
ABSTRACT
Introduction: Inappropriate activation of apoptosis may contribute to neurodegeneration, a multifaceted process that results in various chronic disorders, including Alzheimer’s and Parkinson’s diseases. Several in vitro and in vivo studies demonstrated that neuronal apoptosis is a multi-pathway cell-death program that requires RNA synthesis. Thus, transcriptionally activated genes whose products induce cell death can be triggered by different stimuli and antagonized by neurotrophic factors. Systems biology is now unveiling the series of intracellular signaling pathways and key drug targets at the intersection of neuronal apoptosis and survival.
Areas covered: This review introduces a genomic approach that can be used to elucidate the systems biology of neuronal apoptosis and survival, and to rationally select drug targets, no longer oriented to emulate the action of growth factors at the membrane receptor level, but rather to modulate their downstream signals.
Expert opinion: The advent of genomics is offering an unprecedented opportunity to explore how the delicate balance between apoptosis and survival-inducing signals triggers a transcriptional program. Characterization of this program can be useful to identify potential pharmacological targets for existing drugs. Such knowledge might pave the way towards an innovative pharmacology.
1. Introduction
Selective neuronal loss, accompanied by specific protein aggregation, is the histopathological hallmark of neurodegenerative diseases. Although the abnormal proteins responsible for each disease are different in structure and function, all neurodegenerative disorders share the common process of protein misfolding and aggregation. These aggregates directly and indirectly attack cellular components, leading to neuronal death [Citation1]. For many years, two cell death pathways, namely, apoptosis and necrosis, have been considered as the main forms of cell death.
Apoptosis is a genetically controlled program of cell death by which a cell regulates its natural self-destruction. This intrinsic suicide process involves a well-choreographed sequence of biochemical and morphological events, including fragmentation of nuclear DNA, breakdown of cellular cytoskeleton, cell shrinkage, pyknosis, and blebbing of the plasma membrane, which breaks up into membrane enclosed particles, termed apoptotic bodies [Citation2].
Necrosis is not a regulated form of cell death and is characterized by plasma membrane disruption, organelle and cytoplasm swelling, and release of intracellular contents onto extracellular compartment. Excitotoxicity is a characteristic of the necrotic death and is due to a fast release of Ca2+ from intracellular pools, activating calpains that cleave cytoskeleton proteins [Citation3].
Even though necrosis is characterized by the lack of a clear molecular pathway, a necrosis-like process, partially programmed, seems also to exist. This sort of programmed necrosis, known as necroptosis, has recently been recognized and has the characteristics of apoptosis and necrosis, being a caspase-independent cell death, driven by defined molecular pathways [Citation4]. Necroptosis has been identified as a strong trigger of innate and adaptive immune responses, evolved as a line of defense against intracellular infection [Citation5].
While both necrosis and necroptosis prevalently occur in acute brain injuries, such as ischemic damage [Citation6], there is little doubt that apoptosis plays a major role in neurodegenerative diseases [Citation7].
Apoptosis is intimately linked to sculpting the correct architecture and size of neuronal and glial populations of the nervous system [Citation8]. Following the developmental window, aberrant neuronal apoptosis is thought to contribute to the development of a series of neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases [Citation9]. Owing to this leading role in neurodegenerative disorders, the major interest has traditionally been directed to elucidating the molecular mechanisms underlying neuronal apoptosis and devising strategies aimed at counteracting it.
Neurotrophic factors have been identified as the key factors in antagonizing neuronal apoptosis and promoting life signals in neuronal cells [Citation10,Citation11]. Delivery to the brain of these peptide molecules, however, is normally hindered by the blood–brain barrier (BBB), which limits the therapeutic utility of their exogenous administration [Citation12]. To circumvent this problem, there has been increasing focus on the development of pharmacological strategies no longer oriented to emulate the action of growth factors at the membrane receptor level, but rather to modulate their downstream signals.
The advent of high-throughput technologies has dramatically accelerated efforts in the field of systems biology to understand the transcriptional mechanisms underlying the action of neurotrophic factors under a variety of experimental physiological and pathological conditions [Citation13–Citation16]. Such knowledge is vital in identifying drug targets downstream of the neurotrophic factor receptors and paving the way toward the future aim of transforming genes into pharmaceutical compounds with neurotrophic activity that can interfere with the apoptosis machinery and be effective against untreated neurodegenerative diseases.
This review will specifically focus on recent advancements in the systems biology of neuronal apoptosis and survival, demonstrating the utility of a genomic-based approach to explore pathways and potential drug targets at the crossroad of neuronal apoptosis and neurotrophic factor rescue.
2. Genomic approach
Over the past 20 years, global analysis of mRNA expression has emerged as a powerful strategy for biological discovery. Using the power of parallel processing, robotics, and computer-based informatics, a number of high-throughput genomic technologies have been devised. These include whole-genome DNA microarrays and, most recently, RNA-sequencing (RNA-seq), each one having its own advantages and disadvantages [Citation17]. Before the advent of these two technologies, the majority of studies focused on individual genes or altered pathways and their collective behavior was therefore unknown. Microarray technology was the first to make possible whole-genome transcriptional analysis. Although the cost of microarray analysis is still lower than sequencing, RNA-seq is gaining more users among researchers. Among its major advantages is the ability to have direct access to the RNA sequence, the exon/exon boundaries and, therefore, produce a better picture of alternative splicing isoforms. Following both microarray and RNA-seq analysis (), deregulated genes and enriched pathways can be identified by means of a variety of bioinformatic and statistical tools that, once applied to large-scale genomic and gene expression data, help in interpreting their biological role [Citation18,Citation19].
Figure 1. Genomic approach for drug discovery in CGNs. A drug target discovery pipeline starting from microarray and RNA-seq analysis moves through drug targets identification and ends with their validation in in vitro models.
