Abstract
We report processing and luminescence decay characteristics of Cd1− x Zn x S composite nanocrystals (NCs) conjugated with bovine serum albumin (BSA) proteins. Time-resolved study on unconjugate NCs (with dimensions less than the bulk exciton Bohr radius) suggests that in the radiative emission, the fast (τ 1) and the slow (τ 2) carrier components are equally competitive for a given stoichiometry. Conversely, bioconjugate NCs advocate that the decay component due to the free exciton recombination is ∼9 times faster than the component due to the surface recombination emission. The observation of two distinct decay parameters is due to the fact that the NCs have experienced photostability by way of binding and protecting NC surface with biomolecules (BSA) as binding agents. The occurrence of two decay constants would help in extracting information with regard to the nature of surface recombination, free-exciton relaxation along with the strength of emission. Furthermore, with the increase in % Zn, slow carrier component gets slower owing to the incorporation of extra surface traps due to Zn/Cd incompatibility while making perfect lattice sites in the NCs. As a result, surface emission intensity gets improved compared to the radiative intensity due to core-state direct transitions. Understanding photoluminescence decay of bioconjugated NCs, on a comparative basis, would find scope for biomolecular labelling, sensing and electrophysiology applications.
Keywords:
1. Introduction
The bioconjugated-nanoparticles provide important schemes in nanobiotechnology as they are potential candidates for bioactive fluorescent probes in sensing, imaging, immunoassay and other similar diagnostics applications Citation1–6. An alternative to conventional organic fluorophores (fluorescent dyes), luminescent nanocrystals (NCs) offer many advantages, e.g. enhanced photostability, narrow and symmetric emission spectra without red-tailing, large Stoke's shift etc., apart from the fact that they can be excited at any wavelength shorter than the wavelength of fluorescence Citation1,Citation2,Citation7. In the recent past, the role of biomolecules was realised in nanostructure patterning and nanoscaled building blocks Citation8–10. Previously, it was demonstrated that the core-shell CdSe/ZnS quantum dots can be used as fluorescent labels while conjugated to transferrin, immunoglobulin (IgG) and bovine serum albumin (BSA) Citation11. Also, it was reported that the quantum dots are ideal fluorophores for multiplexed optical encoding of polymeric microbeads Citation12,Citation13. In fact, BSA (Mol. Wt ∼67 kD, hydrodynamic radius ∼7.6 nm) is a well-known protein whose molecular structure and property are known to the research community in great detail. The bioconjugated nanostructures are reported to exhibit fluorescence resonance energy transfer (FRET) owing to adequate spectral overlap between donor emission and acceptor absorption Citation14,Citation15. Following a supercritical fluid processing technique for biofunctionalisation, it was recently shown that the BSA species remain conjugated with CdS NCs over time and the conjugated nanoparticles do not agglomerate via protein–protein interactions Citation16. Whether it is biological imaging/labelling, or application in single molecule spectroscopy, life-time aspect of luminescence patterns provides in-depth knowledge of the radiation process occurring in a given material system. The radiative intensity depends on the number of e–h recombination events, and how fast a recombination event occurs is decided by how good the surface of the nanocrystal is protected from the environment. In other words, surface defects, imperfections arising from the surface non-stoichiometry and unsaturated bonds, etc which obstruct the radiative processes, must be sealed.
In this paper, we report bioconjugation of composite Cd1− x Zn x S-NCs (with x = 0, 0.5 and 0.75) and discuss their radiative features by analysing spectroscopic data. Our time-resolved photoluminescence studies show that there exist two distinct radiative paths for bioconjugate NCs, compared to unconjugate ones both having competitive radiative emissions, due to free exciton relaxation and transition via surface traps. The nature of radiative process is discussed with regard to the stoichiometric variation and BSA conjugation.
2. Experimental: materials and methods
The fabrication procedure of NCs-BSA conjugates involves three successive steps. First, surfactant-based NCs formation in reverse micellar (RM) cages, thiol-stabilisation for making NCs surface hydrophilic and finally, NCs entanglement with BSA. In the first step, we have followed the RM route described by Cizeron and Pileni Citation17. The degree of hydration w 0 = [H2O]/[AOT] = 8.9 was kept constant so that one can obtain definite hydrodynamic size (dia) of the reverse micelles (i.e., d ∼ 4w 0 = 3.6 nm) and thus capable of accommodating NCs of a few atomic clusters only Citation18,Citation19.
