, Bruland ØS, Larsen R. Sterically stabilized liposomes as a carrier for alpha-emitting radium and actinium radionuclides. Nucl Med Biol 2004; 31: 441–9
- Lyng H, Baidee A, Svendsrud DH, Hovig E, Myklebost O, Stokke T. Profound influence of non-linearity in microarray scanners on gene expression ratios. Analysis and procedure for correction. BMC Genomics 2004; 5(1)10
- Meis-Kindblom J, Wejde J, Bjerkehagen B. Diagnosis and tumor response in osteosarcoma and Ewing's sarcoma. Acta Orthop Scand Suppl. 2004; 75(311)72–6
- Mikkelsen IM, Løkke C, Flægstad T, Bruland ØS. Interferon-gamma sensitises human osteosarcoma cells to TRAIL mediated apoptosis. Cancer Genomics & Proteomics 2004; 1: 95–104
- Nilbert M, Meza-Zepeda LA, Francis P, Myklebost O. Lessons from genetic profiling in soft tissue sarcomas. Acta Orthop Scand Suppl 2004; 75(311)35–50
- Nilsson M, Meza-Zepeda L, Mertens M, Forus A, Myklebost O, Mandahl N. Chromosomal distribution of amplified chromosome 1 sequences in soft tissue and bone tumors. Int J Cancer 2004; 109: 363–9
- Nilsson M, Meza-Zepeda LA, Mertens F, Forus A, Myklebost O, Mandahl N. Amplification of chromosome 1 sequences in lipomatous tumors and other sarcomas. Int J Cancer 2004; 109(3)363–9
- Skotheim RI, Kallioniemi A, Bjerkhagen B, Mertens F, Brekke HR, Monni O, Mousses S, Mandahl N, Soeter G, Nesland JM, Smeland S, Kallioniemi OP, Lothe RA. Topoisomerase-II alpha is upregulated in malignant peripheral nerve sheath tumors and associated with clinical outcome. J Clin Oncol 2003; 21(24)4586–91, Erratum in: J Clin Oncol 2004; 22(5): 969
- Smeland S, Wiebe T, Brosjo O, Bøhling T, Alvegård TA. Chemotherapy in Ewing's sarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand Suppl. 2004; 75(311)87–91
- Smeland S, Wiebe T, Bøhling T, Brosjø O, Jonsson K, Alvegård TA. Chemotherapy in osteosarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand Suppl 2004; 75(311)92–8
- Wang J, Meza-Zepeda LA, Kresse SH, Myklebost O. M-CGH. Analysing microarray-based CGH experiments. BMC Bioinformatics 2004; 5: 74
2005
- Berg K, Selbo PK, Weyergang A, Dietze A, Prasmickaite L, Bonsted A, Engesæter BØ, Angell-Petersen E, Warloe T, Frandsen N, Høgset A. Porphyrin-related photosensitizers for cancer imaging and therapeutic applications. J Microscopy 2005; 218: 133–47
- Berg K, Dietze A, Kaalhus O, Høgset A. Site-specific drug delivery by photochemical internalization enhances the antitumor effect of bleomycin. Clin Cancer Res 2005; 11(23)8476–85
- Bielack SS, Marina N, Ferrari S, Helman LJ, Smeland S, Whelan JS, Reaman GH. Osteosarcoma: the same old drugs or more?. J Clin Oncol 2005; 26(18)3102–3
- Bjerkehagen B, Myklebost O. Molekylærbiologisk diagnostikk ved bein- og bløtvevssvulster. Tidsskr Nor Laegeforen 2005; 125(23)3286–9
- Bjørnland K, Flatmark K, Pettersen S, Aasen AO, Fodstad Ø, Mælandsmo GM. Matrix metalloproteinases participate in osteosarcoma invasion. J Surg Res 2005; 127(2)151–6
- Brandal P, Bjerkehagen B, Danielsen H, Heim S. Chromosome 7 abnormalities are common in chordomas. Cancer Genet Cytogenet 2005; 160: 15–21
- Bruland ØS, Høifødt H, Sæter G, Smeland S, Fodstad Ø. Hematogenous micrometastases in osteosarcoma patients. Clin Cancer Res 2005; 11(13)4666–73
- Brustugun OT, Reed W, Poulsen JP, Bruland ØS. Primary osteosarcoma of the breast. Case reports and review of the literature. Acta Oncol 2005; 44(7)767–70
- Dietze A, Peng Q, Selbo PK, Kaalhus O, Müller C, Bown S, Berg K. Enhanced photodynamic destruction of a transplantable fibrosarcoma using photochemical internalisation of gelonin. Br J Cancer 2005; 6: 2004–9
- Dietze A, Engesæter B, Berg K. Photochemical internalisation (PCI) improves the effect of photodynamic therapy (PDT) and gene transduction of fibroblast-like synoviocytes. Photochem Photobiol 2005; 4: 341–7
- Eikenes L, Tari M, Tufto I, Bruland ØS, Davies CdeL. Hyaluronidase improves the distribution and uptake of liposomal doxorubicin (Calyx) in human osteosarcoma xenografts. Br J Cancer 2005; 93: 81–8
- Ferrari S, Smeland S, Mercuri M, Bertoni F, Longhi A, Ruggieri P, Alvegard TA, Picci P, Capanna R, Bernini G, Muller C, Tienghi A, Wiebe T, Comandone A, Bohling T, Del Prever AB, Brosjo O, Bacci G, Saeter G. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of theextremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 2005; 23: 8845–52
- Henriksen G, Bruland ØS, Larsen R. Preparation and preclinical assessment of folate-conjugated radiolabeled antibodies. Anticancer Res 2005; 25: 231–6
- Kresse SH, Berner JM, Meza-Zepeda LA, Gregory SG, Kuo WL, Gray JW, Forus A, Myklebost O. Mapping and characterization of the amplicon near APOA2 in 1q23 in human sarcomas by FISH and array CGH. Mol Cancer 2005; 4: 39
- Müller CR, Smeland S, Bauer HCF, Sæter G, Strander H. Interferon-α as the only adjuvant treatment in high-grade osteosarcoma: Long-term results of the Karolinska Hospital series. Acta Oncol 2005; 44: 475–80
- Norum J, Bruland ØS, Spanne O, Bergmo T, Green T, Olsen DR, Olsen JH, Sjåeng EE, Brukow T. Telemedicine in radiotherapy. A pilot study exploring remote treatment planning and supervision. Telemedicine 2005; 11: 245–50
- Onda M, Bruland ØS, Pastan I. TP-3 immunotoxins improve antitumor activity in mouse bearing osteosarcoma. Clin Orthop Rel Res 2005; 430: 142–8
- Saeter G, Kloke O, Jelic S. ESMO minimum clinical recommendations for diagnosis, treatment and follow-up of osteosarcoma. Ann Oncol 2005; 16(Suppl. 1)71–2