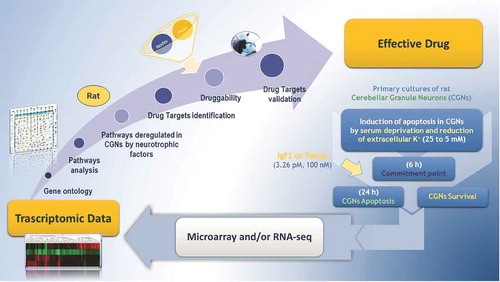
Microarray technology has been the most used approach to assess the systems biology of neuronal apoptosis and survival [Citation20]. In particular, the advent of microarrays has profoundly contributed to elucidating the existence of transcriptional pathways and a wider spectrum of pro- and antiapoptotic mediators then were originally believed to exist. Such knowledge has proved extremely useful in identifying drug targets downstream of the neurotrophic factor receptors and is paving the way toward the transformation of genes into pharmaceutical compounds with neurotrophic activity that can interfere with the apoptosis machinery and be effective against untreated neurodegenerative diseases.
The following part of this review will show some examples of how the genomic approach has been applied to dissecting the gene-based mechanisms underlying neuronal apoptosis and survival, describing the most important studies that have contributed to understanding them.
3. Application of the genomic approach to the study of neuronal apoptosis and survival
Since transcription has emerged as an obligate component of neuronal programmed cell death (PCD) and survival, microarray technology represents an excellent tool to fully understand transcriptional changes occurring during these processes. With the advent of DNA microarray, it has been possible to monitor the global expression changes occurring in numerous in vitro and in vivo neuronal paradigms.
The first paper in the literature that studied the gene expression component of neuronal apoptosis was that of Chiang et al. [Citation21]. By using a brain-biased array, this study profiled the gene expression changes of cerebellar granule neurons (CGNs) induced by different apoptotic signals, including potassium and serum withdrawal (KS-W) [Citation21]. Important progress in understanding the apoptotic program elicited by KCl deprivation derived from genome scale studies based on oligonucleotide microarrays, which identified a great number of temporally coupled genes that delineate functional pathways mobilized during neuronal PCD [Citation14,Citation18]. The limitations of the first studies in encompassing the entire set of deregulated genes in neuronal apoptosis were overcome with the advent of whole-genome microarrays, which allowed the whole apoptotic program to be analyzed [Citation19]. A comparative study between different in vitro paradigms of apoptotic neuronal death has subsequently indicated the existence of common regulatory mechanisms underlying neuronal cell death [Citation13]. Comparison of other gene expression data arising from postmortem brain tissues affected by neurodegenerative disorders has provided the first comprehensive view of the molecular alterations that seem to contribute to human neuropathogenesis [Citation22] and offered an outstanding opportunity not only to look into exclusive disease-specific changes, but also more importantly to look for potential common molecular pathogenic mechanisms [Citation23].
Most of the current knowledge about the transcriptional changes occurring during neuronal death has been obtained using microarray technology. Microarray analysis, however, is capable of measuring only the status of known transcripts. In addition, expression of low-abundance mRNAs is not detected by this hybridization-based approach. Using RNA-seq, a recent study enabled additional genes, deregulated during the process of neuronal death, to be identified [Citation24].
DNA microarray has proved useful for screening cellular targets that govern apoptosis [Citation18,Citation25]. Our most recent studies, in particular, were the first to extend the analysis to survival and led to the identification of key drug targets deregulated in both apoptosis and growth factor rescue [Citation11,Citation15,Citation26]. The following paragraphs will describe how this kind of approach was first utilized to characterize the transcriptional program at the intersection of apoptosis and survival of CGNs and to identify new potential drug targets.
4. Transcriptional programs of neuronal apoptosis and survival in CGNs
One basic feature of neuronal apoptosis in vivo is its sporadic and asynchronous nature. In such a condition, neurons undergo apoptosis at very different times and this may be attributed to a variety of factors, such as date of generation, local environments, and input received from other cells [Citation2]. On the contrary, a synchronous population of neurons can undergo apoptosis in in vitro systems, where most of these variables can be controlled and studied more effectively. In vitro models, therefore, represent an important step in the search for potential drug targets, as they substantially reduce the complexity of in vivo models.
CGNs are the most abundant neuronal type in the mammalian brain and represent the model most commonly used either in vivo or in vitro to study neuronal apoptosis and survival [Citation27]. Although CGNs do not recapitulate all features of human neuropathology, they have been extensively used to evaluate the pathogenic mechanisms underlying neurodegeneration and to validate potential therapeutic compounds.
During early stages of postnatal development, it is assumed that apoptosis of granule cells reflects the failure of these neurons to obtain adequate amounts of specific neurotrophic factors [Citation28]. In the adult, mossy fiber axotomy is followed by apoptosis of CGNs, underlining the crucial role of afferent inputs for the survival of these cells [Citation27]. In vitro, CGNs undergo rapid apoptotic cell death within 24 h after removal of serum and lowering of extracellular potassium from 25 to 5 mM [Citation27,Citation29]. Apoptosis of CGNs requires protein transcription and synthesis, becoming irreversible after the first 6 h following its induction. Before this ‘commitment point,’ CGNs can be rescued by the activation of specific signal transduction pathways or by the treatment with specific neurotrophic factors [Citation27,Citation29].
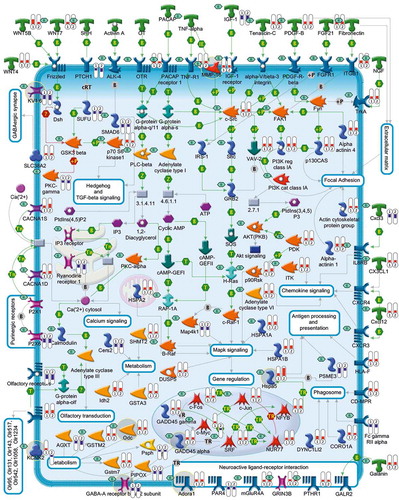
Two of the most effective growth factors capable of preventing apoptosis of CGNs are insulin-like growth factor-1 (Igf1) [Citation29] and pituitary adenylyl cyclase-activating polypeptide (Pacap) [Citation30]. Although the survival effects of these neurotrophic factors are mediated by different receptors and intracellular second messengers [Citation31,Citation32], they are propagated by common transcriptional cascades and show striking similarities [Citation33] ( and ). Indeed, not only a majority of differentially expressed genes, defined as ‘survival related genes’ (SRGs), are common to Igf1 and Pacap (), but also the Pearson correlation coefficient between Igf1 and Pacap for all SRGs is 0.97 [Citation33]. Taken together, these data support the existence of a preserved transcriptional program underlying neuronal survival. shows a comprehensive picture of differently expressed genes and enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with the rescue of CGNs following treatments with Igf1 and Pacap. Several SRGs encode drug targets for existing drugs. In the next section, these targets and their related pharmacological compounds will be described.
