
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Polymeric lipid hybrid nanoparticles (PHLNPs) are core-shell nanoparticle structures made up of polymer cores and lipid shells that have properties similar to both polymeric nanoparticles and liposomes. Methotrexate (MTX) loaded PLHNPs containing tween 80, phosphatidylcholine, poly D, L-lactic-co-glycolic acid (PLGA) and glyceryl tripalmitate prepared using solvent injection and homogenisation method for a glioblastoma treatment option. The MTX-loaded PLHNPs optimised by Box–Behnken design to minimise particle size, higher entrapment efficacy and maximise MTX concentration in the brain at 4 h. The particle size, entrapment efficacy, concentration of drug in the brain at 4 h, zeta potential, and AUC(Brain)/AUC(Plasma) ratio were in the range of 173.51–233.37 nm, 70.56–86.34%, 6.38–12.38 μg/mL, 25.78–36.31 mV and 1.02–5.32. In-vitro drug release studies, cellular internalisation of optimised formulation against U-87 MG shows good anticancer effects.
1. Introduction
Nanotechnology is playing a pivotal role while fighting against cancer. Many drugs, genes and other macromolecules can be used to encapsulate or scaffold within nanoparticles [Citation1,Citation2]. This immune inert nanocarrier system triggers targeted delivery at the site of action, resulting in increased therapeutic activity [Citation3]. Polymeric and lipid nanoparticles are commonly used in such nanocarrier systems, and these nanocarrier systems are tailored using a variety of alteration techniques to achieve higher therapeutic efficacy [Citation4]. This, in turn, aids in improving nanocarrier bio-stability and target specificity [Citation5]. Long-circulating chemotherapeutic agents, for example can be encapsulated within modified nanocarrier systems with active targeting ligands like genes, protein, hormone, folic acid and so on to activate the enhanced permeability and retention (EPR) effect and reticuloendothelial opsonisation [Citation6]. When it comes to polymeric nanoparticles, these nanocarrier systems have good structural integrity and stability during storage; additionally, changes in preparative methods could improve their functionality and target specificity [Citation7,Citation8]. PLGA, PEG and chitosan have been shown to reduce nano-specific interaction and improve the biodegradability and biocompatibility of polymeric nanoparticles in several studies [Citation9]. However, because lipid nanoparticles, such as Liposomes and Nosomes, are nearly identical to biological membranes, they appear to be superior to polymeric nanoparticles in terms of biocompatibility and target specificity [Citation10,Citation11]. Recent research suggests that lipid-PEG liposomes can be combined to create stable nanoparticles. A unique and new generation nano vehicle called polymeric lipid hybrid nanoparticles (PLHNPs) was recently developed to address the limitations of polymeric and lipidic nanoparticles [Citation12]. These hybrid nanoparticles combine the benefits of polymeric and liposomal nanoparticles, as well as the possibility of a stealth coating, to extend in-vitro circulation time and improve biocompatibility and stability [Citation12,Citation13]. Recent research on the preparation of PLHNs has reduced two-step synthesis to one, resulting in increased development throughput. To overcome the difficulties of lipid-based nanoparticles, more research needs to emphasise on polymeric-lipid hydride nanoparticular framework, which could provide strategic advantages by combining core polymeric materials and lipid shells’ ridged structural integrity [Citation14]. The entire drug payload can be retained in these robust polymeric-lipid hydride nanoparticles, and the outer hydrophobic lipid coating can prevent water from entering the central polymer to channelise diffusion [Citation15].
As per Jiayi Gu et al. studies, astaxanthin encapsulated lipid-polymer hybrid nanoparticles have a greater effect against cisplatin-induced toxicity [Citation16]. As per Salam Massadeh et al. research, anastrozole was encapsulated by a polyethylene glycolated (PEGylated) polymer–lipid hybrid nanoparticulate device using a direct emulsification solvent evaporation process [Citation17]. It has a better encapsulation efficacy and superior anticancer effects against MCF-7 cell lines. Similarly, as per Xiangzhao Ai et al.’s studies, in mice cartilage injury, collagen-peptide shell and PLGA core lipid polymer hybrid nanoparticles may be very useful as a control drug delivery [Citation18].
According to multiple research findings and patents, polymeric-lipid hybrid nanoparticles (PLHNPs) could have a sustained release effect on the encapsulated compound [Citation19,Citation20]. Adsorption of a suitable stabiliser or surfactant on the outer layer of the core–shell may also improve nanoparticles permeability through the blood–brain barrier (BBB) [Citation21]. Recent research findings also indicate that during treatment, extracellular serum glycoproteins, such as vitronectin, can adsorb to the surface of PLHNPs, enhancing anticancer activity.
Poly D, L-lactic-co-glycolic acid (PLGA) was used to improve the drug profile in this study [Citation22]. Furthermore, PLGA-encapsulated nanoparticles protect medications from degradation and aid absorptive transcytosis into the endothelial cortex [Citation23]. The reticuloendothelial system (RES) is restrained from PLGA solid lipid nanoparticles below 200 nm [Citation24]. As a result, the primary aim of this study was to make drug-loaded PLHNPs for better stability and rigidity of the formulation [Citation25].
It is often challenging to get the correct medicament for glioblastoma or glioma (GBM) treatment. Methotrexate (MTX) was considered after a thorough analysis of the literature. MTX is a drug used to combat cancers such as non-small cell lung cancer, breast cancer and ovarian cancer [Citation26]. However, only a few experiments have been triggered to treat GBM with PLGA solid lipid nanoparticles filled with MTX [Citation27]. For different cancer therapies, commercial MTX tablets are available: Folitrax®, Mext®, Mexate®, Biotrexate®[Citation28], Oncotrex®, Merex® and Methocel®. However, most tablets are cytotoxic, lack target specificity and degrade more quickly, resulting in poor bioavailability. Polymeric solid lipid drug distribution through the intravenous (IV) route would be the best way to solve both of these issues, as polymeric lipid particles (>200 nm) will penetrate the systematic circulation and in PLGA matrix lipidic shell coating over MTX will shield it from macrophagic absorption and reticuloendothelial opsonisation. As a consequence, MTX could enter the brain and be processed by endothelial cells [Citation29].
As compared to tablets, the key benefits of PLHNPs have increased bioavailability, sustained-release properties and fewer side effects. Therefore, this investigation aimed to emphasise formulation and in-vitro investigation of MTX-loaded PLHNPs using Box–Behnken design for potential GBM treatment options. To obtain this study’s prime objective, a single-step synthesis was performed where MTX-loaded PLHNPs were synthesised using simple solvent injection method. The PLHNPs comprising of PLGA in core and glyceryl tripalmitate & phosphatidylcholine in shell. To stabilise and maintain rigidity, polyvinyl alcohol (PVA) was added into PLHNPs formulation () to strengthen and maintain rigidity. The research also sought to determine the physical compatibility (FTIR, DSC and XRD), particle size, zeta potential, entrapment efficiency (EE), morphology (Transmission electron microscopy [TEM]), in-vitro drug release, brain uptake potential and pharmacokinetics studies, anticancer activity against U-87 MG glioma cells, cellular uptake, biocompatibility (lactate dehydrogenase assay, platelet aggregation and haemolysis) studies of synthesised PLHNPs.
Figure 1. Schematic presentation of methotrexate-loaded poly (lactic-co-glycolic acid) lipid hybrid nanoparticles preparation and drug loading.
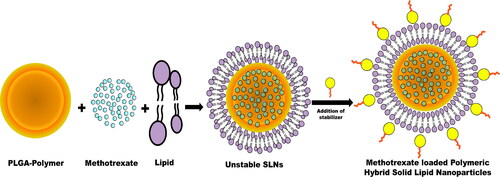
2. Materials and methods
2.1. Materials
The Drug; MTX was a gift sample from Neon Laboratories Ltd, Mumbai, India. The PLGA 75:25 was provided by Nomisma Healthcare, Gujarat, India. The tween 80, phosphatidylcholine, glyceryl tripalmitate, PVA were purchased from Sigma-Aldrich, Bengaluru, India. Analytical grade methanol, ethanol, and other organic chemicals were purchased from Sisco Research Laboratories Pvt. Ltd, Mumbai, India. The Dulbecco’s Modified Eagle Medium (DMEM), human serum albumin (HAS), 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT), 4′,6-diamidino-2-phenylindole (DAPI) was procured from Sigma-Aldrich, Bengaluru, India. The Lactate Dehydrogenase Assay Kit (Colorimetric) was procured from Abcam Laboratories, Fairfield, CA . Rest all the chemicals used in various experiments of this research were of analytical grades.
