
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Introduction: The enthalpic and entropic components of the ligand–protein binding free energy reflect the type and quality of the interactions and relate to the physicochemical properties of the ligands. These findings have significance in medicinal chemistry optimizations since they suggest that the thermodynamic profiling of the binding may help monitor and control the unfavorable size and hydrophobicity increase typically accompanying affinity improvements and leading to suboptimal pharmacokinetic properties.
Areas covered: This review describes the ligand–protein binding event in terms of elementary steps, their associated interactions, and their enthalpic and entropic consequences. The relationships among the breaking and forming interactions, the binding thermodynamic profile, and the physicochemical properties of the ligands are also discussed.
Expert opinion: Analysis of the size dependence of available affinity and favorable enthalpy highlights the limitation of the simultaneous optimization of these quantities. Indeed, moderate, rather than very high affinities can be conciliated with favorable physicochemical and pharmacokinetic profiles as it is supported by the affinity range of historical oral drugs. Although thermodynamic quantities are not suitable endpoints for medicinal chemistry optimizations owing to the complexity of the binding thermodynamics, thermodynamic profiling together with structural studies can be advantageously used to understand the details of the binding process and to optimize it.
1. Introduction
A principal objective of early phase medicinal chemistry optimizations in drug discovery programs is to increase the binding affinity of ligands toward their protein targets. Binding affinity is directly related to the free energy change accompanying ligand–protein binding. The relationship between affinity and the enthalpy and entropy content of binding free energy can be conveniently expressed in terms of the quantities in Eq. (1) [Citation1]
where with
being the dissociation constant of the ligand–protein complex (more precisely it has to be replaced by
), and
and
.
measures the enthalpic and
the entropic contribution to the binding affinity. The transformation of enthalpy (
) and entropy (
) changes into
and
, respectively, has the advantage that affinity (
) and the thermodynamic quantities (
,
) are expressed at comparable scales and higher values corresponds to more favorable contributions.
The recognition that binding thermodynamics relates to the optimization and to the properties of drugs and drug candidates first appears in a series of papers by Freire and coworkers published from the turn of the millennium [Citation2–Citation5]. The enthalpic and entropic contributions to ligand–protein binding were determined for HIV inhibitors, and later for HMG-CoA reductase inhibitors, and it was shown that compounds that come later into the market tend to have increasing enthalpic contributions. This finding can be attributed to the challenge and longer timeframe of optimizing ligand affinity toward its target protein by increasing the favorable enthalpic () rather than the entropic (
) contribution of binding. It was also recognized that compounds with enthalpy dominated binding tend to be more drug like by having favorable physicochemical and pharmacokinetic properties [Citation5–Citation7].
The thermodynamic characterization of ligand–protein binding is most often performed with isothermal titration calorimetry (ITC) measurements [Citation8,Citation9]. They directly provide binding enthalpy and dissociation constants that can be used to obtain free energy and entropy, and thus full thermodynamic profile of the binding. Measuring the temperature dependence of the dissociation constant and using the van’t Hoff equation offers another way to determine thermodynamic quantities [Citation10]. However, ITC is considered to be the preferred method as it gives thermodynamic data from a single titration, it is a label-free technique, and it can be applied at a wide range of binding affinities. Its principal limitation is high protein requirement, however, significant progress has been achieved to decrease protein consumption, increase throughput [Citation11,Citation12] and to make ITC applicable to membrane proteins [Citation13]. It should be noted that measured enthalpies depend on conditions like temperature, pH and buffer, and they may need corrections when ligand–protein binding is linked to protonation or ion binding [Citation14].
Thermodynamic profiling of ligand–protein binding has generated a large amount of data that are useful for recognizing trends in compound property changes in drug discovery programs and to directly support medicinal chemistry optimizations.
