
ABSTRACT
Introduction: Fingolimod, the first oral disease-modifying treatment (DMT) in multiple sclerosis (MS), is a sphingosine 1-phosphate receptor (S1PR) ligand. Approved in 2010, fingolimod has been extensively studied and has been credited with several mechanisms of actions that contribute to its efficacy in MS, among which is the regulation of lymphocyte circulation between the central nervous system and the periphery. Concerns about toxicity, off-target effects, and real-life performance have been raised over time in post-marketing studies of such that next-generation sphingosine-1 phosphate receptor ligands are now being developed.
Areas covered: Herein, the authors expand upon previous systematic reviews obtained via PubMed and through their expert opinion on fingolimod use in clinical practice. Long-term data including long-term efficacy, safety, tolerability, and management especially within growing DMT options and pre-treatment constellation in MS patients are discussed, together with the results of an increased understanding of the chemistry underlying the structure–activity relationship.
Expert opinion: Despite the limitations illustrated in this article, fingolimod still constitutes a paradigm shift in MS treatment. However, although immunomodulation via S1PRs on lymphocytes has represented a major breakthrough in the clinical management of MS, modifying the evolution of progressive MS will likely require the development of approaches other than merely targeting S1PRs.
1. Introduction
Multiple sclerosis (MS) is a demyelinating disorder of the central nervous system (CNS) that typically appears in young adults and continues chronically in older adulthood [Citation1,Citation2]. The long-favored hypothesis in MS implicates autoreactive peripheral T cells that access the CNS, where they persist and induce an inflammatory cascade resulting in myelin destruction, decreased axonal transmission, and eventually neurodegeneration. Clinical symptoms are varied, and can involve any functional neurological system depending on the affected area of the CNS. MS can manifest in different forms, mainly represented by relapsing-remitting MS (RRMS; the most common, affecting about 85% of MS patients at onset and marked by flare-ups of symptoms followed by periods of remission, when symptoms improve or disappear) and primary progressive MS (PPMS; affects approximately 10% of MS patients and continues to worsen gradually from the beginning). Most of the RRMS patients evolve to a secondary progressive course (SPMS) [Citation3].
MS affects approximately more than two million people worldwide (2,221,188 prevalent cases in 2016 [Citation4]). Despite this small global prevalence, MS has been one of the most dynamic segments of the biopharmaceutical market, with the approval of more than five new drugs since 2010 [Citation5]. Among those, fingolimod (FTY720/Gilenya; Novartis) represents the long-searched, disease-modifying drug (DMD) administrable by the oral route. Fingolimod is a highly lipophilic compound that was identified in Japan by a University/industry joint project aimed at improving safety and efficacy of an immunosuppressive metabolite of fungi (). Fingolimod was later on discovered to be a prodrug; in fact, it is metabolized in vivo by the sphingosine kinase (SPHK) enzyme to the active metabolite fingolimod-phosphate (fingolimod-P), a nonselective modulator of sphingosine 1-phosphate receptors (S1PRs). Although not entirely clarified in all molecular events, the S1PR modulatory activity of fingolimod translates into a block of the migration of T lymphocytes from lymph nodes into CNS, thus reducing myelin-specific autoimmune responses. Doing so, fingolimod reduces relapses and delays the progression of disability in RRMS patients.
Figure 1. Timeline of milestones in the discovery and development of fingolimod as disease-modifying drug for MS.
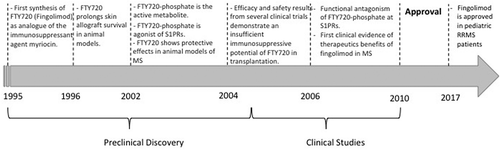
In 2011, when the drug entered the market, fingolimod was considered a true breakthrough in MS therapy, by virtue of the oral administration, the novel mechanism of action, and relative safety. However, the goodness of the fingolimod drug profile has been recently hampered by a more comprehensive analysis of its adverse drug reactions in the post-marketing phase and the discovery of more selective S1PR modulators that may be approved soon.
2. Discovery strategy and preclinical development
2.1. Fingolimod, a myriocin derivative, has a unique mechanism of action
The structure of fingolimod (2-amino-2[2-(4-octylphenyl)ethyl]-1,3-propanediol) was originally designed and synthesized in Japan as a result of joint research efforts among researchers of Kyoto University, Taito and Yoshitomi Pharmaceutical Industries, aimed at developing chemical analogues of the fungal metabolite myriocin (thermozymocidin, ISP-1, 1; ) with reduced toxicity and improved immunosuppressive activity [Citation6]. Early screening protocols were based on the appraisal of the inhibitory effect in experiments of mouse allogeneic mixed lymphocyte reaction (MLR) in vitro and prolonged rat skin graft survival time in vivo [Citation6]. Specifically, a first round of chemical modifications was focused on the simplification of myriocin structure with the removal of side chain functional groups and chiral centers. The first attempt led to investigate the replacement of the carboxylic group with a hydroxymethyl moiety, yielding compound (2a) () that was found to prolong the rat skin survival time and be less toxic than myriocin [Citation7]. Additional simplification of the lead compound 1 led to the synthesis of 2-alkyl-2-amino-l,3-propanediol derivatives (2b-d). These modifications proved successful in increasing immunosuppressive potency as measured by the inhibition of MLR, while slightly reducing toxicity. In particular, the compound bearing an alkyl chain with 14 carbon atoms (2d) proved much more effective both in vitro and in vivo appraisals, and was much less toxic than myriocin. Grounding on these results, a second round of chemical modifications was envisaged to restrict the conformational freedom of the 14-carbon atom side chain by inserting a phenyl ring at different positions, while maintaining the amphiphilicity of the molecule (3a-d; ). The position of the phenyl group in the side chain resulted critical for activity, with the optimal location being at C2 of the side chain. This finding led to disclose the FTY720 compound (fingolimod, 3c) with comparable in vitro activity to 2d, but endowed with a better in vivo immunosuppressive activity. FTY720 (3c) was able to prolong rat skin allograft survival in a dose-dependent manner with no sign of toxicity up to a dose of 10 mg/kg upon intravenous (i.v.) administration.
Figure 2. Discovery of FTY720 (3c) as a result of lead optimization efforts around the chemical structure of myriocin (1). IC50 values are in vitro inhibition data of mouse allogeneic mixed lymphocyte reaction (MLR).
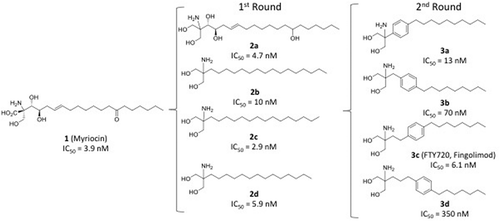
Figure 3. Chemical structures of FTY720 (3c), sphingosine (4) and their phosphate adducts (5, 6).