- Saeter G, Oliveira J, Bergh J. ESMO minimum clinical recommendations for diagnosis, treatment and follow-up of Ewing's sarcoma of bone. Ann Oncol 2005; 16(Suppl 1)73–4
- Teixera MR, Heim S. Multiple numerical chromosome aberrations in cancer: What are their causes and what are their consequences?. Sem Cancer Biol 2005; 15: 3–12
- ågesen TH, Flørenes VA, Molenaar I, Lind GE, Berner JM, Plaat BEC, Komdeur R, Myklebost O, van den Berg E, Lothe RA. Expression patterns of cell cycle components in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol 2005; 64: 74–81
2006
- Aksnes LH, Hall KS, Folleraas G, Stenwig AE, Bjerkehagen B, Taksdal I, Winderen M, Bruland OS, Saeter G. Management of high-grade bone sarcomas over two decades: the Norwegian Radium Hospital experience. Acta Oncol 2006; 45(1)38–46
- Aksnes LH, Bruland ØS. Some musculo-skeletal sequelae in cancer survivors. Acta Oncol 2007; 46(4)490–6
- Berg K, Høgset A, Prasmickaite L, Weyerganga A, Bonsted A, Dietze A, Lou P-J, Bown SG, Norum OJ, Møllergård HMT, Selbo PK. Photochemical internalization (PCI): A novel technology for activation of endocytosed therapeutic agents. Med Laser Application 2006; 21: 239–50
- Berner JM, Müller CR, Holden M, Wang J, Hovig E, Myklebost O. Sampling effects on gene expression data from a human tumor xenograft. Scand J Lab Animal Science 2006; 33: 17–30
- Brandal P, Bjerkehagen B, Heim S. Rearrangement of chromosomal region 8q11-13 in lipomatous tumours: correlation with lipoblastoma morphology. J Pathol 2006; 208(3)388–94
- Bruland OS, Nilsson S, Fisher DR, Larsen RH. High-linear energy transfer irradiation targeted to skeletal metastases by the alpha-emitter 223Ra: adjuvant or alternative to conventional modalities?. Clin Cancer Res 2006; 12(20)6250–7
- Jonasdottir TJ, Fisher DR, Borrebaek J, Bruland OS, Larsen RH. First in vivo evaluation of liposome-encapsulated 223Ra as a potential alpha-particle-emitting cancer therapeutic agent. Anticancer Res 2006; 26(4B)2841–8
- Kaasa S, Brenne E, Lund J-A, Fayers P, Falkmer U, Wallgren A, Glas U, Bruland ØS. Prospective randomised multicenter trial on single fraction radiotherapy (8Gyx1) versus multiple fractions (3Gyx10) in the treatment of painful bone metastases. Radiother Oncol 2006; 79(3)278–84
- Larsen RH, Saxtorph H, Skydsgaard M, Borrebaek J, Jonasdottir TJ, Bruland OS, Klastrup S, Harling R, Ramdahl T. Radiotoxicity of the alpha-emitting bone-seeker 223Ra injected intravenously into mice: histology, clinical chemistry and hematology. In Vivo 2006; 20(3)325–31
- Lloret I, Server A, Bjerkehagen B. Primary spinal chondrosarcoma: radiologic findings with pathologic correlation. Acta Radiol 2006; 47(1)77–84
- Malinen E, Sovik A, Hristov D, Bruland OS, Olsen DR. Adapting radiotherapy to hypoxic tumours. Phys Med Biol 2006; 51(19)4903–21
- Meza-Zepeda LA, Kresse SH, Barragan-Polania AH, Bjerkehagen B, Ohnstad HO, Namlos HM, Wang J, Kristiansen BE, Myklebost O. Array comparative genomic hybridization reveals distinct DNA copy number differences between gastrointestinal stromal tumors and leiomyosarcomas. Cancer Res 2006; 66(18)8984–93
- Micci F, Panagopoulos I, Bjerkehagen B, Heim S. Consistent rearrangement of chromosoma band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res 2006; 66: 107–12
- Micci F, Panagopoulos I, Bjerkehagen B, Heim S. Deregulation of HMGA2 in an aggressive angiomyxoma with t(11;12)(q23;q15). Virchows Arch 2006; 448(6)838–42
- Myklebost O, et al. The MAQC Consortium. The microarray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature Biotech 2006; 24: 1151–1161
- Olsen DR, Bruland B, Frykholm G, Norderhaug I. Protonterapi. Institusjon Nasjonalt kunnskapssenter for helsetjenesten. ISSN 82-8121-067-2. 1890-1298. 2006; 11: 1–63
- Schaad K, Strömbeck B, Mandahl N, Andersen MK, Heim S, Mertens F, Johansson B. FISH mapping of i(7q) in acute leukemias and myxoid liposarcoma reveals clustered breakpoints in 7p11.2: Implications for formation and pathogenetic outcome of the idic(7)(p11.2). Cytogenetic Genome Res 2006; 114(2)126–130
- Serra M, Pasello M, Manara MC, Scotlani K, Ferrari S, Bertoni F, Mercuri M, Alvegaard TA, Picci P, Bacci G, Smeland S. May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int J Oncol 2006; 29: 1459–68
- Storlazzi CT, Brekke HR, Mandahl N, Rydholm A, Brosjo O, Smeland S, Lothe RA, Mertens F. Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumors. J Pathol 2006; 209: 492–500
- Svarvar C, Böhling T, Berlin O, Gustafson P, Follerås G, Bjerkehagen B, Domanski HA, Sundbyhall K, Tukiainen E, Blomqvist C. the Scandinavian Sarcoma Group Leiomyosarcoma Working Group. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian Sarcoma Group. Cancer 2006; 109: 282–91
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA 2006; 103: 1888–93
- The MAQC Consortium [including Myklebost O]. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nature Biotech 2006; 24: 1151–1161
Doctoral thesis
- University of Oslo (2006) by Petter Brandal, Department of Cancer Genetics. The Norwegian Radium Hospital. Molecular Cytogenetic Analysis of Archival Material and Fresh Solid Tumor Samples
Master thesis
- University of Oslo (2006) by Ana H. Barragan Lid, Department of Molecular Biosciences. The Norwegian Radium Hospital. Profiling of DNA copy number in sarcomas by array comparative genomic hybridisation and identification of candidate cancer genes
2007
- Aksnes LH, Bruland ØS. Some musculo-skeletal sequelae in cancer survivors. Acta Oncol 2007; 46(4)490–6
