
Abstract
Chitosan-based drug carriers are being widely exploited for sustained and targeted delivery in cancer, anti-depression and nutritive therapeutics. In this paper, we report the preparation of S-adenosyl-l-methionine (SAMe) drug-loaded nanochitosan-based tablets and the sustained delivery of the drug substance in simulated intestinal conditions through an in vitro study. The convertibility of high molecular weight commercial chitosan to nanoparticles by ionic gelation using potassium pyrophosphate was achieved without employing harsh reaction conditions through an intermediate water-soluble chitosan preparation. The prepared nanochitosan particles with an average size of 85–127 nm showed good drug-loading capacity. In vitro release studies showed a continuous and slow release of the drug over 14 hours. Different kinetics models were applied to drug release data in order to evaluate the releasing mechanism. The drug release data fit well into the Higuchi expression, suggesting a diffusion-controlled drug delivery. The diffusional coefficient of 1.83 indicated that the drug release from the chitosan matrix was through swelling of the matrix. Agreement of the kinetic data with Higuchi and Korsmeyer–Peppas models have led us to conclude that the delivery of the SAMe drug from the nanochitosan drug carrier took place by the diffusion-controlled swelling mechanism described as Super case II transport. The prepared nanochitosan matrix was also found to be an environment-sensitive vehicle suitable for controlled drug delivery.
1. Introduction
Conventional drug delivery systems are often limited in their efficiency with respect to drug delivery site, delivery rate, duration of circulation in blood and the lateral damages caused to other normal cells and organs. New age treatments are swiftly shifting to controlled release technologies, which employ sustained and targeted drug delivery systems. Targeted drug delivery can be achieved using nanomaterials, such as nanoparticles, nanospheres or nanoencapsules, and each has its own characteristic drug-releasing methodology. Drug delivery systems include organic and inorganic nanoparticles, ceramic nanoparticles, micelles, polymeric micelles, dendrimers and liposomes. Nanoparticles can penetrate through smaller capillaries and hence can be absorbed by cells, allowing efficient drug accumulation at the target sites. They also protect the entrapped drug from gastrointestinal interferences. Nanoparticles can be mixed with drug substances in which the active ingredient is dissolved, dispersed, entrapped, encapsulated and absorbed or attached. They can be used to deliver hydrophilic and hydrophobic drugs, vaccines and proteins. These advantages of the nanoparticles allow reduction of dose and dosing frequency, thereby reducing the side effects.[Citation1] The use of biodegradable materials, such as chitosan, poly-L-lactic acid, glycolic acid and polyethylene oxide, for nanoparticle has facilitated targeted and sustained drug release for a wide range of therapeutic applications. Among the drug delivery vehicles, chitosan is one of the most extensively studied because of its biocompatibility, biodegradability, non-toxicity and cost-effectiveness. Furthermore, it possesses antimicrobial properties and absorbs toxic metals such as mercury, cadmium and lead. Chitosan has a positive charge and exhibits an absorption enhancing effect.[Citation2] This characteristic can be adapted to prepare cross-linked chitosan nanoparticles. The preparation of nanoparticles from water-insoluble polymers involves heat, high shear force, organic solvent, emulsion polymerisation, solvent evaporation and a number of other steps, which may be detrimental to drug stability. In contrast, preparation of nanoparticles from water-soluble polymers is fairly simple without the use of organic solvents and high shear force. The mechanism of formation of chitosan nanoparticles from water-soluble chitosan powder is usually based on the electrostatic interaction between the amine group of chitosan and a negatively charged group or polyanion such as tripolyphosphate.[Citation3]
The chosen drug substance, namely, S-adenosyl-l-methionine (SAMe), chemically known as 5′-[(3-amino-3-carboxy-propyl)methylsulphonio]-5′-deoxy adenosine hydroxide, is a naturally occurring, highly hydrophilic substance present in plasma and most tissues. SAMe is the primary methyl donor present in all living organisms involved in the methylation of target molecules such as DNA, proteins, lipids and in polyamine synthesis. SAMe exists in two diastereoisomeric forms, namely, the S,S and the R,S where the designation refers to the stereochemical configurations of sulphur and carbon, respectively. SAMe salts are highly stable when combined with larger anions, such as p-toluensulphonate anion or amidic derivatives of taurine with fatty acids. In addition to its use as a chief dietary supplement, SAMe is finding increased therapeutic application for treatment in liver cirrhosis, hepatitis (for liver cholestasis), osteoarthritis, depression, migraine headaches, Parkinson's disease, attention deficit hyperactivity disorder (ADHD), fibromyalgia, Alzheimer's disease and cardiovascular disease.[Citation4] To the best of our knowledge, reports on efforts to prepare and study SAMe as a nanoparticle-bound sustained release tablet are not available. The present study on the formulation of SAMe-loaded nanochitosan and sustained in vitro release of the drug will hence be helpful in tailoring its dosage level for effective therapeutic use.