Figure 2. Genes differentially expressed in CGNs after induction of apoptosis and rescue by Igf1 and Pacap. (a) Venn Diagram. Figure shows the 1212 genes differentially expressed in apoptotic CGNs (K5 vs K25) defined as ‘Apoptotic related genes’ (ARGs), and the 1535 genes differentially expressed in rescued CGNs (K5 + Igf1 vs K5; K5 + Pacap vs K5) defined as ‘Survival related genes’ (SRGs). (b) Hierarchical cluster. A hierarchical clustering algorithm was used to order the differential expressed genes (ARGs and SRGs) in a dendrogram, in which the pattern and length of the branches reflects the relatedness of the expression levels in the different experimental conditions. Data are presented in a matrix format: each row represents a single gene and each column an experimental condition. As shown in the color bar, the red color indicates highly expressed genes, the blue color down-regulation genes.

Figure 3. A comprehensive picture showing deregulated genes and pathways linked to Igf1 and Pacap rescue of apoptotic CGNs. This figure, illustrates the gene expression profiles and the 14 KEGG pathways that are mainly involved in Igf1 and Pacap neuroprotection of apoptotic CGNs. Each encoded protein is labeled with two thermometers that indicate gene expression ratios under the following experimental conditions: K5+Igf1/K5 (first thermometer) and K5+Pacap/K5 (second thermometer). More specifically, downward thermometers have a blue color and reflect down-regulated expression, whereas upward thermometers have a red color and reflect up-regulated expression following Igf1 or Pacap treatment. Several of the deregulated genes encode drug targets for known pharmacological compounds (indicated by ‘R’ in a small green rhombus). Some of the drug targets discussed in the manuscript are highlighted by a gray circle.
5. Potential drug targets at the crossroad of neuronal apoptosis and survival
As mentioned above, transcriptional analysis of CGNs unveiled a large number of genes and pathways that are commonly deregulated during apoptosis and its rescue by Igf1 and Pacap [Citation11]. Several of these genes encode known drug targets, which include ligands, G-protein-coupled receptors, ion channels, enzymes. and transcription factors ( and ). A comprehensive list of potential drug targets with their pharmacological compounds is shown in supplementary Table S1.
Table 1. Potential drug targets at the intersection of apoptosis and survival following treatment with Igf1 and Pacap.
In the following paragraphs, some of these drug targets will be discussed by grouping them into three main functional clusters: cellular processes, signal transduction, and metabolism. Particular attention will be given to those drug targets that have been already implicated in neurodegenerative diseases and cancer, which may be influenced by common signaling pathways [Citation34].
5.1. Cellular processes
Transcriptional signaling pathways underlying rescue of apoptosis in CGNs by Igf1 and Pacap include genes functionally involved in specific cellular processes: (1) antigen processing and presentation, (2) cytoskeleton remodeling, and (3) cell death. Among potential deregulated therapeutic targets, Hspa5 (i), Limk2 (ii), Mcl1 (iii), and Gja8 were all found to be upregulated by Igf1 and Pacap. Inhibition of Gja8 by 18alpha-glycyrrhetinic acid [Citation35] and activation of Hspa5 by salubrinal, 2DG, and NE-100 [Citation36,Citation37] have already been shown to support cell survival. Similarly, treatment with LIMKi and A-1210477, two selective inhibitors of Limk2 and Mcl1, respectively, prevents apoptosis in cancer [Citation38,Citation39].
5.2. Metabolism
Several enzymes linked to a number of metabolic pathways are deregulated during neuronal apoptosis and survival in vitro. Their additional involvement in neurological disorders has stimulated great interest in their pharmacological modulation. Below, we will discuss some of these potential metabolic drug targets.
5.2.1. Enzymes involved in extracellular matrix remodeling
Mmp16, Adamts2, and Adam1a are members of the matrix metalloproteinase (MMP), a disintegrin, and metalloproteinase with thrombospondin motifs (ADAMTS) and metalloproteinase-disintegrins (ADAMs) family of proteins, participating in the remodeling of the extracellular matrix. Although they are emerging as regulators of nervous system development and adult brain functions, their role in cell death remains to be elucidated [Citation40,Citation41]. Our in vitro data, however, show an increased expression of Mmp16 and Adam1a and the decrease of Adamts2 during CGNs rescue [Citation15]. To clarify the role of these enzymes, additional studies should be performed to investigate the neuroprotective effects of metalloproteinase inhibitors, like BB-94 [Citation42] and marimastat [Citation43], or activators like losartan [Citation44].
5.2.2. Enzymes in anabolic and catabolic chemical reactions
Aco1, Asl, Dio3, Gpld1, Hsd3b5, Odc1, and Nmnat2 are enzymes involved in a series of catabolic reactions that were upregulated in rescued CGNs, while Asah1 was downregulated by Igf1 and Pacap. With the exception of Aco1/Irp1, which is already a known target of neuroprotective drugs like tempol or iron chelators (R-apomorphine) [Citation45,Citation46], the functional effect of pharmacological modulation of the other enzymes remains to be clarified. Some studies have focused on the inactivation of these enzymes in a range of malignancies. Drugs like pegylated arginine deiminase/ADI-PEG20 (Polaris Group) (Als inhibitor) [Citation47], xanthohumol (Dio3 inhibitor) [Citation48], 1,10-phenanthroline (Gpld1 inhibitor) [Citation49], compounds 54, 66, and 67 (inhibitors of Hsd3b5) [Citation50], and alpha-difluoromethylornithine (Odc1 inhibitor) [Citation51] have already been reported to act as sensitizing anticancer agents.
Deregulation of Asah1, an acid ceramidase, and Nmnat2, a nicotinamide mononucleotide adenylyltransferase (NMNAT) enzyme, has been involved in a variety of neurodiseases and/or cancers [Citation52,Citation53]. Recent studies have reported that Asah1 inhibitors, such as B-13, Ceranib 2, or LCL 204, slow the growth of cancer cells, alone or in combination with other established, antioncogenic treatments [Citation54,Citation55]. Although there is no evidence of selective Nmnat2 drugs, palmostatin B is able to boost Nmnat2 palmitoylation that is important for promoting survival of axons and preventing the onset of neurodegenerative disorders [Citation56,Citation57].