About 440 mg of surfactant bis(2-ethylhexyl) sulfosuccinate (aerosol-OT, AOT, Aldrich & Co.) was dissolved in 16.2 mL of n-heptane followed by dropwise addition of 54 µL de-oxygenated water under vigorous stirring. The mixture was stirred for about 3 h until a homogeneous microemulsion was obtained. The resulting precursor was divided into two equal parts A and B. Part A was used for the synthesis of CdS while part B was meant for the synthesis of Cd0.50Zn0.50S.
Next, part A was sub-divided into two equal parts A/ and A//. Then, (7 µL, 1 M) Cd2+ solution was added to A/ so as to receive an optically homogeneous solution. Similarly, (1.5 µL, 1 M) Na2S was transferred to A// by means of a syringe. For complete nucleation of CdS NCs, A/ and A// were then mixed at once and stirring (∼200 rpm) was continued for 10–12 h in a dark chamber. On the other hand, precursor B was sub-divided into part B/ (4.1 mL), B// (2 mL) and B/// (2 mL). Then (7 µL, 1 M) Cd2+ and (7 µL, 1 M) Zn2+ solutions were added to B// and B///, whereas (26 µL, 1 M) S2− solution was transferred to B/. As-received mixtures B// (containing Cd2+) and B/// (containing Zn2+) were added at once under stirring environment. Finally, B/ (containing S2−) was injected drop-wise and the sample was kept under stirring overnight for perfect nucleation of the desired Cd0.50Zn0.50S NCs. In a similar way, Cd0.25Zn0.75S NCs were synthesised by choosing appropriate volumetric concentration of Zn2+ and Cd2+ while mixed with S−2. Note that as the microreactors containing Zn2+ and Cd2+ (with [Cd2+] + [Zn2+]/[S2−] = 1) were continuously in a state of dynamic equilibrium, the favourable possibility of obtaining composite system (Cd1− x Zn x S) was more compared to the existence of independent CdS and ZnS phases. A typical synthesis route of CdS and CdZnS NCs is shown in the flow chart diagram ().
Figure 1. A typical flow chart diagram for obtaining (a) unconjugate CdS- and CdZnS-NCs and (b) thiol-capped and BSA-conjugated CdS NCs.
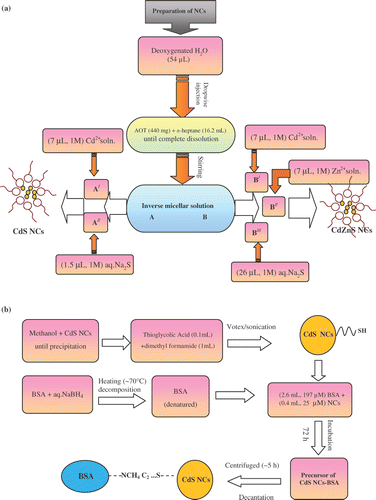
In the second step, for thiol solubilisation, 1 mL of given NC sample was transferred carefully to a 5 mL volumetric flask. Methanol (1 mL) was added dropwise till complete precipitation. The wet precipitate was wobbled well. In another centrifuge, 0.1 mL of mercaptoacetic acid (thioglycolic acid, Loba Chemi) was taken and transferred to a 10 mL volumetric flask containing 1 mL of dimethylformamide (DMF). Subsequently, the NC precursor was added and the whole mixture was vortexed and sonicated for about 45 min. The clear and transparent solutions containing thiol-stabilised NCs were stored for 1–2 days prior to bioconjugation.
Chemically reduced BSA (99.9% pure, Sigma-Aldrich & Co.) was prepared by denaturing BSA in 1 mM sodium borodydride in water at ∼70°C. Excess borohydride was removed by spontaneous decomposition by heating. For effective bioconjugation, (2.6 mL, 197 µM) BSA was treated with (0.4 mL, 25 µM) NCs. After labelling, the test tubes were shaken well and made airtight with teflon. The samples were incubated for 3–5 days at (65–70°C). At last the samples were centrifuged (∼12, 000 rpm) for ∼5 h followed by decantation. The decant was the actual NCs-BSA conjugate specimen, stored for subsequent experimentation. Thiol-treated CdS-NCs including BSA conjugation steps are shown in the block diagram ().