2. Thermodynamics of the ligand–protein binding process
In principle, affinity () optimization can be achieved by increasing
,
, or both. Here, we have to mention enthalpy–entropy compensation [Citation15] that is a general phenomenon and tends to compensate enthalpy and entropy changes resulting in a modest change in
. The basic challenge in affinity optimizations can be formulated as to overcome enthalpy–entropy compensation to achieve increased
. Ligand–protein binding in water is a complex process with various competing enthalpic and entropic contributions [Citation5–Citation7]. Binding requires at least a partial desolvation of both the ligand and the protein, potential conformational changes of both partners, and the formation of ligand–protein interactions. The desolvation of polar groups has a large enthalpic penalty [Citation16] that can only be compensated by good quality ligand–protein interactions. The emphasis here is on the good quality, since especially polar interactions are highly sensitive to the relative geometry of the interacting partners. Therefore, the exchange of ligand–water and protein–water hydrogen bonds to ligand–protein and water–water hydrogen-bonds may eventually be unfavorable and may not yield
increase. In contrast, the desolvation of apolar ligand and protein surfaces that become buried in the ligand–protein complex is typically favorable owing to the
increase attributed to the disappearance of the polar water–apolar solute interactions. Therefore, it is usually a feasible strategy to increase
by building into the ligand nonpolar groups with good shape complementarity to the protein binding pocket. Another optimization technique is to fix the ligand conformation in the geometry of the bound ligand and thus moderating configuration entropy decrease upon ligand binding. These considerations provide rationale for the ease of entropic optimization compared to the enthalpic optimization.
The quantification of the above effects is not straightforward. The association of free energy changes of solvation with surfaces is an approximation that does not take into account atomic details. This is the reason why apolar and polar surface burial give modest correlation with binding free energy and with its components [Citation17]. Moreover, the large variation of free energy changes associated with surface burial as obtained with various models [Citation17–Citation21] suggests model dependence.
A part of conformational entropy decrease upon ligand binding originates from the more restricted variation of torsion angles in the complex compared to the solvated ligand and protein. The loss of conformational entropy is estimated to be around 3 kJ/mol at 300 K for each rotatable bond fixed in the complex [Citation22]. The association of two species, the protein and the ligand, results in a loss of rigid body translational and rotational entropy and its estimated magnitude is around 15–20 kJ/mol at 300 K [Citation23]. These values, however, are crude estimations and the actual magnitude of the entropy loss is affected by the flexibility of the complex; entropy may be associated with the new vibrational modes of the complex.
Ligand–protein binding free energies can be calculated with molecular dynamics based methods with an accuracy of 5–10 kJ/mol in favorable cases [Citation24,Citation25]. Atomic level simulations are best suited for the calculation of binding free energy differences between ligands. The calculation of enthalpic and entropic components, however, faces serious convergence difficulties. Particular attention has been devoted to the calculation of the thermodynamic quantities associated with the perturbation of the binding site water network upon ligand binding. The estimation of the enthalpy and entropy assigned to individual water molecules by the WaterMap method [Citation26] is based on molecular dynamics simulations and the application of the inhomogeneous solvation theory [Citation27]. The complementary WaterFLAP approach [Citation28] assigns enthalpy and entropy by empirical scoring of the water network built using GRID [Citation29] water hotspots and optimized by short molecular dynamics simulations. Computations may support the interpretation of binding thermodynamic data and the design of ligands with improved properties [Citation30–Citation32].
Valuable insight into the binding process can be obtained by combined structural and thermodynamic analyses of a set of related complexes. These analyses are able to reveal how various effects like the water mediated ligand–protein binding [Citation33], the displacement or trapping of water molecules upon ligand binding [Citation34–Citation36], and the interplay between hydrogen-bonding and hydrophobic contacts [Citation37] influence the thermodynamic balance of binding. Ligand binding may be accompanied with protonation/deprotonation processes whose thermodynamic consequences are pH and buffer dependent, and their dissection leads to intrinsic thermodynamic parameters most relevant in drug design [Citation38,Citation39]. These studies highlight the complexity of ligand binding thermodynamics and show that it is often difficult to pinpoint what the origins of the changes in entropy and enthalpy are [Citation40,Citation41]. Owing to their multifactorial character and non-additivity, thermodynamic quantities cannot be used as direct endpoints, but they are useful in analyzing and supporting the optimization of specific systems [Citation42–Citation44].
In spite of the above difficulties in attributing accurate thermodynamic values to the various effects accompanying ligand–protein complex formation, the dissection of the process into elementary steps, and the association of approximate enthalpy and entropy values to these steps supports the identification of useful relationships between thermodynamics and the quality of compounds in the drug discovery process, on one hand, and lead identification and optimization, on the other hand. Both aspects will be discussed in the forthcoming sections.