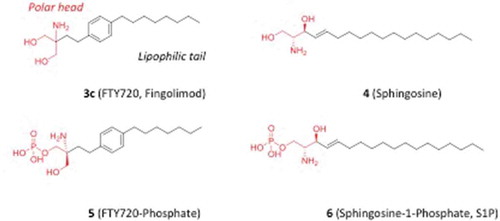
Later on, studies provided first clues on the molecular mechanism of action of these compounds, reporting that myriocin (1) is able to inhibit serine palmitoyltransferase (SPT) [Citation8], an enzyme involved in the biosynthesis of sphingolipids. Strikingly, FTY720 (3c) and compound 2d were found inactive in inhibiting SPT, thereby suggesting a different mechanism of immunosuppressive action for these compounds [Citation9]. The structural similarity between FTY720 (3c) and sphingosine (4) prompted further studies to investigate whether other sphingolipid binding proteins could be targets of FTY720 (3c). As a result, much like sphingosine (4) and the relative product sphingosine 1-phosphate (6, S1P), FTY720 was found to be a substrate for SPHK leading to the formation of FTY720-phosphate (5) [Citation10]. S1P (6) is a nanomolar (nM) agonist of a class of lipid sensing G-protein-coupled receptors (GPCRs), including five members that are termed S1P1–5 receptors. Accordingly, FTY720-phosphate (5) is the active metabolite of FTY720 and a potent agonist of all S1P receptors (S1PR), with the exception of S1P2R () [Citation11–Citation14]. The lack of activity of FTY720-phosphate at the S1P2R is an important feature for its therapeutic benefit. Indeed, S1P2R activation is associated with several unwanted effects, including pathological angiogenesis, vascular leakiness, vasoconstriction, and increased vascular tone [Citation15–Citation18].
Table 1. Binding affinities of FTY720-phosphate (5) and S1P (6) at S1P receptors.
2.2. Pharmacodynamics of fingolimod: from agonist to ‘antagonist’ of SP1Rs
A pioneer work by Brinkmann et al. provided the proof of concept of fingolimod as substrate of SPHK through an in vitro kinase assay and demonstrated that several mouse lymphoid tissues − including Peyer’s patches, spleen, and lymph nodes − contained high level of fingolimod phosphate. Accordingly, the same tissues also matched the RNA localization of SPHK1 [Citation19]. Brinkmann et al. also provided the first evidence that the phosphate metabolite is the active agonist of a family of GPCRs [Citation6] that recognize the endogenous phosphorylated sphingosine 1 (S1P), generated in the intracellular sphingolipid metabolism [Citation11]. Specifically, fingolimod-phosphate (fingolimod-P) − rather than the unphosphorylated molecule − demonstrated high potency agonistic activity at four out of the five members of the S1PR family, expressed in endothelial cells and lymphoid tissues [Citation20]. Competitive binding assay using [33P]-labeled S1P (S133P) on transfected chinese hamster ovary (CHO) cells, expressing each of the five S1PRs, demonstrated that fingolimod-P can displace S1P binding at S1P1R, S1P4R, and S1P5R with an IC50 value in the picomolar and nanomolar range at S1P3, but no activity at S1P2R [Citation12].
Although fingolimod-P acts initially as an agonist at S1PRs, in the longer term its effects are functionally inhibitory, an outcome caused by a prolonged internalization followed by degradation of the receptors. In fact, because of its ability to down-regulate S1P1Rs, FTY720-phosphate has been proposed to act as a functional antagonist rather than classical pharmacological agonist at this class of GPCRs () [Citation21]. This mechanism of action was also proposed to be different from S1P, which does also promote internalization that is, however, followed by recycling and membrane expression of S1P1Rs [Citation22]. Studies in cell lines overexpressing S1PRs demonstrated that treatment with fingolimod caused the receptor to internalize [Citation23]. Similarly, fingolimod causes internalization of S1P1Rs from the cell membrane in lymph node T cells [Citation24]. Moreover, the conditional deletion of S1P1Rs in T lymphocytes and hematopoietic cells mimics in vivo fingolimod effect by decreasing the counts of blood circulating T cells [Citation25]. Therefore, activation of S1PRs by fingolimod-P would alter lymphocyte trafficking by promoting long-term downregulation of S1P1R expression on lymphocytes themselves and inhibition of S1P/S1P1R-dependent lymphocyte egress from secondary lymphoid tissues and thymus [Citation12,Citation25].
Figure 4. Mechanism of action of S1P and fingolimod-P. (a) In physiological conditions, naïve T lymphocytes and memory (both central and effector) T lymphocytes circulate in the blood; the egression from lymph nodes is activated by the signal induced by S1P via S1P1R, overcoming the retention in lymph nodes mediated by the CCR7 receptor (CCR7R) signaling. (b) Fingolimod-P binding induces the internalization, and subsequent degradation, of S1P1R, thus favoring the CCR7R-mediated retention of naïve and central memory T cells in lymph nodes. Effector memory cells lacking CCR7R are not affected by the drug and continue to egress from lymph nodes, ensuring an appropriate immune response, i.e. against microbial and/or danger insults.
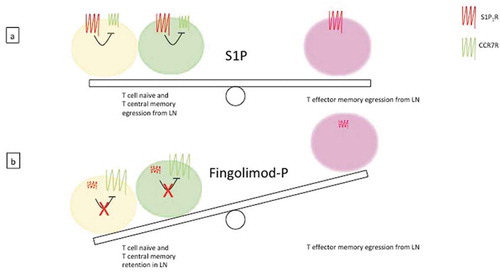
Physiologically, S1P1R and the lymph node-homing CC-chemokine receptor (CCR7) play antagonistic roles. In response to the endogenous ligand SP1, S1P1R signals to counterbalance the retention signals mediated by CCR7 [Citation24]. Therefore, the retention effect of T cells in the lymph node evocated by fingolimod-P treatment may be due to the break of the functional balance between S1P1R and CCR7 activities (). Another mechanism reported for FTY720 involves a sustained activation of S1P1Rs located on the sinus lining the endothelium in the lymph node, which would lead to an increased barrier function and a reduction of lymphocyte transmigration [Citation26–Citation28].
The first phase of in vivo experimentation, conducted between the late 1990s and early 2000s, aimed to demonstrate the efficacy of fingolimod as an immunosuppressant in several animal models of organ transplantation [Citation26,Citation29,Citation30]. However, in the subsequent phases of clinical trials, the drug did not show characteristics that would make its use advisable compared to other well-known immunosuppressants, such as cyclosporine. At the same time, several in vivo preclinical experiments highlighted the efficacy of fingolimod in the management of symptoms and disease progression in animal models of MS. Rat and mouse models of experimental autoimmune encephalomyelitis (EAE) allowed to assess the efficacy of fingolimod both in the prophylaxis and therapy of the disease [Citation31–Citation33], and confirmed the inhibition of T cells recirculating to CNS as one of the mechanism of action of the drug.
