
ABSTRACT
Introduction: CNS drug development has been hampered by inadequate consideration of CNS pharmacokinetic (PK), pharmacodynamics (PD) and disease complexity (reductionist approach). Improvement is required via integrative model-based approaches.
Areas covered: The authors summarize factors that have played a role in the high attrition rate of CNS compounds. Recent advances in CNS research and drug discovery are presented, especially with regard to assessment of relevant neuro-PK parameters. Suggestions for further improvements are also discussed.
Expert opinion: Understanding time- and condition dependent interrelationships between neuro-PK and neuro-PD processes is key to predictions in different conditions. As a first screen, it is suggested to use in silico/in vitro derived molecular properties of candidate compounds and predict concentration-time profiles of compounds in multiple compartments of the human CNS, using time-course based physiology-based (PB) PK models. Then, for selected compounds, one can include in vitro drug-target binding kinetics to predict target occupancy (TO)-time profiles in humans. This will improve neuro-PD prediction. Furthermore, a pharmaco-omics approach is suggested, providing multilevel and paralleled data on systems processes from individuals in a systems-wide manner. Thus, clinical trials will be better informed, using fewer animals, while also, needing fewer individuals and samples per individual for proof of concept in humans.
1. Introduction
The aim of drug discovery and development is to find compounds able to modify body processes by interaction with a target that is related to a disease. It typically consists of a number of phases. These include:
Target identification: aiming to find a molecular target that is involved in the disease (progression);
Target validation: investigating if the target is ‘druggable’;
Lead discovery: identification of small molecules that modulate the target, and transformation of the obtained information into a high content lead series;
Drug candidate selection: identification of a few promising compounds, using in silico and preclinical in vitro and in vivo testing to further understand the compound properties with regard to its pharmacokinetics (PK), pharmacodynamics (PD, the therapeutic effects and side effects), mechanisms of action, and the best dosage and route of administration;
Clinical studies (phase 1–4): investigation of the PK and safety in human, followed by the analysis of the therapeutic effects in healthy subjects and in patients and, if not ‘first in class,’ comparison with similar drugs to obtain a drug that is ‘best in class.’
CNS drug discovery has been hampered by inadequate understanding or consideration of a number of factors. These include the complexity of brain anatomy and function; the neuro-PK with regard to blood–brain barrier (BBB) transport and intra-brain distribution as well as the measures to study these processes; adequate biomarkers of CNS drug effects (neuro-PD), and the complex nature of CNS diseases [Citation1]. Also, the reductionist view and thereby lack of understanding of the interaction and interdependencies of all these factors have contributed to the high attrition rates of CNS drugs. These factors are discussed below, while recent improvements in techniques and approaches to understand neuro-PK and PD will be discussed in the next section.
1.1. The CNS is an organ with complex anatomy, structure, and function
While in many tissues in the body a drug is relatively free to exchange between blood and the extracellular space in the tissues, this is not the case for CNS tissue. The CNS is separated from the blood by BBB and other barriers that have highly specialized properties [Citation2], which has a huge impact on the relationship between plasma PK and neuro-PK. Moreover, the CNS is far from being a homogenous tissue. It has many different tissue structures and fluid cavities (ventricles), while target expression may variate substantially among the different locations [Citation3] and, also, the same target may have distinct functions in different locations. Furthermore, there is fluid flow. The main fluid is the cerebrospinal fluid (CSF) that is produced by the choroid plexus cells of the blood–CSF barrier (BSCFB). Then there is the brain extracellular fluid (brain ECF) produced by the BBB, which flows into the direction of the ventricle (called brain ECF bulk flow). Brain ECF merges with the CSF, and the CSF is eliminated via the arachnoid villi into the blood stream. CSF production and elimination rates determine the CSF turnover rate [Citation4]. Moreover, very recently, it became apparent that also the CNS contains lymphatic vessels, thus also lymph flow needs to be taken into account [Citation5].
All these processes have an impact on (local) neuro-PK. Furthermore, CNS functionality is complicated by the networks of interacting neurotransmission pathways. Neurotransmitter receptors have been the typical target for many (classical) CNS drugs, while ‘single target’ pharmacological intervention and/or the impact of a disease on one target will often influence another one. Last but not least, neuro-PD is typically not directly quantifiable and needs to be assessed in an indirect way, using biomarkers that may be more or less adequate in reflecting the real CNS effect. This makes neuro-PD difficult to measure and to predict.
1.2. The inaccessibility of the human brain for sampling
Since the driving force of CNS drug action is the concentration-time profile of the drug at the target site, it is important for pharmaceutical companies to have effective and cost-efficient tools to measure and predict human brain target site exposure before proceeding to more expensive clinical trials. However, the possibility of direct measurement of human brain concentrations is highly limited. Since information on CNS drug distribution in human brain typically cannot be obtained directly, it must be inferred from in silico, in vitro, and in vivo preclinical experimental approaches.
1.3. Inadequate knowledge on neuro-PK
Neuro-PK results from transport across the BBB and BSCFB, intra-brain distribution, and target interaction. The free drug hypothesis has been around for many years stating that the unbound drug concentration is available for membrane transport and target interaction. Wang and Welty [Citation6] were the first to show the importance of using unbound drug concentrations in proper calculations of BBB transport and intra-brain distribution, and to introduce the term ‘volume of distribution in brain’ as the extent of drug distribution between brain unbound concentrations to total amount of the compound in the brain tissue. However, in CNS drug discovery, the measurement of unbound drug concentrations is typically not applied, not just because of the lack of quick and easy assay methods, but also because of a lack of understanding of the drivers in neuro-PK processes. Instead, total brain drug concentrations are used to determine BBB permeability (rate) of a drug as well as its total brain over total blood concentration ratio (Kp, brain, logBB; extent). The values of these parameters were considered as good indicators of brain penetration and thereby target exposure (the higher the better), often without proper understanding of the difference between rate and extent of BBB transport. This has appeared to be misleading in understanding rate versus extent of BBB transport and intra-brain distribution. With that, also the link to target exposure and interaction could not be made appropriately.
1.4. Incomplete information on and understanding of the complex CNS diseases
CNS diseases are typically multifactorial and complex. They often have genetic, physiological, neurochemical, degenerative, and inflammatory components, which display variation within the patient group [Citation7,Citation8]. For example, for Alzheimer’s disease, Parkinson’s disease, and other neurodegenerative disorders, multiple- and in many cases divergent-disease etiologies are involved. Moreover, CNS diseases often get diagnosed in a late stage of the disease, where the chance of curing the disease is virtually zero, where at best disease progression can be halted and often symptom suppression is the only possibility left. Altogether, this makes that defining a CNS disease, and its state and stage, is very difficult.