3. Thermodynamics and drug-like properties
Achieving affinity improvement is a major objective of early phase medicinal chemistry optimizations; however, the optimization of other properties becomes increasingly important at later stages. Physicochemical properties and the related pharmacokinetic profile are important characteristics of drug candidates, and their simultaneous optimization with affinity is the main challenge in drug discovery programs. Following the early recognition of the relation among affinity, drug-like properties and binding thermodynamics [Citation5–Citation7], significant effort has been devoted to explore, understand and take advantage of this relationship in drug discovery projects.
It is well documented that medicinal chemistry optimizations are typically accompanied with unfavorable changes of physicochemical properties [Citation45–Citation47]. Compounds tend to be larger and increasingly hydrophobic with improving affinity. This is, however, not beneficial, since large and hydrophobic compounds have unfavorable physicochemical properties and are more prone to suboptimal ADME profile as it was shown by the comparison of compounds from various stages of the discovery process [Citation45,Citation48–Citation51]. It was also shown that property inflation is a general phenomenon and is basically independent from the hit identification strategy, rather it could be traced back to optimization practices [Citation7]. It is common to achieve affinity improvements by adding lipophilic groups to facilitate desolvation, and by limiting ligand flexibility using chain to ring replacements typically with the introduction of predominantly apolar groups. Both tactics yield molecular weight and lipophilicity increase, and both contribute to the affinity improvement by increasing binding entropy. Therefore, entropic optimization is often associated with unfavorable ADME properties. In contrast, enthalpic optimization attempts to improve affinity by building into the ligand functional groups with optimal directional interactions to the protein target. This approach can avoid the excessive increase of lipophilicity and the deterioration of ADME properties; therefore, it is considered to be preferable.
Affinity optimization by extending compounds with new groups of atoms is in line with the observation that maximal available affinity increases with the number of heavy atoms. It was shown by Kuntz et al. that for small molecules a sharp increase of available affinity can be achieved with the number of heavy atoms, while the increase is modest for larger molecules [Citation52]. Similar trend was found for compounds with binding thermodynamic data [Citation1]. The maximal values as a function of heavy atom count tend to increase as it is shown in ).
Figure 1. (a) Logarithmic plot of affinity (pKd[max]) of the most potent ligands versus the number of heavy atoms (HAC). (b) Logarithmic plot of maximal favorable enthalpy (pKH[max]; black round markers and straight line), and enthalpy of the most potent ligands (pKH[pKdmax]; red square markers and dashed line) versus the number of heavy atoms (HAC). Data were binned by ΔHAC = 3 and maximal values in each bin are shown. Based on data from Ref. [Citation1].
![Figure 1. (a) Logarithmic plot of affinity (pKd[max]) of the most potent ligands versus the number of heavy atoms (HAC). (b) Logarithmic plot of maximal favorable enthalpy (pKH[max]; black round markers and straight line), and enthalpy of the most potent ligands (pKH[pKdmax]; red square markers and dashed line) versus the number of heavy atoms (HAC). Data were binned by ΔHAC = 3 and maximal values in each bin are shown. Based on data from Ref. [Citation1].](/cms/asset/7bdf36f9-0f32-47f9-a832-081267c7ae76/iedc_a_1691166_f0001_oc.jpg)
A similar plot of the maximal available binding enthalpy as a function of molecular size expressed as heavy atom count shows the opposite trend in ). This plot was obtained with over 800 compounds [Citation1] including natural biological ligands and synthetic compounds from medicinal chemistry programs with a large variety of protein targets. The plot in ) implies that there is a limit for available favorable binding enthalpy, and this limit decreases with ligand size. Moreover, the binding enthalpies of maximal affinity ligands () also show a decreasing trend with ligand size, and they are consistently lower than the available maximal favorable enthalpy (
) values ()). These findings suggest that this is not only the medicinal chemistry optimization practice that hampers enthalpic optimization, but there is an inherent limit for the simultaneous optimization of both affinity and enthalpy. The reconciliation of the dilemma between affinity and enthalpy optimization is suggested by the observation that historical oral drugs show median pXC50 (negative logarithm of IC50 or EC50) value of ˜7.7 (˜50 nM) [Citation53,Citation54] showing that moderate, rather than very high affinity can be combined with favorable ADME properties to produce compounds with high therapeutic value.