In MS, most T cells accumulating in the cerebrospinal fluid (CSF) are ‘central memory’ T cells, endowed with little or no effector functions, but they can be easily restimulated by antigenic stimuli, and can proliferate and differentiate in ‘effector memory’ T cells [Citation34]. It has been demonstrated that fingolimod selectively reduces naïve and central memory T cells, but not effector memory T cells circulating in the blood, irrespective of their T helper (Th) phenotype (i.e. Th1, Th2, or Th17) (). Thus, the selective retention of central memory T cells in lymph nodes by fingolimod seems to be sufficient to control central inflammation and, at the same time, would spare peripheral effector T cells involved in the protection against infections.
Fingolimod has also been shown to affect the functions of B cells in MS patients. More specifically, MS patients treated with fingolimod have reduced numbers of circulating B cells, in which, however, there is an increased proportion of a naïve B-cell subset, i.e. transitional B cells, with anti-inflammatory properties [Citation35]. Recently, the expansion of transitional B cells in MS patients under fingolimod treatment has been ascribed to increased levels of the B cell-activating factor of the tumor necrosis factor family (BAFF) [Citation36]. Because patients treated with fingolimod do not show signs of memory B cell or plasma cell activation, the effects of fingolimod on B cells would be favorable for the treatment of MS.
More in-depth studies further elucidated and explained fingolimod therapeutic effects, involving not only a slowdown of the progression of the pathology, but also mechanisms of repair, remyelination, and reduction of the neuroinflammation in the subjects treated with the drug. It was demonstrated that fingolimod restores the permeability of the blood-brain barrier, and consequently neuronal functions, in rats with EAE [Citation37]. These effects could be explained considering that endothelial barrier permeability can be modulated by S1P1R and that, in an inflammatory context, the expression of those receptors in endothelial cells is upregulated [Citation38]. The S1P1R internalization induced by fingolimod could contribute to the maintenance of the physiologic permeability of the endothelial barriers. A relationship between S1P levels in CSF and the worsening of the disease-related disability in patients with MS has been thoroughly demonstrated [Citation39], as well as an increase in S1P levels in the spinal cord of mice with EAE [Citation40]. Moreover, S1P1R knocked-out mice show reduced EAE symptoms and slow disease progression, and fingolimod is capable to induce the internalization of the receptor in astrocytes [Citation40]. Altogether, these data could give a possible explanation for the therapeutic effect of fingolimod in EAE models and, subsequently, in MS patients and support the evidence of remyelination and restored communication between astrocytes and other neuronal cells found in preclinical animal models [Citation41].
Experimental evidence increasingly supports a direct action of fingolimod on neural cells within the CNS, providing protection against the neurodegenerative component of MS [Citation42]. In fact, S1P receptors, particularly S1P1R, are expressed by astrocytes, oligodendrocytes, neurons, and microglia cells. Fingolimod affects each of these cells in ways relevant to MS pathology. In astrocytes, the drug attenuates astrogliosis, a histopathological feature of EAE, and the production of proinflammatory cytokines. In oligodendrocytes, it promotes cell survival and enhances remyelination. In neurons, fingolimod reduces dendritic spine loss, protects from excitotoxic cell death, and restores functions, whereas, in microglia, it reduces cell activation [Citation42]. Therefore, as a whole, the bulk of available data would suggest that fingolimod restores an appropriate balance between mechanisms of damage and repair in CNS [Citation43].
2.3. Pharmacokinetic profile
The therapeutic effects of fingolimod in animal models were achieved using a dose of 0.3 mg/kg; these data provided fundamental clues for the dose selection in humans, in which similar levels of the phosphorylated and unphosphorylated drug in the blood (7.4 versus 5.4 ng/ml in plasma and 0.23 versus 0.07 ng/ml in CSF, respectively) were reached with a dose of 0.5–1.25 mg of fingolimod/day [Citation44]. A detailed description of the pharmacokinetic features of fingolimod is provided in the report approved by the Food and Drug Administration (FDA; NDA22-527). This submission summarizes the results obtained from in vitro and in vivo pharmacokinetic and pharmacodynamic studies; more in details, 56 human studies, including 31 specific studies of clinical pharmacology, have been considered, collectively involving more than 1,000 subjects. Human studies were conducted in healthy volunteers compared to renal transplanted patients, and patients with moderate or severe hepatic or renal impairment. The overall data allowed to identify the use of a dosage of 0.5 mg fingolimod, administered daily and orally in the form of hard capsules. Notably, the possibility of oral administration represents a relevant feature that differentiates fingolimod from the other DMDs commonly used for the treatment of RRMS. Fingolimod is slowly but efficiently absorbed, the maximal plasma concentration being reached after 12–16 h post-oral dose, irrespective of food intake and with a high bioavailability (93%). Upon daily dosing, steady-state concentrations are reached after 1–2 months. More than 99% of fingolimod binds to plasma proteins, mainly to albumin; the drug has a large volume of distribution (approximatively 20 l/kg), and shows slow blood clearance (6.3 ± 2.3 l/h), with a half-life of 6–9 days [Citation45].
Fingolimod is a prodrug, which requires SPHK to become phosphorylated in its active form, the S-enantiomer fingolimod-P. While both SPHK subtypes, i.e. SPHK1 and SPHK2, can phosphorylate fingolimod in vitro [Citation46], it has very recently been demonstrated that only the SPHK2 enzyme is necessary for the in vivo activation of the drug. In fact, in SPHK2 but not SPHK1 knockout mice, fingolimod fails to protect from disease symptoms and progression in a model of EAE [Citation47]. In the blood, fingolimod and fingolimod-P are found in a stable equilibrium, since they possess similar elimination kinetics. Besides phosphorylation, fingolimod can undergo two different biotransformations, one mediated by the cytochrome P450 isoform CYP4F2 and the other mediated by a still unknown acyltransferase. Because the CYP4F2 oxidative system is not known as being involved in the metabolism of other drugs, the interaction of fingolimod with other drugs seems unlikely.
Fingolimod has a rapid onset, and within hours of its first dose, the peripheral lymphocyte count decreases in a dose-dependent fashion. With daily dosing, a stable blood concentration of the drug as well as a stable reduction of circulating lymphocytes can be observed. The number of circulating peripheral blood lymphocytes slowly increases within few days of fingolimod discontinuation, and returns to normal range within 6 weeks [Citation48].
