
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
As the transistor size approaches nano dimension, short channel effects become dominant in micro devices, leading to an incessant struggle to keep pace with Moore's law. This paved way to development of newer technologies like molecular electronics. The self-assembled bottom-up approach makes molecular switches more prone to defects than micro devices. The widely studied molecular switches are mechanically coupled and this paper deals with bistable rotaxane. Feasibility of using bistable rotaxane as a molecular electronic device is analysed in terms of total molecular energy, energy band gap, ionisation energy and distance between ring and dumbbell. Total molecular energy is the vital criteria that decides the feasibility of using a self-assembled rotaxane as a switch. Rotaxane has a band gap of 1.44 eV at Ground State Co-Conformer (GSCC) and hence acting as a semiconductor. Ionisation energy and position of ring are also important in deciding the switching activity. Due to process variations during self-assembly, ring can localise anywhere over the dumbbell and it may leads to a faulty switch. This work describes a method to verify the switching action of bistable rotaxane through simulation. Testing method described here is carried out before the actual manufacturing of nano crossbar and hence cost effective.
1. Introduction
Probability of achieving cent percent perfect chips during fabrication lifecycle of Integrated Circuits (IC) is next to impossible, without coming across a few faulty ones. The cost of detecting a faulty IC increases by an order of magnitude as dimension of IC decreases. This top down fabrication technology, though until recent years, consistently followed Moore's law; with transistor size on IC's shrinking exponentially, it would never ever be so. Further increase in integration density of IC is becoming a challenge due to major physical limitations [Citation1]. Thus, the implementation of new materials and architectures became a necessity.
Miniaturization down to the size of atoms and molecules can be achieved by using molecular-scale devices [Citation2]. In late 1990, it was experimentally found that individual molecules can conduct electricity which was earlier believed to occur only in bulk semiconductors and this discovery became the heart of molecular electronics. In molecular electronic devices, artificially generated carbon-based molecules behave like diodes or programmable switches [Citation3] and their greatest advantage is their smallest (molecular) size. Molecular electronic devices are a family of devices including molecular diodes, molecular resistors, molecular wires, three terminal devices, and molecular switches [Citation4]. The use of functional molecules as electronic devices offers variability and structural control which cannot be provided by solid state electronic devices [Citation5].
Unlike the conventional Complementary Metal Oxide Semiconductor (CMOS) lithography, nano electronic devices are manufactured through the stochastic self-assembled bottom-up approach which allows the components to guide each other in constructing a structure with no outside intervention. Zhang at.al describes a method for controlling the self assembly of nanoparticles through reversible supra molecular interactions [Citation6]. By utilizing specific intra molecular interactions, regular array like structure are easy to produce with this kind of manufacturing technology [Citation3]. Since two terminal devices like molecular switches in crossbar circuits are more advantageous than three terminal devices in terms of flexible designs and reduced area requirement they become dominant category device [Citation7]. A group of molecules prepared artificially from two or more mechanically coupled components can work as simple switches [Citation8]. Molecular switches remain stable in absence of electrical stimulation while they can turn ON/OFF in its presence. ON/OFF state of a molecular switch corresponds to the two stable states/geometry of the molecule can attain. The state change is normally reversible [Citation3].
The lack of external intervention makes nano scale devices more prone to manufacturing defects and transient faults than micro dimension devices [Citation3]. The major variation in fault behaviour of nano scale and VLSI devises is its higher sensitivity to temperature, background noises, voltage changes, other environmental changes. In current lithography technique, the common treatment employed for defective IC is to summarily reject any chip even if a single defect is found. But this method will not hold good in nano electronics because their defect densities mean that no chip will be totally defect free [Citation9]. This leads to the requirement of new methods for testing molecular electronic devices. In this paper we are presenting a method to verify the proper switching action of rotaxane molecular switch through computational chemistry tools.
2. Molecular electronic devices
Carbon nanotube wires shows transistor like electrical switching properties to act as a gate [Citation10]. Polyphenylene-based molecular wires and switches involve chains of organic aromatic benzene rings. Substituted Polyphenylenes like alkanedithiols are capable of switching small currents [Citation11]. Construction of an electronic device using a single organic molecule is first reported in the paper entitled ‘molecular rectifiers’ by Aviram and Ratner in 1974. This experimental discovery of current conduction through individual molecule lead to all the developments in molecular electronics that we see today [Citation12].
The smallest molecular electronic switch can be formed as a benzene- (1,4)-dithiol (S-C6H4-S, BDT for short) molecule bridging between two Gold (Au) electrodes. The effect of binding site on the conductance of BDT molecules is investigated and found that top-molecule-top (top-top) geometry, (where both S atoms of the molecule occupy the top site of the Au surface) is the OFF state geometry and the top-molecule-hollow (top-hollow) geometry (with one S atom being connected to the threefold hollow site) is the ON state. It is also found that, an isolated BDT molecule cannot used as a molecular switch while it became highly conductive if connected between two gold electrodes [Citation13].
