
1. Introduction
In the last 20 years, Fragment-Based Drug Discovery (FBDD) has established itself as a key approach for finding high-quality lead candidates [Citation1]. Two drugs on the market today originate from fragment-based library screening campaigns, and 40 molecules discovered by FBDD have been advanced to clinical trials. Recent successes in FBDD have given the promise of addressing previously intractable biological targets, such as intracellular protein–protein interactions.
The effectiveness of FBDD depends on the quality of each of the process components () [Citation2]. Desirable fragment library covers a wide range of molecular shapes and pharmacophoric functionalities (e.g., Hydrogen bond donor). Several sensitive biophysical techniques, able to detect low-affinity binding, have been developed as screening methods. The mostly used are ligand-observed nuclear magnetic resonance (NMR), surface plasmon resonance (SPR) and X-ray crystallography (RX). Each method has specific advantages and constraints. Ligand-observed NMR is a label-free technique which requires a large amount of soluble protein. By contrast, SPR requires a lower amount of protein, but protein has to be immobilized on the surface. Only RX accurately shows how the fragment binds, provided that the fragment finds its way into the protein crystal upon soaking. More generally the choice of a screening method is driven by considerations on feasibility, sensitivity, and throughput [Citation2,Citation3]. Structural information on binding advantageously guide fragment elaboration by a merging, linking, or growing approach. In the absence of a crystallographic structure, structure–activity relationships, NMR data, and computational docking solutions have proved to be valuable to advance fragments [Citation4].
Figure 1. The FBDD process. Fragment to lead optimization is exemplified on vemurafenib, the first marketed drug from FBDD. The fragment 7-azaindole is a non-selective kinase inhibitor, here shown in complex with PKA/PKB (PDB code: 2UVX). Vemurafenib is a potent inhibitor of oncogenic B-RAF kinase activity (PDB code: 3OG7).
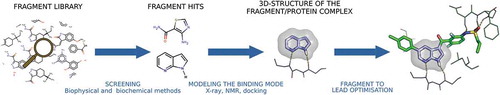
The past decade has seen major advances in library design and screening technologies. Practitioner however still faces the challenge of transforming the fragment hits into a drug lead. Which fragment to elaborate? How to build a relevant synthetic strategy? Recent surveys of 3D-structures in the Protein Data Bank (PDB) as well as retrospective analysis of FBDD campaigns brought material for the discussion and suggested practical guidelines. In parallel, calculation methods have shown that they too can contribute to decision-making in FBDD.
2. A fragment binds to a hotspot, various fragments cover all the interactions of the binding site
Fragments are commonly viewed as part of drug-like molecules. In some cases, fragments of known drugs have been used as starting points to find new inhibitors for different biological targets. More generally, a fragment is a small organic molecule which is very soluble and chemically stable. The most relevant fragment hits show in addition high ligand efficiency (binding free energy per non-hydrogen atom >0.3 kcal/mol) and a well-defined binding mode to their target site. In 2012, an important study of the structural properties and the thermodynamics of fragment binding suggested that fragment tends to bind to protein hot spots, i.e., small regions of the protein site which predominantly contribute to the free energy of ligand binding [Citation5]. Three main results supported this conclusion: fragment binding is generally enthalpy-driven; fragment forms on average two high-quality hydrogen bonds; and two to three hydrogen bonds are mostly conserved when considering six or more fragments bound to the same protein. More recently, the analysis of PDB complexes was extended to all types of non-covalent interactions (hydrophobic contacts, hydrogen, ionic and aromatic bonds, and interaction with a metal cation) and demonstrated that, independently of the protein, interactions patterns defined by the fragments are similar to those defined by the drug-like ligands [Citation6]. Fragments are thus not only able to pick the residues crucial for binding, they also mostly cover the protein pocket polar interactions as well as hydrophobic contacts. In addition, in several proteins, a few directional interactions were fragment-specific, in agreement that fragments more efficiently explore sub-pockets than moieties of larger molecules.
Do all proteins contain hot spots? Are all the hotspots of a protein equivalent? These questions were addressed by examining the ability of protein to bind multiple chemical probes, firstly using biophysical methods, in particular, RX or NMR. Currently, several calculation methods correctly predict, rapidly and at low cost, which regions of the protein are targeted by the molecular probes [Citation7,Citation8]. As an example of actively developed and widely validated method, FTPMap offers valuable help in determining whether a protein is suitable for FBDD and for the prioritization of fragment hits.
3. About fragment binding mode conservation
A common assumption in FBDD is that the binding mode does not vary during the fragment-to-lead optimization. However, deconstruction studies as well as prospective FBDD campaigns have provided examples of ‘flipping’ fragments (see examples referenced in [Citation6,Citation8,Citation9]). How frequent are exceptions? A comprehensive analysis of binding modes in publicly available 3D-structures indicated that conservation is only three times more frequent than variation [Citation6]. Polar interactions, especially hydrogen bonds are mostly preserved and therefore are rightly considered a determining factor for anchoring fragments to a specific binding site. When does a fragment adapt its binding mode? Is variation the consequence of the fragment intrinsic characteristics or, by contrast, is it triggered by external constraints? Comparing the interactions made by PDB fragments and their related drug-like superstructures bound to the same protein site assessed the importance of molecular size, the binding mode being generally conserved if the fragment molecular weight is larger than 150 Da [Citation6]. Changes in interactions are generally accompanied by changes in molecular conformations, particularly in the protein site. Positioning of fragments with several tautomeric states is especially susceptible to the molecular environment.