4. Thermodynamics and fragment-based drug discovery
Fragment-based drug discovery (FBDD) is an approach for identifying chemical starting points for drug discovery programs [Citation55–Citation57]. FBDD starts with the screening of fragment-sized compounds, often defined as molecules with less than 23 heavy atoms or with molecular weight less than 300 Da. Fragment hits typically bind with high ligand efficiency (LE; affinity per heavy atom) and can be optimized into leads and drug candidates with large operational freedom owing to their small size and advantageous physicochemical properties. A further advantage of fragment screening is the better sampling of the chemical space compared to the high throughput screening of lead-like compounds.
The small size and limited number of pharmacophore elements of fragments allow binding with few optimal interactions, albeit with low affinity. A rationale for the success of the fragment approach is given by the complexity model [Citation47,Citation58] claiming that the chance of forming useful interactions falls with increasing ligand complexity. Analyses of fragment interactions in experimental protein complexes show that binding occurs at the hot spot of the protein that is a polar site buried in a lipophilic environment [Citation59–Citation61], where fragments typically bind with few polar interaction [Citation60], frequently with optimal geometry hydrogen-bonds [Citation59]. In contrast, the contribution of apolar desolvation and associated free energy gain with predominantly entropic origin is limited owing to the small size of fragments [Citation59,Citation61]. Therefore, the formation of polar interactions provides favorable enthalpy that is able to compensate for the unfavorable rigid body translational and rotational entropy loss upon binding [Citation23] and results in enthalpy dominated fragment binding as it was shown by the thermodynamic analysis of a large number of fragment–protein complexes [Citation61–Citation63].
The few dominantly polar interactions formed by fragments can be assigned to pharmacophore patterns, and the screening of ultra-low-molecular-weight compounds [Citation64] and minimal pharmacophores [Citation55] has the promise of improved exploration of the binding site and better coverage of the chemical space. Although we are not aware of thermodynamic data for these very small fragments, we anticipate that their binding is enthalpy dominated and they are well suited for enthalpy driven optimization. It has to be noted, however, that in spite of the beneficial properties of fragments, their successful optimization into advantageous leads and drug candidates is not guarantied. Historical data show that fragment and HTS derived lead properties are not necessarily different [Citation7], and the inherent difficulty of enthalpy driven optimization of larger compounds as it is illustrated in ) prevents late stage enthalpic optimization. Nevertheless, early optimizations of fragment hits offer the opportunity for the simultaneous improvement of affinity and binding enthalpy as it was shown by the analysis of the thermodynamic data of Astex drug discovery programs [Citation61]. The combined application of structural and thermodynamic information primarily by X-ray and ITC, respectively, was found to be the key for successful early optimizations [Citation61,Citation65]. These considerations make the use of thermodynamic data particularly fruitful in fragment-based approaches.
5. Case studies for thermodynamics supported fragment optimizations
lists publicly available thermodynamic data of fragment–protein binding. These data provide the basis for analyzing how thermodynamic quantities depend on the properties of the binding partners [Citation61,Citation62]. These data combined with structural information for a set of related complexes are extremely useful in understanding the details of the binding process as it is exemplified with the case studies presented in the forthcoming sections.
Table 1. Reported experimental thermodynamic data for fragment–protein binding.
5.1. Matrix metalloproteinase (MMP12) inhibitors
One of the first studies on the impact of binding thermodynamics on fragment optimization has been published by Bertini et al. [Citation93]. The authors investigated the optimization of low affinity fragments to nanomolar MMP12 inhibitors by evaluating the key compounds along the optimization path by biochemical and ITC measurements, and X-ray crystallography. Matrix metalloproteinases are zinc containing proteases. Most of the high affinity MMP inhibitors form direct interactions with the zinc ion located at the substrate binding groove. Due to the chelating properties of hydroxamic acids, the identification of the low affinity and nonspecific acetohydroxamic acid (1) was not unexpected. In fact, 1 binds to MMP12 with a Kd of 6.18 mM, its binding mode has been confirmed by X-ray crystallography and the subsequent ITC analysis revealed this fragment as a high enthalpy binder. The first optimization step was realized by a linking strategy connecting the acetyl group of 1 to the terminal amino group of the 4-methoxy-benzenesulfonamide (2) that binds to the S1’ pocket of MMP12 (). This modification resulted in a dramatic improvement in the potency that was increased by five orders of magnitude, reaching the Kd of 61 nM. Analyzing the thermodynamic profiles of the linked fragments, the observed improvement in Kd can be traced back to the much improved binding entropy of 3.