2.1. Mechanically interlocked molecular switch
Commonly studied mechanically interlocked molecular switches are Bistable Rotaxanes, Pseudo Rotaxanes and Catenanes. In this paper, we are dealing with bistable rotaxanes. Rotaxanes, an attractive chemical species, capable of performing reversible mechanical motions in response to external stimuli [Citation14]. Bistable rotaxanes are composed of a dumbbell-shaped molecule encircled by a macrocyclic ring. Without breaking the covalent bond, the two components cannot be separated, because the end group molecules of the dumbbell are bulkier than the macrocycle ring cavity [Citation15].
Mechanically interlocked molecules like [Citation1] rotaxane is also become one of the growing interest in the research community. These molecules posses only one molecular component rather than two molecular components in [Citation2] rotaxane. But these molecules can also possess two stable geometries at suitable conditions. Li et.al reported the synthesis of a tri and tetra branched [Citation1] rotaxane with identical rotaxane arms and different cores. [Citation1] rotaxane arm possess a ferrocene unit which connects the macrocycle to thread. The team has investigated the effect of external stimuli over [Citation1] rotaxane with Tri-Fc-1and Tetra-Fc-1 and reported that the macrocycle will have a mechanical movement. They found that the change in structural conformation is strongly dependent on the applied external stimuli [Citation16]. Zhang et. al, reported a bistable [Citation1] rotaxane molecule that responds to some external acid-base stimuli. In this system, the mechanical movement of the ring caused by the external stimuli also generate a fluorescent signal. Once the ferrocene unit is oxidised in [Citation1] rotaxane, the intensity of fluorescence signal is locked and the reduction process cannot result in an obvious fluorescent signal. These kind of rotaxane molecular switches can be used to implement switchable materials with controllable function [Citation17].
Chemical structure of bistable Rotaxane [Citation18], used in this work is shown in . There are two probable sites for the ring tetracationic cyclophane, Cyclobis (paraquat-p-phenylene), CBPQT4+, to localize: the tetrathiafulvalene group (TTF) is to the left and the 1, 5- dioxynaphthalene group (DNP) is to the right of the dumbbell. In this molecule, CBPQT4+ ring act as electron acceptor while TTF and DNP site are the electron donors. The π-election-rich tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) units attracts tetracationic CBPQT4+ ring since it contains two π-electron-deficient bipyridinium units. Under normal conditions, the CBPQT4+ ring in the bistable rotaxane shows a higher preference for encircling the TTF over the DNP unit. The TTF encircled translational isomer of the bistable rotaxane is therefore the ground state co-conformer (GSCC) [Citation19]. A rapidly driven shuttling of CBPQT4+ ring to the DNP site occurs when we remove an electron from the TTF site and it will lead to the first oxidation state of ground state co-conformer. Upon reduction of the TTF site back to its charge-neutral state, the meta state co-conformer, CBPQT4+ encircling DNP site is formed.
Figure 1. Chemical structure of bistable rotaxane. (a) Dumbbell component & CBPQT4+ ring of Rotaxane molecular switch, (b) OFF state of Rotaxane switch (CBPQT4+ ring at TTF site), (c) ON state of rotaxane switch (CBPQT4+ ring at DNP site).
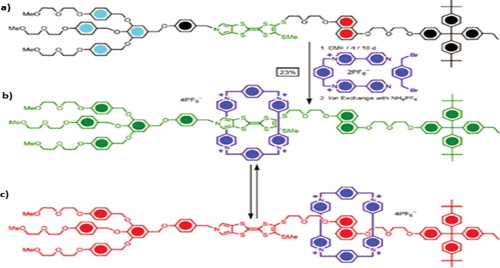
Template directed synthesis of the electron donating species like TTF and DNP with the π electron-accepting tetracationic cyclophane, cyclobis- (paraquat-p-phenylene) (CBPQT4+) is the most common method in rotaxane synthesis. The amphiphilic bistable rotaxane is terminated by a hydrophilic dendritic stopper at one end and a hydrophobic tetraarylmethane stopper at the other end. S. Lee and W. Lu [Citation20] studied a bistable rotaxane molecule with TTF and Hydroquinone(HQ) as ring localising positions for ring shuttling mechanism. But the team does not analyse it in terms of stability or conductivity. The rotaxane structure selected here, has a CBPQT4+ ring and a dumbbell component containing a monopyrrolo-TTF (MPTTF) unit instead of the normal TTF unit and a DNP moiety. This particular structure also has a ‘knob’ in the shape of a SMe group situated between the MPTTF and DNP site. Due to the presence of SMe group, the two stable translational isomers (switching states) become relatively temperature independent. It is also found that, the translational isomers of bistable rotaxane with SMe group between MPTTF and DNP is in 1:1 ratio at 298 K while isomers of the bistable rotaxane without SMe group between MPTTF and DNP is in 3:1 ratio, which is not a desirable property for molecular switch [Citation18]. But this structure has to compromise for lower switching speed due to the hindrance offered by the SMe group. Solution-phase measurements of these molecular switches results in characterizing their structures and functions. Mechanical motions of these mechanically interlocked molecules on a surface are influenced by their interactions with the surface and with neighbouring molecules, as well as by the conformations of the dumbbell components [Citation21].