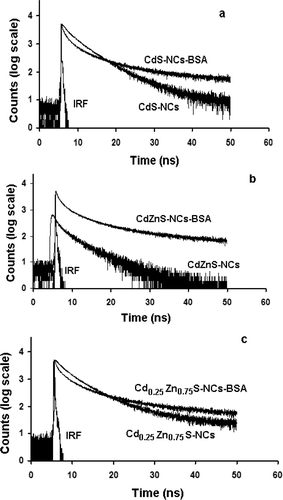
The samples were analysed by optical absorption spectroscopy (OAS), steady-state photoluminescence (PL) spectroscopy and time-resolved photoluminescence (TR-PL) spectroscopy, respectively. Note that a typical BSA molecule (containing two tryptophans) absorbs light at ∼278 nm, but emits within 310–348 nm, depending on the excitation wavelength of light Citation13. In order to isolate NC emission from tryptophans, we had selected λ ex = 375 nm line of excitation, and corresponding emission at ∼430 nm was analysed by a standard picosecond-resolved time correlated single photon counting (TCSPC) system. The commercially available setup was a picosecond diode laser pumped time-resolved fluorescence spectrophotometer (LifeSpec-ps, Edinburgh Instrument, UK).
3. Results and discussion
represents a set of optical absorption spectra of the synthesised NCs samples. The absorption edge (λ e) is the intersection of the sharply decreasing region of the spectrum with the baseline, correlated to the particle size by expression Citation20,Citation21: 2r = 1/(0.1338 − 0.0002345 λe). We have estimated the absorption onsets for CdS, CdZnS and Cd0.25Zn0.75S NCs as 340, 425 and 440 nm, respectively (). Accordingly, NCs (within strong confinement regime (r < a B, Bohr radius)) were obtained with average sizes calculated as 1.8, 2.9 and 3.3 nm. The size of CdS NCs was verified by transmission electron microscopy (TEM), and is shown in . The unconjugate CdS NCs sample displays isolated particles (∼2 nm) with very narrow size distribution (). Bovine serum albumin-conjugated NC structures are shown in . Note that the synthesis steps involved definite NCs concentration, and the microscopic imaging was done at similar magnifications. Statistical analysis on various micrographs has helped us in calculating bioconjugation conversion efficiency i.e., ∼78%. Since a number of biomolecules are attached to individual nanoparticles, clustering effect was seen in , compared to .
Figure 2. Optical absorption spectra (OAS) of (a) CdS, (b) CdZnS and (c) Cd0.25Zn0.75S-NCs systems. Numbers (1), (2) and (3) represent untreated, thiol-stabilised and BSA-NCs conjugates, respectively.
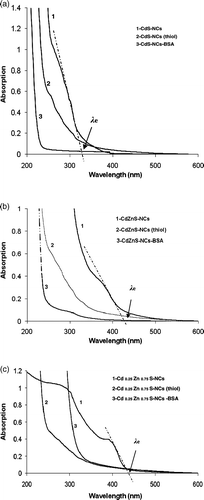
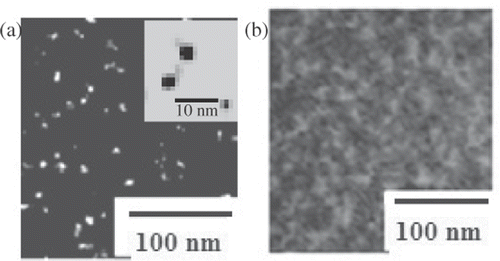
Figure 3. High resolution electron microscopy images of (a) CdS-NCs and (b) CdS-BSA conjugates. A magnified view of isolated, spherical CdS-NCs (∼2 nm) is shown in the figure inset–(a).