2.4. Toxicologic profile
The adverse events seen most frequently in patients with RRMS treated with 0.5 mg fingolimod daily for 24 months are lymphopenia, herpes viral infections, increased liver transaminases, hypertension, initial bradycardia, and first-degree atrioventricular block. The reduction of the peripheral blood lymphocyte count, which is an obvious indication for a positive biological response, is instead considered as adverse event when the count falls below 0.2 × 109 lymphocytes per liter [Citation45]. In a clinical trial known as TRANSFORMS (see below), herpes infections mainly occurred in patients receiving 1.25 mg fingolimod. Nevertheless, those infections might depend on the concomitant use of high-dose steroids to treat relapses and are not easily explained on the basis of the mechanism of action of the drug [Citation49]. As for the higher incidence of herpes infections, the mechanism underlying the increase of hepatic enzymes remains unclear, and this adverse event does not have a correspondence in any animal experiment, even using doses higher than those given to patients with MS [Citation50]. On the contrary, the effects on the cardiovascular system are easily attributable to the fingolimod’s mechanism of action, i.e. modulation of S1P1R. Bradycardia and slowing of the atrioventricular conduction could occur because of a short-term activation of the Gαi protein-gated potassium channel in atrial myocytes, before internalization and/or desensitization of S1P1Rs. Animal data suggested that the transient reduction in heart rate produced by fingolimod may involve mainly S1P3Rs. However, there are species-related differences that may change the relative importance of the individual S1PRs in the cardiovascular effects of fingolimod, particularly on heart rate and atrioventricular conduction, in humans and mice [Citation51]. In fact, there is a higher expression of the S1P1R subtype in human ventricular, septal, and atrial cardiomyocytes, compared to rodents [Citation52,Citation53]. Moreover, a mild prolongation of the QT interval by the drug has also been observed in MS patients treated with the drug [Citation54]. Analyses of pooled safety data from various clinical studies also revealed a potential risk of macular edema in subjects with coexisting diabetes or uveitis [Citation55,Citation56]. Additional adverse effects include reduced lung and renal function, which was noticed in phase II trials with renal transplant recipients treated with fingolimod (5 mg daily) in combination with other immunosuppressive drugs [Citation57]. The combined data from the phase III trials did not suggest any increase of the incidence of malignancies associated with fingolimod treatment [Citation49]. Overall, fingolimod appears to be effective and well tolerated, with a favorable safety profile at the dose of 0.5 mg; the above mentioned adverse events are generally reversible after drug discontinuation. However, fingolimod cessation also requires a careful attention, because it can lead to a ‘rebound syndrome,’ i.e. potent reactivation of lymph node egress of effector T cells. The mechanism underlying this phenomenon is still unclear, but it could be related to the repopulation of the circulating pool of immune cells by the different T cell subsets with different kinetics [Citation58].
3. Clinical development
Long-term data of initial clinical trials and post-marketing evaluations including long-term efficacy, safety, tolerability, and management especially within growing disease-modifying treatment options and pre-treatment constellation in MS patients have recently and critically been discussed in detail in 2017 [Citation59]. An initial evaluation does actually date back to 2016, some six years after its approval for the treatment of patients with RRMS. While it had been designed to reduce the frequency of exacerbations and to delay disability worsening, issues on its safety and efficacy, mainly as compared to other DMDs, were being raised [Citation60]. That initial survey aimed at assessing the safety and benefit of fingolimod versus placebo, or other DMDs, in reducing disease activity in people with RRMS, by searching the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System (CNS) Group’s Specialised Trials Register and US Food and Drug Administration (FDA) reports, as of February 2016. The selection criteria involved randomized controlled trials (RCTs) assessing the beneficial and harmful effects of fingolimod versus placebo or other approved DMDs in people with RRMS, and data were analyzed according to standard methodological procedures as expected by Cochrane [Citation60]. The main conclusions of the study were that treatment with fingolimod compared to placebo in RRMS patients was effective in reducing inflammatory disease activity, but it might lead to little or no difference in preventing disability worsening. The risk of withdrawals due to adverse events would call for careful monitoring of patients over time. The evidence on the risk/benefit profile of fingolimod compared with intramuscular interferon-β1a was uncertain, based on a low number of head-to-head RCTs with short follow-up duration, implicitly requiring the need for further trial results in order to satisfy those issues [Citation60]. On a negative note, the Authors found a likelihood of participants discontinuing fingolimod, as compared to other DMDs, due to adverse events in the short term (6 months) (RR 3.21, 95% CI 1.16 to 8.86), but there was no significant difference versus interferon-β1a at 12 months (RR 1.51, 95% CI 0.81 to 2.80; moderate quality evidence). A higher incidence of adverse events was taken as suggestive of the lower tolerability rate of fingolimod compared to interferon-β1a.
In 2017, a subsequent study conducted a PRISMA-compliant systematic review of the literature (cut-off date: 4 March 2016). Published papers reporting real-world data for fingolimod with regard to clinical outcomes, persistence, adherence, health-care costs, healthcare resource use, treatment patterns, and patient-reported outcomes that met all the eligibility criteria were included for data extraction and quality assessment [Citation61]. Based on 34 included studies, this analysis found that fingolimod treatment improved outcomes compared to the period before treatment initiation and was more effective than interferons or glatiramer acetate (GA). However, among studies comparing fingolimod with natalizumab, overall trends were inconsistent: some reported natalizumab to be more effective than fingolimod and others reported similar effectiveness for natalizumab and fingolimod. These studies were taken as emphasizing the challenges of investigating MS in the real world, including the subjectivity in evaluating some clinical outcomes and the heterogeneity of methodologies used and patient populations investigated, which limit comparisons across studies. Gaps in available real-world evidence for MS were also highlighted, including those relating to patient-reported outcomes, combined clinical outcomes (to measure overall treatment effectiveness), and health-care costs/resource use. Overall, the survey was considered as providing good evidence of the real-world effectiveness of fingolimod and highlighted the diversity of methodologies used to assess treatment benefit in clinical practice, while, again, advocating future studies for addressing the evidence gaps found in the literature and the challenges associated with researching MS when designing real-world studies, assessing data, and comparing evidence across studies [Citation61].
In addition to covering topics as diverse as chemistry, regulatory affairs, pharmacokinetics and metabolism, mechanism of action and S1P receptor modulation, and S1P receptor modulation in CNS, a paper in 2017 provided the most comprehensive update by that time on clinical efficacy/effectiveness of fingolimod for the treatment of RRMS [Citation59]. The study examined three large phase 3 clinical trials () that, overall, demonstrated superiority of fingolimod versus placebo respective fingolimod versus interferon β-1a regarding clinical and magnetic resonance imaging (MRI) disease activity [Citation49,Citation62–Citation64]. These effects on clinical and MRI parameters were observed early after fingolimod initiation. Compared to placebo (FREEDOMS I and II trial) and interferon β-1a (TRANSFORMS trial), 0.5 and 1.2 mg dosing group presented significant lower annualized relapse rate and significant fewer T2 weighted or gadolinium-enhancing lesions on T1 weighted images. However, the secondary end point of confirmed disability progression documented by confirmed EDSS (Expanded Disability Status Scale) change was only met in the FREEDOMS I, but not TRANSFORMS or FREEDOMS II trial. Additional evaluation of FREEDOMS and FREEDOMS II data as to the status of no evidence of disease activity (NEDA) showed that 31.0% of fingolimod-treated patients achieved NEDA-3 status (relapses, MRI activity, disability progression) compared to 9.9% of placebo patients. Using NEDA-4 (additional criteria of 0.4% brain volume loss), 19.7% of fingolimod patients achieved NEDA-4 compared to 5.3% on placebo. Taken as a whole, the major conclusions of the survey were that ‘There is an extensive long-term experience on fingolimod use in clinical practice demonstrating the favorable benefit-risk of this drug. Using a defined risk management approach, experienced MS clinicians should apply fingolimod after critical choice of patients and review of clinical aspects. Further studies are essential to discuss additional benefit in progressive forms in MS’ [Citation59].
