
ABSTRACT
Vesicular systems endow large opportunities for the transdermal delivery of therapeutics. The present study was designed to investigate the potential of a novel class of vesicular system ‘proniosome’ as a carrier for transdermal delivery of bromocriptine (BCT). Proniosome formulations were prepared by the coacervation-phase separation method and the influence of factors like surfactant type and its amount, lipid concentration, cholesterol amount and drug content were studied. Span 60 was the most appropriate surfactant, and yielded vesicle size and percentage encapsulation efficiency of 1.3 µm and 98.9%, respectively. The developed system was characterised w.r.t. morphology, transition temperature, drug release, skin permeation and skin irritancy. Proniosomes exhibited a sustained release pattern of BCT in vitro. Skin permeation study revealed high penetration of proniosomes with sustained release of BCT through rat skin. The optimised proniosomal formulation showed enhanced transdermal flux of 16.15 μg/cm2/h as compared to 3.67 μg/cm2/h for drug dispersion. The developed formulations were observed as non-irritant to the rat skin and were found as quite stable at 4 and 25 °C for 90 days w.r.t. vesicle size and drug content. The dried proniosomal formulation could act as a promising alternative to niosomes and preferably for transdermal delivery of BCT.
1. Introduction
The foremost objective of drug delivery is to provide and maintain the optimal therapeutic levels of drug in the body to produce desired effect. Therapeutic efficacy of drugs depends on the delivery system that is intended to achieve the desired systemic level of drug.[Citation1] In case of conventional dosage forms, a small amount of the drug reaches to the systemic circulation, while the majority of the drug either remains unabsorbed or metabolised by the hepatic system. Transdermal delivery of drugs can eliminate such obstacles and represents a highly suitable alternative to oral or injectable delivery.[Citation2] In comparison to the other routes, transdermal delivery of drugs is of great interest as it evades the first-pass metabolism, eliminates gastrointestinal irritation and improves patient compliance. Also, this is a noninvasive route which does not require experts and medicines can be self-administered on need basis. Additionally, it minimises the dosing frequency by enabling prolonged and constant systemic level of drugs which also facilitates in reducing several side effects.[Citation3]
To achieve the effective transdermal delivery, a carrier should avoid the major skin barrier called ‘stratum corneum’ which restricts the permeation of pathogens and other molecules into systemic circulation. Several approaches have been exploited to improve the skin permeation of drugs like penetration enhancers, iontophoresis, microneedles and thermal ablation. But these methods rely upon damaging the protective barrier function of the skin and may cause irritation or other skin problems.[Citation4] Increasing evidences support the tremendous potential of nanocarriers as transdermal delivery vehicles of therapeutic agents.[Citation1] In this context, vesicular nanocarriers have been widely explored successfully for improving the efficacy of drugs via transdermal route. Liposomes and niosomes have attracted widespread interest in this regard because of their numerous advantages like biodegradability, better skin penetration, amphiphilic drug loading and sustained release properties.[Citation5] Indeed, the drug delivery applications of liposomes is encouraging, but they exhibit some limitations viz. problems of the instability during storage, premature leakage of loaded drugs, high cost and variable purity of the phospholipids; [Citation6] whereas niosomes, the hydrated mixtures of cholesterol and non-ionic surfactant are found as stable vesicular systems.[Citation5] They do not require any special conditions like low temperature or inert atmosphere for their fabrication or storage and relative low cost preparation makes niosomes an encouraging carrier than liposomes. However, they have been reported with physical instability problems e.g. aggregation, fusion, drug leakage or hydrolysis of the active compounds, thus raising concerns over their adoption.[Citation7]
An imperative advancement in the field of vesicular delivery is based on the formulation of dry niosomes named as proniosomes. Upon contact with the skin, these systems get hydrated under occlusion to form niosomes and absorbed to elicit systemic effects.[Citation8] They easily permeate across the skin in the form of niosomes and release the encapsulated drug in controlled manner. The physical stability problems of aggregation, fusion and leaking could be efficiently trounced by this system. The other major advantages of proniosomes include easy transportation, distribution, storage and dosing making them a suitable drug delivery system.[Citation9] Proniosomes have been widely studied for systemic delivery of several classes of compounds and remarkable opportunities have been found in the transdermal delivery of drugs owing to their highly permeable ability.[Citation10] Transdermal delivery of tenoxicam by proniosomes has been reported to significantly enhance the anti-inflammatory and analgesic effects as compared to the marketed tablets.[Citation11] Similarly, the in vivo efficacy of mefenamic acid has been improved with proniosomal gel.[Citation12] A considerable advance in this field has been observed in the delivery of anti-arthritic therapeutics due to the combination of topical and systemic effects. In such a study, the gel mediated delivery of luteolin has been reported to be highly effective in arthritis management.[Citation13] Proniosomes have also been widely reported in the treatment of various skin diseases via efficient and effective topical delivery of drugs. Sandeep et al. demonstrated the high permeation and prolonged release of fluconazole via topical proniosomal gel mediated delivery.[Citation14] The niosomes-derived proniosomes have shown to have good practical anti-allograft rejection efficacy on corneal transplantation rats and also exhibited superior biocompatibility with cornea without any irritation.[Citation15] Thus, the proniosomes have widespread interest as vesicular carrier and safe alternatives for transdermal delivery of therapeutics.