2.2. Preparation of PLGA solid lipid nanoparticles
MTX-containing PLGA PLHNPs were prepared using a solvent injection system with necessary modifications using methanol as organic solvent [Citation30]. The aqueous phase comprising 2.5 mg of MTX dissolved in 0.75% tween 80, phosphatidylcholine, 1.5% w/v PVA (stabiliser) and PLGA was dissolved in the hydroalcoholic mixture; methanol (25%) and double distilled water (75%). The lipid step consisted of various glyceryl tripalmitate quantities (about 99%). The aqueous and lipid process was warmed at 80 °C above the lipid melting point. Using a hydrodynamic needle, in the previously heated water phase, the molten lipid phase eventually fell. When inserting, sustain 15 min of homogenisation speed utilising Ultra Turrax T 25 Digital Homogeniser System.
Though homogenising, an organic residue vaporisation mechanism was left available. The subsequent liquid was prob-sonicated for 15 min after homogenisation (Sonifier® SFX550 Cell Disruptors and Homogenisers, Thomas Scientific, Swedesboro, NJ). The resulting liquid was centrifuged at 20,000 rpm at 4 °C for 30 min (refrigerated centrifuge 5702 R, Thomas Scientific, Swedesboro, NJ). During centrifugation, a white pellet layer was evident at the centrifugation tube rim. The supernatant was analysed for drug quality tests and washed the pellet layer three times using HPLC grade water. Delsa Nano C (Beckman Coulter, Brea, CA) calculated particle size and zeta potential of nanoparticles prepared. In Free Zone® Triad® Benchtop Freeze Dryers (Thomas Science, Swedesboro, NJ), the nanoparticles obtained were further lyophilised considering 2.5% w/w d-trehalose as cryoprotective.
2.3. Optimisation using Box–Behnken design
Preliminary screening’s key objective was to evaluate the various factors influencing particle size, zeta potential and trap efficacy of prepared nanoparticles. The most significant factors were the concentration of phosphatidylcholine, glyceryl tripalmitate and PLGA. The design of Box–Behnken [Citation31]; where quadratic reaction surface design system RSM was introduced, the influence of three factors (phosphatidylcholine, glyceryl tripalmitate and PLGA) on the dependent variables, i.e. particle size (Y1), zeta potential (Y2) and entrapment efficacy (Y3), the concentration of drug in the brain at 4 h interval (Y4), AUC(brain)/AUC(PPC) (Y5) was evaluated. In total, 13 experimental runs were conducted, and nanoparticles were prepared randomly to avoid possible unreliable outcomes. outlines the experimental design data. Model-fitting analysis was performed using program design-expert.
Table 1. Box–Behnken design layout for the optimisation of methotrexate-loaded PLGA lipid hybrid nanoparticles.
3. Evaluation and characterisation of solid lipid nanoparticles
3.1. Particle size analysis and zeta potential determination
The average particle size and zeta potential were determined using Delsa Nano C (Beckman Coulter, Brea, CA) [Citation32], where nanoparticles were suspended in HPLC water and analysed through triplicate photon correlation spectroscopy [Citation33]. Measurement of particle size in Delsa Nano C at a scattering angle of 90° at 25 °C for 120 cycles. For zeta-potential measure, 0.5 mL of the sample was put in the electrophoretic flow cell and measurement was taken for 80 cycles.
3.2. Entrapment efficiency (EE)
The prepared nanosuspension was initially centrifuged at 4 °C for 30 min at 15,000 rpm. During centrifugation, an identical form of pellets can be seen within the centrifuge tubbings at the rim. By means of decantation, the supernatant was obtained and analysed using the Perkin Elmer LAMBDA XLS Spectrometer in UV–visible spectroscopic method in triplicate at 244 nm. The blank was taken as the supernatant nanoparticle, which was prepared without any drug [Citation34]. To obtain regression equation y = 0.056x + 0.18 (co-efficient of correlation R2 = 0.0997), the normal calibration curve of MTX between absorbance (y) and concentration (x) was plotted. The following equation was used to calculate percentage drug entrapment efficacy:
3.3. In-vitro drug release study of the optimised formulation
Studies of the in vitro drug release were performed in the Spectra Por S/P 2 Dialysis Membrane Trial tube (10,000–12,000Dalton 50 mm) [Citation35]. The dialysis tubes were preloaded with nanoparticles that must have 2.5 mg of MTX and 2 mL of phosphate buffer solution (PBS) before being clamped with the USP Type II dissolution apparatus (pH 7.4). The paddle was rotated at 100 rpm, and the dissolution medium was known to be a 500 mL (pH7.4) PBS while retaining 37 ± 2.5 °C. The aliquoted sample quantity (5 mL) was removed from the dissolution medium during perforation of the examination, and the same amount of freshly formulated buffer solution was added to the dissolution medium. The material was analysed spectrophotometrically at 244 nm in triplicate. In addition, triplicate breakup experiments were also carried out, and the results were expressed in terms of percentage of opioid release ± standard deviation. For dissolution purposes, the pH 7.4 PBS was used to maintain the sink condition.
3.4. In vivo studies
Before beginning the experiment, the animals were acclimatised adequately to laboratory environments, and every precaution was taken to avoid any discomfort. The animals were given an adequate diet and free access to water before being placed in a 24-h light/dark cycle [Citation36]. To perform in vitro experiments, Wister rats (250–300 g) were acquired. The Committee for the Monitoring and Supervision of Animal Experiments (CPCSEA) of the Ministry of Environment and Forests (Animal Welfare Division) closely adopted the Government of India guidelines [DPL/1410/c/11/2020/CPCSEANo.458] before any animal experiments. Deshpande Laboratories, Pvt. Ltd., Bhopal, India, received the animal and conducted the tests.
3.5. Fourier -transform infrared (FTIR) spectroscopy
To clarify MTX’s potential interaction with different excipients, Fourier-transform infrared (FTIR) spectroscopy studies were performed [Citation37]. The FTIR spectroscopy was estimated from 400 to 4000 cm−1. The FTIR spectra of lyophilised drug-loaded optimised polymeric nanoparticles, the physical mixture of drug and other excipients, placebo nanoparticles and pure MTX drug were reported. The experiment was conducted in Frontier NIR Spectrometer (Perkin Elmer, Waltham, MA) using the NaBr pellet method to observe chemical interactions inside the formulation between MTX and other carrier composition.
3.6. Differential scanning calorimetry (DSC)
Using a Differential Scanning Calorimeter-800, the DSC thermograph of MTX, optimised formulation with MTX, and placebo formulation without drug were measured (PerkinElmer, Waltham, MA) [Citation38]. DSC experiments can establish the formulation’s physical nature and patterns of drug and polymer behaviour. To perform the DSC thermogram analysis, each individual sample was mounted in crimped aluminium, and heat flow ranged from 25 to 400 °C to 15 °C/min. Nitrogen gas circulated at 150 mL/min flow rate to maintain inert condition. Origin Pro 8.5 version Software analysed the extracted CSV file and analysed data.
3.7. Powder X-ray diffraction study
To recognise the crystal nature of the compound, X-ray Diffraction tests were conducted for pure drug, drug-optimised formulation and optimised formulation with ARLTM EQUINOX 100 X-ray Diffractometer [Citation39]. This experiment used a high-intensity X-ray source. The diffraction pattern was reported at 2ϴ and between 30 and 80 °C with an incremental degree of 0.03° and 3 s per phase.
3.8. Transmission electron microscopy
To investigate the surface morphology and particle size of prepared polymeric nanoparticles, the optimised batch was diluted with HPLC grade water and tested at 120 kV under transmission electron microscopy (JEM-ARM300F GRAND ARM Atomic Resolution Electron Microscope) [Citation40]. The samples were freshly prepared using 2% phosphotungstic acid (pH 7.0) and put on a carbon-coated grid (300 mesh).
3.9. Determination of brain uptake potential
The concentration of drugs in the brain and 4th hour AUC brain/AUC plasma administration must be measured for optimisation purposes [Citation41]. These two were known as response variables in formulation optimisation. For brain absorption trials of pure drug, optimised formulation, placebo, control (normal saline), male Wister rats were divided into four groups of three animals in each group. Before experimenting, the animals were kept for fasting. Water feed was kept accurate; however, pure drug (MTX) test formulation was administered during IV testing. Blood samples (0.5 mL) were obtained in heparin-containing tubes. After extracting blood samples in various time intervals, i.e. 30, 60, 120 and 240 min, the samples were centrifuged for 20 min at 13,000 rpm, and plasma samples were collected and processed at −48 °C. Samples were for analytical purposes.