Table 2. Features of key phase 3 trials of fingolimod.
4. Post-launch of fingolimod
In September 2010, fingolimod (0.5 mg) was approved by FDA as first oral treatment for adults with RRMS. After just 3 months from fingolimod launch, more than 2,000 patients had been treated with the drug with more than 13 million US$ sales. In May 2018, FDA approved the extension of the use of fingolimod to children and adolescents (10–17 years of age) with RRMS. In January 2011, the European Medicines Agency (EMA) expressed a positive opinion for the second-line use of fingolimod in patients with RRMS and first-line use in rapidly evolving, severe RRMS. The drug was eventually approved by the European Commission in March of the same year. In September 2011, fingolimod was approved in Japan to prevent relapses and to delay the progression of physical disability in adults with MS. Fingolimod became available in Canadian pharmacies in April 2011.
4.1. Post-marketing data
The largest real-world data program of fingolimod is PANGAEA, a noninterventional study aimed at assessing prospectively the persistence, effectiveness, and safety of the drug over 36 months in patients with RRMS in Germany. In the most recent analysis [Citation67], patients (N = 2,537) were required to have received fingolimod for the first time in PANGAEA, to have at least 12 months of data, and to have completed each a 12-month follow-up period. For the safety analysis (N = 3,266), patients were additionally allowed to have received fingolimod before enrollment. The results showed sustained effectiveness, high persistence, and manageable safety profile of fingolimod over 36 months. Although the collected data may reflect the use of fingolimod in a specific population (i.e. German) and may not be generalizable to other countries (where fingolimod is mainly used as first-line therapy), the PANGAEA study highlighted the potential of fingolimod as a DMD for the long-term management of patients with RRMS. An ongoing German study, PANGAEA 2.0, aims to assess the benefits of a treatment change to fingolimod in patients (N = 1,500) identified as not responding to or having treatment failure with their current therapy [Citation68]. Therefore, this study may expand the existing safety and efficacy profile of fingolimod. In another real-world study, fingolimod treatment was associated with reduced relapse and MRI activity, and an improved EDSS score (N= 175) [Citation69]. When compared to β-interferons plus GA (PEARL study [Citation70]) or dimethyl fumarate (DMF) [Citation71], fingolimod demonstrated similar efficacy. Further reports presented disease reactivation with severe neurological symptoms and MRI activity following fingolimod withdrawal [Citation72]. However, evaluating the efficacy of natalizumab versus fingolimod in active RRMS leads to controversial results [Citation73,Citation74].
Other groups argued that the results obtained from real-world studies should be interpreted with caution because of the bias introduced by patient dropouts due to adverse events, lack of efficacy, or termination of a study before all patients had reached a defined treatment duration [Citation75]. As a matter of fact, only 60% of the intent-to-treat patients completed the Phase III fingolimod extension studies [Citation59]. Long-term studies (i.e. of at least 10 years) are needed to obtain a more compelling profile of fingolimod performance in the real world.
4.2. Safety and pharmacovigilance
Safety is an important concern in long-term therapy with DMDs in MS. A systematic review and network analysis post-marketing indicated a percentage of 82% of patients that discontinues the fingolimod treatment for adverse events, although the severity of these events appears of a lesser extent as compared to several other MS drugs. The most important safety concern of fingolimod is cardiac toxicity, which includes increased risk of bradycardia and prolonged QT interval. These effects are probably caused by the activation of S1P3R, a receptor associated with heart rate modulation in mice [Citation76,Citation77]. In contrast, S1P1R and S1P5R activation would responsible for the beneficial effects in MS. Although peripheral blood lymphocyte counts decline by 73% from baseline values within 1 month of drug initiation, consistent with the pharmacodynamic action of fingolimod, serious or opportunistic infections have been infrequently observed in the post-marketing setting. However, deaths caused by primary disseminated varicella zoster infection or herpes simplex encephalitis have been reported at the dose of 1.25 mg/day [Citation78]. To date, no correlation has been shown between absolute lymphocyte counts and the incidence of serious or opportunistic infections. Overall, incidence rates of the reported adverse events are consistent with the known safety profile of fingolimod, except for cryptococcal infections and progressive multifocal leukoencephalopathy (PML), which emerged in the postmarketing setting, including pharmacovigilance studies ().
Table 3. Postmarketing reports of fingolimod safety.
PML is a rare opportunistic infection of the CNS caused by reactivation and the presence of mutated forms of a latent John Cunningham virus (JCV) [Citation84]. In 2005, PML was confirmed in three patients participating in natalizumab clinical trials of MS and Crohn disease [Citation85–Citation87], disorders that were not previously associated with PML. In August 2017, hundreds of cases of PML associated with natalizumab were confirmed (Tysabri Safety Update), thus raising the possibility of a general, increased risk of PML in patients with MS treated with immunosuppressive or immunomodulatory agents [Citation88]. Since 2015, PML cases not attributed to prior exposure to immunosuppressants have thus been reported in fingolimod-treated patients. However, a higher risk/occurrence of PML was observed in patients who had switched from natalizumab to fingolimod and/or were positive for anti-JCV antibodies [Citation89]. Although the estimated risk of PML with fingolimod treatment is quite low (i.e. 0.069 per 1,000 patients; 95% confidence interval: 0.039–0.114) [Citation88], vigilance toward PML is required for all patients treated with this drug. The US prescribing information has been in fact updated to include opportunistic infections, specifically mentioning PML. The known mechanism of action of fingolimod does not provide a convincing causal link for the induction of PML. The drug prevents the egress of CCR7+ naive T cells but not of CCR7− effector memory T cells from lymph nodes [Citation90]. Therefore, because of its partial sequestration of T cells, fingolimod may contribute to reduce immune responses to JCV reactivation. However, a full characterization of JCV in fingolimod-associated PML is needed in order to better understand the disease pathogenesis with fingolimod.
