
ABSTRACT
Introduction: The treatment of Mycobacterium abscessus lung disease faces significant challenges due to intrinsic antibiotic resistance. New drugs are needed to cure this incurable disease. The key anti-tubercular rifamycin, rifampicin, suffers from low potency against M. abscessus and is not used clinically. Recently, another member of the rifamycin class, rifabutin, was shown to be active against the opportunistic pathogen.
Areas covered: In this review, the authors discuss the rifamycins as a reemerging drug class for treating M. abscessus infections. The authors focus on the differential potency of rifampicin and rifabutin against M. abscessus in the context of intrinsic antibiotic resistance and bacterial uptake and metabolism. Reports of rifamycin-based drug synergies and rifamycin potentiation by host-directed therapy are evaluated.
Expert opinion: While repurposing rifabutin for M. abscessus lung disease may provide some immediate relief, the repositioning (chemical optimization) of rifamycins offers long-term potential for improving clinical outcomes. Repositioning will require a multifaceted approach involving renewed screening of rifamycin libraries, medicinal chemistry to improve ‘bacterial cell pharmacokinetics’, better models of bacterial pathophysiology and infection, and harnessing of drug synergies and host-directed therapy towards the development of a better drug regimen.
1. Introduction
Non-tuberculous mycobacterial (NTM) disease encompasses a panel of infections caused by relatives of Mycobacterium tuberculosis, the causative agent of tuberculosis (TB) [Citation1]. Pulmonary NTM infections are the most common cause of active disease, and two mycobacterial species account for up to 95% of these lung infections: M. abscessus and M. avium complex (MAC) [Citation2,Citation3]. M. abscessus is the most frequently encountered rapidly growing NTM in human disease [Citation4]. The number of cases is climbing at an alarming rate with an annual increase in NTM isolate prevalence of 8.4% from 1997 through 2003, reaching 50 per 100,000 persons [Citation5–Citation7]. In the US, NTM incidence largely surpasses that of TB, and while TB-related deaths have been decreasing steadily for the past 20 years, NTM-related deaths are on the rise [Citation8]. The annual cost of pulmonary NTM disease in the US was estimated at $815M in 2010 [Citation5].
Among NTM species, the M. abscessus complex – which includes three subspecies, subsp. abscessus, subsp. bolletii and subsp. massiliense – is the ‘ugly duckling’, the most difficult to treat with no reliable cure, similar to extremely drug resistant TB. All NTM species and particularly M. abscessus are intrinsically resistant to many drug classes [Citation9–Citation12]. Among the many factors underlying M. abscessus’ natural drug resistance, the pathogen’s ability to metabolize most antibiotic classes and convert them to inactive derivatives stands as a key mechanism [Citation10,Citation13–Citation17].
Given the limited panel of poorly potent drug classes, standard of care calls for 12 months of negative sputum cultures while on therapy [Citation18], which can result in several years of treatment with a minimum of three antibiotics. Although macrolides remain the pillar of anti-NTM therapy, many clinical isolates of M. abscessus subsp. abscessus and subsp. bolletii harbor the erm41 gene, which encodes an rRNA methylase and confers inducible resistance to macrolides [Citation19,Citation20]. As a result, M. abscessus disease has literally fallen into a ‘post-antibiotic era’ with cure rates ranging from 30 to 50% [Citation21–Citation24] and 5-year cumulative death rates that can exceed 50% in patient populations infected with particularly virulent strains [Citation25,Citation26].
Since the shift in the 1990s from anti-TB regimens toward macrolide-based multidrug therapy for the treatment of NTM lung disease, progress has been slow. Fortunately, drug discovery and development activities for NTM disease are picking up [Citation27,Citation28]. Noteworthy is the recent approval of liposomal amikacin for inhalation for the treatment of MAC disease [Citation29,Citation30]. Whether this therapy is also beneficial for M. abscessus lung disease remains to be determined. However, the current pipeline remains weak and there is an urgent medical need to discover more effective drugs that deliver reliable and faster cure of M.abscessus lung disease [Citation31].
De novo discovery of new drug/target pairs is slow and increasingly costly, plagued by intrinsically high attrition rates. It often takes 10–15 years of research and 0.8–2.5 billion dollars to bring a drug from abstract concept to market-ready product [Citation32]. In the antibiotic field, causes of attrition are multifactorial and include the selection of ‘bad’ bacterial targets. These include functions that are essential in vitro but not in vivo [Citation33], not sufficiently vulnerable, not epidemiologically conserved, or not sufficiently different from the human homolog. Selection of already clinically validated targets, i.e. bacterial targets against which approved drugs are efficacious in patients, eliminates this attrition element. One such target in mycobacterial lung disease is the RNA polymerase. The anti-TB frontline drug rifampicin, targeting transcription via inhibition of RNA polymerase, is bactericidal against growing and non-growing persister bacteria [Citation34]. However, rifampicin is not included in the treatment of M. abscessus lung disease [Citation35] due to insufficient potency.
An alternative approach to de novo antibiotic development is to exploit a clinically validated target via repositioning. We define repositioning as pathogen-specific chemical optimization of clinically used antibiotic classes proven to act against pharmacologically validated targets. Since these drug classes include members that are approved by the US Food and Drug Administration (FDA), attrition rates are lower and the probability of success is higher than incurred through de novo drug discovery [Citation36]. The recent discovery that rifabutin is active against M. abscessus has raised interest in using rifamycins to treat this difficult NTM disease. In this review, we evaluate recent research on rifamycins and M. abscessus, and propose strategies to reposition rifamycins for the treatment of M. abscessus lung disease.
2. Rifampicin vs M.abscessus : a TB killer meets its match
Discovered in 1965 [Citation37,Citation38], rifampicin remains a critical antibiotic in the treatment of TB infections. The drug targets the beta-subunit of bacterial RNA polymerase (RpoB) [Citation39–Citation41] and has bactericidal activity [Citation42–Citation44]. Critically, rifampicin retains bactericidal activity against intra-macrophage [Citation45] and (drug tolerant) non-replicating bacteria [Citation46]. Furthermore, rifamycins are the only drug class known to sterilize caseum, the necrotic material at the center of lung lesions (granulomas), where difficult-to-eradicate mycobacteria reside [Citation34,Citation47]. These properties enable rifampicin’s exceptional potency against M. tuberculosis in vivo. The introduction of rifampicin to the TB drug regimen lowered the relapse rate and shortened TB chemotherapy to 6 months [Citation48].
Despite its exceptional track record as an anti-TB drug, rifampicin usage against NTM infections is less straightforward. While rifampicin is part of established treatment regimens for M. kansasii and MAC infections, it is not recommended against M. abscessus, which is resistant to rifampicin [Citation35,Citation49,Citation50]. Whereas M. tuberculosis can acquire resistance to rifamycins via mutations in the target RpoB, this has not been observed in clinical isolates of M. abscessus [Citation51]. Instead, M. abscessus demonstrates ‘intrinsic’ resistance to rifampicin.
M. abscessus resistance to rifampicin can be partially attributed to its unique cell wall. Mycobacterial cell walls have a higher lipid content, rendering them less permeable than the outer membranes of gram-negative bacteria [Citation52,Citation53]. But even among mycobacteria, cell wall permeability varies dramatically between species. The cell wall of M. chelonae, a species previously undistinguished from M. abscessus, is 10 to 20-fold less permeable than that of M. tuberculosis [Citation54], consistent with M. abscessus being more resistant to antibiotics. Because the low permeability of the mycobacterial cell wall is dependent on its lipid composition, how M. abscessus assembles and maintains this lipid barrier has also been implicated in its drug resistance. M. abscessus’ lipid transport machinery includes homologs of M. tuberculosis major facilitator superfamily transporter Rv1410 and lipoprotein LprG, which work together to transport triacylglycerides and lipoarabinomannans to the mycobacterial outer membrane. Deletion of the Rv1410-LprG operon results in cell wall defects, an increased influx of drugs and greater susceptibility to rifamycins [Citation55]. M. abscessus is also thought to regulate cell wall structure and homeostasis via lipoprotein glycosylation [Citation56]. In the absence of protein-O-mannosyltransferase Pmt (MAB_1122c), M. abscessus lipoprotein glycosylation is lost, resulting in increased cell wall permeability and greater susceptibility to antibiotics like rifampicin [Citation56]. In addition to preventing drug influx, M. abscessus encodes several putative drug efflux systems [Citation57], including a homolog of Rv1258c, which mediates efflux of rifampicin in M. tuberculosis [Citation58,Citation59]. Taken together, these findings reveal how M. abscessus is exceptionally effective at blocking the uptake and intra-bacterial accumulation of rifamycins.