- Aksnes LH, Hall KS, Jebsen N, Fossa SD, Dahl AA. Young survivors of malignant bone tumours in the extremities: a comparative study of quality of life, fatigue and mental distress. Support Cancer Care 2007; 15(9)1087–96
- Berg K, Folini M, Prasmackaite L, Selbo PK, Bonsted A, Engesæther BØ, Zaffaroni N, Weyergang A, Dietze A, Mælandsmoe G, Wagner E, Norum O-J, Høgseth A. Photocemical internalization (PCI): a new tool for drug delivery. Curr Pharm Biotechnol 2007; 8(6)362–72
- Boye K, Andersen K, Tveito S, øyjord T, Mælandsmo GM. Interferon-gamma-induced suppression of S100A4 transcription is mediated by the class II transactivator. Tumour Biol 2007; 28: 27–35
- Cekaite L, Peng Q, Reiner A, Shahzidi S, Tveito S, Furre IE, Hovig E. Mapping of oxidative stress responses of human tumor cells following photodynamic therapy using hexaminolevulinate. BMC Genomics 2007; 8: 273
- Dahle J, Borrebaek J, Jonasdottir TJ, Hjelmerud AK, Melhus KB, Bruland ØS, Press OW, Larsen RH. Targeted cancer therapy with a novel low-dose rate alpha-emitting radioimmunoconjugate. Blood 2007; 110(6)2049–56
- Engellau J, Samuelsson V, Anderson H, Bjerkehagen B, Rissler P, Sundby-Hall K, Rydholm A. Identification of low-risk tumours in histological high-grade soft tissue sarcomas. Eur J Cancer 2007; 43(13)1927–34
- Francis P, Namlos HM, Muller C, Eden P, Fernebro J, Berner JM, Bjerkehagen B, Akerman M, Bendahl PO, Isinger A, Rydholm A, Myklebost O, Nilbert M. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics 2007; 8: 73
- Hallor KH, Micci F, Meis-Kindblom JM, Kindblom LG, Mandahl N, Mertens F, Panagopoulos I. Fusion genes in angiomatoid fibrous histiocytoma. Cancer Letters 2007; 251(1)158–63
- Micci F, Bjerkehagen B, Heim S. Pairwise comparison of genomic imbalances between primary and recurrent well differentiated liposarcomas. Cancer Genetics and Cytogenetics 2007; 15:178(2)163–7
- Micci F, Haugom L, Abeler VM, Bjerkehagen B, Heim S. Trisomy 7 in postoperative spindle cell nodules. Cancer Genet Cytogenet. 2007; 174(2)147–50
- Micci F, Heim S. Pathogenetic Mechanisms in Endometrial Stromal Sarcoma. Cytogenetics and Genome Research 2007; 118(2-4)190–5
- Micci F, Brandal P. Soft Tissue Tumors. Aggressive angiomyxoma. Atlas Genet Cytogenet Oncol Haematol April, 2007
- Müller CR, Paulsen EB, Noordhuis P, Peudeutour F, Sæter G, Myklebost O. Potential For Treatment Of Liposarcomas With The MDM2 Antagonist Nutlin-3a. Int J Cancer 2007; 121: 199–205
- Olsen DR, Bruland OS, Frykholm G, Norderhaug IN. Proton therapy - a systematic review of clinical effectiveness. Radiother Oncol 2007; 83(2)123–32
- Svarvar C, Bohling T, Berlin O, Gustafson P, Folleras G, Bjerkehagen B, Domanski HA, Sundby Hall K, Tukiainen E, Blomqvist C. the Scandinavian Sarcoma Group Leiomyosarcoma Working Group. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian Sarcoma Group. Cancer 2007; 109(2)282–91
- Søvik A, Malinen E, Skogmo HK, Bentzen SM, Bruland OS, Olsen DR. Radiotherapy adapted to spatial and temporal variability in tumor hypoxia. Int J Radiat Oncol Biol Phys 2007; 68(5)1496–504
- Søvik A, Malinen E, Bruland ØS, Bentzen SM, Olsen DR. Optimization of tumour control probability in hypoxic tumours by radiation dose redistribution: a modelling study. Phys Med Biol 2007; 52(2)499–513
2008
- Aksnes LH, Bauer HF, Jensen NL, Follerås G, Allert C, Haugen G, Hall KS. Limb-sparing surgery preserves more function than amputation – A SSG study of 118 extremity bone sarcoma patients. J Bone Joint Surgery (Br) 2008; 90(6)786–94
- Bjerkehagen B, Smeland S, Walberg L, Skjeldal S, Hall KS, Nesland JM, Småstuen MC, Fosså SD, Sæter G. Radiation-induced sarcoma: 25-year experience from The Norwegian Radium Hospital. Acta Oncol. 2008; 7: 1–8
- Boye K, Grotterød I, Aasheim HC, Hovig E, Mælandsmo GM. Activation of NF-κB by extracellular S100A4; analysis of signal transduction mechanisms and identification of target genes. Int J Cancer 2008; 123: 1301–10
- Brandal P, Panagopoulos I, Bjerkehagen B, Gorunova L, Skjeldal S, Micci F, Heim S. Detection of a t(1;22)(q23;q12) translocation leading to an EWSR1-PBX1 fusion gene in a myoepithelioma. Genes Chromosomes Cancer 2008; 47(7)558–64
- Engström K, Bergh P, Gustafson P, Hultborn R, Johansson H, Löfvenberg R, Zaikova O, Trovik C, Wahlström O, Bauer HC. Liposarcoma: outcome based on the Scandinavian Sarcoma Group register. Cancer 2008; 113(7)1649–56
- Jebsen NL, Trovik CS, Bauer HC, Rydholm A, Monge OR, Hall KS, Alvegård T, Bruland OS. Radiotherapy to improve local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma: a Scandinavian sarcoma group study. Int J Radiat Oncol Biol Phys 2008; 71(4)1196–203
- Kresse SH, Skårn M, Ohnstad HO, Namløs HM, Bjerkehagen B, Wang J, Myklebost O, Meza-Zepeda LA. DNA copy number changes in malignant peripheral nerve sheath tumors by array comparative genomic hybridization. Molecular Cancer 2008; 7: 48
- Lybæk H, Meza-Zepeda LA, Kresse SH, Høysæter T, Steen VM, Houge G. Array-CGH fine mapping of minor and cryptic chromosome-CGH detected genomic imbalances in 80 out of 590 patients abnormal development. Eur J Hum Genet 2008, in press
- Lyng H, Lando M, Brøvig RS, Svendsrud DH, Johansen M, Galteland E, Brustugun OT, Meza-Zepeda LA, Myklebost O, Kristensen GB, Hovig E, Stokke T. GeneCount: Genome-wide calculation of absolute tumor DNA copy numbers from array CGH data. Genome Biology 2008; 9: 86