4.3. Studies on market and main competitors
In 2016, worldwide sales of fingolimod were 3.1 billion US$, representing one of the highest percentages (i.e. 14.0%) of the global market for MS drugs together with DMF (18.1%) and GA (19.0%) [Citation5]. However, a consensus forecast for 2022 would indicate a drop of fingolimod sales to 1.3 billion US$ (5.6% of the global MS market), mostly imputable to the approval of new drugs, including ocrelizumab (Ocrevus; Roche/Genentech) and daclizumab (Zinbryta; Biogen and AbbVie), as well as the anticipated introduction of less expensive generic fingolimod [Citation5]. Ocrelizumab, a CD20-specific monoclonal antibody that leads to B-cell depletion, is the first DMD approved for both RRMS and PPMS (March 2017 by FDA and January 2018 by EMA). Daclizumab (a blocker of IL-2 receptors containing the CD25 subunit), however, was withdrawn from the market in 2018, because of mounting concerns about its safety, i.e. severe liver damage and immune-related conditions. Therefore, although very infrequently prescribed, the disappearance of daclizumab as a valid competitor may reset, at least in part, the forecast for 2022 sales of MS drugs, including fingolimod. Regarding novel S1PR-selective drugs that lack the cardiotoxic effects of fingolimod, siponimod (selective for S1P1R; Novartis) recently received regulatory approval, whereas the application for ozanimod (selective agonist of S1P1R and S1P5R) approval has been recently submitted by Celgene to FDA (March 2019). Another selective agonist of S1P1R, i.e. ponesimod (Actelion), is on its way in completing phase 3 clinical studies.
Therefore, although representing a breakthrough in MS therapy as the first oral DMD, fingolimod incomes may considerably decrease over the next years. Nevertheless, the possibility of a forthcoming entrance of several selective S1PR agonists in MS therapy further underlines the importance of S1PRs as valuable target in DMTs of MS.
5. Expert opinion
Given the great complexity of the disease, tackling MS is a very hard task. In fact, MS involves several types of cells, mostly nervous and immune in nature, and multiple pathogenetic mechanisms. The main therapeutic approach in containing MS has been immunosuppression, with the aim of reducing the inflammatory damage provoked by immune cells inside CNS. As per their definition, i.e. drugs that lower adaptive immune responses, immunosuppressive agents impair activation and proliferation of T and of B lymphocytes. However, given their nonselective mode of action, such drugs are unavoidably accompanied by significant adverse effects. As a selective adhesion-molecule inhibitor and thus capable of preventing the entrance of activated lymphocytes in CNS, natalizumab provided a new strategy that could well accommodate the therapeutic needs of a neuroinflammatory, autoimmune disease. In this scenario, the appearance of a small molecule such as fingolimod, whose true mechanism of action was identified after its discovery, was absolutely timely. In fact, fingolimod would act even earlier than natalizumab, i.e. by blocking the lymph node egression of autoreactive lymphocytes on their way to CNS. Thus, although born to be ‘simply’ a better immunosuppressant than its parental molecule (i.e. myriocin), fingolimod was the forerunner in the discovery of a completely new mechanism.
The beneficial effects of fingolimod in MS therapy are currently thought to be due to the action of drug on preventing the egression of lymphocytes from lymphoid tissue into the circulation, thereby sparing the CNS from attack by myelin-reactive lymphocytes. Yet, the data obtained so far are still inconclusive as to its overall mechanisms of action, which appear now to be far more complex than previously anticipated. In vivo, fingolimod exerts – at least in part – immunomodulating effects through binding S1PRs on lymphocytes. Although S1PRs are found with the highest density in leucocytes and lymphoid tissue, they are also widely expressed in many cell types in other organs systems, including the heart, brain, liver, stomach, and probably also in the retina. This ubiquitous nature of the target receptor for fingolimod accounts for the wide range of adverse effects, including hypertension, heart block, bradycardia, and macular edema. Refinement of its clinical use along as well as the search for analogs with improved tolerability are both the subject of ongoing research. Fulminant multifocal relapse in a fingolimod-treated multiple sclerosis patient has also been described.
A recently published primary analysis of severe adverse events in new users of disease-modifying drugs (DMDs) – including fingolimod, natalizumab, glatiramer acetate, and IFNβ1a – did not reveal significant differences in incidence rates among individual DMDs [Citation91]. In general, the observed absolute number of adverse events was low, especially for those with low background incidence in the overall population, such as lymphoma, stroke or PML, as well as for natalizumab and fingolimod, as newer DMDs with small samples. This finding is of particular clinical relevance, since discontinuation of the latter drugs due to safety concerns has been associated with severe rebound effects that have to be balanced against the possible risk of severe adverse events, as mentioned above. The issue of balanced efficacy and tolerability as revealed by drug-use patterns and severe adverse events with DMDs in patients with MS remains critical, with a definite need for appropriate refinement of current therapies.
The FDA has approved siponimod for oral treatment of adults with relapsing forms of MS, including clinically isolated syndrome (initial neurological episode), relapsing-remitting disease, and active SPMS. Siponimod is the second S1PR modulator to be approved in the US; fingolimod, which is approved for oral treatment of RRMS in patients ≥10 years old, was the first. Siponimod is under regulatory review in the EU and Japan for SPMS. Overall, although on the surface the development of new inflammatory CNS lesions in MS may appear consistent with a primary recruitment of peripheral immune cells, questions have been raised as to whether lymphocyte and/or monocyte invasion into the brain are really at the root of inflammatory lesion development. Such a concept is discussed in the context of the EXPAND trial, showing that siponimod exerts anti-inflammatory and neuroprotective activities in SPMS patients. The verification or rejection of such a concept is vital for the development of new therapeutic strategies for progressive MS.
Major therapeutic achievements have been made concerning the relapsing phase but modifying the evolution of progressive MS remains an unmet need. A recent survey led to the conclusion that siponimod may reduce the activity of the disease and has a modest effect on the gradual disability accrual [Citation92]. It is possible that, although immunomodulation through binding S1PRs on lymphocytes has been representing a major breakthrough in the clinical management of MS, modifying the evolution of progressive MS will require the development of approaches other than merely targeting S1PRs.
In conclusion, despite the limitations illustrated in the paragraphs above, fingolimod still constitutes a paradigm shift in treatment optimization for MS. It might represent a starting point for both an improved understanding of the pathogenesis of the disease and the development of new classes of drugs, thus resulting in optimization of therapy in individual patients.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
One referee declares personal compensation for consulting for Alkermes, Biogen, Convelo, EMD Serono, ERT, Gossamer Bio, Novartis, and ProValuate. They are also the recipient of speaker’s fees for Mylan and Synthon and declare that they serve as the editor of a multiple sclerosis themed journal. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Article highlights
Fingolimod is the first disease-modifying drug administrable orally for relapsing-remitting multiple sclerosis.
Fingolimod was instrumental in the discovery of S1PRs.
The molecular basis of the S1PR modulatory effects of fingolimod is still unclear.
Long-term (of at least 10 years) clinical studies are needed to evaluate more carefully the efficacy and safety profile of fingolimod.
In the future, the value of fingolimod may decrease with the entrance in the market of more selective, orally administrable S1PR modulators.
This box summarizes the key points contained in the article.
Additional information
Funding
References
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8(9):913–919.
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517.