In addition to M. abscessus’ ability to ‘keep rifamycins out’, resistance is also mediated by drug metabolism. M. abscessus encodes a homolog of rifampicin ADP-ribosyltransferase (arrMab, MAB_0591), which inactivates rifamycins by catalyzing ADP-ribosylation at the C23 position () [Citation57,Citation60]. Genetic deletion of arrMab in M. abscessus resulted in a 100-fold decrease of rifampicin’s minimum inhibitory concentration (MIC), restoring TB-like potency [Citation14]. Thus, ADP-ribosylation is a major component of rifamycin resistance in M. abscessus.
Figure 1. Impact of known and potential M. abscessus intrinsic antibiotic resistance mechanisms on rifamycin bacterial cell pharmacokinetics. Rifabutin (gold circles) accumulates to higher levels in M. abscessus than rifampicin (purple circles) (our unpublished data). Therefore, rifabutin and rifampicin demonstrate differential bacterial cell pharmacokinetics due to differences in metabolism, uptake or efflux. The low permeability of the M. abscessus outer membrane restricts the uptake of rifamycins which cross this barrier by passive diffusion. Because rifampicin contains a hydroquinone (highlighted red in structure), this compound is prone to autoxidation, which may occur inside or outside of the cell. Rifampicin’s hydroquinone would also be susceptible to inactivation by a Rox monooxygenase (RoxMab, potential resistance mechanism, dashed line). In contrast, rifabutin does not have a hydroquinone and would resist autoxidation or inactivation by RoxMab. The ADP-ribosyltransferase ArrMab catalyzes ADP-ribosylation at the C23 hydroxyl group of both rifamipicin and rifabutin (highlighted blue in structures). Rifamycins are removed from the cell by the putative efflux pump MAB_1409c (homolog of M. tuberculosis Rv1258c).
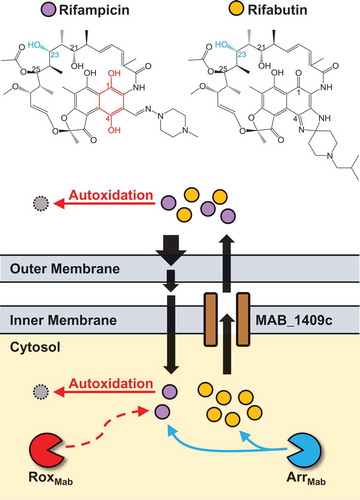
The M. abscessus genome also contains homologs of flavin adenine dinucleotide (FAD)-dependent monooxygenases [Citation57]. This broad class of enzymes includes the Rox monooxygenases, which inactivate rifampicin in other bacteria [Citation61]. So far, only one M. abscessus FAD-dependent monooxygenase has been characterized: MabTetX (MAB_1496c), a TetX monooxygenase family member, catalyzes the inactivation of tetracycline and doxycycline [Citation17]. Whether M. abscessus also has rifampicin-inactivating monooxygenase activity remains to be determined.
The complex nature of M. abscessus rifamycin resistance is representative of greater problems: M. abscessus displays resistance to many antibiotics including all of the first-line anti-TB drugs [Citation10,Citation27], and even for the few drugs that show potency in vitro there is limited correlation between in vitro drug susceptibility and clinical outcomes [Citation62]. Thus, there is no standard, reliable antibiotic regimen for M. abscessus lung disease [Citation18,Citation63]. While the treatment of NTMs is heavily reliant on the use of macrolides (clarithromycin or azithromycin) in combination with other drugs, this is problematic for M. abscessus infections due to the presence of inducible macrolide resistance [Citation19]. With only 50% cure rates observed in the clinic, better outcomes are often only possible when chemotherapy is combined with surgical resection [Citation21]. Given the lack of a sterilizing and bactericidal drug such as rifampicin, and the dire state of the NTM drug pipeline [Citation27], the challenge of treating M. abscessus lung disease remains on par with extremely drug resistant TB.
3. Rifabutin: a new hope
Following a screen of 2,720 FDA approved drugs, rifabutin was identified as a hit against M. abscessus [Citation64]. Further study revealed that rifabutin is active against type strains and clinical isolates from all three subspecies of the M. abscessus complex (subsp. abscessus, subsp. massiliense and subsp. bolletii), and that this activity is bactericidal [Citation64]. Beyond in vitro potency, rifabutin is active against M. abscessus in a macrophage infection model [Citation65]. Considering that other rifamycins (rifampicin and rifapentine) are not active against M. abscessus [Citation64], these results are surprising but suggest that rifamycins may remain a viable drug class for treating M. abscessus lung disease.
As an FDA-approved drug that is already used to treat TB [Citation66] and MAC [Citation67], rifabutin is a compelling repurposing candidate against M. abscessus. Indeed, a rifamycin would be a welcome addition to the M. abscessus treatment regimen, as this drug class is bactericidal and active against mycobacterial persister subpopulations [Citation46]. Rifabutin is orally bioavailable and has excellent pharmacokinetic properties including a long half-life and high cellular penetration, and reaches suitable concentrations in human lung tissue [Citation68,Citation69]. Rifamycins are able to reach the necrotic cores of caseous granulomas at steady state [Citation47], a feature of both TB and M. abscessus lung disease [Citation70]. Rifabutin exhibits reduced induction of cytochrome P450 3A4 (CYP3A4), minimizing adverse rifamycin drug-drug interactions [Citation71–Citation73]. The drug is also better tolerated in patients than rifampicin [Citation74]. Despite these excellent properties, it is important to note that rifabutin is less potent against M. abscessus [Citation64] than against MAC [Citation75] or M. tuberculosis [Citation76]. This suggests that simply repurposing rifabutin for M. abscessus lung disease may not be sufficient to significantly improve clinical outcomes. Animal model and clinical data are needed to assess rifabutin’s pharmacokinetics-pharmacodynamics in the treatment of M. abscessus infections.
4. Bacterial cell pharmacokinetics: an explanation for rifabutin’s potency?
Why does rifabutin but not rifampicin have activity against M. abscessus? Consistent with its greater potency, rifabutin accumulates to higher levels in M. abscessus bacteria than rifampicin (our unpublished data), suggesting that rifamycins experience differential bacterial cell pharmacokinetics in M. abscessus. Rifabutin and rifampicin may be subject to differences in metabolism, uptake or efflux. An examination of the structural differences between rifabutin and rifampicin may help to better understand how M. abscessus responds to rifamycins.
As a derivative of the naturally occurring rifamycin B, the structure of rifampicin features a napthoquinone ring bridged by an aliphatic ansa chain. The hydroquinone of this ring (i.e., hydroxyl groups at C1 and C4 positions) is susceptible to autoxidation in the presence of oxygen and divalent metal cations as a catalyst, generating rifampicin quinone () [Citation77]. Consistent with this, autoxidation of rifampicin results in a loss of anti-mycobacterial activity [Citation78]. Similar results are observed with rifapentine, a rifamycin with the same hydroquinone structure and no activity against M. abscessus [Citation64,Citation78]. Rifabutin, however, does not have a susceptible hydroquinone (). As a result, rifabutin is resistant to autoxidation and does not lose potency under oxidizing conditions [Citation78].
As oxidizing conditions in the cytosol of M. tuberculosis and M. abscessus are likely to be similar, autoxidation cannot explain the differential potency of rifampicin in these species. Rather, M. abscessus may express antibiotic-degrading enzymes that can take advantage of the oxidizable hydroquinone chemistry of rifamycins. M. abscessus encodes putative FAD-monooxygenases [Citation57], a class of enzymes that confer drug resistance by oxidizing rifamycins. Notably, the Rox monooxygenases of Streptomyces venezuelae and Nocardia farcinica conferred resistance to rifampicin and rifapentine, but not rifabutin [Citation61], as these enzymes can only oxidize rifamycins with a susceptible hydroquinone. It is not known if M. abscessus similarly utilizes enzyme-mediated oxidation to resist rifamycins, but rifabutin would be able to avoid such a resistance mechanism ().
A landmark discovery by Sander and colleagues was that M. abscessus expresses an ADP ribosyltransferase that inactivates rifamycins [Citation14]. While ADP ribosyltransferases can use both rifampicin and rifabutin as substrates [Citation60], a lower catalytic efficiency with rifabutin would lead to higher intrabacterial accumulation and greater potency. Rifamycin inactivation by phosphorylation [Citation79] and glycosylation [Citation80] has been described in other bacterial species, but has not been demonstrated in M. abscessus. In Streptomyces speibonae, the rifampicin-inactivating glycosyltransferase exhibited similar kinetic properties with rifampicin and rifabutin as substrates [Citation80]. Similarly, overexpression of rifampicin-inactivating phosphotransferases conferred robust resistance to both rifampicin and rifabutin [Citation79]. Therefore, differential phosphorylation or glycosylation are unlikely to explain the differential potency of rifampicin and rifabutin in M. abscessus.