1.5. Problems in identification and validation of targets for (potential) CNS drugs
Target validation aims to determine whether a biological compound (e.g. receptor, enzyme, DNA/RNA) is directly involved in the disease of interest, and whether it can be modified by a drug or other interventions. For example, if a human disease is the result of a single gene mutation, and that mutation can be corrected therapeutically, the target is validated. An example of this is Huntington’s disease, for which the causative gene huntingtin was identified many years ago [Citation9]. However, to date, no successful therapeutic approach has been demonstrated on the basis of that knowledge, indicating that a valid target does not per se guarantee the development of a drug [Citation10] Moreover, in many cases the etiology of CNS diseases is multifactorial, making target identification and validation difficult if not impossible when focusing on single targets. Indeed, satisfactory treatment of the disease via a single target might be an illusion.
1.6. Reductionist view and fragmented information
Then last, but certainly not least, there has been the tendency to oversimplify the relevant factors underlying disease and drug effects. These factors were evaluated in isolation, neglecting the complex interactions and interrelationships. Parameter values for a very high number of compounds have been assessed, often in high-throughput mode in different systems, without taking into account the context dependency of these values. Such ‘fragmented’ and ‘stand-alone’ data did not lead to increased understanding. Apparently, for the CNS, with its complex PK, PD and disease processes, there has not been an easy way out. To paraphrase Einstein: ‘Everything should be made as simple as possible, but no simpler.’
2. Current advances in approaches and techniques
For a proper CNS effect, the drug should have the ability to access the CNS ‘at the right place, at the right time, and at the right concentration.’ Research advances in chemistry, drug metabolism, pharmacology, and toxicology have provided much insight into understanding the pitfalls of the drug discovery and development, and a number of advances in CNS drug discovery are currently embraced.
2.1. Identification and validation of targets for (potential) CNS drugs
The ‘omics’ and ‘profiling’ research area clearly indicates that neurodegenerative diseases and their associated neuropsychiatric comorbidities are multifactorial in origin. Genomics has provided many molecular targets that provide both opportunities and challenges in the discovery and development of novel medicines for the treatment of human CNS disorders [Citation11–Citation13]. In addition, more is learned about the biology of CNS diseases, and syndromes may be subdivided into more specific categories that are better understood in terms of pathophysiology and patho-etiology, also with regard to early signs of the CNS disease. This is likely to lead to development of more targeted treatments, being focused on the underlying causes of the disease as well as its prevention (life-style, diet, etc.).
In general, the research focus has shifted from single target toward target networks (systems pharmacology), as networks include the compensatory factors that impact the neuro-PD [Citation7,Citation10,Citation11]. These networks are constructed on basis of integrated genomics, proteomics, and metabolomics data, narrowing down the large number of potential genomics-informed targets into a fewer number of relevant disease-causing targets [Citation14,Citation15]. Metabolomics has the advantage that the measured metabolites are accessible from body fluids such as plasma and urine, which provides opportunities for biomarker discovery for clinical application. CNS diseases are often associated with energy substrates, amino acids, neurotransmitters, neurochemicals and structural lipids. Typically, multiple pathways are involved in a single disease, and single pathways are associated with multiple diseases, underlining the need for a systems approach in CNS drug development [Citation15].
Although not being an example in the CNS area, Cho et al. [Citation16] suggested a useful approach that is different from conventional screening systems: the phenotypic screening in combination with multi-omics-based target identification and validation (MOTIV). The phenotypic screening provides information on the effect of small molecule compounds in the cell or at organism level, since small molecules not only affect a single target but the entire cellular mechanism within a cell or organism. The MOTIV approach provides a systematic approach to discover the target protein of a bioactive small molecule. Then, network analysis and validations of these candidates result in identifying the biologically relevant target protein and cellular mechanism. The combination of phenotypic screening and MOTIV may provide an effective approach to discover new bioactive small molecule and their target protein and mechanism of action.
Target identification and validation may be further informed by pharmacometabolomics, which is a new approach to identify changes in the metabolome upon drug action, revealing potentially perturbed biological pathways. A lipidomics analysis revealed a broad range of lipids changing upon treatment with the antipsychotics risperidone and olanzapine, but not aripiprazole [Citation17]. These changes are associated with weight gain as a side effect. Furthermore, an analysis of multiple hormones and biogenic amines after treatment with the dopamine D2 receptor antagonist remoxipride revealed diverse response profiles. With integrated PK–PD analyses, multiple in vivo potencies for these responses were determined, associated with several biological pathways. These included the dopamine metabolism, the adrenaline metabolism, the serine–glycine–threonine metabolism, and the serotonin metabolism [Citation18,Citation19].
2.2. Understanding neuro-PK
Drug distribution into and within the brain is governed by many processes, including plasma PK, plasma protein binding, passive and active transport across the BBB and BCSFB [Citation2], and once within the brain, brain ECF bulk flow, diffusion, passive and active extracellular–intracellular exchange, and CSF turnover. In the preclinical setting, there are several in vitro, ex vivo, and in vivo techniques that provide information on brain target site exposure. Such information can provide either direct or indirect information on bound and/or unbound concentrations, with or without temporal resolution, with or without spatial resolution, and with or without a clear distinction between rate and extent of the processes involved. It is of great importance to understand the mechanisms involved in uptake into and efflux from the brain, on one hand being governed by BBB functionality in terms of passive (paracellular and transcellular) diffusion, facilitated diffusion, active influx, active efflux, and absorptive or receptor-mediated endocytosis, and, on the other hand, by drug physicochemical properties and structure. As only the unbound drug is able to pass through the membranes, it is the unbound concentration difference between brain and plasma that drives BBB transport. Likewise, it is the unbound concentration difference between brain ECF and the cellular cytosol that drives extra-intracellular transport. Also, for drug–target interaction the unbound concentration is the driving factor [Citation20].