Figure 2. (a) Optimization of MMP12 starting fragments using the fragment linking strategy. (b) Thermodynamic profiles of the starting fragments and the linked compound.
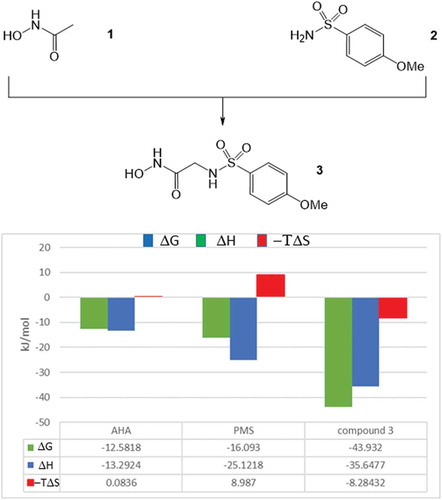
Linking the fragments, they lose their translational and rotational degree of freedom and the related entropy penalty of the linked compound contributes to its increased binding affinity. In this particular case, the improvement in the binding affinity (ΔΔG = −15.2 kJ/mol) was basically due to this effect (−ΔTΔS = −17.4 kJ/mol) while enthalpic components did not contribute significantly (ΔΔH = 2.8 kJ/mol). Based on its binding affinity and thermodynamic profile, compound 3 was selected for further optimization. In the first round the 4-methoxy group of 3 has been replaced to 4-fluoro (4) and 4-phenyl (5) substituents (). The introduction of the 4-fluoro group did not improved the affinity (Kd = 65 nM); however, the 4-phenyl derivative (5) was more potent (Kd = 2 nM) than 3. Comparative analysis of the corresponding X-ray structures revealed that the 4-methoxyphenyl group dives deeper into the S1’ pocket as compared to the 4-fluorophenyl moiety that probably could displace less water molecules at the binding site. On the other hand, the additional phenyl ring of 5 provides new sites for hydrophobic interactions. The thermodynamic profiles of compounds 3–5 are in line with these structural findings ().
Figure 3. Optimization of the aromatic substituent in 3. The diagrams show the change of the thermodynamic quantities (ΔΔG: change in the binding free energy, ΔΔH: change in the binding enthalpy and −ΔTΔS: change in the binding entropy) along the optimization step.
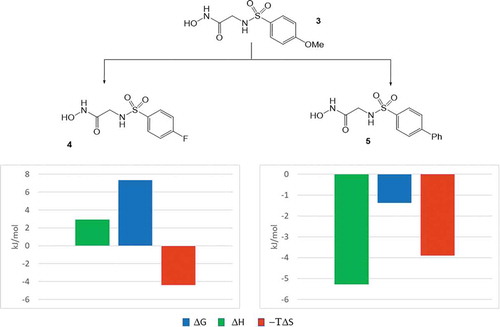
Replacement of the 4-methoxy substituent to 4-fluoro was associated with some improvement in the binding entropy that was compensated by the enthalpic penalty in line with decreased occupancy of the S1’ site. Introducing the apolar phenyl group to position 4; however, resulted in improvements in both the enthalpic and the entropic components. Owing to its larger size, the phenyl group fills the S1’ site better that contributes to the slight improvement to the binding enthalpy. The most significant factor, however, is the increase of entropic reward that may be attributed, at least partly, to improved desolvation.