2. Materials and methods
2.1. Materials
SAMe drug substance was obtained from Orchid Chemicals and Pharmaceuticals Limited, Chennai, India. Chitosan was purchased from Central Marine Fisheries Research Institute, Cochin, India. All other materials and reagents used in the study were of analytical/high performance liquid chromatography (HPLC) grade and used as purchased.
2.2. Preparation of chitosan nanoparticles and loading of SAMe
Commercial chitosan powder was dissolved in 0.1 N hydrochloric acid (in 1:50 ratio) and stirred for 30 minutes. Hydrogen peroxide (30% solution) was added to it (in the ratio of 1:5, 1:7.5, 1:10) and the mixture was heated with constant stirring for an hour at 60 °C. The solution was brought to room temperature. Ethanol was added in the ratio of 1:10 and the solution was kept at 5 °C for 24 hours. Thereafter, ethanol was removed using the Buchi distillation apparatus by keeping the temperature at 70 °C. The remaining solution was lyophilised to obtain water-soluble chitosan powder. The prepared water-soluble chitosan was cross-linked with the chosen polymers, namely, glutaraldehyde, polyvinylalcohol, polyvinylpyrrolidone, polyethyleneglycol 2000, polyethyleneglycol 10,000 and polypropyleneglycol in 100 ml water in the ratio of 1:1.[Citation5]
The water-soluble, polymer cross-linked chitosan powder was converted to nanoparticles and loaded with the SAMe drug by a single-pot ionic gelation method.[Citation6,7] The drug, SAMe, was added to an aqueous solution of chitosan in the ratio of 1:0.5, 1:0.75, 1:1 and 1:2 (polymer to drug ratio). The solution was stirred for an hour at 0 °C and then an aqueous solution of potassium pyrophosphate (polymer to anion ratios 1:0.5, 1:1 and 1:2) was added dropwise into it. The addition was continued till coagulation was observed. The coagulate so obtained was lyophilised to get SAMe-loaded chitosan nanoparticles.
2.3. Formulation of chitosan nanoparticle loaded with SAMe drug substance along with polyvinyl pyrrolidone
A drug formulation in the form of a tablet was prepared by the direct compression method using SAMe-loaded nanochitosan with hydroxypropylmethyl cellulose (HPMC), sodium lauryl sulphate (SLS) and pregelatinised starch in the ratio of 1:1:0.2:1.[Citation8] The tablet was coated with polymethylmethacrylate (0.2% solution in ethanol). Three different formulations were prepared by changing the SAMe-loaded nanochitosan to HPMC ratio of 1:1 (Formulation 1), 1:2 (Formulation 2) and 1:3 (Formulation 3) and keeping all other excipients constant. The in vitro release study was carried out by the paddle method. The dissolution medium for the first two hours was 0.1 N hydrochloric acid and pH 7.2 buffer for further hours. The volume taken for analysis was replaced with the dissolution medium. A blank tablet excluding the drug was also prepared for reference.[Citation9]
2.4. Characterisation studies
2.4.1. Scanning electron microscopy analysis
The particle size of chitosan nanoparticles (chitosan to potassium pyrophosphate ratio 1:0.5, 1:1 and 1:2) was analysed by scanning electron microscopy (SEM). The sample was dispersed on the cover glass and allowed to dry in air. This was mounted directly on the specimen metal disc using a Scotch double adhesive tape (3M, St Paul, Minnesota). Samples were scanned at various scan areas using a Shimadzu SPM 9500-2J scanning probe microscope (Shimadzu, Kyoto, Japan). For high resolution, a contact-mode cantilever was used for all analyses.[Citation10]
2.4.2. FT-IR analysis
Commercial chitosan, nanochitosan and SAMe drug-loaded nanochitosan were analysed using fourier transform infrared spectroscopy (FT-IR) technique. The samples were finely ground and taken as a dispersion in potassium bromide (KBr pellet) which was analysed using a Perkin Elmer Spectrum 65 model instrument (Perkin Elmer, MA, USA).[Citation11–13]
2.4.3. X-ray diffraction analysis