Out of the various artificial molecular machines developed rotaxane is well known for its unique shuttling mechanism and its capability to act as a switch. Various external stimuli like redox, light, chemical input etc are used for achieving this shuttling mechanism. Gao et.al describes a shuttling mechanism based on phototrigger mechanism for a rotaxane which consists of identical ring stabilising stations. A photolabile is introduced between the two stations and covalently bonded with the dumbbell thread. Upon irradiation with UV light, the bulk phototrigger is successfully removed which is followed by a fast ring shuttling movement [Citation22]. For the growth of molecular electronics as future technology, a researcher should focus not only on conductance but also on to the structural stability. In this introductory stage of molecular electronics single molecule experiments are more important than multi molecular experiments [Citation5]. In this work, individual rotaxane molecule is analysed for the various CBPQT4+ ring position over the dumbbell and in terms of structural stability, band gap and ionisation energy.
2.2. Nano crossbar architecture with bistable molecular switch
Bistable rotaxanes are used as the switching element in nano crossbar (NC) architectures. Young Chen et.al proposed a reconfigurable crossbar architecture with molecular switches as the connecting element in a two-dimensional mesh network. Each of the rotaxane molecule act as a resistive element and is reversible, hence they can act as active memory cell. The team, illustrates the use of NC architecture as an 8*8 memory cell with the help of a computer controlled switching matrix [Citation23]. Nano crossbar architectures offer several advantages such as ease-of-programmability, fault-tolerance, low-cost fabrication, and high device-density [Citation24]. A nano scale FPGA (field programmable logic array) using molecular switches as the programming element is proposed [Citation25] which offers reconfigurability. The interconnects are implemented using nanowires and traditional CMOS fuse based logic block is replaced by molecular switch based NC arrays.
But in a NC switch network (NCSN), defects can occur either in switch or in wires. The molecular switch crossbar points may become defective at the time of manufacturing or at later stages because of various imperfections. In literature, various defect tolerant mechanism for NC architectures are existing. In this work, we are identifying the faulty rotaxane molecule through simulation results even before the complete manufacturing of NC architectures. The most common type of switch defect is closed or open switch defect, which results in losing reconfiguration property of the switch [Citation26]. The covalent bond created between the ring and ring stabilising localities makes the structure to be stable with a lower molecular energy. But during the self assembly of rotaxane molecule, there exist a chance of localising ring on any unintended site over the dumbbell. Such a scenario will not provide a normal covalent bond between the electron accepting and electron donating sites and as a result, total molecular energy of the system will be higher or it may damage the mechanical coupling exist in the bistable rotaxane structure.
3. Simulation results
Bistable rotaxane molecule is simulated for various position of CBPQT4+ ring on the dumbbell and the results are analysed in terms of total molecular energy. It is already found in the existing literature that CBPQT4+ ring at TTF and DNP position are the two stable states of the bistable rotaxane molecule [Citation21]. When the bistable rotaxane is used as an electronic device, the stable geometries correspond to proper switching action while unstable/incorrect geometries correspond to a faulty switching action. Hence the structure is optimised for the two stable geometries and single point energy calculations are done for the remaining geometries. In order to find the faulty switching state simulation results are analysed in terms of total molecular energy and band gap energy.
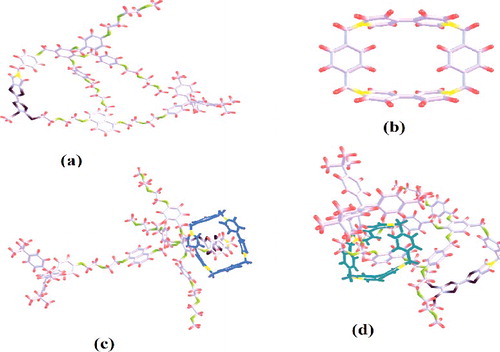
Gaussian 09 W is used for the simulations. The chemical structure of the molecule is identified including the details like type and number of atoms, bond lengths, bond angles, dihedral angles etc. Rotaxane's dumbbell is optimised in various steps and then combined each other. CBPQT4+ ring also optimised for lowest total energy. Optimised dumbbell and ring structure is given (a,b) respectively. Structure corresponding to both ON and OFF position are created on Gaussian software by combining the optimised dumbbell and ring for two different locations and then optimised for minimum energy. Hatree Fock optimisation is used with 6–31 G (d) basis set and the simulation methodology has been previously explained [Citation27]. Ring positioned between the MPTTF and DNP or between one end of rotaxane and switching position is simulated with single point energy calculations. This also use 6–31 G (d) basis set. Mulliken charge based analysis is done for measuring the HOMO and LUMO gap and hence the energy band gap in each case.