By decapitation, animals were executed after extracting blood samples in the presence of anaesthesia. Carefully removed brains and washed blood with bottling sheets. Brain tissues were homogenised with a 0.01 M phosphate buffer (pH 7.4) in tissue homogeniser (f NS-52 Tissue Homogeniser, Microtec, Tokyo, Japan) for 15 min and centrifuged at 13,000 rpm. The resultant supernatant was deposited at −30 °C.
The amount of drug in brain samples was measured using HPLC. During the trial, 100 μg/mL MTX (20 μL) was taken, dissolved in 2 M sodium hydroxide solution and mixed properly. A plasma/brain sample of 300 μL was poured into the mixture. The mixture was thoroughly vortexed, applying ethyl acetate and 30 min shaking at 15 °C. The process was repeated three times, the constituents’ supernatant dried and reconstituted 400 μL mobile phase. To quantify the volume of drugs present in plasma and brain, correctly tested HPLC methods are used. The samples were properly screened using a 0.22 μm syringe filter and the requisite sample quantity was injected into the well-equipped Waters Alliance 2695 HPLC System. MTX HPLC was determined in 303 nm with a flow rate of 1.5 mL/min. The mobile phase consisted of buffer and acetonitrile (98:2). They were dissolving 27.22 g potassium dihydrogen orthophosphate in 1000 mL purified water prepared the Buffer solution. During the HPLC method, the pH with NaOH solution was changed to 6.0 ± 0.05, and the resulting mixture was filtered using a 0.2 μ nylon membrane filter. The propensity for brain and plasma medication was measured by testing the drug in the brain and plasma. Also, AUC brain/AUC plasma was measured using the trapezoidal method.
3.10. Cytotoxicity studies against U-87 MG glioma cells
Using MTT assay in vitro MTX cytotoxicity, drug-loaded optimised formulation, placebo was calculated in U-87 MG cell lines [Citation42,Citation43]. The cells were seeded onto 96-Well Cell Culture Plates (Thermofisher Science, Waltham, MA) at 1 × 104 cells/well in DMEM and incubated for 36 h in a % CO2 incubator while performing the experiment. The cells were subjected to various research conditions when incubating at 37 °C in a 5% CO2 environment for 72 h. In addition, 40 µL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) solution was added to the culture medium and incubated for 8 h at 37 °C. Insoluble formazan crystals form during the reaction phase, which are proportional to the number of viable cells. These insoluble formazan crystals were dissolved in 100 L dimethyl sulfoxide (DMSO), the plates were vigorously agitated for 20 min, and absorption at 580 nm was determined using Multi-Mode and Absorbance Readers (Bio Tek, Winooski, VT). The following equation was used to quantify the percentage of cytotoxicity:
3.11. Cellular uptake
The U-87 MG glioma cells were propagated in a 35 mm petri dish at 1 × 104 cells/cover glass cells, in the presence of DMEM. At 37 °C and further exposed with refined formulation (100 μL/cover glass) in the humidified CO2 (5%) incubator. After 4 h, cells were correctly washed three times using a 7.4 pH PBS, and later the cells were fixed with paraformaldehyde for 40 min. The cells were further washed with PBS three times after 40 min. Furthermore, cells were stained for 20 min using 0.1 μg/mL of DAPI reagent. The developed monolayer was further washed with PBS inside the cover glass, and the slides were examined under a confocal laser microscope (Anamatrix Laser Scanning Microscope).
3.12. Evaluation of haemolysis
On the basis of previous publications, the drug, optimised formulation and placebo were analysed. Since the drug can be absorbed from the blood capillaries and, as a result, possibilities of haemolyse blood cells after ingestion are high. Therefore, a haemolysis analysis is needed to support the product’s biocompatibility. Hemocompatibility tests will also explain how the drug, final product and placebo will respond to blood [Citation44].
Blood was obtained from the nearest blood bank to conduct haemolysis tests [Citation45]. The blood samples (2.5 mL) were centrifuged for 15 min at 1345 × g at room temperature. The graduated centrifuge tubes used in this experiment were previously sterile. Micropipettes were used to scrape the plasma coating during centrifugation. The developed erythrocyte pellets in the bottom of the graduated centrifuge tubes were diluted and suspended further using normal saline solution. At room temperature, the suspended part was centrifuged against 1345 × g for 15 min. Centrifugation was used three times to finish the washing operation. The erythrocyte suspension was further diluted with 10 mL saline water and then 10, 50 and 100 g/mL of pure medication, optimised dosage and placebo were each combined with 2 mL of erythrocyte suspension in sterile tubes separately. The positively regulated examination, which fully lysed erythrocytes, was rendered by dissolving 1% triton X-100 in erythrocyte solutions. The negative control test, in which erythrocytes are not lysed, was made by suspending erythrocytes in regular saline solution in previously sterile Eppendorf tubes. The samples were incubated for at least 15 min at 37 °C. Nearly 200 µL samples were removed at specific pre-determined time intervals, i.e. 0.5, 1, 2, 4–10 h, thus centrifuging at 1345 × g for 15 min. A 100 µL supernatant was incubated for 45 min at room temperature to achieve adequate haemoglobin to oxyhaemoglobin conversion. At 580 nm, the absorbance was measured spectrophotometrically. The following formula is used to calculate the proportion of haemolysis:
where A sample is the absorbance of the test sample containing the drug nanoparticles. A spontaneous control is the absorbance of erythrocytes that are previously incubated with saline water. On the other hand, A positive control was the absorbance of the supernatant of erythrocytes which was exposed with 1% Triton X-100 solution made up of normal saline. The experiment was performed in triplicate, and the data were expressed in mean ± SD (n = 3).
3.13. Evaluation of erythrocyte membrane integrity
Lactate dehydrogenase (LDH) is an enzyme that is produced by erythrocytes and can be determined photometrically with the LDH assay kit/ LDH Assay Kit (Colorimetric) (ab102526) [Citation46,Citation47]. The erythrocyte suspension was prepared according to the protocol previously discussed on haemolysis. About 1 mL of erythrocyte suspension was used to administer the drug’s optimised formulation, the drug and the placebo. The positive control sample (100% lysed erythrocytes) was made by diluting the erythrocyte suspension with 1% Triton-X-100, while the negative control sample was made by diluting the erythrocyte suspension with normal saline solution. To allow LDH natural preparation, 150/UL LDH was incubated with erythrocyte suspension at 37 °C. During specific time periods, such as 2, 4 and 8 h from suspension, 400 L samples were extracted and independently centrifuged at 1345 × g for 20 min. LDH was detected at 500 nm after the supernatant was prepared with a ready-to-use LDH solution. The sum of LDH was calculated using the formula below.
where A sample stands for drug and nanoparticles incubated erythrocytes absorbance, A negative control indicates, erythrocytes absorbance of sample, which was pretreated with normal saline solution. The A standard indicates the absorbance of erythrocytes sustention, which were pretreated 150/UL LDH enzyme. The experiment was performed in triplicate, and the date is expressed as mean ± SD (n = 3).
3.14. Platelet aggregation studies
Platelet aggregation experiments were performed using a haematological counter (Biolinx Lab systems Private Limited, Mumbai, India) [Citation48]. Before platelet counting, 2 mL of blood was incubated with different drug concentrations for 3 h at 37 °C, i.e. 10, 50 and 100 g/mL. After incubation, the required volume of sample was diluted with normal saline water, and additional samples were examined using a triplicate haematological counter (mean SD; n = 3). It is important to do a detailed study of platelet aggregation, which can be performed with an optical microscope. In this experiment, heparinised blood samples were treated with various samples. After treatment with the samples, peripheral blood smears were mounted on a glass slide and air-dried for 5 min. With the aid of Leishman’s dye, the blood smears were pigmented for 10 min. The stain was then washed with purified water and covered with a glass cover. The staining essence of platelets was examined using an optical microscope and high-resolution images were captured.
4. Results and discussion
4.1. Factorial design for optimisation
As seen in , a total of thirteen formulations (F1–F13) were created, and the optimisation method was developed using the Box–Behnken design. The influence of different independent or input variables such as phosphatidylcholine concentration (X1), glyceryl tripalmitate (X2) and PLGA quantity (X3) on dependent or outcome variables, such as particle size (Y1), zeta potential (Y2) and entrapment efficacy (Y3), brain drug concentration at 4 h interval (Y4) and AUC(brain)/AUC(plasma)(Y5) was investigated. applies to the outcome of the results. In the F1–F13 batches, the mean particle size of the prepared nanoparticles ranged from 173.51 (F1) to 233.37 nm (F1) (F7). The range of possible Zeta values was discovered to be between 25.78 (F11) and 36.31 mV (F6). Drug entrapment efficacy (%) ranged from 70.56 (F8) to 86.34% (F9) (F10). The concentration of drugs in the brain was observed to be smaller in F5 (6.38 g/mL) administration at the 4 h time period, while it was higher in F6 (12.38 g/mL) administration. Similarly, the AUC(brain)/AUC(plasma) ratio of the formulation varied from F5 (1.02) to F4 (4.34).