2.2.1. Neuro-PK measurement
Recently, a number of important improvements have been made in the understanding of drug distribution into and within the brain. This has been brought about especially by new approaches which have been developed, that allow a relatively rapid and easy assessment of unbound concentrations in brain tissue (brain homogenate dialysis equilibration and brain slice method) [Citation21,Citation22]. Thus, it is possible to determine the brain over plasma ratio of unbound concentrations (extent of brain equilibration; Kpuu,brain), and extra-intracellular unbound concentration ratios (Kpuu, cell). Furthermore, based on the pH partitioning theory, subcellular distribution into lysosomes can be simulated. With the new Combinatory Mapping Approach [Citation23], the relationship between plasma PK and neuro-PK can be obtained in a more high-throughput mode, which makes it very useful for drug discovery (). These approaches, however, still the focus on (assumed) steady-state conditions.
Figure 1. The combinatory mapping approach. (Redrawn from [Citation23] with permission of Springer).
![Figure 1. The combinatory mapping approach. (Redrawn from [Citation23] with permission of Springer).](/cms/asset/76a631c9-1d13-46ab-b77d-d44a73d5065a/iedc_a_1380623_f0001_oc.jpg)
2.2.2. In silico prediction of BBB transport and brain distribution
In silico approaches to predict BBB permeability in earlier days were typically based on Kp, brain (logBB) relationships and found lipophilicity to be the main driver [Citation24]. As discussed earlier, BBB permeability, which is the rate of BBB transport, was often confused with the extent of brain distribution, which confounded the search for good predictors of brain exposure. Other in silico approaches have focused on quantitative structure–property relationships (QSPR), using the physicochemical properties of a drug as predictors of the rate and extent of BBB transport and brain distribution [Citation25–Citation29].
With unbound concentrations taken into account, it was reported that the main drug property governing the extent of brain distribution is the number of hydrogen bond acceptors. Thus, higher lipophilicity is a helpful property for faster rate of transport across the BBB, while a low number of hydrogen bond acceptors helps in a larger extent of brain distribution. Nevertheless, higher lipophilicity may increase nonspecific binding to brain tissue, while also highly lipophilic compounds have a tendency to be substrates for efflux transport, such as by P-glycoprotein, which leads to a net decrease of brain distribution. Thus, in contrast to earlier guidelines, drugs aimed for the CNS should not be too lipophilic, and have a low number of hydrogen bond acceptors for a more extensive brain distribution of the unbound drug.
It should be noted that in many academic investigations and CNS drug discovery programs, the focus is still on (assumed) steady-state conditions. The argument is that with aiming for a repeated/chronic dosing, a steady-state condition will be reached, so that only steady-state conditions are relevant to investigate. However, fluctuations in drug levels may have important consequences for target occupancy (TO), etc. This will be discussed in the target binding kinetics section below.
2.2.3. In silico prediction of neuro-PK
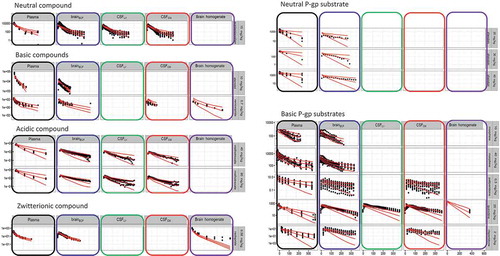
A very useful approach to predict neuro-PK is to make use of physiology-based pharmacokinetic (PBPK) modeling. As in PBPK modeling, the explicit separation is made between drug and systems properties, it is a great translational tool. To date, many more or less complex (semi-)PBPK models have been published for CNS drug distribution [Citation30–Citation34]. Gaohua et al. [Citation33] published a human PBPK model with a few physiological CNS compartments. Other (semi-) PBPK models have been developed using extensive information obtained from preclinical species, with explicit inclusion of unbound drug concentrations [Citation31,Citation32]. To that end, the microdialysis technique [Citation35] provided valuable data that were obtained in parallel in the multiple CNS compartments, to further pave the way toward the generic semi-PBPK CNS drug distribution model. This model has been applied to nine compounds with highly different physicochemical properties and demonstrated excellent description of the rat data for all these compounds, and an adequate prediction of human CNS data that were available for acetaminophen and morphine [Citation34]. Here, it should be noted that one microdialysis experiment in a single freely moving animal can provide lots of serial data points, obtained under the same experimental condition of the animal, and thereby revealing the interrelationships of neuro-PK processes [Citation36,Citation37], which in addition is important also for refinement, reduction, and replacement of animal experiments [Citation38]. Advanced multilevel experiments and mathematical modeling are needed to reveal interrelationships between processes that occur in parallel in the body, and to reduce the use of animals. The ultimate goal is to develop a generic PBPK model by which prediction of CNS drug distribution can be made without the need for animal data. Such a model has recently been developed [Citation39] () and shows good agreement with the observed in vivo data (), and therefore seems suitable to be used as an ‘in silico’ screening method for adequate CNS drug distribution of new chemical entities.
Figure 2. The structure of the generic CNS PBPK model Structure: Black: Plasma PK model, Red: CNS PBPK model. Parameters: Black: estimated plasma PK parameters; Blue: system-specific parameters; Green: drug-specific parameters; Purple: combination of system-specific and drug-specific specific parameters. (Redrawn from [Citation39] with permission of Wiley & Sons).
![Figure 2. The structure of the generic CNS PBPK model Structure: Black: Plasma PK model, Red: CNS PBPK model. Parameters: Black: estimated plasma PK parameters; Blue: system-specific parameters; Green: drug-specific parameters; Purple: combination of system-specific and drug-specific specific parameters. (Redrawn from [Citation39] with permission of Wiley & Sons).](/cms/asset/54b728fa-5f77-400d-94e7-5b81cb78168e/iedc_a_1380623_f0002_oc.jpg)
Figure 3. BPPK model prediction (red lines for median and 95% percentiles) and observed data (black dots) obtained in the five color-filled compartments.
2.3. Target binding kinetics
Target binding kinetics and the resulting induction of signal transduction processes will ultimately lead to a biological effect. Therefore, in current CNS discovery programs, TO is often measured, for which equilibrium is often assumed between free and target-bound drug concentrations [Citation40]. However, this equilibrium is not always reached quickly or maintained continuously after it has been reached [Citation41–Citation43]. The rate of drug-target equilibration (the binding kinetics) is determined by the drug-target association rate constant (kon) and the drug-target dissociation rate constant (koff). Drug-target binding kinetics not only influences the time course of TO, but also affects the local drug concentration around the target for drugs with a high affinity or a high local target concentration [Citation42,Citation44]. This is important as target concentrations (e.g. receptor density) may differ substantially between different CNS regions (). If target concentrations or drug target affinities are high enough, the influence of target binding on drug concentrations can also be observed from plasma concentrations, as illustrated in . Recently, there have been efforts from both academic and industrial research communities to advance the in silico and in vitro assessment of binding kinetics, and the translation of this knowledge into in vivo settings. One of these efforts is the Kinetics for Drug Discovery Consortium, a 5-year public–private partnership which aimed to define the role of binding kinetics in drug discovery [Citation46].