The structure of a compound can also impact its uptake into and efflux from bacteria [Citation81]. Due to their large, bulky structure, rifamycins cannot utilize porins to enter the cell and appear to diffuse across the hydrophobic cell wall [Citation52]; it is plausible that the chemical features of certain rifamycins may favor or hinder this process (). While mycobacteria do use efflux pumps to remove rifamycins from the cytosol [Citation58,Citation59], it is not known whether certain rifamycin scaffolds are better substrates for these pumps than others ().
5. Partners in crime: harnessing antibacterial drug synergies
Treatment of genetically drug-susceptible M. abscessus infections requires combination therapy involving clarithromycin, amikacin and either cefoxitin or tigecycline [Citation18]. In light of this, it is important to consider how inclusion of a rifamycin would affect the potency of other drugs in an M. abscessus treatment regimen. Synergies between drugs in the regimen could improve treatment outcomes by reducing the development of drug resistance and shortening treatment time. Several rifabutin synergies have been reported for M. abscessus () and are discussed below.
Table 1. Summary of rifabutin in vitro synergies against M. abscessus.
Macrolides like clarithromycin are the pillar of many NTM drug treatment regimens. This heavy reliance on macrolides is problematic for treating M. abscessus infections because two of its three subspecies (subsp. abscessus and subsp. bolletii) encode the rRNA methylase gene erm41. Exposure to macrolides induces expression of whiB7, a key regulator of several drug resistance genes including erm41. Expression of erm41 results in methylation of the ribosome, blocking macrolide activity against its target. By targeting transcription, rifamycins could potentially inhibit the induction of erm41 and thereby enhance the activity of macrolides. Consistent with this, synergy between rifabutin and clarithromycin has been observed against M. abscessus [Citation82,Citation83], suggesting the potential for rifamycins to enhance macrolide-based treatment regimens. Pharmacokinetic drug-drug interactions between rifabutin and clarithromycin have been observed [Citation68]. While rifabutin levels doubled when administered with clarithromycin, there was also increased conversion of clarithromycin to 14-OH clarithromycin, an active metabolite. The impact and clinical relevance of this drug-drug interaction is not clear. Ultimately, clinical data will be needed to assess the efficacy of rifabutin-clarithromycin in treating M. abscessus lung disease.
The potency of amikacin is dampened by M. abscessus expression of Eis2, an enzyme that modifies aminoglycosides [Citation16]. It is possible that rifamycin-mediated transcription inhibition also enhances the potency of amikacin by reducing induction of Eis2. Indeed, the combination of rifabutin and amikacin was found to be synergistic against M. abscessus clinical isolates [Citation83]. Although amikacin is often prescribed alongside clarithromycin, this combination is antagonistic because macrolides induce upregulation of Eis2 [Citation84]. Given that both amikacin and clarithromycin synergize with rifabutin, rifamycins may also negate the antagonism between these key M. abscessus drugs.
β-lactams, targeting the cell wall, may augment the potency of rifamycins by aiding their diffusion across this barrier [Citation52]. Due to the broad spectrum β-lactamase activity of M. abscessus, cefoxitin and imipenem are the only β-lactams that can be used to treat M. abscessus infections [Citation85,Citation86]. Rifabutin synergizes with both cefoxitin [Citation83] and imipenem [Citation83,Citation87] against M. abscessus in vitro. In a macrophage infection model, the combination of rifabutin and imipenem was significantly more active against M. abscessus than each drug alone [Citation65].
Tigecycline, the most recently introduced drug for NTM infections [Citation88,Citation89], also synergizes with rifabutin [Citation83,Citation84]. Of note, a triple combination of rifabutin with tigecycline and clarithromycin was also synergistic against M. abscessus [Citation82], putting forth a plausible combination therapy regimen.
Finally, the oxazolidinone linezolid was also reported to synergize with rifabutin [Citation83]. Unfortunately, linezolid’s toxicity profile has limited its use in the clinic, but both tedizolid and LCB01-0371 have activity against M. abscessus and improved safety profiles [Citation90,Citation91]. While synergies between these oxazolidinones and rifabutin have not been determined in vitro, rifabutin addition improved the potency of the imipenem-tedizolid combination against M. abscessus in a macrophage infection model [Citation92].
Drug antagonisms between rifabutin and the major anti-NTM drug classes have not been observed. Rifabutin combinations with clofazimine, moxifloxacin and doxycycline have also been tested and found to be additive [Citation83].
6. The home advantage: applying host-directed therapy
As antibacterial drug discovery is challenged by the emergence of antibiotic resistance, host-directed therapies are likely to be incorporated into future treatment of mycobacterial infections [Citation93,Citation94]. Therefore, the potential to combine rifamycins with host-directed therapies should also be considered.
Imatinib mesylate (STI-571, Gleevec) targets tyrosine kinases of the Abl family, including Abl1 and Abl2, which are required for bacterial entry into host cells [Citation95–Citation97]. Consistent with this, imatinib treatment inhibited entry of M. tuberculosis and M. marinum into macrophages [Citation98]. By blocking mycobacterial entry into macrophage, imatinib could increase the number of extracellular bacteria, which would be more susceptible to antibiotics. Indeed, imatinib treatment enhanced the potency of rifabutin against M. marinum in mice [Citation98]. In humans, however, rifamycins induce CYP3A4, which metabolizes imatinib and would potentially negate the benefits of co-administration. Fortunately, rifabutin is a weaker inducer of CYP3A4 [Citation71,Citation72], making imatinib-rifabutin a plausible treatment regimen for M. abscessus.
Bevacizumab, an anti-Vascular Endothelial Growth Factor (VEGF) neutralizing antibody, was originally developed to halt tumor growth by blocking the formation of blood vessels. Surprisingly, bevacizumab had little anti-tumor effect of its own, but improved survival when co-administered with other chemotherapy or immunotherapy [Citation99]. These results suggest that bevacizumab actually normalizes the tumor’s abnormal vascularization, enabling better drug delivery to malignant cells [Citation100]. Like tumors, granulomas formed by infection with M. tuberculosis and M. abscessus are also characterized by abnormal vasculature that can restrict the access by antibiotics [Citation101]. Indeed, bevacizumab treatment improved the delivery of small molecules to TB granulomas in rabbits [Citation102], and SU5416, an inhibitor of the VEGF receptor, improved rifampicin killing of M. marinum in a zebrafish model [Citation103]. Thus, anti-angiogenic agents may be employed to improve the delivery of rifamycins to M. abscessus infection sites.
Ambroxol, an over the counter expectorant, induces autophagy and promotes mycobacterial killing in macrophages [Citation104], making it a promising candidate for host-directed therapy. Critically, ambroxol potentiates the killing of M. tuberculosis by rifampicin in a mouse infection model [Citation104]. Co-administration of ambroxol also results in elevated levels of rifampicin in the lungs of mice and humans [Citation105], suggesting that the potentiation of rifampicin may be due to improved pharmacokinetics instead of enhanced autophagy. Rifampicin dosages for TB treatment are kept suboptimal due to concerns about side effects [Citation106]. By improving drug levels in the lung, ambroxol could enhance the efficacy of rifamycins without increasing the dosage and exacerbating harmful side effects. While ambroxol is also metabolized by CYP3A4 [Citation107,Citation108], rifabutin induces this mechanism to a lesser degree than rifampicin [Citation71,Citation72]. Notably, co-administration of clarithromycin can enhance ambroxol levels by inhibiting CYP3A4 [Citation109]. As clarithromycin also synergizes with rifabutin against M. abscessus [Citation82,Citation83], a rifabutin-clarithromycin-ambroxol combination therapy may represent a potent treatment option. Similarly, ambroxol may enhance the synergistic rifabutin-clarithromycin-tigecycline combination [Citation82]. The efficacy of a rifabutin-ambroxol combination against mycobacterial infections remains to be determined.
7. Conclusion
Once disregarded for the treatment of M. abscessus, rifamycins have recaptured the attention of NTM researchers in recent years. The marked increase in susceptibility of an ADP ribosyltransferases (Arr)-deficient M. abscessus to rifamycins demonstrates that the bacterium’s resistance to this drug class is largely mediated by metabolic inactivation [Citation14]. As a result, rifamycins suffer from suboptimal bacterial cell pharmacokinetics rather than poor on-target activity. Determination of the structure and substrate specificity of M. abscessus Arr would be valuable in rescuing the activity of rifamycins.
The later revelation that rifabutin has activity against M. abscessus shows that certain rifamycin scaffolds can subvert the bacterium’s intrinsic resistance [Citation64]. The resistance of rifabutin to oxidation may explain its potency, but the presence of a rifampicin-inactivating monooxygenase in M. abscessus remains to be established. Genetic characterization of M. abscessus’s putative monooxygenase genes and MIC profiling of other rifamycin antibiotics could shed light on the molecular basis of rifabutin’s potency against M. abscessus.