Figure 2. Completely optimized bistable rotaxane (a) Dumbbell alone (b) CBPQT4+ Ring alone (c) GSCC/OFF state (CBPQT4+ ring at TTF site) and (d) MSCC/ON state (CBPQT4+ ring at DNP).
3.1. Feasibility of using bistable rotaxane molecule as a molecular electronic switch
3.1.1. Total molecular energy of a successful molecular switch
According to existing literature CBPQT4+ ring on TTF is the Ground State Co Conformer and CBPQT4+ ring on DNP is the Metastable State Co Conformer [Citation28]. Optimisation of molecular structure identifies the feasibility of using any molecule as a molecular electronic device. The higher molecular energy condition or distorted geometry (existence of a chemical bonding rather than the mechanical coupling between ring and dumbbell) condition corresponds to a faulty switch while lower energy with proper geometry corresponds to a true bistable rotaxane molecular switch.
Optimisation of bistable rotaxane shows that, when the ring is situated at a distance of 1.77 Å from the Carbon of Pyrole (taken as origin for the measurement of ring position over dumbbell) of MPTTF structure, total molecular energy will be −9700.538 861 Hartree (−263 971.0635 eV). The negative sign indicates that the structure is highly stable. CBPQT4+ ring is moved across the dumbbell structure for various possible ring localising positions and the stability of bistable rotaxane's switching action is analysed. The MPTTF site is of 7.9Å (5.59Å from the Carbon of pyrole) length and total molecular energy of the structure with ring over MPTTF is calculated for at 4 different positions, each separated by 1–1.5 Å. The optimised structure is analysed in each case. When the ring is positioned at 1.77Å from Carbon of Pyrole, it gives the lowest total molecular energy among all the other 4 positions. Remaining 3 positions are also optimised to a stable geometry while there is a slight increase in total molecular energy and a maximum energy difference of 1.9 eV at a distance of 3.91Å from the origin. Hence, irrespective of the exact location of the ring if it is localised over the MPTTF station during self-assembly then, rotaxane molecule qualifies as a successful electronic molecular switch.
When the ring is positioned at DNP site, the total molecular energy is in the range of −9700 Hartree itself. This indicates that ring localising over the DNP site is also a stable geometry for rotaxane. When the CBPQT4+ ring is located at 15.88 Å from the Carbon of Pyrole (Ring on DNP) total molecular energy of the structure will become −9700.387 199 Hartree (−263 966.93 eV). The DNP structure is of 3.8Å long and two more simulations are done by moving ring 1Å in each trial. But all the 3 optimisation results in the same structure with same total molecular energy. (c) shows the optimised structure of bistable rotaxane in GSCC (ring at MPTTF) state and (d) corresponds to MSCC (ring over DNP) condition. From , it is clear that the ring can easily move across the dumbbell since there is no electrical bond formation between the ring and dumbbell.
3.1.2. Total molecular energy of a faulty molecular switch
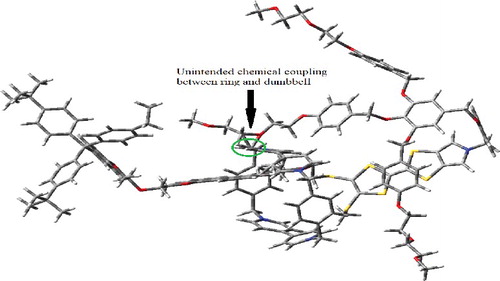
To verify the proper switching action of bistable rotaxane as a molecular electronic switch, we need to compare the stability of the molecular structure by positioning CBPQT4+ ring over dumbbell at various positions other than MPTTF and DNP. , shows the output of a ring position between the MPTTF and DNP unit. Single point energy calculation for the ring between TTF and DNP site, results in creation of one or more additional chemical bond/s between the ring and dumbbell. The type and number of unwanted bond created depends on the position of ring over the dumbbell thread and the distance between the ring and dumbbell. This scenario leads to a chemical coupling between the ring and dumbbell rather than the intended mechanical coupling. Thus, the ring will no longer be able to shuttle between two stations and hence the molecule lost its switching property. The chemical bonding results in an increase in total molecular energy. The total molecular energy difference between the GSCC state's energy and each trial varies from 19 to 96 eV. According to the fundamentals of chemistry a triple bond between carbon and oxygen can't exist in nature and such a condition in our simulation results in a very high total molecular energy difference of around 270 eV. This strongly agrees that ring positioned between the MPTTF and DNP will either yield a theoretically existing geometry but with additional chemical coupling or will result in a geometry that is not possible to exist in nature.