- Meza-Zepeda LA, Noer A, Dahl JA, Micci F, Myklebost O, Collas P. High-resolution analysis of genetic stability of human adipose tissue stem cells cultured to senescence. J Cell Mol Med 2008; 12(2)553–63
- Nakajima G, Patino-Garcia A, Bruheim S, Xi Y, San Julian M, Lecanda F, Sierrasesumaga L, Müller C, Fodstad O, Ju J. CDH11 expression is associated with survival in patients with osteosarcoma. Cancer Genomics Proteomics 2008; 5(1)37–42
- Norum OJ, Gaustad J-V, Angell-Pettersen E, Rofstad EK, Peng Q, Giercksky K-E, et al. Photochemical Internalization of Bleomycin is Superior to Photodynamic Therapy due to the Therapeutic Effect in the Tumor Periphery. Photochem Photobiol 2008, In Press
- Steigen SE, Bjerkehagen B, Haugland HK, Nordrum IS, Løberg EM, Isaksen V, Eide TJ, Nielsen TO. Diagnostic and prognostic markers for gastrointestinal stromal tumors in Norway. Mod Pathol. 2008; 21(1)46–53
Doctoral thesis
- University of Oslo (2008) by Stine H. Kresse, Dept of Tumour Biology, Institute for Cancer Research, The Norwegian Radium Hospital. Chromosomal aberrations in human sarcomas identified using genomic microarrays
Sweden
- Alvegaard TA, Bauer H, Smeland S. The Scandinavian Sarcoma Group–background, organization and the SSG Register–the first 25 years. Acta Orthop Scand Suppl 2004; 75: 1–7
- Bauer HC, Alvegard TA, Berlin O, Erlanson M, Kalen A, Lindholm P, Gustafson P, Smeland S, Trovik CS. Related Articles, The Scandinavian Sarcoma Group Register 1986-2001. Acta Orthop Scand Suppl 2004; 75(311)8–10
- Dahlén A, Fletcher CDM, Mertens F, Fletcher JA, Perez-Atayde AR, Hicks MJ, Debiec-Rychter M, Sciot R, Wejde J, Wedin R, Mandahl N, Panagopoulos I. Activation of the GLI oncogene through fusion with the β-actin gene (ACTB) in a group of distinctive pericytic neoplasms. Am J Pathol 2004; 164: 1645–1653
- Dahlén A, Mertens F, Mandahl N, Panagopoulos I. Molecular genetic characterization of the genomic ACTB-GLI fusion in pericytoma with t(7;12). Biochem Biophys Res Commun 2004; 325: 1318–1323
- Engellau J, Anderson H, Bauer HCF, Hall KS, Gustafson P, Akerman M, Meis-Kindblom J, Alvegård TA, Nilbert M. Time-dependence of prognostic factors for patients with soft tissue sarcomas: A Scandinavian Sarcoma Group Study of 388 malignant fibrous histiocytomas. Cancer 2004; 100(10)2233–9
- Engellau J, Persson A, Bendal P-O, åkerman M, Domanski HA, Bjerkehagen B, Lilleng P, Weide J, Rydholm A, Alvegård TA, Nilbert M. Expression profiling in 211 malignant fibrous histiocytomas confirms the prognostic value of Ki-67. Virchows Arch 2004; 445: 224–30
- Gisselsson D, Pålsson E, Yu C, Mertens F, Mandahl N. Mitotic instability associated with late genomic changes in bone and soft tissue tumours. Cancer Letters 2004; 206: 69–76
- Kuzniacka A, Mertens F, Strömbeck B, Wiegant J, Mandahl N. Combined binary ratio labelling fluorescence in situ hybridization analysis of chordoma. Cancer Genet Cytogenet 2004; 151: 178–181
- Mandahl N, Mertens F, Panagopoulos I, Knuutila S. Genetic characterization of bone and soft tissue tumors. Acta Orthop Scand 2004; 75(Suppl 311)21–28
- Mertens F, Panagopoulos I, Jonson T, Gisselsson D, Isaksson M, Domanski HA, Mandahl N. Retained heterodisomy for chromosome 12 in atypical lipomatous tumors: implications for ring chromosome formation. Cytogenet Genome Res 2004; 106: 33–8
- Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a function of chromosome aberrations in cancer. Nat Genet 2004; 36: 331–334
- Nilsson M, Domanski HA, Mertens F, Mandahl N. Molecular cytogenetic characterization of recurrent translocation breakpoints in bizarre parosteal osteochondromatous proliferation (Nora's lesion). Hum Pathol 2004; 35: 1063–1069
- Nilsson M, Meza-Zepeda LA, Mertens F, Forus A, Myklebost O, Mandahl N. Amplification of chromosome 1 sequences in lipomatous tumors and other sarcomas. Int J Cancer 2004; 109: 363–369
- Panagopoulos I, Mertens F, Isaksson M, Mandahl N. Expression of DOL54 is not restricted to myxoid liposarcomas with the FUS-DDIT3 chimera but is found in various sarcomas. Oncol Reports 2004; 12: 107–110
- Panagopoulos I, Storlazzi CT, Fletcher CDM, Fletcher JA, Nascimento A, Domanski HA, Wejde J, Brosjö O, Rydholm A, Isaksson M, Mandahl N, Mertens F. The chimeric FUS/CREB3L2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chromosomes Cancer 2004; 40: 218–228
- Smeland S, Wiebe T, Brosjo O, Bohling T, Alvegard TA. Chemotherapy in Ewing's sarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand Suppl. 2004; 75: 87–91, Review
- Smeland S, Wiebe T, Bohling T, Brosjo O, Jonsson K, Alvegard TA. Chemotherapy in osteosarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand Suppl. 2004; 75: 92–8, Review
- Surace C, Panagopoulos I, Pålsson E, Rocchi M, Mandahl N, Mertens F. A novel FISH assay for SS18-SSX fusion type in synovial sarcoma. Lab Invest 2004; 84: 1185–1192
- Storlazzi T, Mertens F, Panagopoulos I. CREB3L2. Atlas Genet Cytogenet Oncol Haematol October, 2004
Doctoral thesis
- University of Lund (2004) by Jacob Engellau, Department of Oncology. Prognostic factors in soft tissue sarcoma.Tissue microarray for immunostaining, the importance of whole-tumor sections and time-dependence
2005
- Domanski HA, åkerman M, Carlén B, Engellau J, Gustafson P, Jonsson K, Mertens F, Rydholm A. Core needle biopsy performed by the cytopathologist: A technique to complement fine-needle aspiration of soft tissue and bone lesions. Cancer 2005; 105: 229–239