Figure 2. (A) Particle size, zeta potential and entrapment efficacy of formulations (F1–F13). (B) Concentration in brain and brain/plasma ratio of drug and AUC(brain)/AUC(plasma) at 4 h interval.
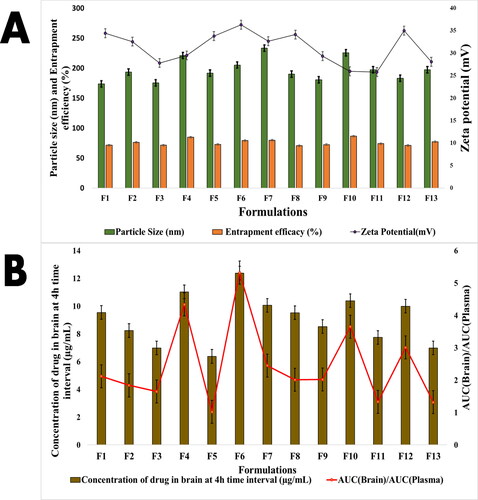
The relationship between a dependent variable (Y) and an independent variable (X) was studied statistically. The Design of Experts software was used to measure the analysis of variance (ANOVA), polynomial equation data, the Fisher’s value (F-value), the degree of freedom (df), the total of the squares and the mean sum of the squares version 11.0.
For particle size (Y1) measurement; the following polynomial equation was obtained:
(1)
(1)
In [I], the influence of all independent variables on particle size (Y1) was addressed (a). The steep slope or curvature of factor ‘C’ or PLGA in [I](a) suggests that concentrated PLGA will have a greater impact on particle size. Increased phosphatidylcholine concentration reduces particle size, as seen in the 3D surface plot of [I](b), indicating a negative coefficient of phosphatidylcholine in Polynomial Equation (1) (−5.41). Reduced PLGA concentration and increased glyceryl tripalmitate concentration result in smaller particles, as seen in [I](c,d).
Figure 3. [I] Effects of particle size (Y1) on perturbation (a). Effects of particle size (Y1) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of particle size (Y1) on PLGA and phosphatidylcholine (c). Effects of particle size (Y1) on glyceryl tripalmitateand PLGA. [II] Effects of zeta potential (Y2) on perturbation (a). Effects of zeta potential (Y2) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of zeta potential (Y2) on PL GA and phosphatidylcholine (c). Effects of zeta potential (Y2) on glyceryl tripalmitate and PLGA(d). [III] Effects of Entrapment efficacy (Y3) on Perturbation (a). Effects of entrapment efficacy (Y3) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of entrapment efficacy (Y3) on PL GA and phosphatidylcholine (c). Effects of entrapment efficacy (Y3) on glyceryl tripalmitate and PLGA. [IV] Effects of concentration of drug in brain at 4 h interval (Y4) on perturbation (a). Effects of concentration of drug in brain at 4 h interval (Y4) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of concentration of drug in brain at 4 h interval (Y4) on PL GA and phosphatidylcholine (c). Effects of concentration of drug in brain at 4 h interval (Y4) on glyceryl tripalmitate and PLGA. [V]. Effects of AUC(brain)/AUC(plasma)(Y5) on perturbation (a). Effects of AUC(brain)/AUC(plasma)(Y5) on glyceryl tripalmitate and phosphatidylcholine (b)., Effects of AUC (brain)/AUC(plasma)(Y5) on PL GA and phosphatidylcholine (c). Effects of AUC(brain)/AUC(plasma) (Y5) on glyceryl tripalmitate and PLGA.
![Figure 3. [I] Effects of particle size (Y1) on perturbation (a). Effects of particle size (Y1) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of particle size (Y1) on PLGA and phosphatidylcholine (c). Effects of particle size (Y1) on glyceryl tripalmitateand PLGA. [II] Effects of zeta potential (Y2) on perturbation (a). Effects of zeta potential (Y2) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of zeta potential (Y2) on PL GA and phosphatidylcholine (c). Effects of zeta potential (Y2) on glyceryl tripalmitate and PLGA(d). [III] Effects of Entrapment efficacy (Y3) on Perturbation (a). Effects of entrapment efficacy (Y3) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of entrapment efficacy (Y3) on PL GA and phosphatidylcholine (c). Effects of entrapment efficacy (Y3) on glyceryl tripalmitate and PLGA. [IV] Effects of concentration of drug in brain at 4 h interval (Y4) on perturbation (a). Effects of concentration of drug in brain at 4 h interval (Y4) on glyceryl tripalmitate and phosphatidylcholine (b). Effects of concentration of drug in brain at 4 h interval (Y4) on PL GA and phosphatidylcholine (c). Effects of concentration of drug in brain at 4 h interval (Y4) on glyceryl tripalmitate and PLGA. [V]. Effects of AUC(brain)/AUC(plasma)(Y5) on perturbation (a). Effects of AUC(brain)/AUC(plasma)(Y5) on glyceryl tripalmitate and phosphatidylcholine (b)., Effects of AUC (brain)/AUC(plasma)(Y5) on PL GA and phosphatidylcholine (c). Effects of AUC(brain)/AUC(plasma) (Y5) on glyceryl tripalmitate and PLGA.](/cms/asset/43e754f8-1bb9-4743-a0c2-5947b96e64d1/tjen_a_1983172_f0003_c.jpg)
For the zeta potential (Y2) measurement; the following polynomial equation was obtained:
(2)
(2)
Phosphatidylcholine (X2); factor ‘A’ has a steep slope, meaning that phosphatidylcholine concentration has a greater effect on zeta potential, according to the perturbation plot ([II](a)). This may be attributed to the fact that phosphatidylcholine is a surfactant. It was verified by [II](b,c) and Polynomial Equation (2) that raising the phosphatidylcholine concentration would increase the cationic nature of the formulation and increase the zeta potential. However, it was also observed from Polynomial Equation (2) that phosphatidylcholine has a noticeable impact on zeta potential as compared to PLGA and glyceryl tripalmitate alone. This may be attributed to the inclusion of amino groups in phosphatidylcholine as well as proper fabrication of the tripalmitate group on the surface of drug-loaded PLHNPs. Where else, it can be estimated from [II](d) that increasing the concentration of glyceryl tripalmitate would have a mild effect on zeta potential.
For effects of entrapment efficacy (Y3); the following polynomial equation obtained:
(3)
(3)
The perturbation plot ([III](a)) reveals that PLGA; factor ‘C’ has a steep slope, suggesting that PLGA concentration has a greater effect on entrapment efficacy. As can be seen in [III](b), glyceryl tripalmitate has little effect on Entrapment efficacy. However, Polynomial Equation (3) and [III](c,d) explicitly show that increasing the concentration of PLGA increased entrapment efficacy; this is attributed to the polymeric gelation of PLGA.
For effects of concentration of drug in the brain at 4 h interval (Y4); following polynomial equation obtained:
(4)
(4)
The perturbation plot ([IV](a)) shows that PLGA; factor ‘C’ has a steep slope, meaning that PLGA concentration has a stronger effect on drug concentration in the brain at the 4 h interval. According to [IV](b), glyceryl tripalmitate has no impact, whereas elevated phosphatidylcholine concentration has an agonistic effect on drug concentration in the brain at a 4 h interval; this may be attributed to the role of phosphatidylcholine enhancing the active transport function of the drug in brain endothelial cells. However, it was clear from [IV](c,d) and Polynomial Equation (4) that an improvement in PLGA and phosphatidylcholine concentration has a significant impact on opioid concentration in the brain at the 4th h interval.
Effects of AUC(brain)/AUC(plasma) (Y5); following polynomial equation obtained:
(5)
(5)
Once again, the perturbation plot ([V](a)) showed that factor ‘C’ has a steep slope, suggesting that PLGA concentration will have a stronger impact on the Effects of AUC(brain)/AUC(plasma) (Y5). According to [V](b), glyceryl tripalmitate has no significant effect on AUC(brain)/AUC(plasma); however, an increase in phosphatidylcholine concentration could increase drug AUC in the brain; this could be due to the presence of the choline group, which helps drugs cross the BBB by speeding up acetylcholine synthesis and boosting cognitive function of the brain. According to [V](c,d) and Polynomial Equation (5), PLGA improves medication transport in the brain and has a better brain-protective impact.