Computational methods for hotspot detection have provided useful insights into binding mode conservation. Recent work on hotspot strength and topology showed that a fixed binding mode is more likely in a primary hotspot, provided that the hotspot dimension matches fragment size and a close secondary hotspot can be reached upon elaboration [Citation8]. In addition, primary hotspots tend to be more rigid than other regions in the binding site. The next breakthrough could come from the analysis of the properties and disposition of the sub-pockets in protein site [Citation10].
4. Towards a unified definition of a fragment
Fragment molecular weight (MW) is now typically <250 Da. Expert guidelines for construction of a fragment library include more physicochemical properties (e.g., less than 16 non-hydrogen atoms, high aqueous solubility, no reactive group) and recommend that each fragment has many real or potential analogues that attest to the synthetic accessibility necessary for optimization [Citation3]. The screening method throughput determines the library maximal size (500–3000). It is important that the library covers as much as possible the chemical space, expressing sufficient structural and pharmacophore diversity for the specific targeting of a large panel of proteins. What level of molecular complexity is suitable for this purpose? The issue is a matter of debate. The 3D character of molecules is widely acknowledged as a good indicator of success in medicinal chemistry programs. Fragment libraries which are largely composed of flat molecules have deservedly been the subject of much criticism. However, three-dimensionality inversely correlates to hit rate, independently of the target type, and fragment planarity is not a presage of a shapeless lead [Citation11]. In FBDD, fragment promiscuity can, therefore, be seen as an advantage, with the possibility of developing a selective lead from a frequent hitter.
Do promiscuous fragments fit similar sub-pockets or are they able to adapt their binding mode? Nature has engendered different proteins with similar binding properties (e.g. ATP-binding). The literature provides many examples of site similarity between unrelated proteins and also reports cases of ligands with versatile binding mode [Citation12]. A next step was recently taken to this issue by analyzing PDB structures of 489 multi-target fragments, including 10% with more than 10 targets (these are mainly natural products, e.g., adenine, salicylic acid, or phosphoenolpyruvate). In this data set, fragment binding mode generally differs in the compared complexes, even when the fragment conformation is conserved and the compared proteins are close homologs [Citation13]. The versatility of binding mode could not be explained by fragment properties alone, yet could be related to an insufficient proportion of groups capable of directional interaction actually engaged in protein binding.
Are fragments more than chemical super-probes? A recent investigation of crystallization additives in the PDB gives arguments in favor of the expected yes [Citation6]. Very simple chemical probes, such as ethanediol, tend to cluster in protein hotpots, unveiling key residues for anchoring of more complex ligands. But, unlike fragments, they often occupy several spots and do not preserve interactions with the protein when incorporated into a larger molecule. Three-dimensionality and MW are suitable descriptors for predicting the ability of a small molecule to retain its binding mode when incorporated into a larger molecule ().
Figure 2. The relationship between binding mode conservation and fragment 3D character. Three-dimensionality is a measure of the average deviation of atom coordinates to a best-fitted plane (detailed method described in reference [Citation11]). Binding mode conservation of a small molecule and its larger superstructure bound to the same protein site is a ratio of interactions counts, with equal contribution of apolar contacts and polar interactions (detailed method described in reference [Citation6]). Comparisons between a fragment and a drug-like ligand are shown in blue, comparisons between a crystallization additive, alone in the site, and a drug-like ligand are shown in red. The data set was selected in the Protein Data Bank as described in [Citation6]. The binding mode conservation is given by the 2D-structure of the small ligand in a selection of pairs (1: acetohydroxamic acid, 2: 1,2-ethanediol, 3: D-alanine, 4: glycerol, 5: hypoxanthine, 6: 2-oxoglutaric acid, 7: bromoacetic acid).
![Figure 2. The relationship between binding mode conservation and fragment 3D character. Three-dimensionality is a measure of the average deviation of atom coordinates to a best-fitted plane (detailed method described in reference [Citation11]). Binding mode conservation of a small molecule and its larger superstructure bound to the same protein site is a ratio of interactions counts, with equal contribution of apolar contacts and polar interactions (detailed method described in reference [Citation6]). Comparisons between a fragment and a drug-like ligand are shown in blue, comparisons between a crystallization additive, alone in the site, and a drug-like ligand are shown in red. The data set was selected in the Protein Data Bank as described in [Citation6]. The binding mode conservation is given by the 2D-structure of the small ligand in a selection of pairs (1: acetohydroxamic acid, 2: 1,2-ethanediol, 3: D-alanine, 4: glycerol, 5: hypoxanthine, 6: 2-oxoglutaric acid, 7: bromoacetic acid).](/cms/asset/a550b79d-135d-409c-9f9e-5b28272c0e44/iedc_a_1583643_f0002_oc.jpg)
5. Conclusion
Thanks to major technological advances, FBDD is becoming a mature method. Example of successful applications as well as large-scale analysis of fragment binding mode has allowed to revisit several general assumptions on fragment selection and elaboration. The better understanding of ligand/protein recognition benefits the development of predictive methods, which help to make the design process more efficient.