- Ferrari S, Smeland S, Mercuri M, Bertoni F, Longhi A, Ruggieri P, Alvegard TA, Picci P, Capanna R, Bernini G, Muller C, Tienghi A, Wiebe T, Comandone A, Bohling T, Del Prever AB, Brosjo O, Bacci G, Saeter G. Italian and Scandinavian Sarcoma Groups. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 2005; 23: 8845–8852
- Hansén Hallor K, Mertens F, Jin Y, Meis-Kindblom JM, Kindblom L-G, Behrendtz M, Kalén A, Mandahl N, Panagopoulos I. Fusion of the EWSR1 and ATF1 genes without expression of the MITF-M transcript in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 2005; 44: 97–102
- Höglund M, Gisselsson M, Mandahl N, Mitelman F. Ewing tumours and synovial sarcomas have critical features of karyotype evolution in common with epithelial tumours. Int J Cancer 2005; 116: 401–406
- Mertens F, Fletcher CDM, Antonescu CR, Coindre J-M, Colecchia M, Domanski HA, Downs-Kelly E, Fisher C, Goldblum JR, Guillou L, Reid R, Rosai J, Sciot R, Mandahl N, Panagopoulos I. Clinicopathologic and molecular genetic characterization of low-grade fibromyxoid sarcoma, and cloning of a novel FUS/CREB3L1 fusion gene. Lab Invest 2005; 85: 408–415
- Mitelman F, Mertens F, Johansson B. Prevalence estimates of recurrent balanced cytogenetic aberrations and fusion genes in unselected patients with neoplastic disorders. Genes Chromosomes Cancer 2005; 43: 350–366
- Nilsson M, Domanski H, Mertens F, Mandahl N. Atypical lipomatous tumor with rare structural rearrangements involving chromosomes 8 and 12. Oncol Reports 2005; 13: 649–652
- Nilsson M, Panagopoulos I, Mertens F, Mandahl N. Fusion of the HMGA2 and NFIB genes in lipoma. Virchows Arch 2005; 447: 855–858
- Panagopoulos I, Mertens F, Isaksson M, Mandahl N. Absence of mutations of the BRAF gene in malignant melanoma of soft parts (clear cell sarcoma of tendons and aponeuroses). Cancer Genet Cytogenet 2005; 156: 74–76
- Surace C, Storlazzi CT, Engellau J, Domanski HA, Gustafson P, Panagopoulos I, D´addabbo P, Rocchi M, Mandahl N, Mertens F. Molecular cytogenetic characterization of an ins(4;X) occurring as the sole abnormality in an aggressive, poorly differentiated soft tissue sarcoma. Virchows Arch 2005; 447: 869–874
- Storlazzi CT, Vult von Steyern F, Domanski HA, Mandahl N, Mertens F. Biallelic somatic inactivation of the NF1 gene through chromosomal translocations in a sporadic neurofibroma. Int J Cancer 2005; 117: 1055–1057
Doctoral thesis
- University of Lund (2005) by Henryk A Domanski, Department of Pathology. Fine needle aspiration diagnosis of spindle cell tumors of soft tissue, including the use of ancillary methods, and correlation with clinical data
2006
- Domanski HA, åkerman M, Engellau J, Gustafson P, Mertens F, Rydholm A. Fine-needle aspiration of neurilemoma (schwannoma). A clinicopathologic study of 116 patients. Diagn Cytopathol 2006; 34: 403–412
- Fernebro J, Francis P, Edén P, Borg å, Panagopoulos I, Mertens F, Vallon-Christersson J, åkerman M, Rydholm A, Bauer HCF, Mandahl N, Nilbert M. Gene expression profiles relate to SS18/SSX fusion type in synovial sarcoma. Int J Cancer 2006; 118: 1165–1172
- Heidenblad M, Hallor KH, Staaf J, Jönsson G, Borg å, Höglund M, Mertens F, Mandahl N. Genomic profiling of bone and soft tissue tumors with supernumerary ring chromosomes using tiling resolution bacterial artificial chromosome microarrays. Oncogene 2006; 25: 7106–7116
- Kaur S, Vauhkonen H, Böhling T, Mertens F, Mandahl N, Knuutila S. Gene copy number changes in dermatofibrosarcoma protuberans – a fine-resolution study using array comparative genomic hybridization. Cytogenet Genome Res 2006; 115: 283–288
- Mertens F, Strömberg U, Rydholm A, Gustafson P, Bauer HCF, Brosjö O, Mandahl N. Prognostic significance of chromosome aberrations in high-grade soft tissue sarcomas. J Clin Oncol 2006; 24: 315–320
- Nilsson M, Mertens F, Höglund M, Mandahl N, Panagopoulos I. Truncation and fusion of HMGA2 in lipomas with rearrangements of 5q32-q33 and 12q14-q15. Cytogenet Genome Res 2006; 112: 60–66
- Panagopoulos I, Nilsson T, Domanski HA, Isaksson M, Lindblom P, Mertens F, Mandahl N. Fusion of the SEC31L1 and ALK genes in an inflammatory myofibroblastic tumor. Int J Cancer 2006; 118: 1181–1186
- Schaad K, Strömbeck B, Mandahl N, Andersen MK, Heim S, Mertens F, Johansson B. FISH mapping of i(7q) in acute leukemias and myxoid liposarcoma reveals clustered breakpoints in 7p11.2: implications for formation and pathogenetic outcome of the idic(7)(p11.2). Cytogenet Genome Res 2006; 114: 126–130
- Storlazzi CT, Brekke HR, Mandahl N, Brosjö O, Smeland S, Lothe RA, Mertens F. Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumours. J Pathol 2006; 209: 492–500
- Storlazzi CT, Wozniak A, Panagopoulos I, Sciot R, Mandahl N, Mertens F, Debiec-Rychter M. Rearrangement of the COL12A1 and COL4A5 genes in subungual exostosis: molecular cytogenetic delineation of the tumor-specific translocation t(X;6)(q13-14;q22). Int J Cancer 2006; 118: 1972–1976
2007
- Bartuma H, Hallor KH, Panagopoulos I, Collin A, Rydholm A, Gustafson P, Bauer HCF, Brosjö O, Domanski HA, Mandahl N, Mertens F. Assessment of the clinical and molecular impact of different cytogenetic subgroups in a series of 272 lipomas with abnormal karyotype. Genes Chromosomes Cancer 2007; 46: 594–606
- Engellau J, Samuelsson V, Anderson H, Bjerkehagen B, Rissler P, Sundby-Hall K, Rydholm A. Identification of low-risk tumours in histological high-grade soft tissue sarcomas. Eur J Cancer. 2007; 43(13)1927–34