5.2.3. Cytochrome enzymes
Transcriptomic analysis of CGNs undergoing apoptosis or growth factor rescue unveiled the deregulated expression of a number of cytochrome b and P450 enzymes, which are some of the most versatile redox proteins known. Among these, Cyp3a23/3a1 and Cyp2d4 were overexpressed, while and Cybb and Cyp2b2 were downregulated in rescued CGNs.
Cyp3a23/3a1 is well recognized for its role in the metabolism of numerous drugs and xenobiotic compounds in several tissue types. Several agents, such as fluconazole [Citation58], myclobutanil, triadimefon [Citation59,Citation60], beta-naphthoflavone, phenobarbital [Citation61], and triadimefon [Citation59], can induce the family of CYP3A isoforms in rat, mouse, and human in a dose-related manner. In contrast, troleandomycin and apatinib are able to selectively inhibit Cyp3a23/3a1 [Citation62]. Cyp2b2 is known to protect neuronal cells from exogenous neurotoxic compounds (i.e. 7-pentoxyresorufin-O-dealkylase) [Citation63] and Cyp2b2 activators have already been demonstrated to have antioxidant effects in rat liver [Citation64]. Cyp2d4 regulates neuronal survival when pharmacologically inhibited by herba erigerontis and fluoxetine drugs [Citation65,Citation66].
5.2.4. Palmitoyltransferase pathway
In rescued CGNs, two proteins containing DHHC-type zinc finger domain, Zdhhc4 and Zdhhc8, were found to be downregulated. These are stable scaffolds that have evolved a palmitoyltransferase activity, which plays a critical function throughout the nervous system [Citation67,Citation68]. Zdhhc8, in particular, influences mitochondria-regulated apoptosis when overexpressed [Citation69]. Chemical inhibitors of palmitoylation, such compound V and 2BP [Citation70], may be used to test the direct involvement of these proteins in neuronal apoptosis and survival.
5.3. Signal transduction
In rescued CGNs, deregulated expression of genes encoding proteins of the neuroactive ligand and receptor interaction pathway, ion channels, chemokine, MAPK, TNF, and transcription factors signaling were observed.
5.3.1. Neuroactive ligand and receptor pathway
A number of G-protein-coupled receptors were differently expressed in CGNs rescue. Overexpression of Adora1, Grk5, and Npr3, as well as downregulation of Bdkrb1, Npbwr1, and Htr3b, is only a few examples of G-protein-coupled receptors linked to Igf1 and Pacap neuroprotection. Several studies have assessed that selective Adora1 agonists, CCPA and CPA, induce neuroprotection [Citation71]. These findings imply that the drug manipulation of Adora1 may facilitate the treatment of Parkinson’s and Alzheimer’s diseases [Citation72,Citation73]. Pharmacological modulation of Bdkrb1 by its agonist R838 is considered a target for the treatment of chronic inflammatory diseases such as multiple sclerosis [Citation74]. Grk5 antagonists, such as amlexanox [Citation75], Npbwr1, (5-chloro-4-(4-methoxyphenoxy)-2-(p-tolyl)pyridazin-3(2H)-one) [Citation76], Htr3b, MDL72222, and Y25130 [Citation77], have protective effects against inflammatory and/or stress-induced apoptosis, highlighting the importance of further investigation in neurodiseases where these genes have been involved [Citation78–Citation80]. Finally, induction of Npr3 by its agonist cANF⁴⁻ [Citation23] exerts cytoprotective effects in nonneuronal cells [Citation81].
5.3.2. Ion channels
Within the genes identified in rescued CGNs, several ion channels are differentially expressed. Among these, Kcna6 was found to be downregulated, while Kcnc1 and Kcnq2 were upregulated. Kcnc1 and Kcna6 are potassium channels, highly expressed in rat cerebellum [Citation82], which are effective in restoring axonal conduction in spinal cord injury and multiple sclerosis when antagonized by voltage-gated K+ channel blockers like 4-AP-3-MeOH and 4-AP [Citation83]. Over-activation of certain potassium channels, like Kcnq2, has been also associated with the early ionic events during the apoptotic cascade. As reported in previous studies, the potent Kcnq2 inhibitor, XE991, suppresses in vitro neuronal cell death [Citation84]. Thus, further pharmacological modulation of Kcnq2 with selective activators like 5-(2,6-dichloro-5-fluoropyridin-3-yl)-3-phenyl-2-(trifluoromethyl) pyrazolo[1,5-a]pyrimidin-7(4H)-one [Citation85] should be tested as an important pharmacological strategy against neurodegeneration.
5.3.3. Chemokine, MAPK, and TNF signaling
Among the chemokine-signaling genes deregulated during CGNs rescue is the overexpressed Ccl2/Mcp1. Accumulating evidence indicates a dual role of Ccl2 in neuronal survival [Citation86,Citation87]. While drugs like bindarit downregulate Ccl2 and protect neurons [Citation88], noradrenaline transporter inhibitors like desipramine elicit neuroprotection by upregulating the expression of Ccl2 [Citation89,Citation90].
Among the potential drug–targets linked to Mapk signaling, we detected the downregulation of Shc1 and the overexpression of Mapk3/Erk1, Map2k3, and Gadd45α in rescued CGNs. Given the significant role Mapk proteins play in cancer development and progression, intense research activities were directed toward pharmacological modulation of Mapk pathway. Potent and selective cell permeable inhibitors of Shc1 (i.e. CGP78850) and Mepk3/Erk1 (i.e. BAY 43-9006 and PD98059) have already been shown to be beneficial in suppression of cancer cell growth [Citation91,Citation92]. Selective inhibitors of Map2k3 have been well characterized by fragment-based drug design and further studies are necessary to validate their potential impact in diseases [Citation93]. Although there is no selective drug for Gadd45, fucoxanthin, docetaxel, peptidylarginine deaminase 4, and histone deacetylase inhibitors are Gadd45 activators that are able to inhibit cancer cell growth [Citation94].
5.3.4. Transcription factor signaling
Among the differentially expressed genes that encode transcription factors involved in CGN rescue are c-Fos, c-Jun, Srf, Nur77, and Myc, which are potential drug targets for several activator and/or inhibitor compounds (). A number of previous, experimental studies show that the pharmacological targeting of these molecules may favor cell survival [Citation95].