Apart from in vitro MICs in standard, nutrient-rich media, the potency of rifabutin has only been determined in a macrophage infection model. While rifabutin is active against M. tuberculosis in hypoxic and nutrient-deprived conditions [Citation46], these properties have not been established for M. abscessus. Rifabutin in vivo efficacy studies need to be performed, especially to validate promising drug synergies and evaluate co-administration with host-directed therapies. As case studies on the clinical outcomes of rifabutin usage for M. abscessus treatment are absent from the literature, additional pre-clinical profiling of rifabutin will be needed to assess its suitability for repurposing.
8. Expert opinion
8.1. The case for repositioning rifamycins
With the discovery that rifabutin has potency against M. abscessus, there is newfound interest in repurposing rifabutin to treat M. abscessus lung disease. While repurposing rifabutin could provide immediate relief to patients with limited treatment options, this strategy may not deliver significant clinical improvement. Rifabutin has a higher MIC against M. abscessusthanagainst M. tuberculosis and MAC, likely because it remains susceptible to ADP-ribosylation [Citation60] – the major mechanism behind M. abscessus’ intrinsic resistance to rifamycins. As is, the rifabutin scaffold will require further optimization to become an effective antibiotic for M. abscessus treatment. In this section, we propose drug discovery strategies to reposition rifamycins for M. abscessus lung disease.
8.2. Screening of rifamycin collections
Since the isolation of rifamycin B, vast numbers of rifamycin analogs have been synthesized in the development of rifampicin and other rifamycins as antibiotics. Screening existing rifamycin libraries may yield additional, non-rifabutin scaffolds with activity against M. abscessus. An analysis of soil metagenomes also revealed that soil bacteria have evolved great diversity in their rifamycin biosynthetic machinery, suggesting an untapped source of rifamycin congeners [Citation110]. For example, the rifamycin congener kanglemycin A inhibits bacterial RNA polymerases via a mechanism that is distinct from rifampicin and retains potency against rifampicin-resistant mycobacteria [Citation110,Citation111].
8.3. Blocking natural rifamycin resistance
Because the activity of rifamycins against M. abscessus is limited by ADP-ribosylation [Citation14], rifamycin repositioning should prioritize the development of rifamycins that are resistant to this metabolic inactivation. The structural requirements for rifamycin activity have been elucidated by structure-activity-relationship studies and structural biology. The C23 hydroxyl group, the site of ADP-ribosylation, is involved in hydrogen bonding interactions with RNA polymerase [Citation112]. As a result, modifications at this position result in loss of activity [Citation113]. As the neighboring C21 hydroxyl is also required for activity [Citation113], the C25 position is best suited for modifications to block ADP-ribosylation. Indeed, several C25 carbamate rifamycin derivatives were found to have Arr-independent activity against M. smegmatis [Citation114]. While the same derivatives were more potent against M. abscessus than rifampicin, they showed even better potency against an Arr-deficient strain, suggesting that these compounds are still susceptible to ADP-ribosylation [Citation14]. Through computational approaches like molecular docking, a better understanding of substrate binding to M. abscessus Arr can be obtained, enabling the rational design of novel C25 modifications. Promising candidates can then be validated by demonstrating no-shift in potency against the Arr-deficient M. abscessus mutant.
Given that rifabutin is resistant to oxidation, novel rifamycin derivatives should also incorporate structural features that block oxidation. Scaffolds in which the napthoquinone has a stable oxidation state may thus exhibit good potency against M. abscessus. For example, rifalazil also lacks a hydroquinone and is resistant to autoxidation [Citation78]; its activity against M. abscessus, however, has yet to be determined. A better understanding of how rifamycin oxidation in M. abscessus (both via autoxidation and enzyme-mediated mechanisms) impacts potency is needed.
8.4. Optimizing bacterial cell pharmacokinetics
As the potency of rifabutin against M. abscessus is likely due to its greater accumulation in the bacterium, rifamycin repositioning efforts should seek to produce a compound with desirable bacterial cell uptake and metabolism. In addition to blocking metabolism (described above), the intra-bacterial levels of rifamycins could be increased by improving uptake or reducing efflux. As recently demonstrated, predictive accumulation assays can inform the addition of structural modifications that improve drug uptake [Citation81]. Bacterial strains that overexpress efflux pump genes can be used to screen and identify compounds that are less prone to efflux [Citation115]. Computational approaches can also be used to compare the binding of compounds to efflux pumps and identify chemical features associated with poor binding [Citation116]. In addition to in vitro potency assays, measurement of intra-bacterial accumulation can help prioritize promising scaffolds and analogs.
8.5. Improved in vitro models for M. abscessus pathophysiology
In the NTM disease field, conventional MIC does not correlate well with clinical outcomes [Citation62]. Identifying a lead rifamycin with good efficacy against M. abscessus in vivo will require in vitro models that better reflect the conditions the bacterium experiences during infection. Similar to M. tuberculosis, M. abscessus likely enters a drug-tolerant state in response to hypoxic and nutrient-deficient conditions in the host [Citation117–Citation120]. The bacterium can infect macrophages [Citation121], which promote drug tolerance by inducing mycobacteria efflux mechanisms [Citation122]. As M. abscessus infection is also characterized by the formation of granulomas, these bacteria may also attain drug tolerance while residing extracellularly in the caseum [Citation34]. Unlike M. tuberculosis, M. abscessus can grow as biofilms in the lung [Citation123,Citation124], and can reside within airway mucus produced by cystic fibrosis and chronic obstructive pulmonary disease patients – these environments may similarly alter the physiology of the bacterium and provide protection from the effects of antibiotics. The development of ‘persister-specific’ assays that model the aforementioned conditions has been reviewed recently [Citation27] and should be applied here. As rifampicin can kill M. tuberculosis persisters [Citation46] in different host environments [Citation34], efforts to reposition rifamycins should similarly aim to develop a lead compound that can eradicate M. abscessus persisters wherever they reside in the host [Citation125].
8.6. Improved in vivo models of M. abscessus infection
Efficacy studies of promising rifamycin lead compounds will require a predictive animal model. However, the current mouse models for M. abscessus have significant drawbacks [Citation126]. Because of the intrinsic mammalian host resistance to NTM and lower virulence of M. abscessus compared to M. tuberculosis, animal models rely on immunocompromised mice and a high dose inoculum, which do not model the natural course of infection or host immune response. Since disease manifestation can vary between models, it is perhaps not surprising that drugs can show differential potency when tested in different models [Citation127,Citation128]. The development of an immunocompetent mouse model that replicates the basic features of human M. abscessus lung disease is greatly needed to advance drug discovery efforts.
8.7. Optimization of non-rifamycin RNA polymerase inhibitors
As the mycobacterial RNA polymerase is a validated drug target, screening for non-rifamycin RNA polymerase inhibitors should also be considered. In a screen of the Medicines for Malaria Venture (MMV) Pathogen Box, Low et al. identified MMV688845 (GSK1729177A) as an NTM hit [Citation129]. This compound has demonstrated activity against M. tuberculosis [Citation130] and analogues of this compound bind the M. tuberculosis RNA polymerase at a different site from the rifamycin binding site [Citation131]. Thus, MMV688845 would circumvent both acquired rifamycin resistance and M. abscessus’ natural resistance to rifamycins. Accordingly, the compound retains bactericidal activity against M. abscessus and MAC [Citation129]. Together, these findings reaffirm the RNA polymerase as a viable drug target for NTM drug discovery and support screening for non-rifamycin RNA polymerase inhibitors. As we have found that screening libraries of whole-cell TB actives improves the chances of finding such a candidate with activity against NTMs [Citation129], this strategy should be pursued in parallel to the repositioning of rifamycins.
8.8. Developing a better drug regimen
Given the many reported rifabutin synergies (), a rifamycin with activity against M. abscessus should be well-suited for combination with existing anti-NTM drugs. Thus, the repurposing of rifamycins represents an opportunity to develop a more efficacious drug regimen for M. abscessus infections. The advancement of promising rifamycin candidates should be based in part on the presence of synergies with other drugs used in M. abscessus treatment. In particular, drug combinations where antibiotic resistance is mitigated (e.g., erm41 inducible resistance to clarithromycin) or the uptake of the rifamycin is enhanced (e.g., cell wall weakening by β-lactams) are desirable. Synergies between rifamycins and new compound classes should be incorporated early in NTM drug discovery cascades. Further potentiation of rifamycin activity by host-directed therapy should be assessed in appropriate in vivo models. At the same time, the potential for detrimental drug-drug interactions should be evaluated. Recently, the first use of bacteriophages to treat a mycobacterial infection resulted in clinical improvement for a CF patient infected with M. abscessus subsp. massiliense [Citation132]; the potential to combine a rifamycin-based drug regimen with phage treatment should also be considered. A critical step in the development of new NTM drug regimens is the ability to recruit sufficiently homogeneous patient cohorts in a timely manner. With the exception of inhaled liposomal amikacin, there has been no recent clinical trial since the 1980’s when the macrolides were evaluated. The NTM community needs to join forces to establish such cohorts and validate adequate read-outs of efficacy, as emphasized in a recent workshop held by the FDA [Citation133].