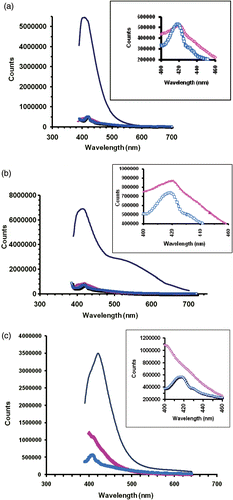
It is worth mentioning here that in case of a bulk, depending on the Cd/Zn stoichiometry, the optical band gap of a CdZnS composite will correspond to a value in between energy gaps of CdS (E g = 2.4 eV) and ZnS (E g = 3.7 eV). Therefore, with the increase of Zn content there can be shifting of the absorption edge towards blue (towards higher energy). Needless to say, such a situation could also have been realised in our case, had composite NCs of exactly identical sizes been produced. Since the energy gap is extremely sensitive to the NC size particularly, when it is comparable to or smaller than the Bohr radius, the discussion on shifting of the absorption edge is beyond the scope of the present study. On the other hand, we have noticed exciton absorption features for all the cases of unconjugate NCs. Thiol-treatment and BSA-conjugation had featureless characteristics owing to the surface treatment of functionalization of NCs due to the functionalization of ligand specific binding agents (. As bioconjugates have at least eight times more concentrated BSA molecules (which absorbs ∼278 nm) with respect to NCs, absorption feature of the former had affected the excitonic feature of the later. Shown in are steady-state photoluminescence of composite Cd1− x Zn x S (x = 0, 0.5, 0.75) NCs that reveal intense emission when they are untreated. However, the intensity of such samples was found to be suppressed with aging (not shown) and might have resulted due to the particle growth within the nanoreactors of the reverse micelles, which were in a state of dynamic Brownian motion. On the other hand, PL is suppressed significantly (11–17%) for thiol-treated and BSA-conjugated samples as a result of suppression of radiative emission due to adequate surface modification and reconstruction while synthesis was in progress. Nevertheless, PL intensity was found to be fairly stable with aging up to 1–2 weeks. For a given stoichiometry, the emission peak for the untreated NCs was found to be close to the bioconjugated NCs. The first prominent peak at ∼410–430 nm was ascribed to the near band edge emission. A broad emission peak at ∼520 nm arising due to surface deep trap states (non-radiative centers) is seen for CdZnS-NCs. The broad spectrum is owing to the unavoidable chemical incompatibility of Cd and Zn realised while forming a NCs lattice. Such a lattice mismatch is presumably minimum when either kind of atom dominates, as noticed for CdS-NCs and Cd0.25Zn0.75 S NCs cases (). Figure insets are shown to compare the emission responses of thiol-treated NCs and NCs-BSA conjugates.
Figure 4. Steady-state PL spectra of (a) CdS, (b) CdZnS and (c) Cd0.25Zn0.75S-NCs systems. Figure insets represent spectral responses in the selective wavelength range (400–460 nm) highlighting prominent peak positions. Symbols (−), (▴) and (□) correspond to untreated, thiol-stabilised and BSA-NCs conjugates, respectively.
In general, the PL decay curve, which has a bi-exponential form, is widely recognised for colloidal NCs and is given by Citation22,Citation23:
Our TR-PL results have predicted time constants ∼2 and 18–20 ns (). Therefore, the contribution due to deep trap centers/impuritiy states with time constant in the ∼ps scale and cases like multiphonon processes can be ignored here. The results are in good agreement with the recent investigations reported by Wang and coworkers who also suggested biexponential response of the ns components Citation29. It is evident that these two ns components are as a result of the two distinct radiative events. First, the shorter life time can be ascribed to the direct radiative transitions of the free excitons (core-state recombination) while the second, relatively slower component should be due to the radiative recombination via surface-trap sites. In contrast, the radiative nature of the fast process (shorter-life time) had been studied both experimentally Citation30 and theoretically Citation31, and was attributed to the initially populated core-state recombination of carriers. A schematic of radiative emission feature is shown in , where |gr⟩, |ex⟩ and |tr⟩ represent ground state, excited state and surface trap state, respectively. As the radiative part in Equation (Equation2) has two components (the first, due to direct recombination emission and the slow due to surface recombination emission), we can rewrite for the radiative time constant as:
Figure 5. Schematic view of the radiative emission process in NCs.
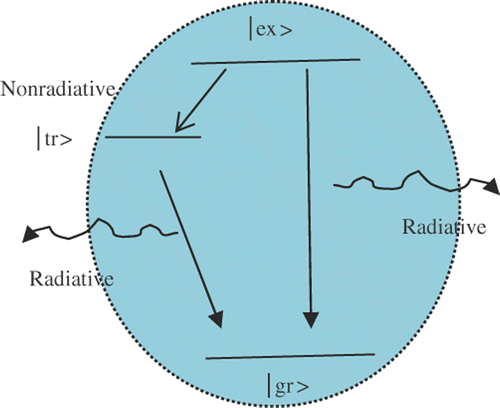
Table 1. PL-decay constants for various Cd1− x Zn x S-NCs and BSA conjugates.