As a result, increasing the PLGA concentration improved the AUC(brain)/AUC(plasma) ratio. Furthermore, the positive and negative signs of the interaction terms, as well as the co-efficient of the key outcomes, were used to denote the synergistic and antagonistic effects of the result variables in the polynomial EquationEquations (1)–(5). The estimation of the p value and F-value () often aids in determining the model’s importance. The p value was calculated at a 95% confidence level (0.05). The R2 value and modified R2 value of a model must be close to 1 in order for it to be relevant. Furthermore, the lowest Bayesian information criterion (BIC) and Akaike information criterion (AIC) must be used; the AIC aids in determining the optimal consistency and goodness of fit standard. In comparison to another quadratic model of dependent variables, shows the medication in the brain at 4 h interval (Y4) is the better fit model, with lower AIC (104.38) and BIC (0.0306) values and important R2 (0.9972) and modified R2 value (0.9887). Y1 (358.78) and Y4 (117.20) have higher F-values, meaning that experimental variation is much greater than error variance.
Table 2. ANOVA responses of prepared PLGA lipid hybrid nanoparticles.
To bring more specification in the optimisation process by Design of Expert version 11.0 software, the process capability index (Cpk) was identified in desirability studies (). The constraint variables were obtained, i.e. particle size at 193.143 nm, zeta potential at 33.7515 mV, EE 74.7937%. The concentration of drugs in the brain at 4 h interval at 8.80 µg/mL, effects of AUC(brain)/AUC(plasma) at 2.37169, respectively, when desirability was 0.988.
Figure 4. Process optimisation by desirability approach (a) and overlay plot of design space and optimum batch (b).
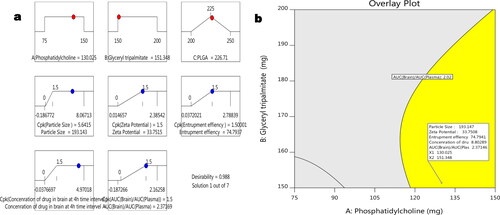
The cumulative disability of this design was 0.988, as shown in . All of the independent factors in this experiment have a disability of one. From the overlay plot () the desire region for constraining variables was identified within the yellow design space. Considering overlay plot’s independent variables (phosphatidylcholine (X1) =130.025 and glyceryl tripalmitate (X2) = 151.348) as a standard, three batches were reproduced. The three batches’ dependent variables or outcome variable’s relative standard error was found to be less than 9.00%. Therefore, the formula obtained from the overlay plot was considered optimum. Characterisation of PLHNPs.
4.1.1. In-vitro drug release study
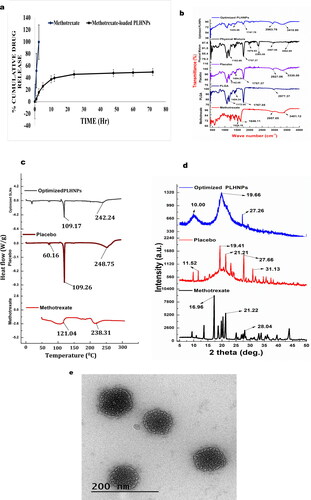
MTX-loaded PLHNPs exhibit unusual transport characteristics, including swelling and regulated release. For in-vitro drug release studies, the optimised batch from the overlay plot was used. Within 10 h, the nanoparticles show initial burst effects and hance; 37.232.35% of the drug was released. The existence of drug particles on the nanoparticle surface could explain the initial drug release phenomenon. In the presence of 0.75% tween 80 and a pH 7.4 PBS, Phosphatidylcholine can facilitate MTX release from the PLGA matrix by forming a canal. The zero-order kinetics was detected as the polymer degraded. After 72 h or 3 d, nearly 49.373.56% cumulative drug release was reported, suggesting a continuous release trend of the nanoparticles, where the correction coefficient (R2) for the zero-order fit model was 0.9954 ().
Figure 5. (a) In-vitro drug release profile of methotrexate and methotrexate-loaded PLGA lipid hybrid nanoparticles. (b) Fourier transform infrared spectroscopy of methotrexate, PLGA, placebo, physical mixture and optimised lipid hybrid nanoparticles. (c). DSC thermograms of methotrexate, placebo and optimised lipid hybrid nanoparticles. (d) XRD patterns of methotrexate, placebo and optimised lipid hybrid nanoparticles. (e) Transmission electron microscopy of optimised methotrexate-loaded PLGA lipid hybrid nanoparticles.
4.1.2. Fourier-transform infrared spectroscopy
To identify the chemical bonds and functional group heterogeneity within the samples, FTIR studies must be performed where the samples absorb selective absorption of infrared radiation. The FTIR studies help us to discover drug-polymer interactions. In , the pure compound, MTX, PLGA, placebo, physical combination and optimised PLHNPs all have a characteristic spectrum peak was mentioned. The N–H stretch of amide R–CO–NH2, carboxylic acid’s C = C–CO–OH stretch, and asymmetric aromatic N–O stretching is all found in the MTX peak at 3401.12, 2957.05, 1646.11 and 1525.18 cm−1, respectively, indicating the existence of N–H stretch of amide R-CO-NH2, carboxylic acid’s C = C−CO–OH stretch, PLGA exhibits a typical peak at 2977.37, 1767.05, 1404.24 and 1172.05 cm−1, indicating the existence of the R–CO–OH group C = O stretch of ester, C–O stretch and carboxylic acid RCO–OH stretch, respectively. One point is evident from the IR spectrum of the placebo and physical mixture: there is no significant molecular activity that could change the chemical composition of the substances. The absence of any chemical reaction during fabrication is confirmed by the FTIR spectra of optimised PLHNPs. The N–H stretch at 3410.80 cm−1 and the ester C = O stretch at 1747.70 cm−1, on the other hand, were observed to have lower intensity and transmittance; this was attributed to the drug’s adequate fabrication with polymer and lipid. This property further establishes MTX’s salt existence within stable PLHNPs.
4.1.3. Thermal characteristics
Differential calorimetry scanning (DSC) was conducted to clarify the drug’s crystalline and amorphous behaviour. Often allows identifying drug association with polymer. shows MTX’s DSC thermogramic peaks at 120.04 and 238.31 °C. The placebo endothermic melting peak showed a glyceryl tripalmitate melting point. PLGA’s transition temperatures (Tg) rose in placebo due to the presence of glyceryl tripalmitate, and two endothermic peaks were found at 109.26 and 248.65 °C. In optimised PLHNPs, the PLGA melting point peak was observed at 109.17 °C, but the peak intensity was poor compared to placebo, confirming optimised PLHNPs amorphous nature. Additionally, the physical condition of the substance was probably altered during encapsulation, so the formulation exhibits amorphous characteristics.
4.1.4. X-ray diffraction study
From diffractogram , the characteristic 2ϴ value for MTX pure drug was observed in 16.96, 21.22 and 28.04 °C. From extremely imminent peaks and some tiny peaks, MTX may be expected to exhibit crystalline form. The diffractogram of placebo reveals the signature 2ϴ value at 11.52, 19.41, 21.21, 27.66 and 31.13 °C. Placebo diffractogram also showing low-intensity partial crystalline nature due to the inclusion of PLGA, phosphatidylcholine and glyceryl tripalmitate in placebo sample. The configured diffractogram of PLHNPs reveals characteristic 2ϴ at 10.0, 19.66 and 27.26 °C. The reduced amplitude of optimised PLHNPs diffractogram peaks indicates the amorphous existence and proper encapsulation of drugs within optimised PLHNPs.
4.1.5. Transmission electron microscopy
With the help of JEM-ARM300F GRAND ARM Atomic Resolution Electron Microscope, it was possible to focus on drug-loaded PLHNPs, which were placed on cooper coated gride (300 mesh). reveals that the projected particle was uniform in size and spherical in nature. The projected nanoparticle in TEM analysis was found to have lesser particle size as compared to particles size, which was obtained from the Delsa Nano C instrument (Beckman Coulter, Brea, CA); this is because Delsa Nano C identifies the apparent volume of distribution of the diluted particles in distilled water, however during TEM measurement, the particle may get dehydrated. Moreover, the chances of aggregated particles while measuring in Delsa Nano C instruments was formidable, as prior measurement, the nanosuspension was exposed to 15 min sonication.
4.1.6. Determination of brain uptake potential
A particular HPLC method was developed from the brain homogenates and plasma samples of experimental animals, i.e. Wister rats, which appeared to have a high degree of specificity and precision at a specific time interval. After 4 h of drug administration, the AUC brain/AUC plasma drug concentration in plasma was measured. The drug concentration in the brain was estimated to be 2.43 µg/g after 4 h of administration, and the AUC brain/AUC plasma of the drug was found to be 2.23 after 4 h of administration. This implies the drug concentration in the brain was 2.23 times greater than the drug concentration in plasma, which was absorbed by the IV route. As a result, MTX may be targeted in the brain in a passive manner.