Figure 4. Cartoon of transversal cross section of human brain with the dopaminergic receptor areas indicated. Receptor density is region specific and receptor subtype specific.
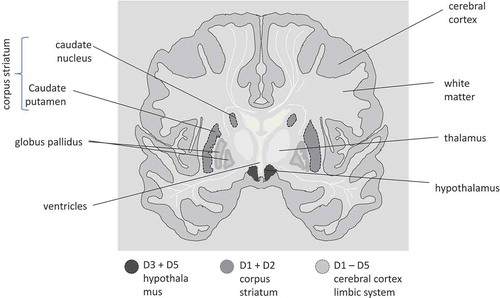
Figure 5. Compared plasma pharmacokinetics of high affinity compounds (top row) and their low affinity analogues (bottom row). The circles represent observed plasma concentrations in rats, the lines represent model predictions. (Reprinted from [Citation45] with permission of ASPET).
![Figure 5. Compared plasma pharmacokinetics of high affinity compounds (top row) and their low affinity analogues (bottom row). The circles represent observed plasma concentrations in rats, the lines represent model predictions. (Reprinted from [Citation45] with permission of ASPET).](/cms/asset/07e7759f-ff48-48c8-8bca-24a066bccd4b/iedc_a_1380623_f0005_b.gif)
For understanding the influence of target binding kinetics on in vivo drug action, it is therefore important to have adequate information on the unbound concentration of the compound in plasma and in brain regions where the target resides. Both ex vivo and in vivo TO of drugs can be evaluated in experimental animals using both invasive and noninvasive methods [Citation47]
The ex vivo approach uses tissue slice autoradiography, or biochemical measures of TO, wherein the fraction of total target binding sites occupied by the drug is inferred from the residual binding capacity of a radiotracer added to the postmortem tissue ex vivo. The in vivo approach, on the other hand, measures the displacement of the radiotracer from the target tissue when both the drug and the radiotracer are administered to the living animal. In the past decade, the use of non-radiolabeled tracers is increasingly adopted in in vivo studies, in which the tissue concentration of the administered non-radiolabeled tracer is quantified by liquid chromatography-mass spectrometry (LC-MS) instead of radioactivity counting [Citation48]. The main advantages of this method are that the parent tracer can be differentiated from its metabolites, and it allows simultaneous quantification of the tracer displacement (TO) and the neuro-PK [Citation49]. The measurement of TO and target site PK can provide valuable insight into the driving factors for the time course of drug action. However, additional factors such as target turnover/desensitization, endogenous ligand binding, and signal transduction can also influence the time course of drug action and need to be taken into account [Citation47].
In humans and animals, TO can be evaluated noninvasively using positron emission tomography (PET) or single-photon emission computed tomography (SPECT). These imaging techniques are powerful because they provide high temporal and spatial resolutions. PET radioligands are now available for more than 40 CNS targets [Citation50]. PET/SPECT is most useful when no easily measurable clinical PD markers are available, or when substantial time is required before an indication of efficacy can be observed clinically. In this respect, PET/SPECT can be used to demonstrate that the drug candidates reach their target and bind to their target, as a proof of concept enabling effective treatment. Moreover, PET/SPECT imaging can also be used to identify whether a delay between plasma unbound drug concentrations and CNS effect is caused by slow target equilibration or by slow transduction processes. Dose (or plasma concentration)-TO curves can be obtained to guide the dosing in clinical trials [Citation50–Citation52].
Drug-target interactions are increasingly incorporated in mechanism-based PK–PD modeling to support drug development. These models allow quantitative assessment of the relation between neuro-PK and TO. As already discussed, PBPK modeling is particularly useful in translating the obtained information from animals to humans by using the available knowledge on species-dependent physiological characteristics such as blood flow and organ volumes [Citation53–Citation55]. Based on the exposure–response relationship in the animal species and using TO as a translational biomarker, the efficacy of a drug candidate in humans could be predicted in early stage development. Systematic implementation of translational modeling can therefore bolster confidence of successfully evaluating proof of mechanism in humans and ultimately improve the success rate in Phase II, which is currently only around 10% [Citation56]. Thus, to predict drug effects based on TO, integration of all determining factors is required, including drug transport between blood and CNS, CNS fluid flows, target site PK profile, drug-target binding kinetics, endogenous ligand-target binding kinetic, and target turnover.
2.4. CNS drug effects
Many times neuro-PK, and/or TO, is studied without measuring associated (biomarkers of the) drug effects. Actually, it would be of great added value if neuro-PK and associated PD would be obtained in a single experimental subject or at least single experimental context, as rate and extent of body processes are context dependent (according to the Mastermind Research Approach [Citation36]). To link (neuro-)PK to the physiologic response, PK–PD modeling is often applied. Depending on the PK, as well as the type of response (inhibition or stimulation), different PK–PD models can be used. With that, it is possible to learn more about factors that play a role in target activation and signal transduction to the ultimate effect or biomarker of the effect [Citation57–Citation61], interspecies differences in concentration–effect relationships [Citation62], tolerance and sensitization [Citation63], and intra- and interindividual variability. Moreover, for studies with only PK and PD observations but not TO data, a delay between the PK and PD is often be explained by a biophase (or effect compartment) distribution model. However, incorporating drug-target binding into the model might explain the same delay and could be more mechanistic [Citation64]. On the other hand, when solving the shortcomings in knowledge on target site distribution of drugs, the principles of the operational model of agonism (receptor theory) will provide the basis for future developments in drug development by classifying drugs and predicting their mechanism of action in pharmacology [Citation65–Citation68]. PK–PD approaches have typically focused on anticipated drug effects. As drug probably has additional, unknown effects, quantitative insights in CNS drug effects should better be obtained for the whole biological system, including the unknown mechanisms of action [Citation69,Citation70].