Bromocriptine (BCT), a semi-synthetic derivative of ergot alkaloid (2-bromo-α-ergocriptine), has been in domination since 30 years in the treatment of several endocrine disorders especially Parkinsonism. It is an agonist of dopamine receptor while inhibitor of prolactin and henceforth used for treating pituitary tumors, breast tumours, hyperprolactinaemia, infertility, menstrual disorders, neuroleptic malignant syndrome and type 2 diabetes.[Citation16] BCT has also been used with other drugs for objective measurements of rigidity, tremor, bradykinesia, gait disorder and total functional disability. It is easily absorbed through the gastroitestinal tract but extensive first pass metabolism accounts for the low bioavailability (6%). The other side effects like headache and dizziness further limits its oral delivery.[Citation17] Nanocarrier-mediated delivery of BCT has been reported to improve its therapeutic effect while minimising the side effects. Recently, BCT loaded in chitosan nanoparticles showed higher concentration in brain as investigated by Shadab and coworkers.[Citation18] Esposito et al. demonstrated the potential of lipid nanocarriers to improve the therapeutic effects of the BCT for Parkinson's disease.[Citation19] Chitosan gel mediated delivery of BCT revealed the effectiveness of transdermal route as an alternate to oral delivery.[Citation20] Literature suggests the application of transdermal delivery system to improve the therapeutic efficacy of BCT by avoiding hepatic metabolism and manitaining optimal plasma concentrations for a long period.[Citation20]
The present work was designed to investigate the feasibility of using proniosomal gel as transdermal vehicle for BCT. The influence of the various surfactants and cosurfactants was evaluated on the characteristics of proniosomes. The prepard formulations were characterised in terms of vesicle size and size distribution, surface charge, entrapment efficiency and morphology. The drug release profile of the different formulations was determined in vitro. Permeation of the proniosomes through rat skin was evaluated and the skin irritation was also assessed to ensure the biocompatibility. The proniosomes were further subjected to stability studies.
2. Materials and methods
2.1. Materials
BCT was received as gift sample from Inga Laboratories Pvt. Ltd, Mumbai (India). Soya Lecithin (Phospholipon® 90H) was kindly provided by Lipoid GmbH, Ludwigshafen (Germany). Cholesterol was procured from Sigma Aldrich (USA). Span 20, span 40, span 60, span 80 were obtained from Loba Chemi Pvt. Ltd., Mumbai (India) and tween 20, tween 60, tween 80 and carbopol - 934 LR were supplied by SD Fine Chem Ltd., Mumbai (India). Dialysis membrane (12 kDa molecular weight cut off) was purchased from Himedia Laboratories Pvt. Ltd., Mumbai, (India). All other chemicals and solvents were of analytical or HPLC grade from Sigma Aldrich (India).
2.2. Animals
Wistar Albino rats (80 ± 20 g) were procured from the disease free small animal house, Lala Lajpat Rai University of Veterinary and Animal Sciences, Hisar (India). Animals were housed in groups of four in colony cages at an ambient temperature of 20–25°C and 45%–55% relative humidity with 12 h light/dark cycles and had free access to diet and water. All research and testing activities were reviewed and approved by the institutional animal ethical committee (approval no.: JCDMCOP/IAEC/08/12/10).