- Hallor KH, Heidenblad M, Brosjö O, Mandahl N, Mertens F. Tiling resolution array comparative genomic hybridization analysis of a fibrosarcoma of bone. Cancer Genet Cytogenet 2007; 172: 80–83
- Hallor KH, Micci F, Meis-Kindblom JM, Kindblom L-G, Bacchini P, Mandahl N, Mertens F, Panagopoulos I. Fusion genes in angiomatoid fibrous histiocytoma. Cancer Letters 2007; 251: 158–163
- Johnsson A, Collin A, Rydholm A, Domanski HA, Mertens F, Mandahl N. Unstable translocation (8;22) in a case of giant cell reparative granuloma. Cancer Genet Cytogenet 2007; 177: 59–63
- Mertens F, Wiebe T, Adlercreutz C, Mandahl N, French CA. Successful treatment of a child with t(15;19)-positive tumor. Pediatr Blood Cancer 2007; 49: 1015–1017
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007; 7: 233–245
- Möller E, Isaksson M, Mandahl N, Mertens F, Panagopoulos I. Comparison of the proximal promoter regions of the PAX3 and PAX7 genes. Cancer Genet Cytogenet 2007; 178: 114–119
- Panagopoulos I, Möller E, Dahlén A, Isaksson M, Mandahl N, Vlamis-Gardikas A, Mertens F. Characterization of the native CREB3L2 transcription factor and the FUS/CREB3L2 chimera. Genes Chromosomes Cancer 2007; 46: 181–191
Doctoral thesis
- University of Gothenburg(2007) by Katarina Engström, Department of Oncology. Clinical and molecular studies of liposarcoma
- University of Lund (2007) by Princy Francis, Department of Oncology. Genetic profiling in soft tissue sarcoma
- University of Lund (2007) by Josefin Fernebro, Department of Oncology. Soft tissue sarcoma patterns multiplicity, heterogeneity and growth characteristics
2008
- Bartuma H, Domanski HA, Vult von Steyern F, Kullendorff C-M, Mandahl N, Mertens F. Cytogenetic and molecular cytogenetic findings in lipoblastoma. Cancer Genet Cytogenet 2008; 183: 60–63
- Billing V, Mertens F, Domanski HD, Rydholm A. Deep-seated ordinary and atypical lipomas; histopathology, cytogenetics, clinical features, and outcome in 215 tumors of the extremity and trunk wall. J Bone Joint Surg Br 2008; 90: 929–933
- Dantonello TM, Int-Veen C, Winkler P, Leuschner I, Schuck A, Schmidt BF, Lochbuehler H, Kirsch S, Veit-Friedrich I, Bielack SS, Niggli F, Kazanowska B, Ladenstein R, Wiebe T, Klingebiel T, Treuner J, Koscielniak E. Initial patient characteristics can predict pattern and risk of relapse in rhabdomyosarcoma. J Clin Oncol 2008; 26: 406–413
- Downs-Kelly E, Goldblum JR, Patel RM, Weiss SW, Folpe AL, Mertens F, Hartke M, Tubbs RR, Skacel M. The utility of fluorescence in situ hybridization (FISH) in the diagnosis of myxoid soft tissue neoplasms. Am J Surg Pathol 2008; 32: 8–13
- Hallor KH, Staaf J, Jönsson G, Heidenblad M, Vult von Steyern F, Bauer HCF, Ijszenga M, Hogendoorn PCW, Mandahl M, Szuhai K, Mertens F. Frequent deletion of the CDKN2A locus in chordoma: analysis of chromosomal imbalances using array comparative genomic hybridisation. Brit J Cancer 2008; 98: 434–442
- Hallor KH, Teixeira MR, Fletcher CD, Bizarro S, Staaf J, Domanski HA, Vonsteyern FV, Panagopoulos I, Mandahl N, Mertens F. Heterogenous genetic profiles in soft tissue myoepitheliomas. Mod Pathol 2008, Epub ahead of print
- Jebsen N, Trovik C, Bauer B, Rydholm A, Monge O, Sundby Hall K, Alvegård T, Bruland ØS. Radiotherapy improves local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma. A study of 1093 patients from the Scandinavian Sarcoma Group. Int J Radiat Oncol Biol Phys 2008; 71(4)1196–203, Epub 2008 Jan 22
- Möller E, Mandahl N, Mertens F, Panagopoulos I. Molecular Identification of COL6A3-CSF1 fusion transcripts in tenosynovial giant cell tumors. Genes Chromosomes Cancer 2008; 47: 21–25
- Panagopoulos I, Mertens F, Löfvenberg R, Mandahl N. Fusion of the COL1A1 and USP6 genes in a benign bone tumor. Cancer Genet Cytogenet 2008; 180: 70–73
- Panagopoulos I, Mertens F, Griffin CA. An endometrial stromal sarcoma cell line with the JAZF1/PHF1 chimera. Cancer Genet Cytogenet 2008; 185: 74–77
Gothenburg
- Bauer HC, Alvegård TA, Berlin O, Erlanson M, Kalén A, Lindholm P, Gustafson P, Smeland S, Trovik CS. The Scandinavian Sarcoma Group Register 1986-2001. Acta Orthop Scand Suppl 2004; 75(311)8–10
- Joensuu H, Kindblom LG. Gastrointestinal stromal tumors–a review. Acta Orthop Scand Suppl 2004; 75(311)62–71
- Klint P, Hellman U, Wernstedt C, Aman P, Ron D, Claesson-Welsh L. Translocated in liposarcoma (TLS) is a substrate for fibroblast growth factor receptor-1. Cell Signal 2004; 16(4)515–20
- Olofsson A, Willén H, Göransson M, Engström K, Meis-Kindblom JM, Stenman G, Kindblom LG, Aman P. Abnormal expression of cell cycle regulators in FUS-CHOP carrying liposarcomas. Int J Oncol 2004; 25(5)1349–55
- Sjögren H, Orndal C, Tingby O, Meis-Kindblom JM, Kindblom LG, Stenman G. Cytogenetic and spectral karyotype analyses of benign and malignant cartilage tumours. Int J Oncol 2004; 24(6)1385–91
2005
- Aman P. Fusion oncogenes. Semin Cancer Biol 2005; 15(3)159–61
- Aman P. Fusion oncogenes in tumor development. Semin Cancer Biol 2005; 15(3)236–43
- Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom LG. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol 2005; 29(9)1170–6
- Asp J, Brantsing C, Lövstedt K, Benassi MS, Inerot S, Gamberi G, Picci P, Lindahl A. Evaluation of p16 and Id1 status and endogenous reference genes in human chondrosarcoma by real-time PCR. Int J Oncol 2005; 27(6)1577–82
- Göransson M, Elias E, Ståhlberg A, Olofsson A, Andersson C, Aman P. Myxoid liposarcoma FUS-DDIT3 fusion oncogene induces C/EBP beta-mediated interleukin 6 expression. Int J Cancer 2005; 115(4)556–60