Figure 3. CBPQT4+ ring positioned between the MPTTF and DNP.
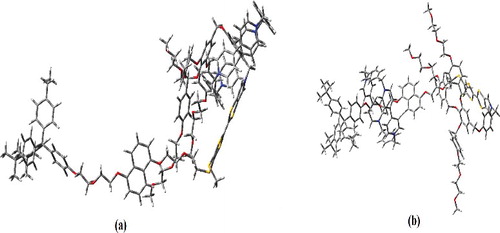
In the next phase of simulation, CBPQT4+ ring is positioned between MPTTF and one end of the dumbbell thread. This results in an undistorted geometry, but the total molecular energy of the system is increased more than 50 eV compared to that of the GSCC state's energy. As the total molecular energy of the system increases, stability of the system decreases. Hence rotaxane cannot give a proper switching action if the ring is located away from MPTTF section. (a) shows the output of ring positioned between MPTTF and one end of the dumbbell. Remaining analysis concentrates in total molecular energy of rotaxane by positioning ring between DNP site and the other end of dumbbell thread. The simulations results in an undistorted geometry but there is an increase in energy of 10–15 eV. (b) depicts the result of ring positioned between DNP and the other end of dumbbell thread.
Figure 4. Ring positioned between (a) MPTTF and one end of the dumbbell (b) DNP and the other end of dumbbell.
shows a graphical representation of change in total molecular energy with respect to the ring position. The Carbon of Pyrole in the MPTTF section is taken as the origin. In , properly optimised/ undistorted geometry of rotaxane molecule is indicated by blue dots while red dots correspond to distorted geometry. Calculated total molecular energies with respect to the ring position over the dumbbell is tabulated in .
Figure 5. Total molecular energy (Hartree) of rotaxane molecule with respect to the distance (Å) moved by the ring along the dumbbell.
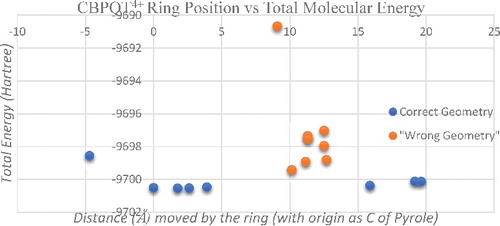
Table 1. Variation in total molecular energy of bistable rotaxane w.r.t position of CBPQT4+ ring.
3.1.3. Distance between ring and dumbbell
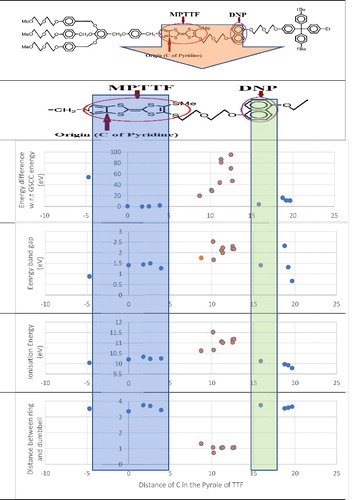
Distance between the ring and dumbbell is the most important factor affecting mechanical coupling. shows the difference in energy from GSCC state's energy with respect to the distance between dumbbell and ring. It is clear that, mechanical coupling is existing in the structure only if the distance is more than 3.4 Å. The ring width is measured to be 7.52 Å across the carbon atoms of Pyridine in the CBPQT4+ ring. The dumbbell and ring distance on one side is 3.75 Å and 3.77 Å on the other side when the ring is positioned over MPTTF. When the ring is localised over the DNP site, these distances are 3.74 Å and 3.78 Å respectively. Thus, the ring is almost equally spanned (less than 1Å difference) into both direction across the dumbbell when it is at GSCC and MSCC states.
Table 2. Variation in different parameters with respect to the position of CBPQT4+ ring.
While, in the all other cases ring is not placed uniformly across the dumbbell and this will alter the force of attraction between the ring and dumbbell and result in an increase in molecular energy. If due to any process variation the ring and dumbbell distance is reduced to be less than 1.4 Å then it leads to unintended bond formation. This results in a chemical coupling and the molecule will lose its switching property. Thus, the possibility of molecular switching can be analysed by calculating the distance between then ring and dumbbell also. This distance does not have effect on the total molecular energy of the system.
3.2. Energy band gap
The stability of the bistable rotaxane molecular switch is analysed based on the total molecular energy of the structure. Any technology or device which is aimed to be developed as a replacement for the Silicon devices must have an energy band gap in the range of semiconductors. Silicon is the most commonly used semiconductor material and it has a band gap energy of 1.14 eV at standard temperature and pressure. Hence energy band gap of bistable rotaxane molecule in his OFF or ON states must be in the range of 1.14 eV to function as a molecular electronic device.