2.3. Formulation of proniosomal gel
Proniosomes were prepared by already reported coacervation-phase separation method with a slight modification.[Citation21] Briefly, the drug (5 mg) and the blend of surfactant, soya lecithin and cholesterol were mixed with 125 µl absolute ethanol in a wide mouth glass tube and covered with lid to prevent loss of solvent. This mixture was heated on a water bath at 60–70 °C until it dissolved completely. To the resultant mixture, phosphate buffer (pH 7.4) was added and heated slightly on a water bath to obtain a clear solution which was allowed to cool at room temperature and then mixed with equal volume of 1% carbopol-934 to obtain clear proniosomal gel. Several grades of spans and tweens were used in the preparation to study their effect on formulation characteristics. For further optimisation of the formulations, different proniosomal formulations were prepared by varying the amount of surfactant, lecithin, cholesterol and BCT, as listed in
Table 1. Entrapment efficiency and vesicle size of different proniosomal formulations.
2.4. Characterisation of proniosomal gel
The size and the size distribution of the niosomes formed after the hydration of proniosomal formulation were measured using Zetasizer Nano-ZS90 (Malvern Instruments, UK). One millilitre of the diluted sample was used for the analysis and size measurements were performed in triplicate at 25 °C using disposable sizing cuvettes.
Proniosomal formulations were also evaluated under optical microscopy to confirm the formation of niosomes by hydration. Briefly, a thin layer of the proniosomal gel was spread on to the glass slide, covered with cover slip and observed under ordinary light microscope (BA-310, Motic, USA) at 100 x magnification.
2.5. BCT encapsulation efficiency (EE)
The percentage EE was calculated by determining the un-encapsulated drug content by HPLC. For this, 0.2 g of proniosomal gel was reconstituted with 10 ml phosphate buffer (pH 7.4) in a glass tube by the application of sonicator (Rolex, India). The un-encapsulated BCT was removed by centrifugation at 14,000 rpm for 30 min at 4 °C (REMI, India). The centrifugation was done in triplicate using fresh medium each time and the supernatant was collected. The supernatant was mixed and assayed for BCT content by HPLC (LC2010C-HT, Shimadzu Corporation, Japan). The percentage drug encapsulation was calculated by following equation.[Citation22]where Ct is the concentration of total BCT, and Cf is the concentration of free BCT.
2.6. HPLC analysis of BCT
The Shimadzu HPLC system (LC-2010C HT, Japan) consisting of LC-20AT gradient pump, an autosampler and UV/VIS detector was used. A Luna 5µ C18 column (100A, 250 × 4.6 mm) was used for the analysis. The mobile phase consisting of acetonitrile and ammonium formate in the ratio of 55:45 v/v was delivered at the rate of 1 ml/min. The aliquots of 20 µl of each sample were injected and analysed at 305 nm.[Citation19] The standard curve was plotted in the concentration range of 0.0078–5µg/ml. The retention time for BCT was 4.5 min and the lowest detection limit was found to be 0.0078 µg/ml.
2.7. Scanning electron microscopy (SEM)
The morphology of the niosomes formed upon hydration of proniosomal formulations and the effect of the amount of cholesterol in vesicle formation was also evaluated using SEM. For this, a small amount of proniosome gel was diluted with phosphate buffer (pH 7.4) in a glass tube. The formed vesicles were dispersed on an adhesive carbon tape mounted on an aluminum stub. The samples were coated with gold using a vacuum evaporator (3 × 10−1 atm.) and examined at 25 kV accelerating voltage under SEM (Jeol, JSM- 6510, Tokyo, Japan) equipped with a digital camera.[Citation22]
2.8. Differential scanning calorimetry (DSC)
DSC analysis was carried out to study the physical state of the drug inside the gel and to determine the possible interactions between BCT and the vesicle ingredients. For this, 6 mg of each sample (BCT, span 60, drug free and drug loaded proniosomes) was sealed in a standard aluminium pan and subjected to analysis using DSC-4000 (Perkin Elmer, USA). The samples were scanned at a heating rate of 10 °C/min. An empty pan was used as a reference. The thermograms were obtained in the temperature range of 40 to 350 °C.