Again, knowing that the two ns components exhibit exponential features, which constitute the second term in Equation (Equation2), in conjunction with Equation (Equation4):
Figure 6. Time-resolved photoluminescence spectra (λ ex = 375 nm; λ em = 430 nm) of (a) CdS, (b) CdZnS and (c) Cd0.25Zn0.75S-NCs systems.
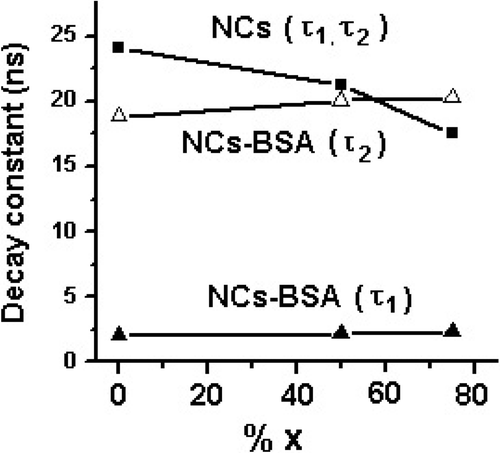
While both the time components are reasonably stable for NC-BSA-conjugated structures, the unconjugate NCs display significant variation in time constants (17.54–24.13 ns) with variation in stoichiometry. In the later case, the variation in time constants could have been due to the stoichiometry dependent electronic structure modification of the NCs. The electronic structure modification could be due to significant chemical incompatibility of Zn (covalent atm. rad. ∼0.131 nm) and Cd (covalent atm. rad. ∼0.148 nm) in Cd1− x Zn x S which had led to lattice contractions and hence, lattice defects on the NC surface. Noting here that for a spherical NC, surface-to-volume ratio ∼3/rn , rn being the NC radius (which actually depends on the covalently bonded atomic radius and the number of atoms therein), there can be a drastic variation in the number of surface defects when there is a variation in stoichiometry. This has affected the radiative recombination time constants at large (). Nevertheless, the most striking feature of albumin is its ability to bind reversibly an incredible variety of ligands and therefore, BSA-conjugated NCs could bring in adequate surface reconstruction and organisation of the surface atoms passivating the surface defects to a great extent. This helps in protecting NCs from photobleaching, keeping distinct radiative pathways intract and, thus, improving environmental stability. For BSA-NCs, the rapid and slow components are estimated to be ∼2 and ∼18–20 ns, respectively (, ). One can say that unconjugated NCs suffer from unwanted photobleaching, whose luminescence decay patterns are affected by alloying (%x) whereas bioconjugated BSA-NCs exhibit rather stable and distinct radiative patterns, independent of stoichiometry while the NCs are close to the strong confinement regime. In the strong confinement regime, electron and hole confine independently in the NCs. Now, since the electron has a smaller effective mass than the hole in a NC, it has a greater chance of going into the surface rather than staying inside the core of the NC Citation32. On the other hand, the heavy hole can remain at the center of mass of the NC Citation33. Compared to the unconjugate NCs, significant decrease in the fast component (τ f) of BSA-NCs might be due to the suppression of screening of the radiation field inside the NCs making ease of the core-state recombination Citation31. Now, since the relative intensities A f and A s are dependent on the number of radiative occurrences due to the free exciton relaxation and relaxation via surface states, the processes can be stoichiometry dependent. In our case, contribution (A f) due to the core-state radiative recombination emission was found to be highest (80.9%) for CdS-NCs-BSA structures. It was, however, suppressed to 74.3% for Cd0.25Zn0.75S-BSA systems. In contrast, the surface-related radiative intensity (A s), was 19.1 (lowest) and 25.7% (highest) for CdS-BSA and Cd0.25Zn0.75S-BSA, respectively (). Even for crystallites of fairly uniform dimension, since there can be additional surface trap states due to Cd/Zn incompatibility, the surface related contribution to the emission intensity is found to be higher in case of Cd0.25Zn0.75S-BSA system. It was believed that, owing to the surface binding capability of BSA, which is more efficient in case of CdS-BSA, the radiative intensity due to core-state recombination emission should be higher. Surface binding would be relatively weak for Cd0.25Zn0.75S-BSA systems and hence there is enhancement in surface emission intensity in comparison to CdS-BSA. The CdZnS-BSA NCs display emission intensities, which is intermediate between above discussed samples. Hence, bioconjugated samples exhibit maximum surface reconstruction for minimum chemical incompatibility and vice-versa.