- Noseworthy JH, Lucchinetti C, Rodriguez M, et al. Multiple sclerosis. N Engl J Med. 2000;343(13):938–952.
- Collaborators GBDMS. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(3):269–285.
- Westad A, Venugopal A, Snyder E. The multiple sclerosis market. Nat Rev Drug Discov. 2017;16(10):675–676.
- Brinkmann V, Pinschewer D, Chiba K, et al. FTY720: a novel transplantation drug that modulates lymphocyte traffic rather than activation. Trends Pharmacol Sci. 2000;21(2):49–52.
- Fujita T, Hamamichi N, Kiuchi M, et al. Determination of absolute configuration and biological activity of new immunosuppressants, mycestericins D, E, F and G. J Antibiot (Tokyo). 1996;49(9):846–853.
- Miyake Y, Kozutsumi Y, Nakamura S, et al. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun. 1995;211(2):396–403.
- Fujita T, Hirose R, Yoneta M, et al. Potent immunosuppressants, 2-alkyl-2-aminopropane-1,3-diols. J Med Chem. 1996;39(22):4451–4459.
- Paugh SW, Payne SG, Barbour SE, et al. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003;554(1–2):189–193.
- Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–21457.
- Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–349.
- Sanchez T, Estrada-Hernandez T, Paik JH, et al. Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability. J Biol Chem. 2003;278(47):47281–47290.
- Hale JJ, Lynch CL, Neway W, et al. A rational utilization of high-throughput screening affords selective, orally bioavailable 1-benzyl-3-carboxyazetidine sphingosine-1-phosphate-1 receptor agonists. J Med Chem. 2004;47(27):6662–6665.
- Skoura A, Hla T. Regulation of vascular physiology and pathology by the S1P2 receptor subtype. Cardiovasc Res. 2009;82(2):221–228.
- Skoura A, Sanchez T, Claffey K, et al. Essential role of sphingosine 1-phosphate receptor 2 in pathological angiogenesis of the mouse retina. J Clin Invest. 2007;117(9):2506–2516.
- Ohmori T, Yatomi Y, Osada M, et al. Sphingosine 1-phosphate induces contraction of coronary artery smooth muscle cells via S1P2. Cardiovasc Res. 2003;58(1):170–177.
- Peter BF, Lidington D, Harada A, et al. Role of sphingosine-1-phosphate phosphohydrolase 1 in the regulation of resistance artery tone. Circ Res. 2008;103(3):315–324.
- Puneet P, Yap CT, Wong L, et al. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328(5983):1290–1294.
- Hla T, Lee MJ, Ancellin N, et al. Lysophospholipids–receptor revelations. Science. 2001;294(5548):1875–1878.
- Sanna MG, Wang SK, Gonzalez-Cabrera PJ, et al. Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat Chem Biol. 2006;2(8):434–441.
- Jo E, Sanna MG, Gonzalez-Cabrera PJ, et al. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12(6):703–715.
- Graler MH, Goetzl EJ. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. Faseb J. 2004;18(3):551–553.
- Pham TH, Okada T, Matloubian M, et al. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity. 2008;28(1):122–133.
- Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360.
- Brinkmann V, Lynch KR. FTY720: targeting G-protein-coupled receptors for sphingosine 1-phosphate in transplantation and autoimmunity. Curr Opin Immunol. 2002;14(5):569–575.
- Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4(7):1019–1025.
- Baumruker T, Billich A, Brinkmann V. FTY720, an immunomodulatory sphingolipid mimetic: translation of a novel mechanism into clinical benefit in multiple sclerosis. Expert Opin Investig Drugs. 2007;16(3):283–289.
- Suzuki S, Li XK, Enosawa S, et al. A new immunosuppressant, FTY720, induces bcl-2-associated apoptotic cell death in human lymphocytes. Immunology. 1996;89(4):518–523.
- Yanagawa Y, Sugahara K, Kataoka H, et al. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. II. FTY720 prolongs skin allograft survival by decreasing T cell infiltration into grafts but not cytokine production in vivo. J Immunol. 1998;160(11):5493–5499.
- Fujino M, Funeshima N, Kitazawa Y, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305(1):70–77.
- Webb M, Tham CS, Lin FF, et al. Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. J Neuroimmunol. 2004;153(1–2):108–121.
- Kataoka H, Sugahara K, Shimano K, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2(6):439–448.
- Kivisakk P, Mahad DJ, Callahan MK, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55(5):627–638.
- Grutzke B, Hucke S, Gross CC, et al. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann Clin Transl Neurol. 2015;2(2):119–130.
- Miyazaki Y, Niino M, Takahashi E, et al. Fingolimod induces BAFF and expands circulating transitional B cells without activating memory B cells and plasma cells in multiple sclerosis. Clin Immunol. 2018;187:95–101.
- Balatoni B, Storch MK, Swoboda EM, et al. FTY720 sustains and restores neuronal function in the DA rat model of MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull. 2007;74(5):307–316.
- Igarashi J, Erwin PA, Dantas AP, et al. VEGF induces S1P1 receptors in endothelial cells: implications for cross-talk between sphingolipid and growth factor receptors. Proc Natl Acad Sci U S A. 2003;100(19):10664–10669.
- Kulakowska A, Zendzian-Piotrowska M, Baranowski M, et al. Intrathecal increase of sphingosine 1-phosphate at early stage multiple sclerosis. Neurosci Lett. 2010;477(3):149–152.
- Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108(2):751–756.
- Rouach N, Pebay A, Meme W, et al. S1P inhibits gap junctions in astrocytes: involvement of G and Rho GTPase/ROCK. Eur J Neurosci. 2006;23(6):1453–1464.
- Hunter SF, Bowen JD, Reder AT. The direct effects of fingolimod in the central nervous system: implications for relapsing multiple sclerosis. CNS Drugs. 2016;30(2):135–147.
- Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158(5):1173–1182.
- Foster CA, Howard LM, Schweitzer A, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323(2):469–475.
- Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: recent findings and new perspectives. Pharmacol Ther. 2018;185:34–49.
- Billich A, Bornancin F, Devay P, et al. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278(48):47408–47415.
- Imeri F, Schwalm S, Lyck R, et al. Sphingosine kinase 2 deficient mice exhibit reduced experimental autoimmune encephalomyelitis: resistance to FTY720 but not ST-968 treatments. Neuropharmacology. 2016;105:341–350.
- Tedesco-Silva H, Mourad G, Kahan BD, et al. FTY720, a novel immunomodulator: efficacy and safety results from the first phase 2A study in de novo renal transplantation. Transplantation. 2004;77(12):1826–1833.
- Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415.
- Tawadrous MN, Mabuchi A, Zimmermann A, et al. Effects of immunosuppressant FTY720 on renal and hepatic hemodynamics in the rat. Transplantation. 2002;74(5):602–610.
- Snelder N, Ploeger BA, Luttringer O, et al. Characterization and prediction of cardiovascular effects of fingolimod and siponimod using a systems pharmacology modeling approach. J Pharmacol Exp Ther. 2017;360(2):356–367.