2.9. In vitro drug release study
The in vitro release profile of BCT from the proniosomes was determined using dialysis technique and compared with control (ethanolic solution of BCT). Proniosomal gel equivalent of 5 mg of BCT was placed in a pre-swelled dialysis bag (MWCO 12 kDa) and immersed in 100 ml mixture of citrate buffer and ethanol (70:30% v/v) adjusted to a pH of 5.6 to mimic the skin pH. The outer phase was maintained at 32 ± 0.5 °C and stirred continuously over a magnetic stirrer (100 rpm). Owing to the poor solubility of BCT in water, ethanol was used to maintain the sink conditions.[Citation23] The beaker was closed during the analysis to minimise the solvent evaporation. The samples were withdrawn at predetermined time intervals of 1, 2, 3, 4, 6, 8, 12, 24 and 48 h, followed by replenishment with 5 ml fresh medium to maintain a constant volume. The samples were further centrifuged at 12,000 rpm at 4 °C and the supernatant was analysed using the HPLC. For comparison, the in vitro release study of control was also carried out in similar conditions. The data collected were subjected to various release kinetic models treatment i.e. zero, first, Higuchi and Korsmeyer-Peppas models.[Citation24] All the measurements were done in triplicates.
2.10. Skin permeation studies
After the abdominal hair of the animals were shaved using a hand razor with care, they were sacrificed. The abdominal skins were excised and stored in a deep freezer (−20°C) until further use. The skin was properly cleaned to remove the subcutaneous fat and other blood vessels. Further, it was equilibrated in phosphate buffer solution (pH 7.4) for 1 h prior to the experiment and sandwiched between the two compartments such that the stratum corneum side and the dermis of the skin were towards the donor and receptor compartments of the Franz diffusion cell (diffusion area: 2.26 cm2 and volume: 25 ml), respectively.[Citation11] The receptor medium consisted of 25 ml mixture of ethanol: phosphate buffer (30:70 v/v).[Citation23] To simulate the in vivo conditions, the proniosomal gel (eq. to 2 mg of BCT) was spread evenly onto the skin surface and covered tightly with an occlusive film. Paraffin wax was applied on the circular rim of the dosing site to ensure occlusion and also to minimise the formulation leakage during study.[Citation25]
Finally, the Franz cell compartments were clamped together and maintained at the temperature of 37 ± 0.5 °C with circulator water bath. The receiver medium was continuously stirred (100 rpm) with a small magnetic bar in order to prevent any boundary layer effects. One ml sample was taken at predetermined time intervals of 1, 2, 3, 4, 6, 8, 12, 24 and 48 h, from the receptor compartment and replaced with the equivalent amount of fresh receptor solution. Each experiment was replicated three times and the samples were analysed by HPLC. The amount permeated through the skin membrane was calculated from the concentration of BCT in the receiving solution.
2.11. Skin irritation study
The safety of skin is the prime importance for topical administration. This test was carried out to determine the consequential localised skin reaction of the formulation. Wistar Albino rats weighing 80–85 g were used. The formulation was applied to the left ear of the animals and the right ear was used as a control. The grades of the erythema developed were visually monitored for six days and reported as: 0 (no erythema development), 1 (very slight erythema), 2 (barely visible few blood vessels), 3 (light erythema development), 4 (main blood vessels visible), 5 (slight erythema development) and 6 (main blood vessels more obvious and slight erythema development).[Citation26] Equation Equation(1)(1) was used to calculate the irritation potential.
(1) where, A represented the erythema value and B the corresponding day.
2.12. Stability studies
The optimised proniosomal formulation was stored in a closed amber coloured covered glass tube at 4 ± 2 °C, 25 ± 2 °C/60 ± 5% RH and 40 ± 2 °C/75 ± 5% RH, respectively, as per International Conference on Harmonisation (ICH) guidelines. The particle size distribution and the percentage EE were determined after 30, 60 and 90 days. The results were reported as the mean of three separate measurements of each sample. Data were statistically analysed by two-way ANOVA (followed by Bonferroni's multiple comparison test) using GraphPad Prism software-5.0 and P value of 0.05 was considered as significant.
3. Results and discussion
3.1. Optimisation of the proniosomal formulations
The formulation variables greatly influence the characteristics as well as performance of the obtained formulation. The proniosomal formulations were optimised by ‘one factor at one time.’ First, a different grade of the commonly used surfactants i.e. spans or tweens were screened to choose the suitable surfactant for the formulations. The amount of the selected surfactant was varied thus at high and low levels. Similarly, the amount of the lecithin and the cholesterol was also varied to high levels to study their influence on the formulation characteristics. Finally, the drug amount was optimised.