Figure 7. Decay time constant vs. stoichiometry (% x, Zn) for Cd1− x Zn x S-NCs and BSA conjugates.
On the other hand, two possibilities are invoked for the origin of non-radiative processes: (i) defect concentration changes due to the variation in Cd and Zn composition to form NCs and (ii) surface defects due to the large s/v ratio for NCs. It is expected that the non-radiative channels are due to lattice mismatch as a result of difference in atomic sizes of Zn and Cd as described above. The amount of lattice mismatch depends on the stoichiometric ratio Zn:Cd, which in turn results in strain-induced surface states. Thus, surface recombination emission is affected by such surface defects. ZnS in the bulk form is considered as a phosphorescent material capable of giving out light up to several hundred seconds while CdS is a good fluorescent candidate. A relative change in Cd to Zn stoichiometry would be very much desired while looking at physiological activities like bioimaging and sensing. For instance, in an electrophysiology experiment, the helical peptides, e.g. alamethicin Citation34, gramicidin Citation35 etc can form voltage/ligand gated ion channels across phospholipid bilayers. On the other hand, the importance of ion channel mechanism has already been experienced in vivo real cells. Ion channel activity due to the non-selective ion transport was reported in human red blood cell (RBC) that can explain the nature of blood clotting Citation36, while the importance of light sensitivity to the ion channels was realised for neuronal firing Citation37. Due to the limited advantage and very often chemical reactivity of the organic fluorophors/dyes they can not be used in these experiments, especially while going for simultaneous optical and electrical measurements. Since the surface emission is highly controlled with Zn/Cd variation, one can use NCs-entangled ion channel peptides in order to understand dynamic nature of pore formation, reactivity of peptides with the bilayer in the process of making ion channels. In addition, opening and closing states of the ion channels can be related with the radiative emission that would occur in a given composite NCs, tagged to peptides. Moreover, NCs being efficient and alternative to conventional fluorophors can open up an area of study where the peptides do not undergo voltage gated expression but display either ligand gated or mechanically gated ion channel response. Apart from the unconjugate cases, where radiative emission is equally competitive for slow and fast decay components, a large incremental % of Zn, resulted in slow decay of the surface emission with a decay time τ s ∼ 20.31 ns (for a conjugated system with Zn : Cd = 3 : 1). It clearly indicates that though the surface reconstruction takes place in BSA conjugates, the contribution to the lattice mismatch cannot be ignored especially when the stoichiometry variation is very high. In addition, BSA-NCs system provides an ideal scheme where the core-state radiative emission can contribute ∼9 times faster than the surface trap recombination emission.
4. Summary
In summary, we have produced stable composite semiconductor NCs adopting a RM route. Upon thiol-stabilisation, BSA was conjugated independently with CdS, CdZnS and Cd0.25Zn0.75S-NCs systems. We have also addressed the light-emission process by studying steady-state and time-resolved PL measurements. We found that unconjugate NCs exhibit competitive radiative processes while bioconjugated NCs reveal that the luminescence decay proceeds through distinct paths with fast component about nine times faster than the slow component. The fast component is found to be fairly stable irrespective of stoichiometry variation. With reference to the stoichiometry variation and bioconjugation, various decay constants are estimated and discussed on a comparative basis. We believe that BSA conjugation could help in surface reconstruction, to the extent chemical incompatibility of Cd and Zn realised on the NC lattice. Hence, NC-BSA proteins are advantageous in the sense that they can protect and preserve fluorescence, especially when NCs are in the strong confinement regime. Understanding dynamics of NC surface emission pattern would provide electronic and optical transduction of the biological phenomena in many electrophysiological experiments including intracellular imaging with high selectivity and specificity.
Acknowledgements
The authors would like to thank colleagues for TR-PL measurements and D. Mohanta would like to thank the Indian Academy of Science for sponsoring the work carried out under the teacher fellow scheme during 2006–2007.
References
- Bruchez , M , Moronne , M , Gin , P , Weiss , S and Alivisatos , AP . 1998 . Semiconductor nanocrystals as fluorescent biological labels . Science , 281 : 2013 – 2016 .