8.9. Outlook
The repositioning of rifamycins/rifabutin for M. abscessus lung disease will require a multifaceted approach. The screening of rifamycin libraries will provide additional starting points for chemistry. Given M. abscessus’ intrinsic antibiotic resistance, medicinal chemistry projects will be aimed at blocking metabolism, improving uptake, and reducing efflux, thereby optimizing bacterial cell pharmacokinetics. The repositioned rifamycin candidate must then be placed within a treatment regimen that harnesses drug synergies and host-directed therapies. Critically, the success of repositioning will hinge on the development of appropriate in vitro and in vivo models, which remain significant limitations to NTM drug discovery in general. In parallel to rifamycin repositioning efforts, we propose to follow up non-rifamycins such as MMV688845 as another way to take advantage of the mycobacterial RNA polymerase as a validated drug target.
While more challenging than repurposing rifabutin, repositioning holds the potential to develop a rifamycin that is better equipped to tackle one of the world’s most drug-resistant bacterial infections. A repositioned rifamycin would have greater potency against M. abscessus by avoiding the bacterium’s intrinsic antibiotic resistance mechanisms. This novel rifamycin would synergize with existing antibiotics and retain activity in different host microenvironments, curbing the development of antibiotic resistance and preventing treatment relapse. In this way, repositioning rifamycins for M. abscessus lung disease could produce a next-generation rifamycin that may also benefit the treatment of TB and other NTM diseases.
Article highlights
The differential potency of rifampicin and rifabutin against M. abscessus reflects the critical importance of bacterial uptake and metabolism in the pathogen’s intrinsic antibiotic resistance.
Rifamycins are poised to synergize with key drugs involved in current M. abscessus treatment and stand to benefit from co-administration with host-directed therapies.
The repositioning of rifamycins for M. abscessus lung disease will require a multipronged approach and presents an opportunity to develop an improved drug regimen against this dreadful disease.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewers Disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Acknowledgments
The authors thank Courtney Aldrich (University of Minnesota), Joel Freundlich (Rutgers University, New Jersey), Adrian Richter (Halle-Wittenberg University, Germany) and Graham Timmins (University of New Mexico) for their helpful discussions. T.D. holds a Toh Chin Chye Visiting Professorship at the Department of Microbiology and Immunology, Yong Loo Lin School of Medicine, National University of Singapore.
Additional information
Funding
References
- Tortoli E. Chapter 1 - The Taxonomy of the Genus Mycobacterium. In: Velayati AA, Farnia P, editors. Nontuberculous Mycobacteria (NTM). Cambridge (MA): Academic Press; 2019. p. 1–10.
- Park IK, Olivier KN. Nontuberculous mycobacteria in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin Respir Crit Care Med. 2015 Apr;36(2):217–224. . PubMed PMID: 25826589
- Daniel-Wayman S, Adjemian J, Rebecca Prevots D. Epidemiology of Nontuberculous Mycobacterial Pulmonary Disease (NTM PD) in the USA. In: Griffith DE, editor. Nontuberculous Mycobacterial Disease: A Comprehensive Approach to Diagnosis and Management. Cham: Springer International Publishing; 2019. p. 145–161.
- Wassilew N, Hoffmann H, Andrejak C, et al. Pulmonary disease caused by non-tuberculous mycobacteria. Respiration. 2016;91(5):386–402. . PubMed PMID: 27207809
- Strollo SE, Adjemian J, Adjemian MK, et al. The Burden of pulmonary nontuberculous mycobacterial disease in the United States. Ann Am Thorac Soc. 2015 Oct;12(10):1458–1464. PubMed PMID: 26214350; PubMed Central PMCID: PMCPMC4627421.
- Adjemian J, Frankland TB, Daida YG, et al. Epidemiology of nontuberculous mycobacterial lung disease and tuberculosis, Hawaii, USA. Emerg Infect Dis. 2017 Mar;23(3):439–447. PubMed PMID: 28221128; PubMed Central PMCID: PMCPMC5382761.
- Mirsaeidi M, Vu A, Leitman P, et al. A patient-based analysis of the geographic distribution of mycobacterium avium complex, mycobacterium abscessus, and mycobacterium kansasii infections in the United States. Chest. 2017 Apr;151(4):947–950. PubMed PMID: 28390637.
- Vinnard C, Longworth S, Mezochow A, et al. Deaths related to nontuberculous mycobacterial infections in the United States, 1999-2014. Ann Am Thorac Soc. 2016 Nov;13(11):1951–1955. PubMed PMID: 27607541; PubMed Central PMCID: PMCPMC5122483.
- Maurer FP, Bruderer VL, Ritter C, et al. Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob Agents Chemother. 2014 Jul;58(7):3828–3836. PubMed PMID: 24752273; PubMed Central PMCID: PMCPMC4068550.
- Luthra S, Rominski A, Sander P. The role of antibiotic-target-modifying and antibiotic-modifying enzymes in mycobacterium abscessus drug resistance. Front Microbiol. 2018;9:2179. PubMed PMID: 30258428; PubMed Central PMCID: PMCPMC6143652.
- Falkinham JO 3rd. Challenges of NTM Drug Development. Front Microbiol. 2018;9:1613. . PubMed PMID: 30072975; PubMed Central PMCID: PMCPMC6058048
- Philley JV, Griffith DE. Disease Caused by Mycobacterium Abscessus and Other Rapidly Growing Mycobacteria (RGM). In: Griffith DE, editor. Nontuberculous Mycobacterial Disease: A Comprehensive Approach to Diagnosis and Management. Cham: Springer International Publishing; 2019. p. 369–399.
- Dal Molin M, Gut M, Rominski A, et al. Molecular mechanisms of intrinsic streptomycin resistance in mycobacterium abscessus. Antimicrob Agents Chemother. 2018 Jan;62(1). PubMed PMID: 29061744; PubMed Central PMCID: PMCPMC5740355. DOI:10.1128/AAC.01427-17.
- Rominski A, Roditscheff A, Selchow P, et al. Intrinsic rifamycin resistance of Mycobacterium abscessus is mediated by ADP-ribosyltransferase MAB_0591. J Antimicrob Chemother. 2017 Feb;72(2):376–384. . PubMed PMID: 27999011.
- Rominski A, Schulthess B, Muller DM, et al. Effect of beta-lactamase production and beta-lactam instability on MIC testing results for Mycobacterium abscessus. J Antimicrob Chemother. 2017 Nov 1;72(11):3070–3078. PubMed PMID: 28961987.
- Rominski A, Selchow P, Becker K, et al. Elucidation of Mycobacterium abscessus aminoglycoside and capreomycin resistance by targeted deletion of three putative resistance genes. J Antimicrob Chemother. 2017 May 09. DOI:10.1093/jac/dkx125. [ PubMed PMID: 28486671].
- Rudra P, Hurst-Hess K, Lappierre P, et al. High levels of intrinsic tetracycline resistance in mycobacterium abscessus are conferred by a tetracycline-modifying monooxygenase. Antimicrob Agents Chemother. 2018 Jun;62(6). PubMed PMID: 29632012; PubMed Central PMCID: PMCPMC5971581. DOI:10.1128/AAC.00119-18.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007 Feb 15;175(4):367–416. PubMed PMID: 17277290.
- Nash KA, Brown-Elliott BA, Wallace RJ Jr. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother. 2009 Apr;53(4):1367–1376. . PubMed PMID: 19171799; PubMed Central PMCID: PMCPMC2663066
- Griffith DE, Brown-Elliott BA, Benwill JL, et al. Mycobacterium abscessus. “Pleased to meet you, hope you guess my name”. Ann Am Thorac Soc. 2015 Mar;12(3):436–439. PubMed PMID: 25643064.
- Jarand J, Levin A, Zhang L, et al. Clinical and microbiologic outcomes in patients receiving treatment for Mycobacterium abscessus pulmonary disease. Clin Infect Dis. 2011 Mar 1;52(5):565–571. PubMed PMID: 21292659.
- van Ingen J, Bendien SA, de Lange WC, et al. Clinical relevance of non-tuberculous mycobacteria isolated in the Nijmegen-Arnhem region, The Netherlands. Thorax. 2009 Jun;64(6):502–506. PubMed PMID: 19213773.
- Koh WJ, Jeon K, Lee NY, et al. Clinical significance of differentiation of Mycobacterium massiliense from Mycobacterium abscessus. Am J Respir Crit Care Med. 2011 Feb 1;183(3):405–410. PubMed PMID: 20833823.
- Kasperbauer SH, De Groote MA. The treatment of rapidly growing mycobacterial infections. Clin Chest Med. 2015 Mar;36(1):67–78. . PubMed PMID: 25676520
- Andrejak C, Thomsen VO, Johansen IS, et al. Nontuberculous pulmonary mycobacteriosis in Denmark: incidence and prognostic factors. Am J Respir Crit Care Med. 2010 Mar 1;181(5):514–521. PubMed PMID: 20007929.