Tweens and spans are the popular non-ionic surfactants used as vesicles forming agent in the preparation of niosomes and proniosomes. On evaluating different grades of spans e.g. 20, 40, 60 or 80 and tweens e.g. 20, 60 or 80, it was found that their nature considerably affected the drug entrapment and the final vesicles size (). Percentage EE of the proniosomes was found to be higher with spans than with tweens which might be attributed to the high hydrophobicity of spans than tweens. Spans also have low hydrophile-lipophile balance (HLB) value and higher phase transition temperature (Tc), thus greater amount of the drug was expected to be encapsulated in the lipid layers of the vesicles. Higher TC further aids in the formation of ordered gel with less leaky lipid bilayers. Among all the formulations, proniosomes prepared with span 60 (F3S60) showed highest EE (98.9%) with a vesicles size of 1.3 µm. Tweens not only showed the low EE but the vesicles size was also very high e.g. 2.6 µm with tween 60, because of their longer alkyl chain. Thus, the 90 mg of span 60 was chosen as the optimised amount for the formation of proniosomes.
The amount of the selected surfactant ‘span 60’ was further varied at low level (45 mg) and high level (135 mg). The surfactant concentration was found to be an important variable in the formation of niosomal vesicles and encapsulation of drug therein. The variation in the concentration greatly affected the entrapment efficiency with a predominant effect on vesicles size also. Proniosomes with high amount of span 60 were found to increase the vesicle size to 2.6 µm with negligible effect on EE. An increase in surfactant concentration after an appropriate level facilitate the formation of the mixed micelles which can adhere along with the niosomal vesicles, thus show increased size and lowered drug encapsulation. While the decrease in surfactant concentration from 90 mg to 45 mg led to the significant decrement in EE (87.9%) with large vesicles of 2.0 µm.
Lecithin acts as co-surfactant in the formation of smaller size proniosomal vesicles and also improves the drug permeation into vesicles by increasing hydrophobicity. A significant increment in EE (99%) was observed by increasing lecithin content to 135 mg. The un-hydrogenated lecithin used in this study allowed the chains to bend, loosening the adjacent molecule that assembled to form the niosomal membrane. The loosely packed membrane was thus formed which showed high permeability.[Citation27]
The concentration of cholesterol also plays an important role in the formation and drug entrapment in the vesicles because these molecules house themselves as ‘vesicular cement’ into the bilayers of the surfactant membranes.[Citation28,Citation29] This mechanism is responsible for the permeability, rigidity and stability of the niosomes vesicles. In this study, an increase in the cholesterol content from 10 to 15 mg was accompanied by a significant decrease in EE% as well as vesicles size. The larger vesicles of 2.3 µm were observed at low cholesterol amount (5 mg). Increasing the cholesterol content beyond a certain limit, it competes with drug to get entrapped in the vesicles bilayers, hence displace the drug and results in low EE. Vesicular membranes with high level of cholesterol molecules further limit the water intake therein and result in decreased vesicle size.[Citation30]
Finally, the amount of drug was also optimised to attain the optimal drug loading. Increasing the drug content from 5 to 7 mg resulted in reduced EE of 90.9% while high EE of 99.1% was observed at low drug content (3 mg). The fact lies in the formation of a fixed number of niosomes at a given amount of vesicle components (surfactant, cholesterol and lecithin), thus having definite entrapment efficiency and the excess drug gets precipitated.[Citation27,Citation31]
3.2. Morphology of proniosomes
The formation of proniosomal vesicles was observed under light microscope with 100x magnification to validate their formation. The niosomes were found to be unilamellar spherical vesicles with smooth surface and were discrete and separate with no aggregation or agglomeration ((A)).
Figure 1. Photomicrograph of proniosomal formulation F3S60 (A). SEM image of niosomes formed after hydration of F3S60 (B), F10SCL (C) and F11SCH (D).

The surface morphology of the proniosomes was evaluated by SEM. The micrographs revealed that the vesicles were almost spherical and clearly identifiable with sharp boundaries and large internal aqueous space. A clear difference was observed by varying the cholesterol content in the formulation and an increasing cholesterol amount revealed the higher rigidity in the vesicle walls ().