For the OFF state (GSCC condition), ie ring located at 1.77 Å from the origin, energy band gap is 1.44 eV. From the simulation results, it is found that some ring position over dumbbell with a mechanically coupled geometry results in a much lower energy band gap. But those states are not acceptable since the total molecular energy for them is considerably high. For the ON state (MSCC state), a band gap energy of 1.414 eV is obtained when the ring is positioned at 15.88Å from the origin. This result shows an improvement [Citation27] in the band gap energy of bistable rotaxane in its ON state and is now comparable to that of silicon's band gap energy. It is also found that, the energy band gap is always greater than 1.44 eV in the case of chemically coupled (wrong) geometries. These results are concluded in .
3.3. Ionisation energy
According to the fundamentals of chemistry, a lower value of ionisation energy indicates that the molecular structure is more conducting. But ionisation energy cannot give a quantitative analysis on the energy required for obtaining conductivity. Koopman's theorem [Citation29] gives a measure of ionisation energy (I) and it can be calculated from the highest occupied molecular orbital energy (EHOMO). This is given by EquationEquation (1)(1)
(1) ;
(1)
(1)
From the analysis of ring positioned over MPTTF site, we can find that ionisation energy of the most stable position is 10.334 eV and the similar geometries has a slight in change in ionisation energy. This shows that OFF state of the rotaxane molecule has the lowest conductivity. For the MSCC or ON state the ionisation energy is slightly lower and it is 10.119 eV, and it is only 0.215 eV less than that of the GSCC state. Change in ionisation energy with respect to the various position of ring over the dumbbell are tabulated and given in . From , it can be found that when the ring is kept midway between MPTTF and DNP unit the ionisation energy is always greater than 10.5 eV. This is because the ring is positioned between two electron donor regions. While, when the ring is kept between MPTTF and one end of the dumbbell or between DNP and other end of dumbbell, ionisation energy is less always greater than that of GSCC state which indicates a decrease in conductivity.
All these results, are summarised and plotted into a graphical form as given in . Here all energies are in electron volt and the distance is measured in Å. It is observed that when the ring is located at 9.07 Å right to the origin, the total molecular energy is jumped to −263 702.73 eV creating an increase in energy of 268.34 eV from the GSCC state's energy. The sharp increase in energy makes the molecule highly unstable and this is due to the formation a triple bond between Carbon and Oxygen, which is impossible to exist according to the fundamentals of chemistry. This point is eliminated from the graphical representation, so that the change in energy values become clearer.
Figure 6. Comparison of Ionisation energy, Energy band gap and Difference in molecular energy (from GSCC state's energy) at various CBPQT4+ ring over dumbbell of bistable rotaxane.
4. Conclusions
Nano crossbar architectures are one of the dominant nanotechnology research area. Since the assembly of NCSN is through self-assembled bottom up approach, fault rate is higher compared to that of the traditional semiconductor based devices. In this work, a method for identifying faults in molecular switch is exclusively described. Current molecular electronic research is mostly concentrated on to the chemical aspects and/or conductivity of molecular electronic devices especially onto bistable molecular switches. Feasibility of using bistable rotaxane molecule as a molecular electronic device is analysed in terms of total molecular energy, energy band gap, ionisation energy and the distance between the ring and dumbbell of the mechanically coupled molecule. According to the fundamentals of chemistry, total molecular energy of a stable system is always negative and the conformation with lowest molecular energy will be the ground state. Also as the total energy increases stability of the system decreases.
The total molecular energy of bistable rotaxane molecular switch obtained from these simulation results show that, the energy is lowest when the ring is located over MPTTF and is −263 971.0635 eV. Hence the GSCC site is always taken as the position with ring located over MPTTF site and which satisfies the existing literature [Citation19]. When the ring is positioned over the DNP site, total molecular energy is slightly higher than that of GSCC and is −263 966.93 eV. It is clear that the energy difference between the two states is only 4 eV and this makes ring localised over DNP site as the meta stable state (MSCC), hence satisfies the literature [Citation19]. When the ring is positioned over the dumbbell between the two electron donating sites (MPTTF and DNP), then the total molecular energy of the system is increased more than 25 eV and hence the stability decreases. At these positions, molecule forms a chemical coupling instead of the intended mechanical coupling. When the ring is positioned anywhere between one end of the rotaxane and any one electron donating unit, then the total molecular energy is increased by around 15 eV and this results in decreased stability for the system.