- Jenkins PA, Campbell IA. Research committee of the british thoracic s. pulmonary disease caused by Mycobacterium xenopi in HIV-negative patients: five year follow-up of patients receiving standardised treatment. Respir Med. 2003 Apr;97(4):439–444. PubMed PMID: 12693807
- Wu ML, Aziz DB, Dartois V, et al. NTM drug discovery: status, gaps and the way forward. Drug Discov Today. 2018 Apr 7. PubMed PMID: 29635026. DOI:10.1016/j.drudis.2018.04.001
- ClinicalTrials.gov: U.S. national library of medicine; [cited 2019 April 24]. Available from: https://clinicaltrials.gov
- Daley CL, Olivier KN. ALIS (Amikacin Liposome Inhalation Suspension): the Beginning of a Wonderland? Am J Respir Crit Care Med. 2018 Oct 26. DOI:10.1164/rccm.201810-1901ED. PubMed PMID: 30365392.
- Griffith DE, Eagle G, Thomson R, et al. amikacin liposome inhalation suspension for treatment-refractory lung disease caused by mycobacterium avium complex (CONVERT): a prospective, open-label, randomized study. Am J Respir Crit Care Med. 2018 Sep 14. DOI:10.1164/rccm.201807-1318OC. [ PubMed PMID: 30216086].
- Daniel-Wayman S, Abate G, Barber DL, et al. Advancing translational science for pulmonary nontuberculous mycobacterial infections. a road map for research. Am J Respir Crit Care Med. 2019 Apr 15;199(8):947–951. PubMed PMID: 30428263.
- DiMasi JA, Grabowski HG, Hansen RA. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33.
- Pethe K, Sequeira PC, Agarwalla S, et al. A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010;1:57. . PubMed PMID: 20975714; PubMed Central PMCID: PMC3220188
- Sarathy JP, Via LE, Weiner D, et al. Extreme Drug Tolerance of Mycobacterium tuberculosis in Caseum. Antimicrob Agents Chemother. 2018 Feb;62(2). PubMed PMID: 29203492; PubMed Central PMCID: PMCPMC5786764. DOI:10.1128/AAC.02266-17.
- Griffith DE, Aksamit TR. Therapy of refractory nontuberculous mycobacterial lung disease. Curr Opin Infect Dis. 2012 Apr;25(2):218–227. . PubMed PMID: 22327466
- An antibiotic comeback? Nat Rev Drug Discov. 2014 02/28/online; 13:165. .
- Sensi P. History of the development of rifampin. Rev Infect Dis. 1983;Jul-Aug;5(Suppl 3):S402–6. PubMed PMID: 6635432.
- Maggi N, Pasqualucci CR, Ballotta R, et al. Rifampicin: a new orally active rifamycin. Chemotherapy. 1966;11(5):285–292. PubMed PMID: 5958716.
- Calvori C, Frontali L, Leoni L, et al. Effect of rifamycin on protein synthesis. Nature. 1965 Jul 24;207(995):417–418. PubMed PMID: 4957347
- Hartmann G, Honikel KO, Knusel F, et al. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochim Biophys Acta. 1967;145(3):843–844. PubMed PMID: 4863911
- Yarbrough LR, Wu FY, Wu CW. Molecular mechanism of the rifampicin -RNA polymerase interaction. Biochemistry. 1976 Jun 15;15(12):2669–2676. PubMed PMID: 779828
- Arioli V, Pallanza R, Furesz S, et al. Rifampicin: a new rifamycin. I. Bacteriological studies. Arzneimittelforschung. 1967 May;17(5):523–529. PubMed PMID: 4385644
- Pallanza R, Arioli V, Furesz S, et al. Rifampicin: a new rifamycin. II. Laboratory studies on the antituberculous activity and preliminary clinical observations. Arzneimittelforschung. 1967 May;17(5):529–534. PubMed PMID: 5631580
- Lancini G, Pallanza R, Silvestri LG. Relationships between bactericidal effect and inhibition of ribonucleic acid nucleotidyltransferase by rifampicin in Escherichia coli K-12. J Bacteriol. 1969 Feb;97(2):761–768. PubMed PMID: 4886292; PubMed Central PMCID: PMCPMC249757
- Kenny MT, Strates B. Metabolism and pharmacokinetics of the antibiotic rifampin. Drug Metab Rev. 1981;12(1):159–218. . PubMed PMID: 7028436
- Lakshminarayana SB, Huat TB, Ho PC, et al. Comprehensive physicochemical, pharmacokinetic and activity profiling of anti-TB agents. J Antimicrob Chemother. 2015 Mar;70(3):857–867. PubMed PMID: 25587994.
- Prideaux B, Via LE, Zimmerman MD, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med. 2015 Oct;21(10):1223–1227. PubMed PMID: 26343800; PubMed Central PMCID: PMCPMC4598290.
- Fox W, Ellard GA, Mitchison DA. Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946-1986, with relevant subsequent publications. Int J Tuberc Lung Dis. 1999 Oct;3(10 Suppl 2):S231–79. PubMed PMID: 10529902
- Chopra S, Matsuyama K, Hutson C, et al. Identification of antimicrobial activity among FDA-approved drugs for combating Mycobacterium abscessus and Mycobacterium chelonae. J Antimicrob Chemother. 2011 Jul;66(7):1533–1536. PubMed PMID: 21486854.
- Pang H, Li G, Zhao X, et al. Drug susceptibility testing of 31 antimicrobial agents on rapidly growing mycobacteria isolates from China. Biomed Res Int. 2015;2015:419392. . PubMed PMID: 26351633; PubMed Central PMCID: PMCPMC4550772
- Sha W, Weng XH, Xiao HP, et al. [Investigation of drug-resistance to rifampin and rpoB gene sequence analysis of Mycobacterium abscessus]. Zhonghua Jie He He Hu Xi Za Zhi. 2003 Sep;26(9):544–547. PubMed PMID: 14521758
- Lambert PA. Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria. J Appl Microbiol. 2002;92(Suppl):46S–54S. PubMed PMID: 12000612
- Brennan PJ, Nikaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. PubMed PMID: 7574484.
- Jarlier V, Nikaido H. Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol Lett. 1994 Oct 15;123(1–2):11–18. . PubMed PMID: 7988876.
- Hohl M, Remm S, Eskandarian HA, et al. Increased drug permeability of a stiffened mycobacterial outer membrane in cells lacking MFS transporter Rv1410 and lipoprotein LprG. Mol Microbiol. 2019 Feb 11. DOI:10.1111/mmi.14220. [ PubMed PMID: 30742339].
- Becker K, Haldimann K, Selchow P, et al. Lipoprotein glycosylation by protein-o-mannosyltransferase (MAB_1122c) contributes to low cell envelope permeability and antibiotic resistance of mycobacterium abscessus. Front Microbiol. 2017;8:2123. . PubMed PMID: 29163413; PubMed Central PMCID: PMCPMC5673659
- Ripoll F, Pasek S, Schenowitz C, et al. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One. 2009 Jun 19;4(6):e5660. PubMed PMID: 19543527; PubMed Central PMCID: PMCPMC2694998.
- Balganesh M, Dinesh N, Sharma S, et al. Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob Agents Chemother. 2012 May;56(5):2643–2651. PubMed PMID: 22314527; PubMed Central PMCID: PMCPMC3346595.
- Szumowski JD, Adams KN, Edelstein PH, et al. Antimicrobial efflux pumps and Mycobacterium tuberculosis drug tolerance: evolutionary considerations. Curr Top Microbiol Immunol. 2013;374:81–108. . PubMed PMID: 23242857; PubMed Central PMCID: PMCPMC3859842
- Baysarowich J, Koteva K, Hughes DW, et al. Rifamycin antibiotic resistance by ADP-ribosylation: structure and diversity of Arr. Proc Natl Acad Sci U S A. 2008 Mar 25;105(12):4886–4891. PubMed PMID: 18349144; PubMed Central PMCID: PMCPMC2290778.
- Koteva K, Cox G, Kelso JK, et al. Rox, a rifamycin resistance enzyme with an unprecedented mechanism of action. Cell Chem Biol. 2018 Apr 19;25(4):403–412 e5. PubMed PMID: 29398560.
- van Ingen J, Boeree MJ, van Soolingen D, et al. Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist Updat. 2012 Jun;15(3):149–161. PubMed PMID: 22525524.