3.3. DSC analysis
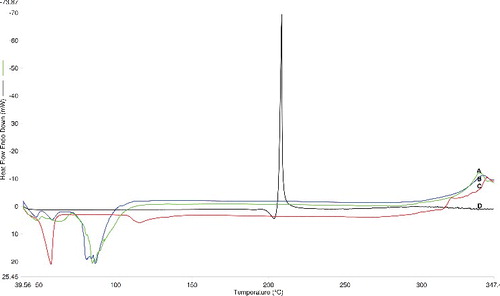
DSC thermograms of BCT, span 60, drug free and drug loaded niosomes are illustrated in . BCT, span 60 depicted endotherms at 197 and 58 °C, respectively, corresponding to their melting temperatures. DSC thermogram of the proniosomes showed a sharp endothermic peak at 84.98 °C indicating the interaction between the molecules of span 60, cholesterol and lecithin. This confirms the formation of the double layer structure of the vesicle, as reported previously.[Citation32] No peak of BCT was observed in the drug-loaded formulation, indicating the amorphous nature of the drug. No major difference was observed in the thermograms of drug-free and drug-loaded proniosomes. However, the endothermic peak of the niosomal bilayer was shifted from 84.98 °C to a broad wide peak at 90.74 °C after drug loading, because some cholesterol molecules were displaced by the drug. These results suggest the formation of bilayers of niosomal vesicles and the complete encapsulation of BCT therein.
Figure 2. DSC thermograms of bromocriptine proniosomes (A), blank proniosomes (B), span 60 (C) and bromocriptine (D).
3.4. In vitro release studies
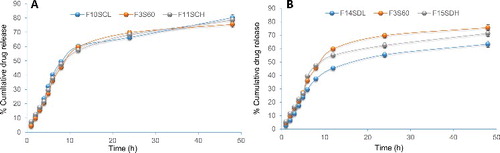
It was apparent that the incorporation of a drug into vesicular systems resulted in sustaining the drug release profile for a longer period of time. BCT was released from the proniosomes with an initial fast release phase followed by a sustained release pattern. The initial fast release was due to desorption of the drug molecules that were present on the surface of formed niosomes. In later phase, the BCT release was regulated by diffusion through the inflated bilayers and exhibited sustained release pattern.[Citation33] The optimised proniosomes (F3S60) showed a significant sustained drug release which was 75.53% after 48 h, in comparison to the control which released 97.76% of BCT within 5 h (). The formulation prepared with high cholesterol content (FCH) showed a more sustained release of BCT with reduced initial burst release effect. Because the high cholesterol content facilitated the formation of compact niosomal bilayers with reduced permeability and thus hindered the release of drug entrapped.[Citation34,Citation35] When the release data were fitted into various kinetic models, it was found that BCT release from the proniosomal formulations followed Higuchi kinetics. This model suggested the diffusion controlled release of the drug.
Figure 3. In vitro drug release from proniosomes prepared using different cholesterol (A) and drug (B) content (n = 3). Amount of cholesterol was 5 mg (F10SCL), 10 mg (F3S60) and 15 mg (F11SCH), and the amount of bromocriptine was 3 mg (F14SDL), 5 mg (F3S60) and 7 mg (F15SDH).
3.5. Skin permeation studies
Ex vivo permeation studies predicted the in vivo permeation behaviour of the product when it was subjected to the topical applications. The skin permeation of BCT from the optimised formulation (F3S60) and control was performed using skins obtained from the Wistar Albino rats using vertical Franz diffusion cell.[Citation36] BCT dispersed in carbopol 934 gel was used as a control. After 48 h of the study period, only 33.034% of BCT of the control permeated thorough the rat skin while it was 86.191% in case of proniosomal formulation (). It became clear that proniosomes possess higher skin penetration ability. The skin permeation parameters of proniosomes as well as drug suspension at the equal drug concentration were also determined. The flux value obtained from proniosomal gel (16.15 μg/cm2/h) was found to be 4.4 times higher than that of the drug suspension (3.67 μg/cm2/h) (). The comparative low skin permeability of BCT in suspension was due to its lower aqueous solubility, while the high permeability of proniosomes resulted in improved skin penetration of BCT. Better transdermal flux without lag phase of proniosomal gel might be the result of one or more of the wing mechanisms as reported previously i.e. increased solubility of BCT, high association of drugs with vesicle bilayers, increased partitioning of vesicles into the stratum corneum and elasticity of vesicle membrane.
Figure 4. Ex vivo release of bromocriptine from proniosomes and drug dispersion (n = 3).
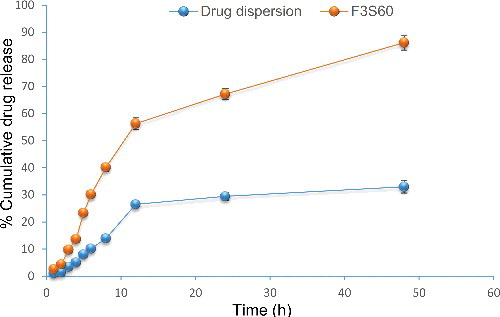
Table 2. Permeation parameters of drug dispersion and proniosomal formulation.