2.5. Biomarkers of drug effects and disease
To predict the drug effects in human on the basis of translational animal and mathematical models, specific expressions are needed to quantitatively characterize the processes on the causal path between drug administration and effect. These include target site distribution, target binding and activation, transduction, PD interactions, and homeostatic feedback mechanisms. Ultimately also the effects on and of disease processes and disease progression have to be considered. These can be characterized by biomarkers according to the biomarker classification system [Citation71] ().
Table 1. Biomarker classification [Citation71] and approaches to assess quantitative information.
Obtaining combined information on a number of biomarker types (preferable in parallel, within a single biological system) will allow the development of better models, with increased accuracy and predictability. The better we will be able to develop predictive models in preclinical studies, the more the number of often extremely costly clinical studies can be reduced. The focus should therefore be on the design of quantitative in vivo animal studies such that translational pharmacology approaches can be applied [Citation25,Citation36,Citation37,Citation61,Citation89], which will be discussed below. In refined animal models, the biomarkers of the effect that can be measured in both animals and human will be particularly useful.
Multimodal neuroimaging that combines PET/SPECT with techniques like magnetic resonance imaging, computed tomography, and electroencephalography can simultaneously provide anatomic, functional, biochemical, and metabolic information alongside TO assessment. These techniques are increasingly employed in assessing the pathophysiological changes in different disease states, such as target expression, release of endogenous ligands that bind the drug target (e.g. neurotransmitters), neuroinflammation/glial activation, cerebral blood flow, and BBB integrity, all of which could alter the drug PK, TO, and biomarker profiles [Citation50–Citation52].
To obtain insight in the multiple processes in the biological system, increasing efforts are made to show the utility of a multi-biomarker approach, both in disease conditions and upon drug administration [Citation17,Citation90]. With that, the system-wide pathophysiological and pharmacological influences are reflected by a multi-biomarker response. To that end, it is important to connect such data to information on drug distribution to target sites, target binding kinetics, signal transduction, and homeostatic feedback mechanisms. Such insight is obtained by integration of data obtained from multilevel studies, that is, measurement of different biomarker types in a time-dependent manner [Citation19,Citation71,Citation91,Citation92,Citation93]. In all cases, the experimental approach should be such that a distinction can be made between drug-specific and system-specific properties, to allow for scaling between drugs and/or scaling between species [Citation61].
3. Conclusion
For a proper CNS effect, the drug should have the ability to access the CNS ‘at the right place, at the right time, and at the right concentration.’ To develop treatments with improved safety and efficacy, one of the scientific challenges is to understand the biological mechanisms underlying the PK–PD relationships of CNS drugs. The currently applied simplistic approach to produce data on multiple processes in isolation is not informative as processes are context dependent and interdependent. The knowledge on heterogeneity (variability) in rate and extent of processes between drug dosing and CNS effects is needed to predict the impact of drug-induced and disease-induced perturbations in the biological system. To that end, the integrative ‘Mastermind Research Approach’ [Citation36] is needed to decipher the interrelationships of processes that govern plasma PK, BBB transport, intra-brain distribution, as well as CNS effects in different conditions.
4. Expert opinion
One of the major weaknesses in CNS drug discovery and development has been the tendency to oversimplify relevant factors underlying CNS disease and drug effects. Typically, parameter values of PK, PD, and disease processes were obtained in isolation, in different systems, thereby ignoring their interrelationships and systems dependencies. Simple decision trees based on such isolated parameter values were used to drive decisions on taking compounds further in development or not. However, this has not led to knowledge and understanding the system, in terms of interdependencies and condition dependent rate and extent of multiple processes between drug dosing, neuro-PK, and neuro-PD, as needed for predictions.
In the relationship between plasma PK and neuro-PK, a first improvement was by the notion for the need for unbound drug concentration-time profiles in plasma and brain to inform on rate and extent of BBB transport, which can only be provided by the microdialysis technique. A second improvement was on understanding of extra-intracellular drug distribution within the brain (at presumed steady-state conditions), using the brain slice technique. Then, knowledge on (steady state) distribution of bound and unbound drug between plasma, brain ECF, brain intracellular space, and cellular compartments, was integrated in the Combinatory Mapping Approach [Citation22]. This approach has recently been embraced by a number of pharmaceutical companies.
A first left-over issue is, however, to understand time-dependency. Even in chronic dosing, fluctuations in plasma and CNS drug levels may occur and may have important consequences for CNS TO [Citation44]. Also, relationships between concentration–time profiles in brainECF and intracellular space (drug target sites) and between those target-site concentration–time profiles and the ones in CSF compartments are important, as mostly only CSF can be obtained in the clinical setting and should be relied on for having information about drug concentrations at the target site [Citation4]. A number of CNS PBPK models have been developed, however, only addressing steady-state conditions [Citation30,Citation33]. The use of extensive series data sets on unbound drug concentrations, obtained in parallel in brainECF and different CSF locations in the rat, a generic semi-PBPK brain rat model has been developed [Citation34] followed by the full-PBPK brain rat model [Citation39]. This model can scale between species, and allows prediction of concentration–time profiles of compounds in multiple compartments of the human brain on the basis of solely physicochemical properties of compounds [Citation39]
This indicates the following future perspective for prediction of neuro PK and TO profiles: In a very early stage of drug development, physicochemical properties of drugs can be measured in vitro and/or predicted by in silico models. Also, plasma PK profiles can be predicted by currently available full PBPK models. This information on plasma PK and physicochemical drug properties can inform the CNS PBPK model to predict neuro-PK of individual candidate compounds, in either rat or human. Then, in vitro drug-target binding kinetics can be included in the model to predict TO-time profiles in the specific species selected. TO predictions could be validated by actual measurements. With this approach fewer animals are needed (replacement by using in silico models). Also, clinical trials will be better informed, needing fewer individuals and samples per individual for first-in human studies.
A next, important challenge is prediction of drug effects, especially in disease states. Currently, to link (neuro-)PK to the systems response, PK–PD modeling is often applied. These models can include target activation (receptor theory [Citation65–Citation68]), signal transduction [Citation57–Citation61], interspecies differences [Citation62], tolerance and sensitization [Citation63], and intra- and interindividual variability. Also here, it should be noted that parameter values of PK, PD, and disease processes should not be obtained in isolation, and in different systems, because in such manner interrelationships and systems dependencies of processes cannot be assessed. Moreover, so far, PK–PD approaches have typically focused on a single anticipated drug effects. As a drug probably has additional, unknown effects, quantitative insights in CNS drug effects should better be obtained for the whole biological system (systems-wide approach), thereby including the unknown mechanisms of action [Citation69,Citation70]. Here, also, time-course data, obtained at different biomarker levels () under multiple conditions, is key to be able to apply mathematical modeling for unraveling interrelationships and condition dependencies. Where it concerns body fluids, serial sampling, and where possible microdialysis, is very useful. Such time-course samples can be subjected to ‘omics’ approaches that will broaden our understanding of (changes in) the networks that compose the biological system, in health and disease. Especially, a pharmacometabolomics approach is suggested for potentially multi-target neuropharmacodynamics. In addition, the use of imaging techniques should be considered, as those can be applied to animals as well as human subjects, providing translational insights. Though ‘omic’ approaches are expensive, it is the only way to go if we wish to have better insights into condition-dependency of biological systems processes.