- Nilsson B, Bümming P, Meis-Kindblom JM, Odén A, Dortok A, Gustavsson B, Sablinska K, Kindblom LG. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era–a population-based study in western Sweden. Cancer 2005; 103(4)821–9
2006
- Andersson J, Bümming P, Meis-Kindblom JM, Sihto H, Nupponen N, Joensuu H, Odén A, Gustavsson B, Kindblom LG, Nilsson B. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology 2006; 130(6)1573–81
- Bümming P, Ahlman H, Andersson J, Meis-Kindblom JM, Kindblom LG, Nilsson B. Population-based study of the diagnosis and treatment of gastrointestinal stromal tumours. Br J Surg 2006; 93(7)836–43
- Bümming P, Nilsson B, Sörensen J, Nilsson O, Ahlman H. Use of 2-tracer PET to diagnose gastrointestinal stromal tumour and pheochromocytoma in patients with Carney triad and neurofibromatosis type 1. Scand J Gastroenterol. 2006; 41(5)626–30
- Dalén BP, Geijer M, Kvist H, Bergh PM, Gunterberg BU. Clinical and imaging observations of desmoid tumors left without treatment. Acta Orthop 2006; 77(6)932–7
- Dalén BP, Meis-Kindblom JM, Sumathi VP, Ryd W, Kindblom LG. Fine-needle aspiration cytology and core needle biopsy in the preoperative diagnosis of desmoid tumors. Acta Orthop 2006; 77(6)926–31
- Ekeblad S, Nilsson B, Lejonklou MH, Johansson T, Stålberg P, Nilsson O, Ahlman H, Skogseid B. Gastrointestinal stromal tumors express the orexigen ghrelin. Endocr Relat Cancer 2006; 13(3)963–70
- Engström K, Willén H, Kåbjörn-gustafsson C, Andersson C, Olsson M, Göransson M, Järnum S, Olofsson A, Warnhammar E, Aman P. The myxoid/round cell liposarcoma fusion oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma phenotype in transfected human fibrosarcoma cells. Am J Pathol 2006; 168(5)1642–53
- Kindblom LG. Lipomatous tumors-how we have reached our present views, what controversies remain and why we still face diagnostic problems: a tribute to Dr Franz Enzinger. Adv Anat Pathol 2006; 13(6)279–85, Review
- Meis-Kindblom JM. Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 2006; 13(6)286–92
- Tostar U, Malm CJ, Meis-Kindblom JM, Kindblom LG, Toftgård R, Undén AB. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol 2006; 208(1)17–25
2007
- Bümming P, Nilsson O, Ahlman H, Welbencer A, Andersson MK, Sjölund K, Nilsson B. Gastrointestinal stromal tumors regularly express synaptic vesicle proteins: evidence of a neuroendocrine phenotype. Endocr Relat Cancer 2007; 14(3)853–63
- Engström K, Bergh P, Cederlund CG, Hultborn R, Willen H, Aman P, Kindblom LG, Meis-Kindblom JM. Irradiation of myxoid/round cell liposarcoma induces volume reduction and lipoma-like morphology. Acta Oncol 2007; 46(6)838–45
- Hallor KH, Micci F, Meis-Kindblom JM, Kindblom LG, Bacchini P, Mandahl N, Mertens F, Panagopoulos I. Fusion genes in angiomatoid fibrous histiocytoma. Cancer Lett 2007; 251(1)158–63
- Nilsson B, Sjölund K, Kindblom LG, Meis-Kindblom JM, Bümming P, Nilsson O, Andersson J, Ahlman H. Adjuvant imatinib treatment improves recurrence-free survival in patients with high-risk gastrointestinal stromal tumours (GIST). Br J Cancer 2007; 96(11)1656–8
- Svarvar C, Böhling T, Berlin O, Gustafson P, Follerås G, Bjerkehagen B, Domanski HA, Sundby Hall K, Tukiainen E, Blomqvist C. the Scandinavian Sarcoma Group Leiomyosarcoma Working Group. Clinical course of nonvisceral soft tissue leiomyosarcoma in 225 patients from the Scandinavian Sarcoma Group. Cancer 2007; 109(2)282–91
2008
- Andersson MK, Aman P. Proliferation of Ewing sarcoma cell lines is suppressed by the receptor tyrosine kinase inhibitors gefitinib and vandetanib. Cancer Cell Int 2008; 8: 1
- Andersson MK, Ståhlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O, Aman P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol 2008; 9: 37
- Engström K, Bergh P, Gustafson P, Hultborn R, Johansson H, Löfvenberg R, Zaikova O, Trovik C, Wahlström O, Bauer HC. Liposarcoma: outcome based on the Scandinavian Sarcoma Group register. Cancer 2008; 113(7)1649–56
- Hagberg K, Brånemark R, Gunterberg B, Rydevik B. Osseointegrated trans-femoral amputation prostheses: prospective results of general and condition-specific quality of life in 18 patients at 2-year follow-up. Prosthet Orthot Int 2008; 32(1)29–41
- Kivioja AH, Blomqvist C, Hietaniemi K, Trovik C, Walloe A, Bauer HC, Jorgensen PH, Bergh P, Follerås G. Cement is recommended in intralesional surgery of giant cell tumors: a Scandinavian Sarcoma Group study of 294 patients followed for a median time of 5 years. Acta Orthop 2008; 79(1)86–93
- Nilsson B, Andersson A, Ahlman H. Adjuvant and down-staging treatment with imatinib in gastrointestinal stromal tumors. J Surg Oncol 2008; 98(3)145–6
- Persson F, Olofsson A, Sjögren H, Chebbo N, Nilsson B, Stenman G, Aman P. Characterization of the 12q amplicons by high-resolution, oligonucleotide array CGH and expression analyses of a novel liposarcoma cell line. Cancer Lett 2008; 260(1-2)37–47
Stockholm
- Brosjö O, Bauer HCF. Diagnostic procedures and surgical treatment of bone sarcomas: Experience from the Scandinavian Sarcoma Group and Karolinska Hospital. Acta Orthop Scand Suppl 2004; 75(311)57–61
- Dahlén A, Fletcher CDM, Mertens F, Fletcher JA, Perez-Atayde AR, Hicks MJ, Debiec, Rychter M, Sciot R, Wejde J, Wedin R, Mandahl N, Panagopoulos I. Activation of the GLI oncogene through fusion with the Beta-Actin (ACTB) gene in a group of distinctive pericytic neoplasms. American J Pathol 2004; 164(5)164–53