6. Pharmacological modulation of myc, a potential drug target downstream of growth factors
Transcription factors have recently received considerable attention as potential targets for counteracting inappropriate activation of neuronal apoptosis due to their significant role to act downstream of signaling cascades and control global gene expression in response to a specific stimulus. Although this interest has produced encouraging results, much still remains to be investigated, especially concerning the effective targeting of these molecules with therapeutic compounds. Among the groups of differentially expressed genes encoding for transcription factors during CGNs rescue by growth factors, we focused on Myc. This transcription factor is known to play a key role in neuronal apoptosis by regulating the expression of many genes through binding enhancer box sequences (E-boxes) and recruiting histone acetyltransferases. Based on these observations, we decided to investigate the role of Myc in neuronal survival by studying the effects of its inhibitor, 10058-F4. As shown in our previous work [Citation26], treatment with this compound was able to reduce the rescue effect of growth factors. Although preliminary, these experiments support a new pharmacological strategy based on the systems biology of neuronal apoptosis and survival, no longer oriented to emulate the neuroprotective action of growth factors at their membrane receptor level but, rather, to modulate their downstream signals.
7. Conclusion
Although this review demonstrates the utility of DNA microarray technology as a mean for dissecting the multigenic programs at the crossroad of neuronal apoptosis and survival, the presented data represent just a glimpse of their complexity. In light of the recent description of the involvement of necroptosis in neurodegeneration, it is conceivable that the large number of genes and pathways that are commonly deregulated during apoptosis could also influence regulated necroptosis, since they may be both not mutually exclusive but proceed simultaneously. Indeed, rather than a ‘switch’ operating between the two modes of cell death, the final mechanism may depend on levels of the respective signaling and effector proteins. Understanding how the brain is self-programmed to counteract neurodegeneration in a concerted and structured manner, with the interplay of different cell death processes, represents a future key for the development of appropriate and effective therapeutic approaches. Therefore, monitoring of gene expression, in different neuronal types, remains the next challenge and will require further bioinformatic advancements. The comprehensive understanding of the genes or sets of genes that play critical roles in neuronal death and survival will represent the starting point to elaborate new pharmacological therapies against neurodegenerative disorders.
8. Expert opinion
There is a vast amount of evidence indicating that neurotrophic factors are diffusible peptides that play a key role in modulating the development, maintenance, and survival of neurons and neuron-supporting cells. In addition, it is well known that alterations in levels of neurotrophic factors can lead to a dramatic cell loss through a transcription-dependent PCD mechanism and contribute to the pathogenesis of neurodegenerative diseases such as PD and AD [Citation96]. Recently, researchers have directed their attention to the action of neurotrophic factors in promoting neuronal survival and preventing neurodegenerative diseases. Studies in in vitro models suggest that pretreatment with neurotrophic factors induces the activation of antiapoptotic pathways [Citation33], while tests in in vivo models conclude that administration of neurotrophic factors limits their neuroprotective action due to their poor stability and minimal BBB permeation. Although approaches, such as intraventricular infusion of nerve growth factor in rats after traumatic brain injury, or using IGF1 in clinical trials in patients with amyotrophic lateral sclerosis, resulted in a reduction of neuronal loss, there is not yet an effective treatment for neurodiseases [Citation9].
Working on the hypothesis that the limited survival effects of neurotrophic factors resulting from direct delivery to the brain could be improved by modulating their downstream signaling pathways, various molecular biological approaches have been developed. The advent of microarray technology has led to the development of a genomic approach that has dramatically accelerated efforts to understand the signaling pathways underlying the pro- and antiapoptotic role of neurotrophic factor withdrawal in several in vitro models [Citation19,Citation97]. Microarray technology is supported by a number of bioinformatic and statistical tools applied to genomic data. These tools allow to translate genomic data into information that can assist in understanding the delicate balance between neuronal apoptosis and survival and in identifying genes that showing an altered expression in the promotion and/or protection from apoptosis may represent potential pharmacological targets for already existing drugs. This new therapeutic perspective paves the way toward an innovative pharmacology based on drug targets downstream rather than upstream of membrane receptors, allowing the use of substances acting on key pathways activated by neurotrophic factors.
Despite the considerable progress that has been made in this area, there are still some limitations inherent to microarray technology, as described in the dedicated Section 3. In particular, since there are considerable issues regarding sensitivity and reproducibility of microarray studies, which inevitably cause an high number of false positives, confirmatory techniques, such as proteomic methods, are necessary to test and validate data inferred through the genomic analysis. Proteomics can help to decipher the protein products of genes of interest and to get a clearer picture of the complex regulatory networks that control fundamental biological processes.
The simultaneous monitoring of both RNA and protein expression levels, therefore, may strongly contribute to increasing reliability in drug target discovery and improving the pharmacological approach for counteracting neurodegeneration. In the future, comparing further public transcriptomic and proteomic data inferred across more complex biological models and phenomena will help to deduce useful information about the spectrum of potential drug targets that may give more effective answers for the therapy of neurodegenerative diseases.
Article highlights box
Neuronal apoptosis is a normal physiological process that is an essential and integral part of neurogenesis. However, its inappropriate activation may contribute to neurodegeneration, a progressive and multifaceted process that results in various chronic disorders, such as Alzheimer’s and Parkinson’s diseases.
Growth factors are known to be potent agents that counteract neuronal cell death in in vitro models. However, the peptidergic structure of these molecules limits their ability to cross the blood brain barrier (BBB) and exert effective neuroprotection.
In recent years, the advent of high-throughput technologies has offered a new approach to decipher the transcriptional mechanisms underlying the molecular framework (genes and pathways) of growth factors, unraveling a systems biology based perspective. In particular, in vitro models have been extensively used to study the neuronal apoptotic and/or survival signaling events regulated by growth factors.
By combining whole genome expression analyses of apoptotic neurons rescued by two very effective growth factors, Igf1 and Pacap, a variety of common genes and functional pathways were found to be statistically deregulated. Several of these deregulated genes encode known drug targets for pharmacological compounds.