- Matssoussi , H , Mauro , JM , Goldman , ER , Anderson , GP , Sundar , VC , Milkulec , FV and Bawendi , MG . 2000 . Self-assembly of CdSe−ZnS quantum dot bioconjugates using an engineered recombinant protein . J. Am. Chem. Soc. , 122 : 12142 – 12150 .
- Jaiswal , JK , Mattoussi , H , Mauro , JM and Simon , SM . 2003 . Long-term multiple color imaging of live cells using quantum dot bioconjugates . Nat. Biotechnol. , 21 : 47 – 51 .
- Medintz , IL , Uyeda , HT , Goldman , ER and Mattoussi , H . 2005 . Quantum dot bioconjugates for imaging, labelling and sensing . Nat. Mater. , 4 : 435 – 446 .
- Goldman , ER , Medintz , IL , Whitley , JL , Hayhurst , A , Clapp , AR , Uyeda , HT , Deschamps , JR , Lassman , ME and Mattoussi , H . 2005 . A hybrid quantum dot−antibody fragment fluorescence resonance energy transfer-based TNT Sensor . J. Am. Chem. Soc. , 127 : 6744 – 6751 .
- Zhang , C-Y , Yeh , H-C , Kuroki , MT and Wang , T-H . 2005 . Single-quantum-dot-based DNA nanosensor . Nat. Mater. , 4 : 826 – 831 .
- Wang , F , Tan , WB , Zhang , Y , Fan , X and Wang , M . 2006 . Luminescent nanomaterials for biological labeling . Nanotechnology , 17 : R1 – R13 .
- Parak , WJ , Gerion , D , Pellegrino , T , Zanchet , D , Micheel , C , Williams , SC , Boudreau , R , Le Gros , MA , Larabell , CA and Alivisatos , AP . 2003 . Biological applications of colloidal nanocrystals . Nanotechnology , 14 : R15 – R27 .
- Weiss , PS . 2001 . Nanotechnology: molecules join the assembly line . Nature , 413 : 585
- Yokoyama , T , Yokoyama , S , Kamikado , T , Okuno , Y and Mashiko , S . 2001 . Selective assembly on a surface of supramolecular aggregates with controlled size and shape . Nature , 413 : 619 – 621 .
- Chan , WC and Nie , S . 1998 . Quantum dot bioconjugates for ultrasensitive nonisotopic detection . Science , 281 : 2016 – 2018 .
- Han , M , Gao , X , Su , JZ and Nie , S . 2001 . Quantum-dot-tagged microbeads for multiplexed optical coding of biomolecules . Nat. Biotechnol. , 19 : 631 – 635 .
- Gao , X , Chan , WCW and Nie , S . 2002 . Quantum-dot nanocrystals for ultrasensitive biological labeling and multicolor optical encoding . J. Biomed. Optics , 7 : 532 – 537 .
- Mamedova , NN , Kotov , NA , Rogach , AL and Studer , J . 2001 . Albumin−CdTe nanoparticle bioconjugates: preparation, structure, and interunit energy transfer with antenna Effect . Nano Lett. , 1 : 281 – 286 .
- Wang , S , Mamedova , NN , Kotov , NA , Chen , W and Studer , J . 2002 . Antigen/Antibody immunocomplex from CdTe nanoparticle bioconjugates . Nano Lett. , 2 : 817 – 822 .
- Meziani , MJ , Pathak , P , Harruff , BA , Hurezeanu , R and Sun , Y-P . 2005 . Direct conjugation of semiconductor nanoparticles with proteins . Langmuir , 21 : 2008 – 2011 .
- Cizeron , J and Pileni , MP . 1995 . Solid Solution of CdyZn1−yS nanosize particles made in reverse micelles . J. Phys. Chem. , 99 : 17410 – 17416 .
- Luisi , PL , Giomini , M , Pileni , MP and Robinson , BH . 1988 . Reverse micelles as hosts for proteins and small molecules . Biochim. Biophys. Acta , 947 : 209 – 246 .
- Majumder , P , Sarkar , R , Shaw , AK , Chakraborty , A and Pal , SK . 2005 . Ultrafast dynamics in a nanocage of enzymes: solvation and fluorescence resonance energy transfer in reverse micelles . J. Coll. and Interf. Sci. , 290 : 462 – 474 .