3.6. Skin irritancy test
The optimised proniosomal formulation (F3S60) showed an irritation potential of 0.66 which was in an acceptable range, as previously reported by Van-Abbe N. et al., which depicted that a value between 0 and 9 indicate the applied formulation to be non-irritant to human skin.[Citation37] Any erythema, edema or inflammation were absent on rat skin after six days of application of the formulation. Thus, the developed formulation was validated to have a completely non-irritant effect.
3.7. Stability study
As evident from the results, BCT leakage from the vesicles may be due to the phase transition of the surfactant and lipid at 40 °C (). However, no significant change was observed in the vesicular size or EE of the formulations when stored at 4 ± 2 °C and 25 ± 2 °C/60 ± 5% RH for the period of 90 days.
Table 3. Stability study of the optimised proniosomal formulation (F3S60).
4. Conclusion
The study reports proniosomes as a potential carrier for transdermal delivery of BCT. The dry proniosomal gel exhibited the formation of 1.3 µm sized smooth spherical niosome vesicles after hydration. Span 60 was found to be most appropriate surfactant for the preparation of proniosomes. The influence of amount of lecithin, cholesterol and drug was also studied and optimised. Proniosomes exhibited 98.9% of entrapment efficiency and facilitated a sustained release of encapsulated drug. The proniosomal formulation improved the transdermal flux of drug dispersion by 4.4 times when evaluated using ex vivo skin permeation study. The developed formulation was found to be non-irritant to the rat skin. The high stability of system further favours the potential of proniosomes for systemic delivery of therapeutics.
Acknowledgments
The writing assistance from Mr Sandeep Kumar is gratefully acknowledged. The authors would also like to thank Lipoid for kind support with materials.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Tomoda K, Makino K. Chapter 7 – Nanoparticles for transdermal drug delivery system (TDDS). In: Makino HO, editor. Colloid and interface science in pharmaceutical research and development. Amsterdam: Elsevier; 2014. p. 131–147.
- Prausnitz MR, Langer R. Transdermal drug delivery. Nat Biotechnol. 2008;26:1261–1268.
- Ita K. Transdermal delivery of drugs with microneedles: Strategies and outcomes. J Drug Deliv Sci Technol. 2015;29:16–23.
- Wiedersberg S, Guy RH. Transdermal drug delivery: 30+ years of war and still fighting! J Control Release. 2014;190:150–156.
- Moghassemi S, Hadjizadeh A. Nano-niosomes as nanoscale drug delivery systems: an illustrated review. J Control Release. 2014;185:22–36.
- Lauterbach A, Muller-Goymann CC. Applications and limitations of lipid nanoparticles in dermal and transdermal drug delivery via the follicular route. Eur J Pharm Biopharm. 2015;97:152–163.
- Abdelkader H, Alani AW, Alany RG. Recent advances in non-ionic surfactant vesicles (niosomes): self-assembly, fabrication, characterization, drug delivery applications and limitations. Drug Deliv. 2014;21:87–100.
- Yasam VR, Jakki SL, Natarajan J, et al. A review on novel vesicular drug delivery: proniosomes. Drug Deliv. 2014;21:243–249.
- Rahimpour Y, Kouhsoltani M, Hamishehkar H. Proniosomes in transdermal drug delivery. Curr Pharm Des. 2015;21:2883–2891.
- Mokale VJ, Patil HI, Patil AP, et al. Formulation and optimisation of famotidine proniosomes: an in vitro and ex vivo study. J Exp Nanosci. 2016;11:97–110.
- Ammar HO, Ghorab M, El-Nahhas SA, et al. Proniosomes as a carrier system for transdermal delivery of tenoxicam. Int J Pharm. 2011;405:142–152.
- Wen MM, Farid RM, Kassem AA. Nano-proniosomes enhancing the transdermal delivery of mefenamic acid. J Liposome Res. 2014;24:280–289.
- Abidin L, Mujeeb M, Imam SS, et al. Enhanced transdermal delivery of luteolin via non-ionic surfactant-based vesicle: quality evaluation and anti-arthritic assessment. Drug Deliv. 2015:1–6.
- Sandeep G, Reddy V, Devireddy SR. Formulation and evaluation of fluconazole pro-niosomal gel for topical administration. J Appl Pharm Sci. 2014;4:098–104.