Commercial chitosan, nanochitosan and SAMe drug-loaded nanochitosan were analysed using powder X-ray diffraction (XRD) method. The samples were finely ground and placed in an aluminium sample holder. Bruker D8 advance model instrument (Bruker, Billerica, MA, USA) was used to scan the samples.[Citation14]
2.5. HPLC method development and validation
The HPLC system, used for method development and method validation, was a Waters 2695 series HPLC system with a 2996-photo diode array (PDA) detector (Waters, Milford, MA, USA). The output signal was monitored and processed using Empower2 software. Chromatographic separation was achieved on a 50 mm × 4.6 mm id, 5 μm particle, Zorbax SB C-18 reverse phase column. The mobile phase A used was 0.01 M potassium dihydrogenorthophosphate monohydrate along with 0.01 M octane sulphonic acid sodium salt buffer, adjusted to pH 3 and the mobile phase B was methanol. The flow rate was maintained at 1 ml/min and the wavelength of detection was performed at 254 nm. The HPLC gradient program was set as: 0–2/30, 2–3/30–40, 3–8/40–45, 8–10/45–95, 10–12/95, 12–13/95–30, 13–15/30 (time divided by percentage of mobile phase B).
The prepared tablet was tested for the repeatability analysis by analysing six different sample preparations and checked for its per cent relative standard deviation. The linearity experiment was established in the concentration range of 70%–130% of the drug composition in the tablet. The specificity was also established by injecting all its process- and degradation-related impurities and the peak purity was evaluated for the separation of impurities from the main peak. The recovery study was also performed by deliberately adding 70%, 100% and 130% of the drug in the final composition of the tablet. Limit of detection (LOD) and limit of quantitation (LOQ) values were also established.
2.6. In vitro release studies
The in vitro release study for the formulation was carried out using USP XXIII dissolution rate test apparatus type II by paddle method (Model Disso 2000; Lab India Dissolution Apparatus) at a rotating speed of 50 rpm, 37.5 °C using 0.1 N hydrochloric acid (900 ml) for two hours. The dissolution medium was then replaced with pH 7.2 phosphate buffer (900 ml) and tested for drug release for up to 14 hours. Ten millilitres of the sample was withdrawn at regular time intervals and the same volume was replaced with fresh dissolution medium. The amount of drug released was calculated at 254 nm measured using HPLC (Waters 2690 with PDA Detector).[Citation15,16]
3. Results and discussion
3.1. Optimisation studies on the preparation of drug-loaded chitosan
Preliminary experiments were performed to determine the optimum ratio of hydrogen peroxide for producing freely water-soluble chitosan, the ratio of potassium pyrophosphate for obtaining the desired average size of nanoparticles and the most suitable cross-linking agent with respect to drug release.
3.1.1. Optimisation of hydrogen peroxide ratio
The molecular weight of commercial as well as synthesised water-soluble chitosan samples was determined using Ostwald's viscometer at 25 °C. 1 g/l samples (treated with different ratios of hydrogen peroxide) dissolved in 0.1 M acetic acid and 0.2 M NaCl solution were taken for analysis and the molecular weight was calculated using the following Mark–Houwink equation:(1) where K = 1.81 × 10−3 cm3/g and α = 0.93.
After peroxide treatment, the viscosity and molecular weight of chitosan decreased and continued to decrease with the addition of peroxide (). This was due to the degradation of the chitosan molecular chain by peroxide. As can be seen from the results, chitosan treated with hydrogen peroxide in a 1:10 ratio was freely soluble in water. It is evident that the dramatic decrease in the molecular weight below 8000 was due to extensive chain cleavage in chitosan, which in turn has resulted in the desired solubility in water.