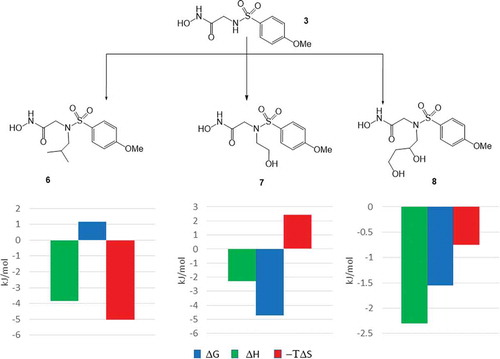
In the second round of the optimization, the authors investigated the effect of substituents introduced to the sulfonamide nitrogen atom. Having the best balance between its potency and the physico-chemical and thermodynamic profiles, the sulfonamide nitrogen of the 4-methoxy analogue (3) was substituted by an isobutyl (6), a 2-hydroxyethyl (7), and a 2,3-dihydroxypropyl (8) groups (). All of the N-substituents resulted in improvements in the binding affinity, Kd values reached the single digit nanomolar range (Kd = 4 nM, 8 nM, and 8 nM, respectively). The highest affinity has been achieved by alkyl group (iBu in compound 6) while alcohols (7 and 8) showed modest improvement. Analysis of the X-ray structures suggests that except for the isobutyl group, the terminal hydroxyl group of the alcohols forms interactions with water molecules located at the entrance of the substrate binding site. Thermodynamic profiling of compounds 6–8 provided more details on the energetics of ligand binding. ITC measurements showed that the N-alkyl derivative (6) gains binding free energy from the entropic reward potentially originate from its improved desolvation. Contrary, the introduction of hydroxyl substituents provided more polar interactions and therefore improved the affinity via the corresponding enthalpic reward. Comparing, however, the thermodynamic profiles of the alcohol (7) and the diol (8), it seems that the enthalpic reward achieved in 7 has been largely compensated by an entropic penalty. Interestingly, the profile of the diol (8) suggests much less gain in the binding enthalpy and some reward in the binding entropy. The decreased enthalpy contribution can be rationalized by the less favored desolvation of the diol.
Figure 4. Optimization of the sulfonamide nitrogen substituent in 3. The diagrams show the change of the thermodynamic quantities (ΔΔG: change in the binding free energy, ΔΔH: change in the binding enthalpy and −ΔTΔS: change in the binding entropy) along the optimization step.
In summary, the optimization of MMP12 inhibitors provided a good example for analyzing the thermodynamic character of the two most important fragment optimization strategies, linking and growing. While linking is mostly entropically driven, there are multiple opportunities for fragment growing. Structural studies and the parallel analysis of the thermodynamic profiles allow understanding the changes in the binding mode and energetics along the optimization path. These data together might help optimizing protein–ligand interactions while keeping the physico-chemical properties under control.
5.2. Carbonic anhydrase (CA) inhibitors
Thermodynamic characterization is now a routine label-free methodology for the investigation of protein ligand interactions in industry settings. One of the first studies of this type has been published by a Pfizer team on carbonic anhydrase ligands [Citation42]. Profiling 17 substituted benzene sulfonamides () the authors identified compounds with better affinity than the unsubstituted core. In addition to the 4-chloro (Kd = 141 nM) and 4-cyano (Kd = 142 nM) derivatives they found that both the 2-fluoro (Kd = 314 nM) and 3-fluoro (Kd = 118 nM) analogues showed submicromolar affinity.
Figure 5. Thermodynamic profiling of benzene sulfonamides on carbonic anhydrase.
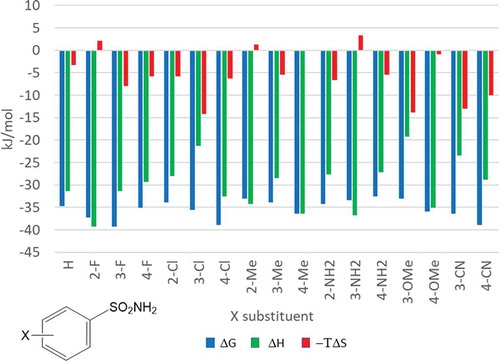
Comparing the thermodynamic profiles of the most active compounds it has been suggested that the significantly improved potency observed for the 4-Cl and 3-F compounds is driven by a favorable entropic component. Interestingly, the thermodynamic profile of the 2-fluoro-benzenesulfonamide showed significant improvement in the binding enthalpy. Considering only potency data, the 2-fluoro derivative is inferior; however, the more enthalpic character of this compound suggests improved polar interactions with the protein binding site. In fact, X-ray crystallography of the 2-fluoro analogue cocrystallized with CA revealed a specific interaction formed between the 2-fluoro group and the backbone NH of Thr200. In contrast, 3-fluoro, 4-chloro and 4-cyano substituents are all located close to the hydrophobic part of the binding site ().