- Spanhel , L , Hasse , M , Weller , H and Henglein , A . 1987 . Photochemistry of colloidal semiconductors: surface modification and stability of strong luminescing CdS particles . J. Am. Chem. Soc. , 109 : 5649 – 5655 .
- Moffitt , M and Eisenberg , A . 1997 . Size control of nanoparticles in semiconductor-polymer composites: control via multiplet aggregation numbers in styrene-based random ionomers . Chem. Mater. , 7 : 1478 – 1184 .
- GaO , LY , Lu , Y-J , Zheng , JS , Cai , Z-G , Sang , H-Y and Zeng , X-R . 2002 . Time-resolved photoluminescence study of Ga0.52In0.48P alloys . Euro. Phys. J. B , 28 : 145 – 148 .
- Li , CP , Guo , L , Wu , ZY , Ren , LR , Ai , XC , Zhang , JP , Lv , YZ , Xu , HB and Yu , DP . 2006 . Photoluminescence and time-resolved photoluminescence of star-shaped ZnO nanostructures . Solid State Commun. , 139 : 355 – 359 .
- Schlegel , G , Bohnenberger , J , Potapova , I and Mews , A . 2002 . Fluorescence decay time of single semiconductor nanocrystals . Phys. Rev. Lett. , 88 : 137401-1 – 137401-4 .
- Hong , S , Joo , T , Park , W , Jun , YH and Yia , G-C . 2003 . Time-resolved photoluminescence of the size-controlled ZnO nanorods . Appl. Phys. Lett. , 83 : 4157 – 4159 .
- Klimov , VI , Schwarz , CJ , McBranch , DW , Leatherdale , CA and Bawendi , MG . 1999 . Ultrafast dynamics of inter- and intraband transitions in semiconductor nanocrystals: implications for quantum-dot lasers . Phys. Rev. B. , 60 : R2177 – R2180 .
- Henry , CH and Nassau , KK . 1970 . Lifetimes of bound excitons in CdS . Phys. Rev. B , 1 : 1628 – 1634 .
- p’t Hooft , GW , van der Poel , WAJA and Molenkamp , LW . 1987 . Giant oscillator strength of free excitons in GaAs . Phys. Rev. B , 35 : 8281 – 8284 .
- Wang , X , Qu , L , Zhang , J , Peng , X and Xiao , M . 2003 . Surface-related emission in highly luminescent CdSe quantum dots . Nano Lett. , 3 : 1103 – 1106 .
- Wang , X , Zhang , J , Nazal , A , Darragh , M and Xiao , M . 2002 . Electronic structure transformation from a quantum-dot to a quantum-wire system: photoluminescence decay and polarization of colloidal CdSe quantum rods . Appl. Phys. Lett. , 81 : 4829 – 4831 .
- Weherenberg , BL , Wang , C and Guyot-Sionnest , P . 2002 . Interband and intraband optical studies of PbSe colloidal quantum dots . J. Phys. Chem. B , 106 : 10634 – 10640 .
- O’Neil , M , Marhon , J and McLendon , G . 1990 . Dynamics of electron-hole pair recombination in semiconductor clusters . J. Phys. Chem. , 94 : 4356 – 4363 .
- Woggon , U , ed. 1997 . Optical Properties of Semiconductor Quantum Dots , Vol. 136 , Berlin : Springer . Springer Tracts in Modern Physics
- Woolley , GA , Biggin , PC , Schultz , A , Lien , L , Jaikaran , DC , Breed , J , Crowhurst , K and Sansom , MS . 1997 . Intrinsic rectification of ion flux in alamethicin channels: studies with an alamethicin dimmer . Bio. Phys. J. , 73 : 770 – 778 .
- Harms , GS , Orr , G , Montal , M , Thrall , BD , Colson , SD and Lu , HP . 2003 . Probing conformational changes of gramicidin ion Channels by single-molecule patch-clamp fluorescence microscopy . Bio. Phys. J. , 85 : 1826 – 1838 .
- Kaestner , L and Bernhardt , I . 2002 . Ion channels in the human red blood cell membrane: their further investigation and physiological relevance . Bioelectrochemistry , 55 : 71 – 74 .
- Banghart , M , Borges , K , Isacoff , E , Trauner , D and Kramer , RH . 2004 . Light-activated ion channels for remote control of neuronal firing . Nature Neurosci. , 7 : 1381 – 1386 .