From the analysis of rotaxane molecule in terms of energy band gap shows that when the ring is positioned exactly either on MPTTF or DNP, then the energy band gap falls on the semiconductor energy band gap range. Traditional integrated circuits are based on the semiconductor material Silicon, which has a band gap energy of 1.14 eV. It is calculated that at standard temperature and pressure band gap energy of rotaxane bistable molecular switch at GSCC state is 1.44 eV while at MSCC state is 1.41 eV. If the ring is situated between the two electron donating regions, then the energy band gap is slightly higher and is still in the semiconductor region. But since the structure lost its mechanical coupling in this region, rotaxane molecule cannot be used as a molecular electronic switch. If the ring is moved to a considerable distance away from the MPTTF site then energy band gap becomes less than one and it becomes a conductor. When the ring is positioned away from DNP site, energy band gap increases with increases in total molecular energy rather than the distance moved by ring. Hence, in these positions either the energy band gap can fall into a conductive region (less than 1 eV) or the energy band gap can remain in semiconductor range (1–2 eV) [Citation30]. But, when the ring is positioned between DNP and one end of rotaxane, then absence of two stable conformations near the ring makes rotaxane unable to switch between ON and OFF position even if the band gap energy falls in semiconductor range.
Ionisation energy will give a qualitative measure on the conductivity of a molecule in various conformations or states. A lower value of ionisation energy indicates higher conductance. The GSCC states corresponds to an ionisation energy of 10.334 eV. In the existing literature [Citation19] it was found that MSCC state is more conductive than GSCC and MSCC states ionisation energy from the HF simulation is 10.119 eV, hence validated. From the calculated results, it can be found that when the ring is positioned between the two electron donating sites ionisation energy is more (0.4–1.5 eV) compared to that of GSCC or MSCC states ionisation energy. This indicates that these positions are less conductive and hence bistable rotaxane cannot act as an electronic molecular switch if the ring is positioned between MPTTF and DNP. As the ring is positioned between electron donating species and one end of the rotaxane, it can be seen that ionisation energy is less compared (0.1–0.5 eV) to that of MSCC. Hence these positions are more conductive. But since there is only one electron donating species near the ring, it cannot shuttle between stable states and hence can't use as an electronic molecular switch.
When the distance between the ring and dumbbell is greater than 3.4 Å molecule can function as a molecular switch because of its mechanical coupling. When the ring is localised over MPTTF site then the separation between ring and dumbbell is almost equal on both sides (3.75 and 3.77 Å) while the ring is on DNP site these distances corresponds to 3.74 and 3.78 Å. Thus the difference between the separation of ring over dumbbell on both sides is less than 1 Å. While if the distance between ring and dumbbell becomes less than 1.4 Å onto any side then a chemical coupling is formed, and the molecule will lose its switching ability. From these results, we can confirm that bistable rotaxane molecule can function as a proper molecular electronic switch only if the CBPQT4+ ring is located either at MPTTF or at DNP site. If the ring gets localised anywhere other than MPTTF or DNP site due to any variations in rotaxane's self-assembly process, then the molecule cannot be used as an electronic device.
Due to the process variations that can occur during the self-assembly of rotaxane synthesis the CBPQT4+ ring can localise over any location on the dumbbell. This scenario will lead to a faulty switch if the ring gets fixed over any location other than MPTTF or DNP site. A faulty molecular switch will lead to a defective Nano crossbar architecture. Since the manufacturing technology is self-assembled, the rate of defect formation is unpredictable and an exhaustive testing on all NCSN becomes a necessity. An exhaustive testing on NCSN after its complete manufacturing becomes tedious and this leads to the requirement of identification of faulty/defective switch before the actual manufacturing of nano crossbar architecture. This work describes a method to verify the switching action of bistable rotaxane molecule through simulation. Based on the results of this work, it can be concluded that a bistable rotaxane molecule will lose its switching ability, 1. if the difference between total molecular energy of the synthesised rotaxane and GSCC states energy (−263 966.93 eV) is more than 10 eV, 2. If the measured distance between ring and dumbbell in the synthesised molecule is less than 1.4 Å, or 3. If the energy band gap is more than 2 eV. If all these results are leading to a successful switching, then with the help of spectroscopic measurements identify the ring position over dumbbell and verify the number of electron donating units near the ring. If there is only such site near the ring, then the molecule cannot shuttle between OFF/ON position. In brief, a bistable rotaxane molecule can act as a proper electronic molecular switch only if there are two electron donating sites near the ring and it satisfies the above mentioned three criteria. Testing method described here for molecular switches are cost effective since it is carried out before the actual manufacturing of NCSN. A fault aware/fault avoided logic mapping can be then done for nano crossbar structure and thus minimising the error percentage in a fully manufactured NCSN
Acknowledgement
This work has been carried out as a part of doctoral research work of Devisree S at Birla Instiute of Technology and Science Pilani, Dubai Campus, UAE.
References
- Lu W, Lieber CM. Nanoelctronics from bottom up. Nat Mat. 2007;6:841–850.
- Zahir A, Zaidi S, Pulimeno A, et al. Molecular transistor circuits: from device model to circuit simulation. In: IEEE/ACM International Symposium on nanoscale architectures; Paris, France, 2014. p. 129–134.