- Hansen BH, Keller J, Laitinen M, Berg P, Skjeldal S, Trovik C, Nilsson J, Walloe A, Kalén A, Wedin R. The Scandinavian sarcoma group skeletal metastasis registry. Survival after surgery for pathological fractures. Acta Orthop Scand Suppl 2004; 75(311)11–15
- Panagopoulos I, Strorlazzi CT, Fletcher CDM, Fletcher JA, Nascimento A, Domanski HA, Wejde J, Brosjö O, Rydholm A, Isaksson M, Mandahl N, Mertens F. The chimeric FUS/CREB3L2 gene is specific for low-grade fibromyxoid sarcoma. Genes Chrom Cancer 2004; 40: 218–228
- Smeland S, Wiebe T, Brosjö O, Böhling T, Alvegård TA. Chemotherapy in Ewing's sarcoma. The Scandinavian sarcoma Group experience. Acta Orthop Scand Suppl 2004; 75(311)87–91
- Smeland S, Wiebe T, Böhling T, Brosjö O, Jonsson K, Alvegård TA. Chemotherapy in osteosarcoma. The Scandinavian Sarcoma group experience. Acta Orthop Scand Suppl 2004; 75(311)92–98
- Söderlund V, Skoog L, Unni KK, Bertoni F, Brosjö O, Kreicbergs A. Diagnosis of high-grade osteosarcoma by radiology and cytology. A retrospective study of 52 cases. Sarcoma 2004; 8(1)31–36
- Wedin R, Skoog L, Bauer H. Proliferaton rate, hormone receptor status and p53 expression in skeletal metastasis of breast carcinoma. Acta Oncologica 2004; 43(5)460–466
2005
- Ahlen J, Wejde J, Brosjö O, Vonrosen A, Weng WH, Girnita L, Larsson O, Larsson C. Insulin-like growth factor type 1 receptor expression correlates to good prognosis in highly malignant soft tissue sarcoma. Clin Cancer Res 2005; 11: 206–16
- Bauer HC. Controversies in the surgical management of skeletal metastases. J Bone Joint Surg Br 2005; 87(5)608–17
- Ferrari S, Smeland S, Mercuri M, Bertoni F, Longhi A, Ruggeri P, Alvegard TA, Picci P, Capanna R, Bernini G, Muller C, Tienghi A, Wiebe T, Comadone A, Boehling T, Delprever AB, Brosjö O, Bacci G, Saeter G. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxyrubicin for patients with localized osteosarcoma of the xtremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 2005; 34: 8845–52
- Muller CR, Smeland S, Bauer HC, Saeter G, Strander H. Interferon-alpha as the only adjuvant treatment in high-grade osteosarcoma: long term results of the Karolinska Hospital series. Acta Oncol 2005; 44(5)475–80
- Wedin R, Bauer HC. Surgical treatment of skeletal metastatic lesions of the proximal femur: endoprosthesis or reconstruction nail?. J Bone Joint Surg Br 2005; 87(12)1653–7
2006
- Ekström W, Söderlund V, Brosjö O. Osteoid osteoma in a 1-year old boy – a case report. Acta Orthop 2006; 77: 786–788
- Jansson Kå, Bauer HCF. Survival, complications and outcome in 282 patients operated for neurological deficit due to thoracic or lumbar spinal metastases. Eur Spine J 2006; 15(2)196–202
- Mertens F, Stromberg U, Rydholm A, Gustafson P, Bauer HC, Brosjo O, Mandahl N. Prognostic significance of chromosome aberrations in high-grade soft tissue sarcomas. J Clin Oncol 2006; 24(2)315–20
- Storlazzi CT, Brekke HR, Mandahl N, Brosjö O, Smeland S, Lothe RA, Mertens F. Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheet tumours. J Pathol 2006; 209: 492–500
- Vultvonsteyern F, Bauer HC, Trovik C, Kivioja A, Bergh P. Treatment of local recurrences of giant cell tumour in long bones after curettage and cementing. J Bone Joint Surg Br 2006; 88(4)531–5
2007
- Bartuma H, Hallor KH, Panagopoulos I, Collin A, Rydholm A, Gustafson P, Bauer HC, Brosjö O, Domanski HA, Mandahl N, Mertens F. Assessment of the clinical and molecular impact of different cytogenetic subgroups in a series of 272 lipomas with abnormal karyotype. Genes Chromosomes Cancer 2007; 46(6)594–606
- Hallor KH, Heidenblad M, Brosjö O, Mandahl N, Mertens F. Tilling resolution array comparative genomic hybridization analysis of a fibrosarcoma of bone. Cancer Genet Cytogenet 2007; 172(1)80–3
- Widhe B, Widhe T, Bauer HC. Ewing sarcoma of the rib – initial symptoms and clinical features: tumor missed at the first visit in 21 of 26 patients. Acta Orthop 2007; 78(6)840–4
2008
- Aksnes LH, Bauer HC, Jebsen NL, Follerås G, Allert C, Haugen GS, Hall KS. Limb-sparing surgery preserves more function than amputation: a Scandinavian sarcoma group study of 118 patients. J Bone Joint Surg Br 2008; 90(6)786–94
- Engström K, Bergh P, Gustafson P, Hultborn R, Johansson H, Löfvenberg R, Zaikova O, Trovik C, Wahlström O, Bauer HC. Liposarcoma: outcome based on the Scandinavian Sarcoma Group register. Cancer 2008; 113(7)1649–56
- Hallor KH, Staaf J, Jönsson G, Heidenblad M, Vultvonsteyern F, Bauer HC, Ijszenga M, Hogendoorn PC, Mandahl N, Szuhai K, Mertens F. Frequent deletion of the CDKN2A locus in cordoma: analysis of chromosomal imbalance using array comparative genomic hybridization. Br J Cancer 2008; 98(2)434–42
- Jebsen NL, Trovik CS, Bauer HC, Rydholm A, Monge OR, Hall KS, Alvegård T, Bruland OS. Radiotherapy to improve local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma: a Scandinavian sarcoma group study. Int J Radiat Oncol Biol Phys 2008; 71(4)1196–203
- Kivioja AH, Blomqvist C, Hietaniemi K, Trovik C, Walloe A, Bauer HC, Jorgensen PH, Bergh P, Follerås G. Cement is recommended in intralesional surgery of giant cell tumors: a Scandinavian sarcoma group study of 294 patients followed for a median time of 5 years. Acta Orthop 2008; 79(1)86–93
- Möller E, Stenman G, Mandahl N, Hamberg H, Mölne L, Vandenoord JJ, Brosjö O, Mertens F, Panagopoulos I. POU5F1, encoding a key regulator of stem cell pluripotency, is fused to EWSR1 in hidradenoma of the skin and mucoepidermoid carcinoma of the salivary glands. J Pathol 2008; 215(1)78–86