6. Expert opinion
Has FBDD become a new standard? Over a short period of time, FBDD delivered valuable starting points for medicinal chemistry programs while holding remarkable promise for innovative therapeutics. Fragment screening has suggested original chemical entities, has detected unexplored secondary sites, and has provided allosteric modulators and hits for difficult targets [Citation14,Citation15]. Just like the classical design of drug from high-throughput screening hits, FBDD is not the perfect way to reach the magic bullet. Specific shortcomings have nevertheless prompted continuous technical and methodological developments. Knowledge of the fragment/protein binding mode is a key element of the success of FBDD but can be difficult to obtain. To that respect, predictive methods have great potential. The calculation of hotspots and the comparison of binding modes are already able to give relevant information for the selection and the development of fragment hits. Computing may also rescue FBDD projects when physical screening is not possible. Docking and site comparison methods may both suit virtual screening. A few examples of fragments identified by docking are already present in the literature [Citation16]. There is still work to do to improve scoring in fragment docking, and similarly, a robust metric is needed to match hotspots in pocket comparison. The future of FBDD will also rely on the development of sophisticated chemistry for the synthesis of novel, diverse and easy-to-grow fragments [Citation17]. Here again, computing can help, merging synthetic rules and inspiration from pocketome. The advantageous interplay between computing and automated experimental methods was nicely illustrated in the exemplary design of potent bromodomain inhibitors using virtual screening of a focused-chemical-library, robotic diversity-oriented de novo synthesis, and automated in vitro evaluation [Citation18].
Declaration of interest
E Kellenberger has been funded via the Lilly Research Award Program (LRAP) for her study on PDB fragments back in 2016-17. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Erlanson DA, Fesik SW, Hubbard RE, et al. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016;15:605–619.
- Lamoree B, Hubbard RE. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017;61:453–464.
- Keserű GM, Erlanson DA, Ferenczy GG, et al. Design principles for fragment libraries: maximizing the value of learnings from pharma Fragment-Based Drug Discovery (FBDD) programs for use in academia. J Med Chem. 2016;59:8189–8206.
- Erlanson DA, Davis BJ, Jahnke W. Fragment-based drug discovery: advancing fragments in the absence of crystal structures. Cell Chem Biol. 2019;26:9–15.
- Ferenczy GG, Keserű GM. Thermodynamics of fragment binding. J Chem Inf Model. 2012;52:1039–1045.
- Drwal MN, Bret G, Perez C, et al. Structural insights on fragment binding mode conservation. J Med Chem. 2018;61:5963–5973.
- Radoux CJ, Olsson TSG, Pitt WR, et al. Identifying interactions that determine fragment binding at protein hotspots. J Med Chem. 2016;59:4314–4325.
- Hall DR, Kozakov D, Whitty A, et al. Lessons from hot spot analysis for fragment-based drug discovery. Trends Pharmacol Sci. 2015;36:724–736.
- Malhotra S, Karanicolas J. Correction to when does chemical elaboration induce a ligand to change its binding mode? J Med Chem. 2017;60:5940.
- Bartolowits M, Davisson VJ. Considerations of protein subpockets in fragment-based drug design. Chem Biol Drug Des. 2016;87:5–20.
- Hall RJ, Mortenson PN, Murray CW. Efficient exploration of chemical space by fragment-based screening. Prog Biophys Mol Biol. 2014;116:82–91.
- Ehrt C, Brinkjost T, Koch O. Impact of binding site comparisons on medicinal chemistry and rational molecular design. J Med Chem. 2016;59:4121–4151.
- Drwal MN, Bret G, Kellenberger E. Multi-target fragments display versatile binding modes. Mol Inform. 2017;36. doi: 10.1002/minf.201700042.
- Ludlow RF, Verdonk ML, Saini HK, et al. Detection of secondary binding sites in proteins using fragment screening. Proc Nat Acad Sci. 2015;112:15910–15915.
- Roca C, Requena C, Sebastián-Pérez V, et al. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J Enzyme Inhib Med Chem. 2018;33:1034–1047.
- Chen D, Ranganathan A, IJzerman AP, et al. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A 2A adenosine receptor. J Chem Inf Model. 2013;53:2701–2714.
- Kidd SL, Osberger TJ, Mateu N, et al. Recent applications of diversity-oriented synthesis toward novel, 3-dimensional fragment collections. Front Chem. 2018;6:460.
- Hoffer L, Voitovich YV, Raux B, et al. Integrated strategy for lead optimization based on fragment growing: the diversity-oriented-target-focused-synthesis approach. J Med Chem. 2018;61:5719–5732.