Pharmacological modulation of drug targets in neuroprotective-signaling pathways could be a key to antagonize neurodegeneration. Such a strategy, may focus on the development of pharmacological compounds no longer oriented to emulate the action of growth factors at the membrane receptor level but rather, to modulate their downstream signals.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgements
The authors gratefully acknowledge Cristina Calì, Alfia Corsino, Maria Patrizia D’Angelo and Francesco Marino for their administrative and technical assistance.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Bourdenx M, Koulakiotis NS, Sanoudou D, et al. Protein aggregation and neurodegeneration in prototypical neurodegenerative diseases: examples of amyloidopathies, tauopathies and synucleinopathies. Prog Neurobiol. 2015 Jul;21.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007 Jun;35(4):495–516.
- Bianchi L, Gerstbrein B, Frokjaer-Jensen C, et al. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci. 2004 Dec;7(12):1337–1344.
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014 Jan 30;370(5):455–465.
- Kaiser WJ, Upton JW, Mocarski ES. Viral modulation of programmed necrosis. Curr Opin Virol. 2013 Jun;3(3):296–306.
- Vieira M, Fernandes J, Carreto L, et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis. 2014;68:26–36.
- Honig LS, Rosenberg RN. Apoptosis and neurologic disease. Am J Med. 2000 Mar;108(4):317–330.
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501.
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Bio. 2000 Nov;1(2):120–129.
- Aloe L. Rita Levi-Montalcini and the discovery of NGF, the first nerve cell growth factor. Arch Ital Biol. 2011 Jun;149(2):175–181.
- Maino B, Ciotti MT, Calissano P, et al. Transcriptional analysis of apoptotic cerebellar granule neurons following rescue by gastric inhibitory polypeptide. Int J Mol Sci. 2014;15(4):5596–5622.
- Thorne RG, Frey WH 2nd. Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet. 2001;40(12):907–946.
- Paratore S, Parenti R, Torrisi A, et al. Genomic profiling of cortical neurons following exposure to beta-amyloid. Genomics. 2006 Oct;88(4):468–479.
- Cavallaro S, D’Agata V, Alessi E, et al. Gene expression profiles of apoptotic neurons. Genomics. 2004 Sep;84(3):485–496.
- Maino B, D’Agata V, Severini C, et al. Igf1 and pacap rescue cerebellar granule neurons from apoptosis via a common transcriptional program. Cell Death. 2015;1.
- Cavallaro S, Calissano P. A genomic approach to investigate neuronal apoptosis. Curr Alzheimer Res. 2006 Sep;3(4):285–296.
- Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011 May;31(9):34.
- Desagher S, Severac D, Lipkin A, et al. Genes regulated in neurons undergoing transcription-dependent apoptosis belong to signaling pathways rather than the apoptotic machinery. J Biol Chem. 2005 Feb 18;280(7):5693–5702.
- Kristiansen M, Menghi F, Hughes R, et al. Global analysis of gene expression in NGF-deprived sympathetic neurons identifies molecular pathways associated with cell death. BMC Genomics. 2011;12:551.
- Murphy D. Gene expression studies using microarrays: principles, problems, and prospects. Adv Physiol Educ. 2002 Dec;26(1–4):256–270.
- Chiang LW, Grenier JM, Ettwiller L, et al. An orchestrated gene expression component of neuronal programmed cell death revealed by cDNA array analysis. Proc Natl Acad Sci U S A. 2001 Feb 27;98(5):2814–2819.
- Ritz MF, Grond-Ginsbach C, Kloss M, et al. Identification of inflammatory, metabolic, and cell survival pathways contributing to cerebral small vessel disease by postmortem gene expression microarray. Curr Neurovasc Res. 2016;13(1):58–67.
- Durrenberger PF, Fernando FS, Kashefi SN, et al. Common mechanisms in neurodegeneration and neuroinflammation: a brainnet Europe gene expression microarray study. J Neural Transm. 2015 Jul;122(7):1055–1068.
- Sharma D, Kim MS, D’Mello SR. Transcriptome profiling of expression changes during neuronal death by RNA-Seq. Exp Biol Med. 2015 Feb;240(2):242–251.
- Yap YW, Chen MJ, Choy MS, et al. Temporal transcriptomic profiling reveals cellular targets that govern survival in HOCl-mediated neuronal apoptosis. Life Sci. 2010 Oct 9;87(15–16):457–467.
- Paparone S, Severini C, Ciotti MT, et al. Transcriptional landscapes at the intersection of neuronal apoptosis and substance P-induced survival: exploring pathways and drug targets. Cell Death Discov. 2016;2:16050.
- Contestabile A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum. 2002 Jan-Mar;1(1):41–55.
- Tanaka M, Marunouchi T. Immunohistochemical analysis of developmental stage of external granular layer neurons which undergo apoptosis in postnatal rat cerebellum. Neurosci Lett. 1998 Feb 13;242(2):85–88.
- D’Mello SR, Galli C, Ciotti T, et al. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci U S A. 1993 Dec 1;90(23):10989–10993.
- Canonico PL, Copani A, D’Agata V, et al. Activation of pituitary adenylate cyclase-activating polypeptide receptors prevents apoptotic cell death in cultured cerebellar granule cells. AnnNYAcadSci. 1996;805:470–472.
- Cavallaro S, Copani A, D’Agata V, et al. Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol Pharmacol. 1996 Jul;50(1):60–66.
- Zhong J, Deng J, Huang S, et al. High K+ and IGF-1 protect cerebellar granule neurons via distinct signaling pathways. J Neurosci Res. 2004 Mar 15;75(6):794–806.
- Maino B, D’Agata V, Severini C, et al. Igf1 and Pacap rescue cerebellar granule neurons from apoptosis via a common transcriptional program. Cell Death Discov. 2015;1:15029. 09/07/online
- Lanni C, Racchi M, Memo M, et al. p53 at the crossroads between cancer and neurodegeneration. Free Radic Biol Med. 2012 May 1;52(9):1727–1733.
- Lin D, Shanks D, Prakash O, et al. Protein kinase C gamma mutations in the C1B domain cause caspase-3-linked apoptosis in lens epithelial cells through gap junctions. Exp Eye Res. 2007 Jul;85(1):113–122.
- Ono Y, Tanaka H, Tsuruma K, et al. A sigma-1 receptor antagonist (NE-100) prevents tunicamycin-induced cell death via GRP78 induction in hippocampal cells. Biochem Biophys Res Commun. 2013 May 17;434(4):904–909.