- Li Q, Li Z, Zeng W, et al. Proniosome-derived niosomes for tacrolimus topical ocular delivery: in vitro cornea permeation, ocular irritation, and in vivo anti-allograft rejection. Eur J Pharm Sci. 2014;62:115–123.
- Desplantie O, Tremblay-Gravel M, Avram R, et al. The Medical treatment of new-onset peripartum cardiomyopathy: a systematic review of prospective studies. Can J Cardiol. 2015;31:1421–1426.
- Thongrangsalit S, Phaechamud T, Lipipun V, et al. Bromocriptine tablet of self-microemulsifying system adsorbed onto porous carrier to stimulate lipoproteins secretion for brain cellular uptake. Colloids Surf B Biointerfaces. 2015;131:162–169.
- Md S, Haque S, Fazil M, et al. Optimised nanoformulation of bromocriptine for direct nose-to-brain delivery: biodistribution, pharmacokinetic and dopamine estimation by ultra-HPLC/mass spectrometry method. Expert Opin Drug Deliv. 2014;11:827–842.
- Esposito E, Mariani P, Ravani L, et al. Nanoparticulate lipid dispersions for bromocriptine delivery: characterization and in vivo study. Eur J Pharm Biopharm. 2012;80:306–314.
- Degim IT, Acarturk F, Erdogan D, et al. Transdermal administration of bromocriptine. Biol Pharm Bull. 2003;26:501–505.
- Abd El-Alim SH, Kassem AA, Basha M. Proniosomes as a novel drug carrier system for buccal delivery of benzocaine. J Drug Deliv Sci Technol. 2014;24:452–458.
- Alsarra IA, Bosela AA, Ahmed SM, et al. Proniosomes as a drug carrier for transdermal delivery of ketorolac. Eur J Pharm Biopharm. 2005;59:485–490.
- Fang JY, Yu SY, Wu PC, et al. In vitro skin permeation of estradiol from various proniosome formulations. Int J Pharm. 2001;215:91–99.
- Higuchi T. Mechanism of sustained-action medication. Theoretical Analysis of Rate of Release of Solid Drugs Dispersed in Solid Matrices. J Pharm Sci. 1963;52:1145–1149.
- Hwang B-Y, Jung B-H, Chung S-J, et al. In vitro skin permeation of nicotine from proliposomes. J Control Release. 1997;49:177–184.
- Goyal G, Garg T, Malik B, et al. Development and characterization of niosomal gel for topical delivery of benzoyl peroxide. Drug Deliv. 2015:1–16.
- Singh VS, Gaur RC. Dispersion of cholesterol in aqueous surfactant solutions: interpretation of viscosity data. J Dispers Sci Technol. 1983;4:347–359.
- Uchegbu IF, Florence AT. Non-ionic surfactant vesicles (niosomes): physical and pharmaceutical chemistry. Adv Colloid Interface Sci. 1995;58:1–55.
- Agarwal R, Katare OP, Vyas SP. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm. 2001;228:43–52.
- Mokhtar M, Sammour OA, Hammad MA, et al. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361:104–111.
- Honeywell-Nguyen PL, Bouwstra JA. Vesicles as a tool for transdermal and dermal delivery. Drug Discov Today Technol. 2005;2:67–74.
- Hathout RM, Mansour S, Mortada ND, et al. Liposomes as an ocular delivery system for acetazolamide: in vitro and in vivo studies. AAPS PharmSciTech. 2007;8:E1–E12.
- Subczynski WK, Wisniewska A. Physical properties of lipid bilayer membranes: relevance to membrane biological functions. Acta Biochim Pol. 2000;47:613–625.
- Abd-Elbary A, El-laithy HM, Tadros MI. Sucrose stearate-based proniosome-derived niosomes for the nebulisable delivery of cromolyn sodium. Int J Pharm. 2008;357:189–198.
- Guinedi AS, Mortada ND, Mansour S, et al. Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm. 2005;306:71–82.
- El-Laithy HM, Shoukry O, Mahran LG. Novel sugar esters proniosomes for transdermal delivery of vinpocetine: preclinical and clinical studies. Eur J Pharm Biopharm. 2011;77:43–55.
- Van-Abbe N, Nicholas P, Boon E. Exaggerated exposure in topical irritancy and sensitization testing. J Soc Cosmet Chem. 1975;26:173.