Table 1. Viscosity, molecular weight and solubility analysis of chitosan.
3.1.2. Optimisation of ratio of potassium pyrophosphate
The effect of varying the ratio of potassium pyrophosphate on the average particle size of nanochitosan was studied through SEM analysis. It was observed that at a chitosan to potassium pyrophosphate ratio of 1:0.5, large lumps were formed. While chitosan to pyrophosphate in the ratio of 1:2 resulted in clusters of more than 300 nm size, a 1:1 ratio of chitosan to the gelating agent yielded nanochitosan of 85–127 nm mean particle size ((a) and (b) and ). Drug loading and release generally require matrices consisting of 100 nm sized particles or less, as these particles can have prolonged circulation in blood and be active for longer durations with increased targeted drug delivery. Hence, a ratio of 1:1 for chitosan to potassium pyrophosphate was fixed to be optimum.
Table 2. Average particle size of chitosan obtained with different ratio of water soluble chitosan to potassium pyrophosphate ratio.
Figure 1. (a) SEM image of water soluble chitosan to potassium pyrophosphate (1:1) ratio. (b) SEM image of water soluble chitosan to potassium pyrophosphate (1:2) ratio.

3.1.3. Choice of cross-linking agent with respect to drug release
The choice of cross-linking agent to prepare drug-loaded nanochitosan by the ionic gelation method was determined through the drug release efficiency analysis with different cross-linking agents. SAMe drug, nanochitosan, hydroxypropyl methyl cellulose, SLS and pregelatinised starch along with each one of the cross-linking agents listed in were considered for the study in the ratio of 1:1:1:0.2:0.5:1. From , it is evident that except polyvinylpyrrolidone, all other cross-linking agents released the drug in <5 hours and the percentage of drug release was also below 85, indicating that drug–matrix attachment efficiencies were less than the desired release. Drug release duration is an indication of the entrapment efficiency of the drug-carrier matrix, which in turn is influenced by the choice of cross-linking agent used. Hence, it can be concluded that polyvinylpyrrolidone was the best choice among the selected cross-linking agents for the delivery of SAMe drug from the nanochitosan matrix.
Table 3. Drug release analysis of different cross-linking agents.
3.2. FT-IR analysis
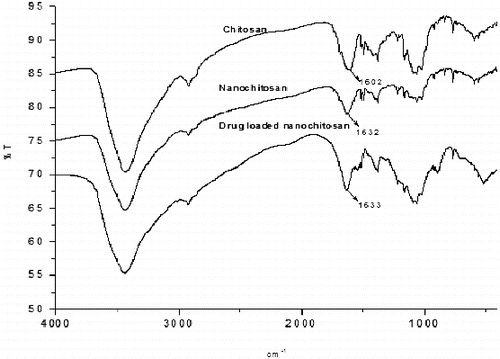
In , IR absorption spectra of chitosan, nanochitosan and SAMe drug-loaded nanochitosan can be seen. Except the peak due to NH2 stretching vibration, all the peaks, their location and intensities are similar. The shifting of the NH2 stretching vibration from 1602 cm−1 in commercial chitosan to 1632 cm−1 in nanochitosan indicates the formation of phosphate group linkage between potassium pyrophosphate and the amino group of water-soluble chitosan.
Figure 2. FT-IR spectrum of chitosan, nanochitosan and drug-loaded nanochitosan.
3.3. X-ray diffraction study
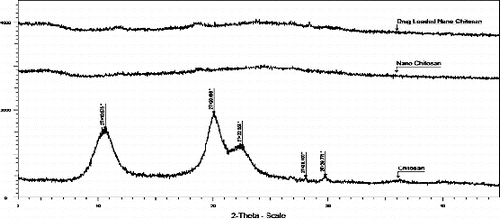
The semi-crystalline nature of the commercial chitosan is evident from its XRD pattern shown in . The conversion of semicrystalline chitosan to the amorphous form confirmed the formation of nanochitosan on ionic gelation with potassium pyrophosate.
Figure 3. XRD pattern of chitosan, nanochitosan and drug-loaded nanochitosan.