- Lee J, Bruce-Keller AJ, Kruman Y, et al. 2-Deoxy-D-glucose protects hippocampal neurons against excitotoxic and oxidative injury: evidence for the involvement of stress proteins. J Neurosci Res. 1999 Jul 1;57(1):48–61.
- Croft DR, Crighton D, Samuel MS, et al. p53-mediated transcriptional regulation and activation of the actin cytoskeleton regulatory RhoC to LIMK2 signaling pathway promotes cell survival. Cell Res. 2011 Apr;21(4):666–682.
- Leverson JD, Zhang H, Chen J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015;6:e1590.
- Abdullah Thani NA, Sallis B, Nuttall R, et al. Induction of apoptosis and reduction of MMP gene expression in the U373 cell line by polyphenolics in Aronia melanocarpa and by curcumin. Oncol Rep. 2012 Oct;28(4):1435–1442.
- Lee SR, Tsuji K, Lee SR, et al. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neuroscience. 2004 Jan 21;24(3):671–678.
- Walker EJ, Rosenberg GA. TIMP-3 and MMP-3 contribute to delayed inflammation and hippocampal neuronal death following global ischemia. Exp Neurol. 2009 Mar;216(1):122–131.
- Sinno M, Biagioni S, Ajmone-Cat MA, et al. The matrix metalloproteinase inhibitor marimastat promotes neural progenitor cell differentiation into neurons by gelatinase-independent TIMP-2-dependent mechanisms. Stem Cells Dev. 2013 Feb 1;22(3):345–358.
- Ding HL, Xu MT, Guo Y, et al. Effect of losartan on the mRNA expressions of MT3-MMP and TIMP-2 in diabetic kidneys. Rev Diabet Stud. 2005 Winter;2(4):216–220.
- Ghosh MC, Tong WH, Zhang D, et al. Tempol-mediated activation of latent iron regulatory protein activity prevents symptoms of neurodegenerative disease in IRP2 knockout mice. Proc Natl Acad Sci U S A. 2008 Aug 19;105(33):12028–12033.
- Youdim MB. What have we learnt from CDNA microarray gene expression studies about the role of iron in MPTP induced neurodegeneration and Parkinson’s disease?. J Neural Transm. 2003(65):73–88.
- Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat. 2013 Dec;45(4):251–262.
- Renko K, Schache S, Hoefig CS, et al. An improved nonradioactive screening method identifies genistein and xanthohumol as potent inhibitors of iodothyronine deiodinases. Thyroid. 2015 Aug;25(8):962–968.
- Naghibalhossaini F, Ebadi P. Evidence for CEA release from human colon cancer cells by an endogenous GPI-PLD enzyme. Cancer Lett. 2006 Mar 28;234(2):158–167.
- Frye SV, Haffner CD, Maloney PR, et al. 6-Azasteroids: structure-activity relationships for inhibition of type 1 and 2 human 5 alpha-reductase and human adrenal 3 beta-hydroxy-delta 5-steroid dehydrogenase/3-keto-delta 5-steroid isomerase. J Med Chem. 1994 Jul 22;37(15):2352–2360.
- Selamnia M, Mayeur C, Robert V, et al. Alpha-difluoromethylornithine (DFMO) as a potent arginase activity inhibitor in human colon carcinoma cells. Biochem Pharmacol. 1998 Apr 15;55(8):1241–1245.
- Ljungberg MC, Ali YO, Zhu J, et al. CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum Mol Genet. 2012 Jan 15;21(2):251–267.
- Vethakanraj HS, Babu TA, Sudarsanan GB, et al. Targeting ceramide metabolic pathway induces apoptosis in human breast cancer cell lines. Biochem Biophys Res Commun. 2015 Aug 28;464(3):833–839.
- Proksch D, Klein JJ, Arenz C. Potent inhibition of Acid ceramidase by novel B-13 analogues. J Lipids. 2011;2011:971618.
- Elojeimy S, Liu X, McKillop JC, et al. Role of acid ceramidase in resistance to FasL: therapeutic approaches based on acid ceramidase inhibitors and FasL gene therapy. Mol Therapy: Journal Am Soc Gene Ther. 2007 Jul;15(7):1259–1263.
- Milde S, Coleman MP. Identification of palmitoyltransferase and thioesterase enzymes that control the subcellular localization of axon survival factor nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2). J Biol Chem. 2014 Nov 21;289(47):32858–32870.
- Coleman MP. The challenges of axon survival: introduction to the special issue on axonal degeneration. Exp Neurol. 2013 Aug;246:1–5.
- Sun G, Thai SF, Lambert GR, et al. Fluconazole-induced hepatic cytochrome P450 gene expression and enzymatic activities in rats and mice. Toxicol Lett. 2006 Jun 20;164(1):44–53.
- Sun G, Grindstaff RD, Thai SF, et al. Induction of cytochrome P450 enzymes in rat liver by two conazoles, myclobutanil and triadimefon. Xenobiotica. 2007 Feb;37(2):180–193.
- Barton HA, Tang J, Sey YM, et al. Metabolism of myclobutanil and triadimefon by human and rat cytochrome P450 enzymes and liver microsomes. Xenobiotica. 2006 Sep;36(9):793–806.
- Fujimura H, Murakami N, Miwa S, et al. The suitability of rat hepatoma cell line H4IIE for evaluating the potentials of compounds to induce CYP3A23 expression. Exp Toxicol Pathol. 2012 Jul;64(5):527–533.
- Zhou Y, Wang S, Ding T, et al. Evaluation of the effect of apatinib (YN968D1) on cytochrome P450 enzymes with cocktail probe drugs in rats by UPLC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2014 Oct;22;973C:68-75.
- Kapoor N, Pant AB, Dhawan A, et al. Differences in the expression and inducibility of cytochrome P450 2B isoenzymes in cultured rat brain neuronal and glial cells. Mol Cell Biochem. 2007 Nov;305(1–2):199–207.
- Nishimura J, Saegusa Y, Dewa Y, et al. Antioxidant enzymatically modified isoquercitrin or melatonin supplementation reduces oxidative stress-mediated hepatocellular tumor promotion of oxfendazole in rats. Arch Toxicol. 2010 Feb;84(2):143–153.
- Han YL, Li D, Ren B, et al. Evaluation of impact of herba erigerontis injection, a Chinese herbal prescription, on rat hepatic cytochrome P450 enzymes by cocktail probe drugs. J Ethnopharmacol. 2012 Jan 6;139(1):104–109.