4.1.7. Cytotoxicity studies against U-87 MG glioma cells
To compare the efficacy of MTX against U-87 MG glioma cell lines, cytotoxicity tests were carried out. MTX, placebo-PLHNPs and optimised-PLHNPs cytotoxicity against 87 MG glioma cell lines can be seen in . In all concentrations, the cellular cytotoxicity of MTX-loaded optimised-PLHNPs was found to be significantly higher than that of free MTX (p < .05). The cytotoxicity study’s most unexpected result was that placebo-PLHNPs exhibit cytotoxicity when concentration rises. According to Sima Rezvantalab et al., this may be attributed to the existence of PLGA in the formulation. According to the article, PLGA-based nanoparticles have promising anticancer properties.
Figure 6. (a) In-vitro cytotoxicity of methotrexate, placebo-lipid hybrid nanoparticles and optimised lipid hybrid nanoparticles. Values are represented as mean ± SD (n = 3) p < .05 (b) By using confocal laser scanning microscopy (CLSM) the cellular internalisation of optimised lipid hybrid nanoparticles in U-87 MG glioma cell lines were evaluated; where 4′,6-diamidino-2-phenylindole (DAPI) channel was used to stain nucleus.
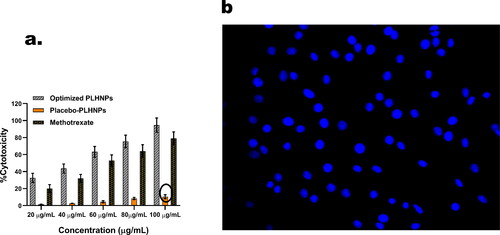
4.1.8. Cellular uptake results
The U-87 MG glioma cells were picked up with optimised-PLHNPs, and stained with DAPI reagent, as shown in . Suitable filters were used to capture the CLSM representation of DAPI reagent. After 4 h of incubation with U-87 MG glioma cells, the image indicates extensive internalisation of optimised-PLHNPs, It is possible that nanoparticles can clump together in the cytoplasm. Many apoptotic processes, such as the expression of caspase-3 mRNA, the downregulation of metalloproteinases (MMPs), the upregulation of p53 and the deactivation of IAP5, are believed to take place in the cytoplasm, according to various reports. As a consequence, optimised-PLHNPs, may be an useful tool to induce cytotoxicity in the cytoplasm.
4.1.9. Evaluation of haemolysis
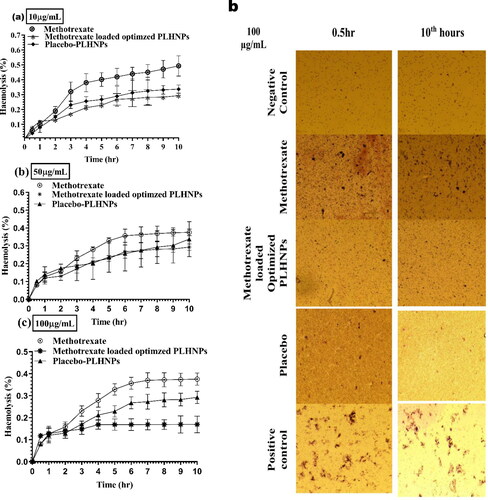
The most important study to learn about the behaviour of prepared PLHNPs with blood was an in-vivo toxicity study. The association of PLHNPs with blood and its substances must be investigated. The PLHNPs of PLGA, which are made up of lactide (LA) and glycolide (GA) monomers, have a minor impact on haemolyses. The findings of the analysis suggest that PLHNPs can be used with care. The profile of percentage haemolysis absorbance with 0.5th and 10th-h shifts of erythrocytes with different treatments (10, 50 and 100 g/mL) containing MTX, placebo-PLHNPs and MTX primed optimised PLHNPs is depicted in . It can be inferred from that placebo-PLHNPs and MTX-loaded optimised PLHNPs cause fewer haemolysis than MTX. Regardless of Drug and PLHNPs concentrations of 10, 50 and 100 g/mL, the placebo-PLHNPs and MTX-loaded optimised PLHNPs displayed fewer haemocompatibility (>1%) and were found to be safe to RBC membrane integrity. Most notably, MTX-loaded optimised PLHNPs formulations display fewer haemolysis; this is attributed to adequate MTX encapsulation inside the PLHNPs, which prevents red blood cells from MTX-induced haemolysis. One thing that can be proven from the haemolysis studies is that i.v. administration of MTX primed optimised PLHNPs can have a beneficial impact on erythrocytes. As a consequence, there is a decent risk of lowering malondialdehyde (MDA) levels and carbonyl group content. As a consequence, MTX-loaded optimised PLHNPs can prevent erythrocyte degradation during glioma care when delivered intravenously.
Figure 7. (a) Haemolysis (%) profile of methotrexate, methotrexate loaded optimised lipid hybrid nanoparticles, placebo-lipid hybrid nanoparticles after 10 µg/mL treatment; (b) Haemolysis (%) profile of methotrexate, methotrexate-loaded optimised lipid hybrid nanoparticles, placebo-lipid hybrid nanoparticles after 50 µg/mL treatment; (c) Haemolysis (%) profile of methotrexate, methotrexate loaded optimised lipid hybrid nanoparticles, placebo-lipid hybrid nanoparticles after 100 µg/mL treatment; (D) High contrast microscopic images of erythrocytes during haemolysis (%) studies after 100 µg/mL treatment of methotrexate loaded optimised lipid hybrid nanoparticles, placebo-lipid hybrid nanoparticles during 0.5 and 10 h.
4.1.10. Evaluation of erythrocyte membrane integrity
White blood cells and blood plasma were separated using centrifugation, and natural saline comprising erythrocytes was used instead. Enumerating the LDH enzyme is expected to determine membrane integrity. Only when the structural integrity of the erythrocytes was disrupted was a hyper or elevated degree of LDH recorded. shows the sum of LDH released after treatment with 2 mL of erythrocyte suspension with MTX, MTX-loaded optimised PLHNPs (equivalent to 10, 50 and 100 g/mL of MTX), and placebo-PLHNPs. By matching placebo-loaded optimised PLHNPs of 10, 50 and 100 g/mL concentration at 8th-h intervals to MTX-loaded MTX-loaded optimised PLHNPs of 10, 50 and 100 g/mL concentration, the LDH release was not as conspicuous (20%). Triton X 100, on the other side, displays approximately 290% LDH release, suggesting total erythrocyte annihilation. As a result, MTX-loaded optimised PLHNPs are unlikely to damage erythrocyte membrane integrity. Thus, MTX-loaded optimised PLHNPs will be suitable for IV administration.
Figure 8. Amount of LDH release after treating with PBS, methotrexate, placebo-lipid hybrid nanoparticles and methotrexate loaded optimised lipid hybrid nanoparticles at 1 h (a), 4 h (b) and 8 h (c).

4.1.11. Platelet aggregation study results
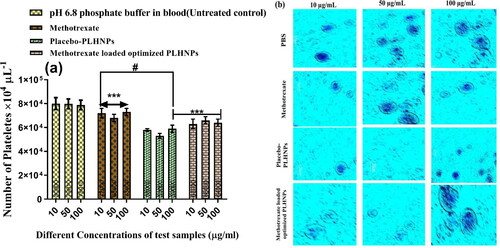
Patients can experience myocardial infarction, ischemia, or stroke after receiving nanoparticles from i.v. routs; these problems can occur as a result of platelet aggregation that leads to thrombus formation. As a result, platelet aggregation must be assessed after i.v. administration of MTX, placebo-PLHNPs, and MTX-loaded optimised PLHNPs. The quantitative estimation of platelets was measured by haematological counter after incubating with MTX, placebo-PLHNPs and MTX-loaded optimised PLHNPs at 10, 50 and100 µg/mL concentration for 3 h at 37 °C. The pH 6.4 phosphate buffer was considered a stand-alone control group throughout the study. At all concentration, the MTX did not show any significant differences in platelet count as compared to the control pH 6.4 PBS, which postulating MTX did not show any platelet aggregation (). The placebo-PLHNPs showed significantly lower (p < .005) platelet count than that of MTX and MTX-loaded optimised PLHNPs treated samples. However, 100 µg/mL of MTX-loaded optimised PLHNPs shows higher platelet aggregation compare to placebo-PLHNPs, which indicates a non-significance (p > .05) relationship. Lower platelet aggregation for placebo-PLHNPs was due to the absence of MTX, which may influence platelet aggregation. The light microscopy images of Leishman’s-stained whole blood samples after treating with different Concentration of pH 6.4 PBS, MTX, placebo-PLHNPs and MTX-loaded optimised PLHNPs is shown in .