3.4. Effect of drug substance ratio to nanochitosan
In order to optimise the concentration of drug over the nanochitosan matrix, the drug was loaded in different concentrations. The effect was studied by considering different nanochitosan to drug ratios of 1:0.5, 1:0.75, 1:1 and 1:2. The percentage process yield along with drug content was calculated and is presented in . Percentage process yield is calculated as follows:(2)
Table 4. Process yield and drug recovery for drug to polymer ratio.
3.5. HPLC method development and validation for SAMe
A simple and robust method was developed in HPLC for the analysis of the content of SAMe drug substance in the chitosan matrix. The drug was found to be associated with five different impurities (designated as A, B, C, D and E in ). The separation of impurity E from the main peak was found to be critical. Different trials were carried out and the separation was achieved only when the ion paired reagent octane sulphonic acid was used in the mobile phase. When the pH was neutral or in alkaline condition, impurities D, C and E were co-eluted. Only when the pH was about 3, the separation was optimum. When acetonitrile was used in the mobile phase, these three impurities merged with the main peak. Methanol was mainly helpful for the separation of all these impurities from the main peak. Different gradient trials were also carried out in order to achieve a good separation for all impurities from the main peak and also in the less run time of about 15 minutes.
Figure 4. A typical chromatogram showing the peak of SAMe at a retention time of about 6.6 minutes obtained from drug-loaded nanoparticles.
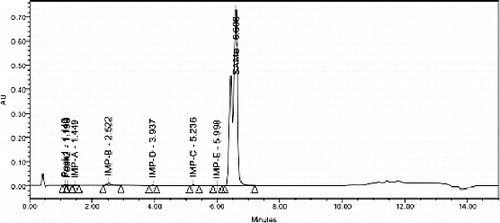
The specificity experiment was carried out by injecting individual isolated impurities (A–E) along with the SAMe drug substance. All these individual impurities were spiked at the 0.2% level to the drug substance and injected into the HPLC system with a PDA detector. The purity angle and purity threshold values confirmed the purity of the main peak and impurities. The results of repeatability, linearity, LOQ, LOD and recovery of excess drug substance present on the chitosan matrix presented in prove that the devised validation method is precise and accurate.
Table 5. HPLC method validation of SAMe drug.
The HPLC method was developed and validated so as to ensure the accuracy of data with respect to drug release. It can be concluded from the results of the above studies that only the SAMe drug release was measured in all the analyses, as all the impurities have been shown to be completely separated under the specified conditions of the HPLC analysis.
3.6. In vitro drug release studies: kinetics and mechanism
shows the release profile of the drug in the formulations. The in vitro release profile of 1:2 of drug to HPMC had a slow drug release for about 14 hours. This result proved that the prepared nanochitosan acted as an effective carrier for the controlled drug delivery of SAMe drug substance.
Table 6. Dissolution data of SAMe-based formulation from nanochitosan matrix.
The rate and mechanism of release of SAMe from the prepared coated tablets were analysed by fitting the release data into a series of kinetic equations:
Zero-order equation
(3)
where Qt is the amount of drug released at time t and K0 is the zero-order release rate constant.
First-order equation
where K is the first-order release rate constant.
Higuchi's equation
where Q is the amount of drug released at time t and K is the diffusion rate constant.
Hixson–Crowell cube root law
where Qt is the amount of drug released in time t, Q0 is the initial amount of the drug in the tablet and KHC is the rate constant. The plots were used to evaluate the drug release with changes in the surface area and the diameter of the tablet.
Korsmeyer–Peppas equation,
The results obtained from were plotted in the kinetic expressions and the plots are given in . The drug release constant was calculated from the slope of the appropriate plots and the regression coefficient (R2) was determined. The model that best fitted the release data was selected based on the regression coefficient value of various models.
Figure 5. Zero-order release kinetics model.
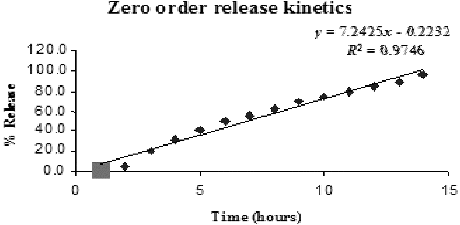
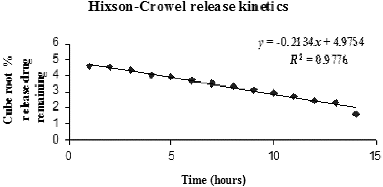
Figure 6. First-order release kinetics model.