- Forrest M, Sun SY, Hajdu R, et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309(2):758–768.
- Mazurais D, Robert P, Gout B, et al. Cell type-specific localization of human cardiac S1P receptors. J Histochem Cytochem. 2002;50(5):661–670.
- Pilote S, Simard C, Drolet B. Fingolimod (Gilenya((R))) in multiple sclerosis: bradycardia, atrioventricular blocks, and mild effect on the QTc interval. Something to do with the L-type calcium channel? Fundam Clin Pharmacol. 2017;31(4):392–402.
- Moss HE. Visual consequences of medications for multiple sclerosis: the good, the bad, the ugly, and the unknown. Eye Brain. 2017;9:13–21.
- Zarbin MA, Jampol LM, Jager RD, et al. Ophthalmic evaluations in clinical studies of fingolimod (FTY720) in multiple sclerosis. Ophthalmology. 2013;120(7):1432–1439.
- Budde K, Schutz M, Glander P, et al. FTY720 (fingolimod) in renal transplantation. Clin Transplant. 2006;20(Suppl 17):17–24.
- Hatcher SE, Waubant E, Graves JS. Rebound syndrome in multiple sclerosis after fingolimod cessation-reply. JAMA Neurol. 2016;73(11):1376.
- Thomas K, Proschmann U, Ziemssen T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert Opin Pharmacother. 2017;18(15):1649–1660.
- La Mantia L, Tramacere I, Firwana B, et al. Fingolimod for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev. 2016;4:CD009371.
- Ziemssen T, Medin J, Couto CA, et al. Multiple sclerosis in the real world: A systematic review of fingolimod as a case study. Autoimmun Rev. 2017;16(4):355–376.
- Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
- Khatri B, Barkhof F, Comi G, et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol. 2011;10(6):520–529.
- Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–556.
- Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–1084.
- Chitnis T, Arnold DL, Banwell B, et al. Trial of fingolimod versus interferon beta-1a in pediatric multiple sclerosis. N Engl J Med. 2018;379(11):1017–1027.
- Ziemssen T, Lang M, Tackenberg B, et al. Real-world persistence and benefit-risk profile of fingolimod over 36 months in Germany. Neurol Neuroimmunol Neuroinflamm. 2019;6(3):e548.
- Ziemssen T, Kern R, Cornelissen C. Study design of PANGAEA 2.0, a non-interventional study on RRMS patients to be switched to fingolimod. BMC Neurol. 2016;16:129.
- Al-Hashel J, Ahmed SF, Behbehani R, et al. Real-world use of fingolimod in patients with relapsing remitting multiple sclerosis: a retrospective study using the national multiple sclerosis registry in Kuwait. CNS Drugs. 2014;28(9):817–824.
- Alsop J, Medin J, Cornelissen C, et al. Two studies in one: A propensity-score-matched comparison of fingolimod versus interferons and glatiramer acetate using real-world data from the independent German studies, PANGAEA and PEARL. PloS One. 2017;12(5):e0173353.
- Braune S, Grimm S, van Hovell P, et al. Comparative effectiveness of delayed-release dimethyl fumarate versus interferon, glatiramer acetate, teriflunomide, or fingolimod: results from the German neuroTransData registry. J Neurol. 2018;265(12):2980–2992.
- Andrade C. Rebound syndrome in multiple sclerosis after fingolimod cessation. JAMA Neurol. 2016;73(11):1375–1376.
- Bergvall N, Lahoz R, Reynolds T, et al. Healthcare resource use and relapses with fingolimod versus natalizumab for treating multiple sclerosis: a retrospective US claims database analysis. Curr Med Res Opin. 2014;30(8):1461–1471.
- Kalincik T, Brown JWL, Robertson N, et al. Treatment effectiveness of alemtuzumab compared with natalizumab, fingolimod, and interferon beta in relapsing-remitting multiple sclerosis: a cohort study. Lancet Neurol. 2017;16(4):271–281.
- Druart C, El Sankari S, van Pesch V. Long-term safety and real-world effectiveness of fingolimod in relapsing multiple sclerosis. Patient Relat Outcome Meas. 2018;9:1–10.
- Koyrakh L, Roman MI, Brinkmann V, et al. The heart rate decrease caused by acute FTY720 administration is mediated by the G protein-gated potassium channel I. Am J Transplant. 2005;5(3):529–536.
- Camm J, Hla T, Bakshi R, et al. Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am Heart J. 2014;168(5):632–644.
- Yoshii F, Moriya Y, Ohnuki T, et al. Neurological safety of fingolimod: an updated review. Clin Exp Neuroimmunol. 2017;8(3):233–243.
- Mancano MA. Ziprosidone-induced transient agranulocytosis; delayed onset of cardiac adverse effects with fingolimod; telmisartan-induced myotoxicity; severe dilated cardiomyopathy induced by adalimumab and ustekinumab. Hosp Pharm. 2015;50(10):855–858.
- Gyang TV, Hamel J, Goodman AD, et al. Fingolimod-associated PML in a patient with prior immunosuppression. Neurology. 2016;86(19):1843–1845.
- Van Schependom J, Gielen J, Laton J, et al. Assessing PML risk under immunotherapy: if all you have is a hammer, everything looks like a nail. Mult Scler. 2016;22(3):389–392.
- Minuk A, Belliveau MJ, Almeida DR, et al. Fingolimod-associated macular edema: resolution by sub-tenon injection of triamcinolone with continued fingolimod use. JAMA Ophthalmol. 2013;131(6):802–804.
- Pul R, Osmanovic A, Schmalstieg H, et al. Fingolimod associated bilateral cystoid macular edema-wait and see? Int J Mol Sci. 2016;17(12).
- Palazzo E, Yahia SA. Progressive multifocal leukoencephalopathy in autoimmune diseases. Joint Bone Spine. 2012;79(4):351–355.
- Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353(4):369–374.
- Langer-Gould A, Atlas SW, Green AJ, et al. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381.
- Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–368.
- Berger JR, Cree BA, Greenberg B, et al. Progressive multifocal leukoencephalopathy after fingolimod treatment. Neurology. 2018;90(20):e1815–e1821.
- Berger JR. Classifying PML risk with disease modifying therapies. Mult Scler Relat Disord. 2017;12:59–63.
- Mehling M, Brinkmann V, Antel J, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology. 2008;71(16):1261–1267.
- Simbrich A, Thibaut J, Khil L, et al. Drug-use patterns and severe adverse events with disease-modifying drugs in patients with multiple sclerosis: a cohort study based on German claims data. Neuropsychiatr Dis Treat. 2019;15:1439–1457.
- Dumitrescu L, Constantinescu CS, Tanasescu R. Siponimod for the treatment of secondary progressive multiple sclerosis. Expert Opin Pharmacother. 2019;20(2):143–150.