- Haselman M, Hauck S. The future of integrated circuits: A survey of nanoelectronics. Proc IEEE. 2010;98:11–38.
- Ruhee OS, Randhawa DKK. Visions for a molecular future. Int J Adv Res Comput Commun Eng. 2015;4:260–264.
- Scheer E. Visions for a molecular future. Nat Nanotechnol. 2013;8:385–389.
- Zhang Q, Qu D-H, Wang Q-CTH. Dual-mode controlled self-assembly of TiO2 nanoparticles through a Cucurbit[8]uril-Enhanced radical cation dimerization interaction. Angew Chemie Int Ed. 2015;54:15789–15793.
- Heath JR, Ratner MA. Molecular electronics. Phys Today. 2003;56:43–49.
- Chen J, Wang W, Reed M, et al. Room-temperature negative differential resistance in nanoscale molecular junctions. Appl Phys Lett. 2000;77:1224–1226.
- Mishra M, Goldstein SC. Defect tolerance at the end of roadmap. In: Proceedings of IEEE International test conference; Charlotte, USA. 2003.
- Ellenbogen JC, Love JC. Architectures for molecular electronic computers: Logic structures and an adder designed from molecular electronic diodes. Proc IEEE. 2000;88:386–426.
- Hornyak G, Moore JJ, Tibbals HF, et al. Fundamentals of nanotechnology. London, New York: CRC Press; 2008.
- Aviram A, Ratner MA. Molecular rectifiers. Chem Phys Lett. 1974;29:277–283.
- Koydeen TR, Devisree S, Kumar A, et al. Simulation and Analysis of BDT Molecule with Au Electrodes as a Molecular Switch. In: UKSim-AMSS 18th International Conference on Computer Modelling and Simulation; UK: IEEE; 2016.
- Coskun A, Banaszak M, Astumian R, et al. Great expectations: can artificial molecular machines deliver on their promise ? Chem Soc Rev. 2012;41:19–30.
- Sauvage J, Dietrich-Buchecker C. Molecular catenanes, rotaxanes and knots: a journey through the world of molecular topology. Weinheim,Germany: John Wiley & Sons; 2008.
- Li H, Li X, Agren H, et al. Two switchable star-shaped [1](n) rotaxanes with different multibranched cores. Org Lett. 2014;16:4940–4943.
- Li H, Zhang J-N, Zhou W, et al. Dual-mode operation of a Bistable [1]Rotaxane with a flourescence signal. Org Lett. 2013;15:3070–3073.
- Jeppesen JO, Nielsen KA, Perkins J, et al. Amphiphilic bistable rotaxanes. Chem-A European J. 2003;9:2982–3007.
- Dichtel W, Heath JR, Stoddart JF. Designing bistable [2]rotaxanes for molecular electronic devices. Philosophycal Trans Royal Soc A. 2007;365:1607–1625.
- Lee S, Lu W. The switching of rotaxane based motors. Nanotechnology. 2011;22:205501–205507.
- Ye T, Kumar AS, Saha S, et al. Changing stations in single bistable rotaxane molecules under electrochemical control. ACS Nano. 2010;4:3697–3701.
- Gao C, Luan Z-L, Zhang Q, et al. Triggering a [2]Rotaxane molecular shuttle by a photochemical bond-cleavage strategy. Org Lett. 2017;19:1618–1621.
- Chen Y, Jung G-Y, Ohlberg DAA, et al. Nanoscale molecular-switch crossbar circuits. Nanotechnology. 2003;14:462–468.
- Kule M, Hafizur R, Bhattacharya BB. On finding a defect-free component in nanoscale crossbar circuits. Procedia Comput Sci. 2015;70:421–427.
- Devisree S, Kumar A, Raj R. Nanoscale FPGA with reconfigurability. In: 18th Mediterranean Electrotechnical Conference (MELECON); Cyprus: IEEE; 2016.
- Ghavami B, Tajary A, Raji M, et al. Defect and variation issues on design mapping of reconfigurable nanoscale crossbars. In: IEEE Computer Society Annual Symposium on VLSI; Greece. 2010. p. 173–178.
- Steuerman DW, Tseng HR, Peters AJ, et al. Molecular mechanical switch based solid state electrochromic devices. Angew Chemie Int Ed. 2004;43:6486–6491.
- Devisree S, Kumar A, Koyadeen RT. A methedology to simulate and analyse molecules as molecular switches. Int J Simul-Syst, Sci Technol. 2016;17:22.1–22.11.
- Pantelides ST, Ventra DM, Lang ND, et al. Molecular electronics by the numbers. IEEE Trans Nanotechnol. 2002;1:86–90.
- Kittel C. Introduction to solid state physics. Berkeley: John Wiley & Sons; 2005.