Article highlights
Factors that govern neuro-PK and CNS target exposure profiles plasma PK, BBB transport, brain extracellular-intracellular and brain-intracellular-subcellular distribution, as well as brainECF bulk flow and CSF turnover. Information on these factors should include unbound and bound drug concentrations, and integration of these factors is needed for understanding neuro-PK, as the underlying processes occur simultaneously and are interdependent.
Experimental approaches should be such that a distinction can be made between drug specific and system specific properties. With such information the recently developed CNS PBPK model can be further informed, to allow for scaling between drugs and/or scaling between species to predict CNS target exposure profiles.
CNS target exposure profiles can be used to predict CNS TO profiles, by incorporation of drug-target binding kinetics, endogenous ligand-target binding kinetic and target turnover.
Typically, multiple biological pathways are involved in a single CNS disease, and single pathways are associated with multiple diseases. This indicates the need for a systems approach for identification and validation of targets for (potential) CNS drugs
This box summarizes key points contained in the article.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- de Lange ECM, Hammarlund-Udenaes M. Translational aspects of blood-brain barrier transport and central nervous system effects of drugs: from discovery to patients. Clin Pharmacol Ther. 2015;97(4):380–394.
- Abbott NJ, Patabendige AAK, Dolman DEM, et al. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25.
- Sawada Y, Kawai R, McManaway M, et al. Kinetic analysis of transport and opioid receptor binding of [3H](-)-cyclofoxy in rat brain in vivo: implications for human studies. J Cereb Blood Flow Metab. 1991;11(2):183–203.
- De Lange ECM. Utility of CSF in translational neuroscience. J Pharmacokinet Pharmacodyn. 2013;40(3):315–326.
- Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–341.
- Wang Y, Welty DF. The simultaneous estimation of the influx and efflux blood-brain barrier permeability of gabapentin using a microdialysis-pharmacokinetic approach. Pharm Res. 1996;13(3):398–403.
- Van der Schyf CJ. Psychotropic drug development strategies that target neuropsychiatric etiologies in Alzheimer’s and Parkinson’s diseases. Drug Dev Res. 2016;77(8):458–468.
- De Lange ECM. Disease influence on BBB transport in neurodegenerative disorders. In: Hammarlund-Udenaes M, De Lange ECM, Thorne RG, editors. Drug delivery to the brain: physiological concepts, methodologies and approaches. New York, NY:AAPSpress & Springer; 2014. p. 591–634.
- MacDonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983.
- Hutson PH, Clark JA, Cross AJ. CNS target identification and validation: avoiding the valley of death or naive optimism? Annu Rev Pharmacol Toxicol. 2017;57(1):171–187.
- Krause JE, Chenard BL. Opportunities and challenges in the discovery of new central nervous system drugs. Ann N Y Acad Sci. 2008;1144(1):243–250.
- Preskorn SH. CNS drug development: part III: future directions. J Psychiatr Pract. 2011;17(1):49–52.
- Karamanos Y, Pottiez G. Proteomics and the blood–brain barrier: how recent findings help drug development. Expert Rev Proteomics. 2016;13(3):251–258.
- Xia J, Wishart DS. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010;38(Web Server):W71–7.
- Dumas M-E, Davidovic L. Metabolic profiling and phenotyping of central nervous system diseases: metabolites bring insights into brain dysfunctions. J Neuroimmune Pharmacol. 2015;10(3):402–424.
- Cho YS, Kwon HJ. Identification and validation of bioactive small molecule target through phenotypic screening. Bioorganic Med Chem. 2012;20(6):1922–1928.
- Kaddurah-Daouk R, Kristal BS, Weinshilboum RM. Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol. 2008;48(1):653–683.
- van den Brink WJ, Wong YC, Gülave B, et al. Revealing the neuroendocrine response after remoxipride treatment using multi-biomarker discovery and quantifying it by PK/PD modeling. AAPS J. 2017;19(1):274–285.
- Van den Brink W, Elassais-Schaap J, Gonzales-Amoros B, et al. Multivariate pharmacokinetic/pharmacodynamic (PKPD) of remoxipride effects in rats using metabolomics. Eur J Pharm Sci. 2017;109:431–490.
- Watson J, Wright S, Lucas A, et al. Receptor occupancy and brain free fraction. Drug Metab Dispos. 2009;37(4):753–760.
- Fridén M, Gupta A, Antonsson M, et al. In vitro methods for estimating unbound drug concentrations in. Drug Metab Dispos. 2007;35(9):1711–1719.
- Loryan I, Fridén M, Hammarlund-Udenaes M. The brain slice method for studying drug distribution in the CNS. Fluids Barriers CNS. 2013;10(1):6.
- Loryan I, Sinha V, Mackie C, et al. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm Res. 2014;23:1–17.
- Bendels S, Kansy M, Wagner B, et al. In silico prediction of brain and CSF permeation of small molecules using PLS regression models. Eur J Med Chem. 2008;43(8):1581–1592.
- Fridén M, Winiwarter S, Jerndal G, et al. Structure-brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J Med Chem. 2009;52(20):6233–6243.
- Varadharajan S, Winiwarter S, Carlsson L, et al. Exploring in silico prediction of the unbound brain-to-plasma drug concentration ratio: model validation, renewal, and interpretation. J Pharm Sci. 2015;104(3):1197–1206.
- Loryan I, Sinha V, Mackie C, et al. Molecular properties determining unbound intracellular and extracellular brain exposure of CNS drug candidates. Mol Pharm. 2015;12(2):520–532.
- Grumetto L, Russo G, Barbato F. Immobilized artificial membrane HPLC derived parameters vs PAMPA-BBB data in estimating in situ measured blood-brain barrier permeation of drugs. Mol Pharm. 2016;13(8):2808–2816.