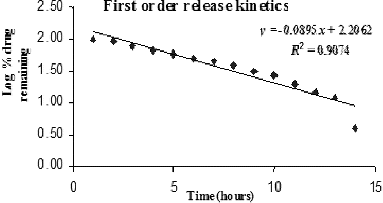
Figure 7. Higuchi release kinetics model.
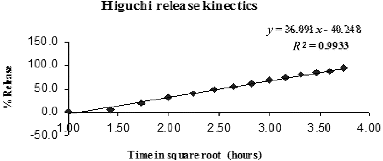
Figure 8. Korsmeyer–Peppas release kinetics model.
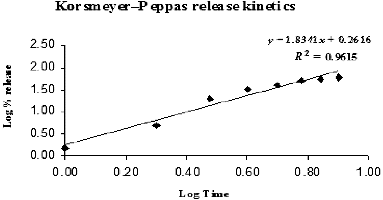
Figure 9. Hixson–Crowell cube root release kinetics model.
Derivative data from were compared to various kinetic models and the results are listed in and . The resulting data fitted well into the Higuchi kinetic model. The regression coefficient value of 0.9933 obtained in the Higuchi model indicated that the release of the SAMe drug from nanochitosan carrier was diffusion controlled. This corroborates with the observation that the drug release was prolonged over 14 hours. Thus, the release of the SAMe drug from the nanochitosan matrix is proposed to fit the Higuchi model, which best describes the release of water-soluble drugs from polymeric matrices.
Table 7. R2 values from zero-order, first-order and Higuchi kinetics models for SAMe drug release from nanochitosan matrix.
Table 8. R2 values from Korsmeyer–Peppas and Hixson–Crowell kinetics models for SAMe drug release from nanochitosan matrix.
In the controlled-release matrix tablet, which comprises drug and hydrophilic polymers, the drug release may follow a three-step mechanism, namely: (1) penetration of dissolution medium in the tablet matrix (hydration), (2) swelling with concomitant or subsequent dissolution or erosion of the matrix and (3) the transport of the dissolved drug either through the hydrated matrix or from the parts of the eroded tablet to the surrounding dissolution medium.[Citation17] The magnitude of the diffusional exponent (n) calculated from the Korsmeyer–Peppas plot provides information about the mechanism of drug release ().
Table 9. Diffusion exponent and solute release mechanism proposed by Korsmeyer–Peppas.
In the present study, the value of n for the release of SAMe from the nanochitosan matrix was calculated to be 1.83. This value indicated a Super case II transport mechanism for the drug release, in which the swelling and the resultant relaxation of the nanochitosan matrix was responsible for the release of the SAMe drug. The highest R2 for the Higuchi model data-fit and the n value of 1.83 together suggest that the drug release mechanisms for SAMe from the nanochitosan matrix were diffusion and swelling controlled.
A closer analysis of the above results suggested that the drug release pattern was sensitive to the dissolution environment. During the first two hours, when the dissolution medium was an acidic environment similar to that in the stomach, the matrix swelling was minimal and the drug release was very slow (5%). When the medium was changed to pH 7.2 buffer, resembling the intestinal environment, the polymeric swelling occurred rapidly and the drug release rate increased, but was still controlled and persisted over a long duration. These facts lead to the conclusion that the nanochitosan polymeric matrix prepared from water-soluble chitosan behaved as an environmentally responsive drug delivery system.
4. Conclusion
SAMe drug substance-loaded chitosan nanoparticles were successfully prepared by the ionic gelation method using potassium pyrophosphate and polyvinylpyrrolidone as cross-linking agents. The developed chitosan matrix possessed nanoparticles of about 100 nm size. The sustained-release drug was formulated using HPMC and SLS. In vitro release of the drug product was studied using a newly developed and validated HPLC method. The HPLC method developed in this study was found to be successful in separating all the related impurities and yielded accurate results with respect to the SAMe drug. The in vitro drug release data were fit into different release models. Drug release kinetics corresponded best to Higuchi's model of diffusion-controlled release. The diffusional coefficient value suggested the mechanism to be Super case II transport proposed by Korsmeyer and Peppas. The prepared nanochitosan drug delivery matrix was found to be an excellent drug carrier, capable of sustained release of SAMe drug under conditions similar to that of the intestinal environment. These results indicate that the environmentally responsive nanochitosan drug delivery vehicle prepared in the present study can be explored for sustained and targeted release of SAMe-based formulations for nutritive as well as therapeutic applications.