- Strnad L, Winthrop KL. Treatment of Mycobacterium abscessus Complex. Semin Respir Crit Care Med. 2018 Jun;39(3):362–376. . PubMed PMID: 30071551
- Aziz DB, Low JL, Wu ML, et al. Rifabutin is active against mycobacterium abscessus complex. Antimicrob Agents Chemother. 2017 Apr 10. PubMed PMID: 28396540. DOI:10.1128/AAC.00155-17
- Le Run E, Arthur M, Mainardi JL. In vitro and intracellular activity of imipenem combined with rifabutin and avibactam against mycobacterium abscessus. Antimicrob Agents Chemother. 2018 Aug;62(8). DOI:10.1128/AAC.00623-18. PubMed PMID: 29866869; PubMed Central PMCID: PMCPMC6105861.
- Lee H, Ahn S, Hwang NY, et al. Treatment outcomes of rifabutin-containing regimens for rifabutin-sensitive multidrug-resistant pulmonary tuberculosis. Int J Infect Dis. 2017 Dec;65:135–141. PubMed PMID: 29224631.
- Griffith DE. Treatment of Mycobacterium avium Complex (MAC). Semin Respir Crit Care Med. 2018 Jun;39(3):351–361. . PubMed PMID: 30071550
- Blaschke TF, Skinner MH. The clinical pharmacokinetics of rifabutin. Clin Infect Dis. 1996 Apr;22(Suppl 1):S15-21; discussion S21-2. PubMed PMID: 8785251
- Van der Auwera P, Matsumoto T, Husson M. Intraphagocytic penetration of antibiotics. J Antimicrob Chemother. 1988 Aug;22(2):185–192. PubMed PMID: 2846489
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. This official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997 Aug;156(2 Pt 2):S1–25. PubMed PMID: 9279284.
- Li AP, Reith MK, Rasmussen A, et al. Primary human hepatocytes as a tool for the evaluation of structure-activity relationship in cytochrome P450 induction potential of xenobiotics: evaluation of rifampin, rifapentine and rifabutin. Chem Biol Interact. 1997 Nov 6;107(1–2):17–30. PubMed PMID: 9402947
- Reinach B, de Sousa G, Dostert P, et al. Comparative effects of rifabutin and rifampicin on cytochromes P450 and UDP-glucuronosyl-transferases expression in fresh and cryopreserved human hepatocytes. Chem Biol Interact. 1999 Jun 1;121(1):37–48. PubMed PMID: 10418969
- Baciewicz AM, Chrisman CR, Finch CK, et al. Update on rifampin, rifabutin, and rifapentine drug interactions. Curr Med Res Opin. 2013 Jan;29(1):1–12. PubMed PMID: 23136913.
- Horne DJ, Spitters C, Narita M. Experience with rifabutin replacing rifampin in the treatment of tuberculosis. Int J Tuberc Lung Dis. 2011 Nov;15(11):1485–9, i. . PubMed PMID: 22008761; PubMed Central PMCID: PMCPMC3290133
- van Ingen J, van der Laan T, Dekhuijzen R, et al. In vitro drug susceptibility of 2275 clinical non-tuberculous Mycobacterium isolates of 49 species in The Netherlands. Int J Antimicrob Agents. 2010 Feb;35(2):169–173. PubMed PMID: 20006470.
- Kunin CM. Antimicrobial activity of rifabutin. Clin Infect Dis. 1996 Apr;22(Suppl 1):S3-13; discussion S13-4. PubMed PMID: 8785253
- Buss WC, Reyes E, Barela TD Metal ion catalyzed oxidation of the antibiotic rifampicin. Res Commun Chem Pathol Pharmacol. 1977 Jul;17(3):547–550. PubMed PMID: 19824.
- Staudinger T, Redl B, Glasgow BJ. Antibacterial activity of rifamycins for M. smegmatis with comparison of oxidation and binding to tear lipocalin. Biochim Biophys Acta. 2014 Apr;1844(4):750–758. . PubMed PMID: 24530503; PubMed Central PMCID: PMCPMC3992280
- Spanogiannopoulos P, Waglechner N, Koteva K, et al. A rifamycin inactivating phosphotransferase family shared by environmental and pathogenic bacteria. Proc Natl Acad Sci U S A. 2014 May 13;111(19):7102–7107. PubMed PMID: 24778229; PubMed Central PMCID: PMCPMC4024899.
- Spanogiannopoulos P, Thaker M, Koteva K, et al. Characterization of a rifampin-inactivating glycosyltransferase from a screen of environmental actinomycetes. Antimicrob Agents Chemother. 2012 Oct;56(10):5061–5069. PubMed PMID: 22802246; PubMed Central PMCID: PMCPMC3457401.
- Richter MF, Drown BS, Riley AP, et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature. 2017 May 18;545(7654):299–304. PubMed PMID: 28489819; PubMed Central PMCID: PMCPMC5737020.
- Pryjma M, Burian J, Thompson CJ. Rifabutin acts in synergy and is bactericidal with frontline mycobacterium abscessus antibiotics clarithromycin and tigecycline, suggesting a potent treatment combination. Antimicrob Agents Chemother. 2018 Aug;62(8). DOI:10.1128/AAC.00283-18. PubMed PMID: 29760147; PubMed Central PMCID: PMCPMC6105836.
- Cheng A, Tsai YT, Chang SY, et al. In vitro synergism of rifabutin with clarithromycin, imipenem, and tigecycline against the mycobacterium abscessus complex. Antimicrob Agents Chemother. 2019 Apr;63(4). DOI:10.1128/AAC.02234-18. PubMed PMID: 30670428.
- Pryjma M, Burian J, Kuchinski K, et al. Antagonism between front-line antibiotics clarithromycin and amikacin in the treatment of mycobacterium abscessus infections is mediated by the whiB7 gene. Antimicrob Agents Chemother. 2017 Nov;61(11). PubMed PMID: 28874379; PubMed Central PMCID: PMCPMC5655113. DOI:10.1128/AAC.01353-17.
- Soroka D, Dubee V, Soulier-Escrihuela O, et al. Characterization of broad-spectrum Mycobacterium abscessus class A beta-lactamase. J Antimicrob Chemother. 2014 Mar;69(3):691–696. PubMed PMID: 24132992.
- Lavollay M, Dubee V, Heym B, et al. In vitro activity of cefoxitin and imipenem against Mycobacterium abscessus complex. Clin Microbiol Infect. 2014 May;20(5):O297–300. PubMed PMID: 24112243.
- Story-Roller E, Maggioncalda EC, Lamichhane G. Select beta-lactam combinations exhibit synergy against Mycobacterium abscessus in vitro. Antimicrob Agents Chemother. 2019 Feb 11. DOI:10.1128/AAC.02613-18. PubMed PMID: 30745389.
- Ferro BE, Srivastava S, Deshpande D, et al. Tigecycline is highly efficacious against mycobacterium abscessus pulmonary disease. Antimicrob Agents Chemother. 2016 May;60(5):2895–2900. PubMed PMID: 26926649; PubMed Central PMCID: PMCPMC4862465.
- Wallace RJ Jr., Dukart G, Brown-Elliott BA, et al. Clinical experience in 52 patients with tigecycline-containing regimens for salvage treatment of Mycobacterium abscessus and Mycobacterium chelonae infections. J Antimicrob Chemother. 2014 Jul;69(7):1945–1953. PubMed PMID: 24633206; PubMed Central PMCID: PMCPMC4054987.
- Brown-Elliott BA, Wallace RJ Jr. In vitro susceptibility testing of tedizolid against nontuberculous mycobacteria. J Clin Microbiol. 2017 Jun;55(6):1747–1754. . PubMed PMID: 28330892; PubMed Central PMCID: PMCPMC5442531
- Kim TS, Choe JH, Kim YJ, et al. Activity of LCB01-0371, a novel oxazolidinone, against mycobacterium abscessus. Antimicrob Agents Chemother. 2017 Sep;61(9). PubMed PMID: 28674049; PubMed Central PMCID: PMCPMC5571369. DOI:10.1128/AAC.02752-16.
- Le Run E, Arthur M, Mainardi JL. In vitro and intracellular activity of imipenem combined with tedizolid, rifabutin, and avibactam against mycobacterium abscessus. Antimicrob Agents Chemother. 2019 Apr;63(4). DOI:10.1128/AAC.01915-18. PubMed PMID: 30745387.
- Hawn TR, Matheson AI, Maley SN, et al. Host-directed therapeutics for tuberculosis: can we harness the host? Microbiol Mol Biol Rev. 2013 Dec;77(4):608–627. PubMed PMID: 24296574; PubMed Central PMCID: PMCPMC3973381.
- Kolloli A, Subbian S. Host-directed therapeutic strategies for tuberculosis. Front Med (Lausanne). 2017;4:171. . PubMed PMID: 29094039; PubMed Central PMCID: PMCPMC5651239
- Burton EA, Plattner R, Pendergast AM. Abl tyrosine kinases are required for infection by Shigella flexneri. Embo J. 2003 Oct 15;22(20):5471–5479. . PubMed PMID: 14532119; PubMed Central PMCID: PMCPMC213767.