- Chen H, Winiwarter S, Fridén M, et al. In silico prediction of unbound brain-to-plasma concentration ratio using machine learning algorithms. J Mol Graph Model. 2011;29(8):985–995.
- Ball K. A physiologically based modeling strategy during preclinical CNS drug development. Mol Pharm. 2014;11(3):836–848.
- Westerhout J, Ploeger B, Smeets J, et al. Physiologically based pharmacokinetic modeling to investigate regional brain distribution kinetics in rats. AAPS J. 2012;14(3):543–553.
- Westerhout J, Van Den Berg DJ, Hartman R, et al. Prediction of methotrexate CNS distribution in different species - influence of disease conditions. Eur J Pharm Sci. 2014;57(1):11–24.
- Gaohua L, Neuhoff S, Johnson TN, et al. Development of a permeability-limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab Pharmacokinet. 2016;31(3):224–233.
- Yamamoto Y, Välitalo PA, Van Den Berg D-J, et al. A generic multi-compartmental CNS distribution model structure for 9 drugs allows prediction of human brain target site concentrations. Pharm Res. 2017;34(2):333–351.
- Yamamoto Y, Danhof M, De Lange ECM. Microdialysis: the key to physiologically based model prediction of human CNS target site concentrations. AAPS J. 2017;19:891–909.
- de Lange EC. The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS. 2013;10(1):12.
- de Lange ECM. PBPK modeling approach for predictions of human CNS drug brain distribution. Li Di and Edward H. Kerns (eds). Blood-Brain Barrier in Drug Discovery. Wiley. 2015;296–323.
- Kilkenny C, Browne W, Cuthill IC, et al. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160(7):1577–1579.
- Yamamoto Y, Välitalo P, Huntjens D, et al. Predicting drug concentration-time profiles in multiple relevant CNS compartments using a comprehensive physiologically-based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2017 Sep 11. doi: 10.1002/psp4.12250. [Epub ahead of print].
- Liefaard CL, Tagawa Y, Danhof M, et al. Population pharmacokinetic/pharmacodynamic analysis of different subunit selective GABAergic ligands in an animal model of epilepsy. 2002;2:2002.
- Mukherjee J, Yang Z-Y, Lew R, et al. Evaluation of d -amphetamine effects on the binding of dopamine D-2 receptor radioligand, 18F-fallypride in nonhuman primates using positron emission tomography. Synapse. 1997;27(1):1–13.
- Perry DC, Mullis KB, Oie S, et al. Opiate antagonist receptor binding in vivo: evidence for a new receptor binding model. Brain Res. 1980;199(1):49–61.
- Yassen A, Olofsen E, Dahan A, et al. Pharmacokinetic-pharmacodynamic modeling of the antinociceptive effect of buprenorphine and fentanyl in rats: role of receptor equilibration kinetics. J Pharmacol Exp Ther. 2005;313(3):1136–1149.
- de Witte WEA, Danhof M, van der Graaf PH, et al. In vivo target residence time and kinetic selectivity: the association rate constant as determinant. Trends Pharmacol Sci. 2016;37(10):831–842.
- Yamazaki S, Shen Z, Jiang Y, et al. Application of target-mediated drug disposition model to small molecule heat shock protein 90 inhibitors. Drug Metab Dispos. 2013;41(6):1285–1294.
- Schuetz DA, de Witte WEA, Wong YC, et al. Kinetics for drug discovery: an industry-driven effort to target drug residence time. Drug Discov Today. 2017;22(6):896–911.
- de Witte WEA, Wong YC, Nederpelt I, et al. Mechanistic models enable the rational use of in vitro drug-target binding kinetics for better drug effects in patients. Expert Opin Drug Discov. 2016;11(1):45–63.
- Wong YC, Ilkova T, van Wijk R, et al. Development of a population pharmacokinetic model to predict brain distribution and dopamine D2 receptor occupancy of raclopride in non-anesthetized rat. 2017. Manuscript under revision
- Joshi EM, Need A, Schaus J, et al. Efficiency gains in tracer identification for nuclear imaging: can in vivo LC-MS/MS evaluation of small molecules screen for successful PET tracers? ACS Chem Neurosci. 2014;5(12):1154–1163.
- Finnema SJ, Scheinin M, Shahid M, et al. Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology (Berl). 2015;232(21–22):4129–4157.
- Liu S, Cai W, Liu S, et al. Multimodal neuroimaging computing: a review of the applications in neuropsychiatric disorders. Brain Inform. 2015;2(3):167–180.
- Slifstein M, Abi-Dargham A. Recent developments in molecular brain imaging of neuropsychiatric disorders. Semin Nucl Med. 2017;47(1):54–63.
- Espié P, Tytgat D, Sargentini-Maier M-L, et al. Physiologically based pharmacokinetics (PBPK). Drug Metab Rev. 2009;41(3):391–407.
- Johnson M, Kozielska M, Pilla Reddy V, et al. Erratum to: translational modeling in schizophrenia: predicting human dopamine D2 receptor occupancy. Pharm Res. 2016;33(5):1305–1306.
- Brochot A, Zamacona M, Stockis A. Physiologically based pharmacokinetic/pharmacodynamic animal-to-man prediction of therapeutic dose in a model of epilepsy. Basic Clin Pharmacol Toxicol. 2010;106(3):256–262.
- Lavé T, Caruso A, Parrott N, et al. Translational PK/PD modeling to increase probability of success in drug discovery and early development. Drug Discov Today Technol. 2016;21–22:27–34.
- Groenendaal D, Freijer J, de Mik D, et al. Influence of biophase distribution and P-glycoprotein interaction on pharmacokinetic-pharmacodynamic modelling of the effects of morphine on the EEG. Br J Pharmacol. 2007;151(5):713–720.
- Groenendaal D, Freijer J, Rosier A, et al. Pharmacokinetic/pharmacodynamic modelling of the EEG effects of opioids: the role of complex biophase distribution kinetics. Eur J Pharm Sci. 2008;34(2–3):149–163.
- De Lange ECM. Pharmacometrics in psychiatric diseases. In: Schmidt S, Derendorf H, editors. Applied pharmacometrics. New York, NY:AAPSpress & Springer; 2014. p. 407–449.
- van Schaick EA, Tukker HE, Roelen HCPF, et al. Selectivity of action of 8-alkylamino analogues of N6-cyclopentyladenosine in vivo: haemodynamic versus anti-lipolytic responses in rats. Br J Pharmacol. 1998;124(3):607–618.