Figure 9. (a) Number of platelets counts after treatment with PBS, methotrexate, placebo-lipid hybrid nanoparticles and methotrexate-loaded optimised lipid hybrid nanoparticles at different concentrations (10, 50 and 100 µg/mL). As per Bonferroni’s multiple comparisons test (values are represented as mean ± SD (n = 3)), ***indicates high statistical significance (p < .001) and # indicates insignificance (p = .4037) as compared to the different concentrations of methotrexate; all the formulations were compared against with treated controlled group (methotrexate treatment). (b) Light microscopy images of platelets with different treatments at prescribed concentration.
5. Discussion
This study demonstrated the in-vitro investigation and systematic optimisation of MTX loaded PLHNPs using Box–Behnken design. The optimisation process seeks to provide adequate particle size, zeta potential and entrapment efficacy with maximum drug ratio in the brain. From the optimisation process, it was also understood that the increased concentration of PLGA and glycerol tripalmitate helps to increase the particle size. On the other hand, increase the concentration of phosphatidylcholine could increase the zeta potential (cationic) of the formulation, which is due to the presence of the L-alpha-phosphatidylcholine group. Noticeably, increase concentration of PLGA improvises drug entrapment efficacy (%), and increase concentration of PLGA and phosphatidylcholine increases drug concentration in the brain at 4th interval; as per Sandeep Godaraa et al. while preparing paclitaxel PLGA PLHNPs, it was observed that coating of lipid over nanoparticles could enhance paclitaxel blood circulation time as compared to uncoated nanoparticles. This is due to the restriction of plasma protein adsorption over lipid corona coating [Citation49].
As per recent research done by Vanesa Noz et al., PLGA nanoparticles improve dopamine transmission to Parkinson’s disease rats and shield dopamine from fast cortical metabolism [Citation50].
In this research, increased concentration of PLGA could improve the effects of the AUC(brain)/AUC(plasma) ratio. Therefore, optimisation of PLGA concentration was essential. Upon optimisation, the resultant formulation shows excellent sustain release property in in-vitro drug release studies for 72 h, which would be beneficial for prolonged action. From the FTIR, DSC and XRD analysis, it was confirmed that no such potential interaction took place between MTX and other components, and the MTX loaded optimised PLHNPs show amorphous nature.
From the observation of transmission electron microscopy, it was concluded that MTX loaded optimised PLHNPs showed good spherical shape with the absence of mushroom and Ostwald ripening effect, indicating higher stability as formulation.
From the cytotoxicity studies, it was confirmed that MTX-loaded optimised PLHNPs produces significantly higher cytotoxicity in U-87 MG glioma cell lines than that of free MTX (p < .05); This may be attributed to the combined effects of MTX and PLGA, but more importantly due to the improved stable structure of PLHNPs after absorption of PVA, as the drug diffusion through the cellular matrix is well regulated and stable, and therefore indirectly this phenomenon could improve the anticancer activity of PLHNPs. As per Iliyas Khan et al., PLGA [Citation51,Citation52] is a biodegradable polymer used widely to produce NPs for drug delivery [Citation53]. The extensive application of PLGA NPs promises cancer treatment with better efficacy and with fewer side effects.
A confocal laser scanning microscopy (CLSM) confirmed that MTX-loaded optimised PLHNPs show excellent internalisation in U-87 MG glioma cell lines, which was approved by observing highly concentrated cytoplasm in DAPI reagent. Since the cytoplasm was an established site of action for any chemotherapeutic medicaments, hance, free MTX rising from MTX loaded optimised PLHNPs would exert higher anticancer efficiency. The brain uptake studies confirmed that passive brain targeting is possible with the MTX-loaded optimised PLHNPs. From the haemolysis studies, it was witnessed that, MTX-loaded optimised PLHNPs exerts less than 1% haemolysis; as per Justin M. Zook et al., the percentage of haemolysis decreases when prepared stable AgNP agglomerate size increases [Citation54]. Since, in existing research, the optimised PLHNPs show much less than 200 nm particles size, hence chances of haemolysis would be negligible. The platelet aggregation studies confirmed that platelets were less aggregated in the presence of MTX-loaded optimised PLHNPs. The optimised PLHNPs established themselves as the most promising candidate for cancer targeting; therefore, optimised PLHNPs would be a reliable drug delivery system for glioblastoma treatment. However, in vivo anticancer potential of MTX-loaded optimised PLHNPs will need to be evaluated in future studies.
6. Conclusion
The solvent injection method is used to create MTX-loaded PLHNPs, which are synthesised in a single step. PLHNPs with a lipid shell and a polymeric core exhibit less haemolysis and platelet aggregation when there is a higher concentration of the drug in the brain. It can be concluded that the use of phosphatidylcholine and glyceryl tripalmitate coated with a PLGA core matrix can provide a novel method of delivering lipophilic constituents with increased potential for glioma treatment.
| Abbreviations | ||
| %EE | = | Percentage entrapment efficiency |
| ANOVA | = | Analysis of variance |
| AUC | = | Area under the curve |
| BBB | = | blood-brain barrier |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DCM | = | Dichloromethane |
| DLS | = | Dynamic light scattering |
| DSC | = | Differential scanning calorimetry |
| GBM | = | Glioblastoma multiform |
| KBr | = | Potassium bromide |
| LDH | = | Lactate dehydrogenase |
| MDA | = | Malondialdehyde |
| MTT | = | 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| MTX | = | Methotrexate |
| PBS | = | Phosphate buffered saline |
| PLGA | = | Poly(lactic-co-glycolic acid) |
| PLHNPs | = | Polymeric-lipid hybrid nanoparticles |
| PVA | = | Polyvinyl alcohol |
| TEM | = | Transmission electron microscopy |
Acknowledgements
I would like to acknowledge Dr. R.S. Gaud, Director, SVKM’s NMIMS Deemed-to-be University, Shirpur Campus, for providing me with excellent research facilities and deep encouragement while pursuing this project. I would also like to acknowledge Bharathidasan University Tiruchirappalli Tamil Nadu-620024, India, for studying Confocal Laser Scanning Microscope (CLSM). I express my sincere gratitude to CSIR North East Institute of Science and Technology (NEIST) Jorhat-785006 Assam, India, for supporting me in my studies on Atomic Force Microscope (AFM).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Shi J, Votruba AR, Farokhzad OC, et al. Nanotechnology in drug delivery and tissue engineering: from discovery to applications. Nano Lett. 2010;10(9):3223–3230.
- Li W, Szoka FC. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24(3):438–449.
- Mirnezami SMS, Heydarinasab A, Akbarzadeh A, et al. Investigation of characterization and cytotoxic effect of PEGylated nanoliposomal containing melphalan on ovarian cancer: an in vitro study. J Exp Nanosci. 2021;16(1):102–116.
- Batrakova EV, Kabanov AV. Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Control Release. 2008;130(2):98–106.
- Senthilkumar S, Dhivya V, Sathya M, et al. Synthesis and characterization of magnetite/hydroxyapatite nanoparticles for biomedical applications. J Exp Nanosci. 2021;16(1):160–180.
- Jasim A, Abdelghany S, Greish K. Current update on the role of enhanced permeability and retention effect in cancer nanomedicine. Nanotechnology-based approaches for targeting and delivery of drugs and genes. Amsterdam, Netherlands: Elsevier; 2017. p. 62–109.
- Sur S, Rathore A, Dave V, et al. Recent developments in functionalized polymer nanoparticles for efficient drug delivery system. Nano-Struct Nano-Objects. 2019;20:100397.
- Abbas S, Chang D, Riaz N, et al. In-vitro stress stability, digestibility and bioaccessibility of curcumin-loaded polymeric nanocapsules. J Exp Nanosci. 2021;16(1):230–246.
- Jesus S, Schmutz M, Som C, et al. Hazard assessment of polymeric nanobiomaterials for drug delivery: what can we learn from literature so far. Front Bioeng Biotechnol. 2019;7:261.
- Rangsimawong W, Opanasopit P, Rojanarata T, et al. Skin transport of hydrophilic compound-loaded PEGylated lipid nanocarriers: comparative study of liposomes, niosomes, and solid lipid nanoparticles. Biol Pharm Bull. 2016;39(8):1254–1262.
- Puri A, Loomis K, Smith B, et al. Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst. 2009;26:523–580.
- Mandal B, Bhattacharjee H, Mittal N, et al. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine. 2013;9(4):474–491.
- Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. Acs Nano. 2008;2(8):1696–1702.
- Bertoni S, Passerini N, Albertini B. Nanomaterials for oral drug administration. Nanotechnology for oral drug delivery. Amsterdam, Netherlands: Elsevier; 2020. p. 27–76.