- Elwell CA, Ceesay A, Kim JH, et al. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008 Mar 7;4(3):e1000021. PubMed PMID: 18369471; PubMed Central PMCID: PMCPMC2267011.
- Pielage JF, Powell KR, Kalman D, et al. RNAi screen reveals an Abl kinase-dependent host cell pathway involved in Pseudomonas aeruginosa internalization. PLoS Pathog. 2008 Mar 21;4(3):e1000031. PubMed PMID: 18369477; PubMed Central PMCID: PMCPMC2265438.
- Napier RJ, Rafi W, Cheruvu M, et al. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. Cell Host Microbe. 2011 Nov 17;10(5):475–485. PubMed PMID: 22100163; PubMed Central PMCID: PMCPMC3222875.
- Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005 Jan 7;307(5706):58–62. . PubMed PMID: 15637262.
- Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014 Nov 10;26(5):605–622. . PubMed PMID: 25517747; PubMed Central PMCID: PMCPMC4269830.
- Dartois V. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Microbiol. 2014 Mar;12(3):159–167. . PubMed PMID: 24487820; PubMed Central PMCID: PMC4341982
- Datta M, Via LE, Kamoun WS, et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc Natl Acad Sci U S A. 2015 Feb 10;112(6):1827–1832. PubMed PMID: 25624495; PubMed Central PMCID: PMCPMC4330784.
- Oehlers SH, Cronan MR, Scott NR, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. 2015 Jan 29;517(7536):612–615. PubMed PMID: 25470057; PubMed Central PMCID: PMCPMC4312197.
- Choi SW, Gu Y, Peters RS, et al. Ambroxol induces autophagy and potentiates rifampin antimycobacterial activity. Antimicrob Agents Chemother. 2018 Sep;62(9). DOI:10.1128/AAC.01019-18. PubMed PMID: 30012752; PubMed Central PMCID: PMCPMC6125555.
- Imaoka M. [Preclinical and clinical investigation about combination effects of expectorants in chemotherapy of infectious respiratory diseases]. Chemotherapy. 1986;34(3):262–270.
- van Ingen J, Aarnoutse RE, Donald PR, et al. Why do we use 600 mg of rifampicin in tuberculosis treatment? Clin Infect Dis. 2011 May;52(9):e194–9. . PubMed PMID: 21467012.
- Ishiguro N, Senda C, Kishimoto W, et al. Identification of CYP3A4 as the predominant isoform responsible for the metabolism of ambroxol in human liver microsomes. Xenobiotica. 2000 Jan;30(1):71–80. PubMed PMID: 10659952
- Lehmann JM, McKee DD, Watson MA, et al. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998 Sep 1;102(5):1016–1023. PubMed PMID: 9727070; PubMed Central PMCID: PMCPMC508967.
- Lee C-K, Choi J-S. Effect of clarithromycin on the pharmacokinetics of ambroxol in rats. J Pharm Invest. 2006;36(3):157–160.
- Peek J, Lilic M, Montiel D, et al. Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism. Nat Commun. 2018 Oct 8;9(1):4147. 10.1038/s41467-018-06587-2. PubMed PMID: 30297823; PubMed Central PMCID: PMCPMC6175910.
- Mosaei H, Molodtsov V, Kepplinger B, et al. Mode of action of kanglemycin a, an ansamycin natural product that is active against rifampicin-resistant mycobacterium tuberculosis. Mol Cell. 2018 Oct 18;72(2):263–274 e5. PubMed PMID: 30244835; PubMed Central PMCID: PMCPMC6202310.
- Campbell EA, Korzheva N, Mustaev A, et al. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell. 2001 Mar 23;104(6):901–912. PubMed PMID: 11290327
- Sensi P, Maggi N, Furesz S, et al. Chemical modifications and biological properties of rifamycins. Antimicrob Agents Chemother (Bethesda). 1966;6:699–714. PubMed PMID: 4862170.
- Combrink KD, Denton DA, Harran S, et al. New C25 carbamate rifamycin derivatives are resistant to inactivation by ADP-ribosyl transferases. Bioorg Med Chem Lett. 2007 Jan 15;17(2):522–526. PubMed PMID: 17070048.
- Petersen PJ, Jacobus NV, Weiss WJ, et al. In vitro and in vivo antibacterial activities of a novel glycylcycline, the 9-t-butylglycylamido derivative of minocycline (GAR-936). Antimicrob Agents Chemother. 1999 Apr;43(4):738–744. PubMed PMID: 10103174; PubMed Central PMCID: PMCPMC89200.
- Collu F, Vargiu AV, Dreier J, et al. Recognition of imipenem and meropenem by the RND-transporter MexB studied by computer simulations. J Am Chem Soc. 2012 Nov 21;134(46):19146–19158. PubMed PMID: 23146101.
- Dick T, Lee BH, Murugasu-Oei B. Oxygen depletion induced dormancy in Mycobacterium smegmatis. FEMS Microbiol Lett. 1998 Jun 15;163(2):159–164. PubMed PMID: 9673018
- Wu ML, Gengenbacher M, Dick T. Mild nutrient starvation triggers the development of a small-cell survival morphotype in mycobacteria. Front Microbiol. 2016;7:947. . PubMed PMID: 27379076; PubMed Central PMCID: PMCPMC4909757
- Boon C, Dick T. How Mycobacterium tuberculosis goes to sleep: the dormancy survival regulator DosR a decade later [Research Support, Non-U.S. Gov’t Review]. Future Microbiol. 2012 Apr;7(4):513–518. . PubMed PMID: 22439727; eng
- Gerasimova A, Kazakov AE, Arkin AP, et al. Comparative genomics of the dormancy regulons in mycobacteria. J Bacteriol. 2011 Jul;193(14):3446–3452. PubMed PMID: 21602344; PubMed Central PMCID: PMCPMC3133309.
- Roux AL, Viljoen A, Bah A, et al. The distinct fate of smooth and rough Mycobacterium abscessus variants inside macrophages. Open Biol. 2016 Nov;6(11). PubMed PMID: 27906132; PubMed Central PMCID: PMCPMC5133439. DOI:10.1098/rsob.160185.
- Adams KN, Takaki K, Connolly LE, et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011 Apr 1;145(1):39–53. PubMed PMID: 21376383; PubMed Central PMCID: PMC3117281.
- Qvist T, Eickhardt S, Kragh KN, et al. Chronic pulmonary disease with Mycobacterium abscessus complex is a biofilm infection. Eur Respir J. 2015 Dec;46(6):1823–1826. 10.1183/13993003.01102-2015. PubMed PMID: 26493807
- Fennelly KP, Ojano-Dirain C, Yang Q, et al. Biofilm Formation by Mycobacterium abscessus in a Lung Cavity. Am J Respir Crit Care Med. 2016 Mar 15;193(6):692–693. PubMed PMID: 26731090.
- Dick T. Dormant tubercle bacilli: the key to more effective TB chemotherapy? J Antimicrob Chemother. 2001 Jan;47(1):117–118. PubMed PMID: 11152444
- Bernut A, Herrmann JL, Ordway D, et al. The diverse cellular and animal models to decipher the physiopathological traits of mycobacterium abscessus infection. Front Cell Infect Microbiol. 2017;7:100. PubMed PMID: 28421165; PubMed Central PMCID: PMCPMC5378707.
- Lerat I, Cambau E, Roth Dit Bettoni R, et al. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis. 2014 Mar;209(6):905–912. PubMed PMID: 24253289.
- Obregon-Henao A, Arnett KA, Henao-Tamayo M, et al. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob Agents Chemother. 2015 Nov;59(11):6904–6912. PubMed PMID: 26303795; PubMed Central PMCID: PMCPMC4604395.
- Low JL, Wu ML, Aziz DB, et al. Screening of TB actives for activity against nontuberculous mycobacteria delivers high hit rates. Front Microbiol. 2017;8:1539. . PubMed PMID: 28861054; PubMed Central PMCID: PMCPMC5559473
- Ballell L, Bates RH, Young RJ, et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. Chem Med Chem. 2013 Feb;8(2):313–321. PubMed PMID: 23307663; PubMed Central PMCID: PMCPMC3743164.
- Lin W, Mandal S, Degen D, et al. Structural basis of mycobacterium tuberculosis transcription and transcription inhibition. Mol Cell. 2017 Apr 20;66(2):169–179 e8. PubMed PMID: 28392175; PubMed Central PMCID: PMCPMC5438085.
- Dedrick RM, Guerrero-Bustamante CA, Garlena RA, et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat Med. 2019 May;25(5):730–733. PubMed PMID: 31068712.
- Development of antibacterial drugs for the treatment of nontuberculous mycobacterial disease: U.S. food and drug administration; [cited 2019 April 24]. Available from: https://www.fda.gov/drugs/development-antibacterial-drugs-treatment-nontuberculous-mycobacterial-disease-04082019-04082019