- Danhof M, De Lange ECM, Della Pasqua OE, et al. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29(4):186–191.
- Stevens J, Ploeger BA, Hammarlund-Udenaes M, et al. Mechanism-based PK-PD model for the prolactin biological system response following an acute dopamine inhibition challenge: quantitative extrapolation to humans. J Pharmacokinet Pharmacodyn. 2012;39(5):463–477.
- Cleton A, Odman J, Van der Graaf PH, et al. Mechanism-based modeling of functional adaptation upon chronic treatment with midazolam. Pharm Res. 2000;17(3):321–327.
- Shimada S, Nakajima Y, Yamamoto K, et al. Comparative pharmacodynamics of eight calcium channel blocking agents in Japanese essential hypertensive patients. Biol Pharm Bull. 1996;19(3):430–437.
- van der Graaf PH, Stam WB. Analysis of receptor inactivation experiments with the operational model of agonism yields correlated estimates of agonist affinity and efficacy. J Pharmacol Toxicol Methods. 1999;41(2–3):117–125.
- Garrido M, Gubbens-Stibbe J, Tukker E, et al. Pharmacokinetic-pharmacodynamic analysis of the EEG effect of alfentanil in rats following beta-funaltrexamine-induced mu-opioid receptor “knockdown” in vivo. Pharm Res. 2000;17(6):653–659.
- Jonker DM, Vermeij DAC, Edelbroek PM, et al. Pharmacodynamic analysis of the interaction between tiagabine and midazolam with an allosteric model that incorporates signal transduction. Epilepsia. 2003;44(3):329–338.
- Ploeger BA, van der Graaf PH, Danhof M. Incorporating receptor theory in mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling. Drug Metab Pharmacokinet. 2009;24(1):3–15.
- Van der Greef J, Martin S, Juhasz P, et al. The art and practice of systems biology in medicine: mapping patterns of relationships. J Proteome Res. 2007;6(4):1540–1559.
- Quinones MP, Kaddurah-Daouk R. Metabolomics tools for identifying biomarkers for neuropsychiatric diseases. Neurobiol Dis. 2009;35(2):165–176.
- Danhof M, Alvan G, Dahl SG, et al. Mechanism-based pharmacokinetic–pharmacodynamic modeling—a new classification of biomarkers. Pharm Res. 2005;22(9):1432–1437.
- Uchida Y, Tachikawa M, Obuchi W, et al. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood–brain barrier in ddY, FVB, an. Fluids Barriers CNS. 2013;10(1):21.
- Uchida Y, Zhang Z, Tachikawa M, et al. Quantitative targeted absolute proteomics of rat blood-cerebrospinal fluid barrier transporters: comparison with a human specimen. J Neurochem. 2015;134(6):1104–1115.
- Huntley MA, Bien-Ly N, Daneman R, et al. Dissecting gene expression at the blood-brain barrier. Front Neurosci. 2014;8(OCT):1–14.
- Bergen AA, Kaing S, Ten Brink JB, et al. Gene expression and functional annotation of human choroid plexus epithelium failure in Alzheimer’s disease. BMC Genomics. 2015;16(1):956.
- Kubo Y, Ohtsuki S, Uchida Y, et al. Quantitative determination of luminal and abluminal membrane distributions of transporters in porcine brain capillaries by plasma membrane fractionation and quantitative targeted proteomics. J Pharm Sci. 2015;104(9):3060–3068.
- Bettler B, Fakler B. Ionotropic AMPA-type glutamate and metabotropic GABAB receptors: determining cellular physiology by proteomes. Curr Opin Neurobiol. 2017;45:16–23.
- Borroto-Escuela DO, Carlsson J, Ambrogini P, et al. Understanding the role of GPCR heteroreceptor complexes in modulating the brain networks in health and disease. Front Cell Neurosci. 2017;11(February):1–20.
- Becker G, Bolbos R, Costes N, et al. Selective serotonin 5-HT1A receptor biased agonists elicitdistinct brain activation patterns: a pharmacoMRI study. Sci Rep. 2016;6(1):26633.
- Bagli M, Süverkrüp R, Quadflieg R, et al. Pharmacokinetic-pharmacodynamic modeling of tolerance to the prolactin- secreting effect of chlorprothixene after different modes of drug administration. J Pharmacol Exp Ther. 1999;291(2):547–554.
- Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523–1631.
- Borsook D, Becerra L, Fava M. Use of functional imaging across clinical phases in CNS drug development. Transl Psychiatry. 2013;3(7):e282.
- Clement P, Mutsaerts H-J, Václavů L, et al. Variability of physiological brain perfusion in healthy subjects – a systematic review of modifiers. Considerations for multi-center ASL studies. J Cereb Blood Flow Metab. 2017. Epub ahead of print
- Brawek B, Garaschuk O. Monitoring in vivo function of cortical microglia. Cell Calcium. 2017;64:109–117.
- Chan PLS, Nutt JG, Holford NHG. Modeling the short- and long-duration responses to exogenous levodopa and to endogenous levodopa production in Parkinson’s disease. J Pharmacokinet Pharmacodyn. 2004;31(3):243–268.
- Erdő F, Denes L, de Lange E. Age-associated physiological and pathological changes at the blood–brain barrier: a review. J Cereb Blood Flow Metab. 2017;37(1):4–24.
- Müller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18(5):281–298.
- Holford N, Nutt JG. Disease progression, drug action and Parkinson’s disease: why time cannot be ignored. Eur J Clin Pharmacol. 2008;64(2):207–216.
- Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm. 1982;10(2):201–227.
- van der Greef J, McBurney RN. Innovation: rescuing drug discovery: in vivo systems pathology and systems pharmacology. Nat Rev Drug Discov. 2005;4(12):961–967.
- de Lange ECM, Ravenstijn PGM, Groenendaal D, et al. Toward the prediction of CNS drug-effect profiles in physiological and pathological conditions using microdialysis and mechanism-based pharmacokinetic-pharmacodynamic modeling. AAPS J. 2005;7(3):E532–43.
- Morgan P, Van der Graaf PH, Arrowsmith J, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today. 2012;17(9–10):419–424.
- Groeneveld GJ, Hay JL, Van Gerven JM. Measuring blood–brain barrier penetration using the NeuroCart, a CNS test battery. Drug Discov Today Technol. 2016;20:27–34.