Acknowledgements
The authors B. Magesh and P.Y. Naidu sincerely thank the management of Orchid Chemicals and Pharmaceuticals Limited for supporting the research.
References
- Viswanathan B. Nanomaterials as therapeutic agent. 2009;3.7–3.8. Oxford (UK): Alpha Science International Ltd.
- Bernkop-Schnurch A, Dunnhaupt S. Chitosan-based drug delivery systems. Eur J Pharm Biopharm. 2012;81:463–469.
- Saha P, Goyal AK, Rath G. Formulation and evaluation of chitosan-based ampicillin trihydrate nanoparticles. Trop J Pharm Res. 2010;9:483–488.
- Magesh B, Chandrasekhar M, Sastry PVSA, Murugan S, Sivakumar B. A validated stability indicating RP-LC method for the determination of related impurities in S-adenosyl-L-methionine (SAMe) API. Der Pharma Chemica. 2012;4:2327–2332.
- Wilson B, Samanta MK, Santhi K, Kumar KPS, Ramasamy M, Suresh B. Chitosan nanoparticles as a new delivery system for the anti-Alzheimer drug tacrine. Nanomed: Nanotechnol, Biol Med. 2010;6:144–152.
- Agnihotri SA, Mallikarjuna NN, Aminabhavi Tejraj M. Recent advances on chitosan-based micro and nanoparticles in drug delivery. J Controlled Release. 2004;100:5–28.
- Dustgani A, Farahani EV, Imani M. Preparation of chitosan nanoparticles loaded by dexamethasone sodium phosphate. Iranian J Pharm Sci. 2008;4:111–114.
- Islam MS, Khan F, Jalil R-ul. Sustained release theophylline matrix tablets prepared by direct compression II: effect of polymers. Bangladesh Pharm J. 2010;13:42–48.
- Amish AD, Rai MK, Shukla TM. Formulation and optimization of extended release metformin hydrochloride tablet, effects of polymers and additives on drug release mechanism. Asian J Biochem Pharm Res. 2011;1:480–499.
- Muhammed RPE, Junise V, Saraswathi R, Krishnan PN, Dilip C. Development and characterization of chitosan nanoparticles loaded with isoniazid for the treatment of Tuberculosis. Res J Pharm, Biol Chem Sci. 2010;1:383–390.
- Zhang H-l, Wu S-h, Tao Y, Zang L-q, Su Z-q. Preparation and characterization of water-soluble chitosan nanoparticles as protein delivery system. J Nanomater. 2010;2010:1–5 .
- Brugnerotto J, Lizardi J, Goycoolea FM, Arguelles-Monal W, Desbrieres J, Rinaudo M. An infrared investigation in relation with chitin and chitosan characterization. Polymer. 2001;42:3569–3580.
- Wu S. Preparation of water soluble chitosan by hydrolysis with commercial α-amylase containing chitosanase activity. Food Chem. 2011;128:769–772.
- Bathool A, Vishakante GD, Khan MS, Shivakumar HG. Development and characterization of atorvastatin calcium loaded chitosan nanoparticles for sustain drug delivery. Adv Mater Lett. 2012;3:466–470.
- Razzak MSMI, Khan F, Hossain M, Anika T, Moon SA. Impact of sodium lauryl sulphate on the release of carbamazepine from methocel k15m cr based matrix tablets. Bangladesh Pharm J. 2012;15(1):79–82.
- Abirami A, Halith SM, Jayaprakash S, Prabhu RS, Karthikeyini C, Pillai KK. Fabrication and in vitro evaluation of Chitosan matrix tablets of diclofenac on colon drug delivery system (codes). Int J ChemTech Res. 2009;1:920–924.
- Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modelling on drug release from controlled drug delivery systems. Acta Poloniae Pharm – Drug Res. 2010;67:217–223.