- Shi J, Xiao Z, Votruba AR, et al. Differentially charged hollow core/shell lipid-polymer-lipid hybrid nanoparticles for small interfering RNA delivery. Angew Chem Int Ed Engl. 2011;50(31):7027–7031.
- Gu J, Chen Y, Tong L, et al. Astaxanthin-loaded polymer-lipid hybrid nanoparticles (ATX-LPN): assessment of potential otoprotective effects. J Nanobiotechnol. 2020;18:1–17.
- Massadeh S, Omer ME, Alterawi A, et al. Optimized polyethylene glycolylated polymer–lipid hybrid nanoparticles as a potential breast cancer treatment. Pharmaceutics. 2020;12:666.
- Ai X, Duan Y, Zhang Q, et al. Cartilage-targeting ultrasmall lipid-polymer hybrid nanoparticles for the prevention of cartilage degradation. Bioeng Transl Med. 2021;6(1):e10187.
- Tahir N, Madni A, Balasubramanian V, et al. Development and optimization of methotrexate-loaded lipid-polymer hybrid nanoparticles for controlled drug delivery applications. Int J Pharm. 2017;533(1):156–168.
- Gardouh AR, Attia MA, Enan ET, et al. Synthesis and antitumor activity of doxycycline polymeric nanoparticles: effect on tumor apoptosis in solid Ehrlich carcinoma. Molecules. 2020;25:3230.
- Panday R, Poudel AJ, Li X, et al. Amphiphilic core-shell nanoparticles: synthesis, biophysical properties, and applications. Colloids Surf B Biointerfaces. 2018;172:68–81.
- Sah AK, Suresh PK, Verma VK. PLGA nanoparticles for ocular delivery of loteprednol etabonate: a corneal penetration study. Artif Cells Nanomed Biotechnol. 2017;45(6):1–64.
- Luo Y, Yang H, Zhou YF, et al. Dual and multi-targeted nanoparticles for site-specific brain drug delivery. J Control Release. 2020;317:195–215.
- Tapeinos C, Battaglini M, Ciofani G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J Control Release. 2017;264:306–332.
- Haggag YA, Ibrahim RR, Hafiz AA. Design, formulation and in vivo evaluation of novel honokiol-loaded PEGylated PLGA nanocapsules for treatment of breast cancer. Int J Nanomedicine. 2020;15:1625–1642.
- Yang V, Gouveia MJ, Santos J, et al. Breast cancer: insights in disease and influence of drug methotrexate. RSC Med Chem. 2020;11(6):646–664.
- Jang JH, Jeong SH, Lee YB. Preparation and in vitro/in vivo characterization of polymeric nanoparticles containing methotrexate to improve lymphatic delivery. Int J Mol Sci. 2019;20(13):3312.
- Bhaskarabhatla A. Is the 2013 price control regulation necessary? Regulating pharmaceutical prices in India. Berlin/Heidelberg, Germany: Springer; 2018. p. 33–67.
- Kim KT, Lee JY, Kim DD, et al. Recent progress in the development of poly (lactic-co-glycolic acid)-based nanostructures for cancer imaging and therapy. Pharmaceutics. 2019;11:280.
- Dave V, Tak K, Sohgaura A, et al. Lipid-polymer hybrid nanoparticles: synthesis strategies and biomedical applications. J Microbiol Methods. 2019;160:130–142.
- Shaikh MV, Kala M, Nivsarkar M. Formulation and optimization of doxorubicin loaded polymeric nanoparticles using Box-Behnken design: ex-vivo stability and in-vitro activity. Eur J Pharm Sci. 2017;100:262–272.
- Phalguna Y, Pothula S, Sarepalli RK, et al. Design, characterization and in vitro evaluation of polymeric nanoparticles containing decitabine. Pharm Innov. 2020;9:494–499.
- Surve DH, Jindal AB. Development and validation of reverse-phase high-performance liquid chromatographic (RP-HPLC) method for quantification of efavirenz in Efavirenz-Enfuvirtide co-loaded polymer-lipid hybrid nanoparticles. J Pharm Biomed Anal. 2019;175:112765.
- Behbahani ES, Ghaedi M, Abbaspour M, et al. Optimization and characterization of ultrasound assisted preparation of curcumin-loaded solid lipid nanoparticles: application of central composite design, thermal analysis and X-ray diffraction techniques. Ultrason Sonochem. 2017;38:271–280.
- Prajatelistia E, Sanandiya ND, Nurrochman A, et al. Biomimetic janus chitin nanofiber membrane for potential guided bone regeneration application. Carbohydr Polym. 2021;251:117032.
- de Almeida Moura C, da Silva Lima JP, Silveira VAM, et al. Time place learning and activity profile under constant light and constant dark in zebrafish (Danio rerio). Behav Processes. 2017;138:49–57.
- Bhattacharya S. Fabrication and characterization of chitosan-based polymeric nanoparticles of imatinib for colorectal cancer targeting application. Int J Biol Macromol. 2020;151:104–115.
- Bhattacharya S. Design and development of docetaxel solid Self-Microemulsifying drug delivery system using principal component analysis and D-Optimal design. Asian J Pharm. 2018;12:S1–S23.
- Tan Y, Li XC, Liu XY. Chemoselective hydrogenation of 3-nitrostyrene over supported gold catalysts: effect of loadings of gold. J Chin Chem Soc Taipei. 2020;68:444–450.
- Tamura K, Kubo T, Nagaoki I, et al. A 120 kV transmission electron microscope series for both life science and material science fields. Microsc Microanal. 2018;24(S1):1156–1157.
- Hady MA, Sayed OM, Akl MA. Brain uptake and accumulation of new levofloxacin-doxycycline combination through the use of solid lipid nanoparticles: formulation; optimization and in-vivo evaluation. Colloids Surf B Biointerfaces. 2020;193:111076.
- Caetano-Pinto P, Jamalpoor A, Ham J, et al. Cetuximab prevents methotrexate-induced cytotoxicity in vitro through epidermal growth factor dependent regulation of renal drug transporters. Mol Pharm. 2017;14(6):2147–2157.
- Li T, Yan G, Bai Y, et al. Papain bioinspired gold nanoparticles augmented the anticancer potency of 5-FU against lung cancer. J Exp Nanosci. 2020;15(1):109–128.
- Moll G, Ankrum JA, Kamhieh-Milz J, et al. Intravascular mesenchymal stromal/stem cell therapy product diversification: time for new clinical guidelines. Trends Mol Med. 2019;25(2):149–163.
- Madani F, Esnaashari SS, Bergonzi MC, et al. Paclitaxel/methotrexate co-loaded PLGA nanoparticles in glioblastoma treatment: formulation development and in vitro antitumor activity evaluation. Life Sci. 2020;256:117943.
- Su J, Liu G, Lian Y, et al. Preparation and characterization of erythrocyte membrane cloaked PLGA/arsenic trioxide nanoparticles and evaluation of their in vitro anti-tumor effect. RSC Adv. 2018;8(36):20068–20076.
- Gurel A, Armutçu F, Damatoglu S, et al. Evaluation of erythrocyte Na+, K+-ATPase and superoxide dismutase activities and malondialdehyde level alteration in coal miners. Eur J Gen Med. 2004;1:22–28.
- Lee BJ, Jo IY, Bu Y, et al. Antiplatelet effects of spatholobus suberectus via inhibition of the glycoprotein IIb/IIIa receptor. J Ethnopharmacol. 2011;134(2):460–467.
- Godara S, Lather V, Kirthanashri S, et al. Lipid-PLGA hybrid nanoparticles of paclitaxel: preparation, characterization, in vitro and in vivo evaluation. Mater Sci Eng C Mater Biol Appl. 2020;109:110576.
- Nozal V, Rojas-Prats E, Maestro I, et al. Improved controlled release and brain penetration of the small molecule S14 using PLGA nanoparticles. Int J Mol Sci. 2021;22:3206.
- Zhu R, Lei Z. COSMO-based models for predicting the gas solubility in polymers. Green Energy Environ. 2021;6(3):311–313.
- He R, Zhang G, Yang J, et al. Investigating dual drug loaded PLGA nanocarriers for improved efficacy in endometritis therapeutics. J Exp Nanosci. 2021;16(1):117–132.
- Khan I, Gothwal A, Sharma AK, et al. PLGA nanoparticles and their versatile role in anticancer drug delivery. Crit Rev Ther Drug Carrier Syst. 2016;33:159–193.
- Zook JM, MacCuspie RI, Locascio LE, et al. Stable nanoparticle aggregates/agglomerates of different sizes and the effect of their size on hemolytic cytotoxicity. Nanotoxicology. 2011;5(4):517–530.