- Niwa T, Okada K, Hiroi T, et al. Effect of psychotropic drugs on the 21-hydroxylation of neurosteroids, progesterone and allopregnanolone, catalyzed by rat CYP2D4 and human CYP2D6 in the brain. Biol Pharm Bull. 2008 Mar;31(3):348–351.
- Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001 Feb;11(1):39–46.
- Fukata M, Fukata Y, Adesnik H, et al. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004 Dec 16;44(6):987–996.
- Maynard TM, Meechan DW, Dudevoir ML, et al. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 2008 Nov;39(3):439–451.
- Jennings BC, Nadolski MJ, Ling Y, et al. 2-Bromopalmitate and 2-(2-hydroxy-5-nitro-benzylidene)-benzo[b]thiophen-3-one inhibit DHHC-mediated palmitoylation in vitro. J Lipid Res. 2009 Feb;50(2):233–242.
- Bynoe MS, Viret C, Yan A, et al. Adenosine receptor signaling: a key to opening the blood-brain door. Fluids Barriers CNS. 2015;12:20.
- Morin N, Di Paolo T. Interaction of adenosine receptors with other receptors from therapeutic perspective in Parkinson’s disease. Int Rev Neurobiol. 2014;119:151–167.
- Yan R, Hu ZY, Zhou WX, et al. [Roles of adenosine receptors in Alzheimer’s disease]. Yao Xue Xue Bao. 2014 Jun;49(6):751–756.
- Schulze-Topphoff U, Prat A, Prozorovski T, et al. Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat Med. 2009 Jul;15(7):788–793.
- Homan KT, Wu E, Cannavo A, et al. Identification and characterization of amlexanox as a G protein-coupled receptor kinase 5 inhibitor. Molecules. 2014;19(10):16937–16949.
- Urbano M, Guerrero M, Zhao J, et al. Design, synthesis and SAR analysis of novel potent and selective small molecule antagonists of NPBWR1 (GPR7). Bioorg Med Chem Lett. 2012 Dec 1;22(23):7135–7141.
- Lee HJ, Ban JY, Seong YH. Blockade of 5-HT(3) receptor with MDL7222 and Y25130 reduces hydrogen peroxide-induced neurotoxicity in cultured rat cortical cells. Life Sci. 2005 Dec 5;78(3):294–300.
- Zhang Y, Shen GL, Shangguan LJ, et al. Involvement of NFkappaB signaling in mediating the effects of GRK5 on neural stem cells. Brain Res. 2015 May 22;1608:31–39.
- Zaratin PF, Quattrini A, Previtali SC, et al. Schwann cell overexpression of the GPR7 receptor in inflammatory and painful neuropathies. Mol Cell Neurosci. 2005 Jan;28(1):55–63.
- Walstab J, Hammer C, Bonisch H, et al. Naturally occurring variants in the HTR3B gene significantly alter properties of human heteromeric 5-hydroxytryptamine-3A/B receptors. Pharmacogenet Genomics. 2008 Sep;18(9):793–802.
- Peake N, Su N, Ramachandran M, et al. Natriuretic peptide receptors regulate cytoprotective effects in a human ex vivo 3D/bioreactor model. Arthritis Res Ther. 2013;15(4):R76.
- Muller YL, Reitstetter R, Yool AJ. Regulation of Ca2+-dependent K+ channel expression in rat cerebellum during postnatal development. J Neuroscience. 1998 Jan 1;18(1):16–25.
- Yan R, Page JC, Shi R. Acrolein-mediated conduction loss is partially restored by potassium channel blockers. J Neurophysiol. 2015 [Nov];18(jn):00467.
- Zhou X, Wei J, Song M, et al. Novel role of KCNQ2/3 channels in regulating neuronal cell viability. Cell Death Differ. 2011 Mar;18(3):493–505.
- Qi J, Zhang F, Mi Y, et al. Design, synthesis and biological activity of pyrazolo[1,5-a]pyrimidin-7(4H)-ones as novel Kv7/KCNQ potassium channel activators. Eur J Med Chem. 2011 Mar;46(3):934–943.
- Yao H, Peng F, Dhillon N, et al. Involvement of TRPC channels in CCL2-mediated neuroprotection against tat toxicity. J Neuroscience. 2009 Feb 11;29(6):1657–1669.
- Liu S, Zhang L, Wu Q, et al. Chemokine CCL2 induces apoptosis in cortex following traumatic brain injury. J Mol Neurosci. 2013 Nov;51(3):1021–1029.
- Severini C, Passeri PP, Ciotti M, et al. Bindarit, inhibitor of CCL2 synthesis, protects neurons against amyloid-beta-induced toxicity. J Alzheimers Dis. 2014;38(2):281–293.
- Hinojosa AE, Garcia-Bueno B, Leza JC, et al. Regulation of CCL2/MCP-1 production in astrocytes by desipramine and atomoxetine: involvement of alpha2 adrenergic receptors. Brain Res Bull. 2011 Nov 25;86(5–6):326–333.
- Lin HY, Yeh WL, Huang BR, et al. Desipramine protects neuronal cell death and induces heme oxygenase-1 expression in Mes23.5 dopaminergic neurons. Plos One. 2012;7(11):e50138.
- Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004 Oct 1;64(19):7099–7109.
- Suthiphongchai T, Phimsen S, Sakulkhu U, et al. PD98059-inhibited invasion of Dunning rat prostate cancer cells involves suppression of motility but not MMP-2 or uPA secretion. Oncol Rep. 2006 Jun;15(6):1605–1610.
- Adams M, Kobayashi T, Lawson JD, et al. Fragment-based drug discovery of potent and selective MKK3/6 inhibitors. Bioorg Med Chem Lett. 2015 Nov;17.
- Tamura RE, De Vasconcellos JF, Sarkar D, et al. GADD45 proteins: central players in tumorigenesis. Curr Mol Med. 2012 Jun;12(5):634–651.
- Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999 May 13;18(19):2967–2987.
- Oppenheim RW, Prevette D, Tytell M, et al. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev Biol. 1990 Mar;138(1):104–113.
- Kristiansen M, Ham J. Programmed cell death during neuronal development: the sympathetic neuron model. Cell Death Differ. 2014 Jul;21(7):1025–1035.