Figure 6. Experimental binding modes of the 2-fluoro and 3-fluoro benzenesulfonamides at the binding site of human carbonic anhydrase. The 2-fluoro group points toward the hydrophilic wall (green) of the binding site. Contrary, the 3-fluoro group is located in a dominantly hydrophobic subpocket that is preferred by all the other halogens and also the nitriles used at different positions. Copyright: Royal Society of Chemistry [Citation94] https://pubs.rsc.org/en/content/chapter/bk9781849733533-00023/978-1-84973-353-3.
![Figure 6. Experimental binding modes of the 2-fluoro and 3-fluoro benzenesulfonamides at the binding site of human carbonic anhydrase. The 2-fluoro group points toward the hydrophilic wall (green) of the binding site. Contrary, the 3-fluoro group is located in a dominantly hydrophobic subpocket that is preferred by all the other halogens and also the nitriles used at different positions. Copyright: Royal Society of Chemistry [Citation94] https://pubs.rsc.org/en/content/chapter/bk9781849733533-00023/978-1-84973-353-3.](/cms/asset/7c52232e-1a5a-46ce-9977-f47c29ef3960/iedc_a_1691166_f0006_oc.jpg)
Based on the crystallographic and thermodynamic analysis, the 2-fluoro derivative forms more directed interactions with the target that provides a stable binding mode necessary for further optimization of the initial fragment. Growing the 2-fluoro and 3-fluoro derivatives with a 4-benzylamide moiety () resulted in a significant increase in the binding affinity of the former fragment (55 fold, Kd = 5.7 nM). The affinity improvement was much less for the 3-fluoro analogue (6 fold, Kd = 20 nM).
Figure 7. Fragment growing applied for the optimization of 2-fluoro and 3-fluoro benzenesulfonamides against human carbonic anhydrase. The thermodynamic profile of the optimized 2-fluoro fragment preserved its enthalpic character.
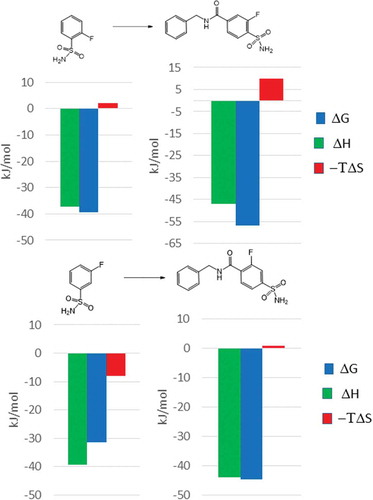
Focusing to the thermodynamic profiles of the optimized fragments, both bindings are enthalpy driven. The addition of the p-benzylamide group increased the enthalpy content of the 2-fluoro derivative more significantly than that observed for the corresponding 3-fluoro analogue. Furthermore, the same optimization strategy turned the originally less active 2 fluoro fragment into a more potent lead than that obtained from the 3-fluoro-benzenesulfonamide fragment.
In summary, this study shows that evaluation of the thermodynamic profiles of prioritized fragment sets would provide useful information to pick up the most promising fragment hits for further optimizations. Structural and thermodynamic characterization of the fragment ligand complex might help to understand the driving forces behind the binding that points farther than the affinity alone. Highly enthalpic fragments form good quality polar interactions at the binding site including H-bonds with the target and structural waters. Thus, the stable binding mode might provide better starting point for further optimization.
5.3. Heat shock protein 90 (HSP90) inhibitors
One of the first Astex program entered into the clinical phase aimed the discovery of HSP90 inhibitors for oncology indications. This program was initiated by the X-ray and NMR screening of the corporate fragment library that provided two series of hits. One of these series was aminopyrimidines exemplified by 9 that was picked up as a potential starting point [Citation95]. Despite of its moderate affinity (Kd = 240 μM), 9 has a stable binding mode in a deep pocket of HSP90 and forming good quality H-bonds with Asp93 and four structural waters located at the binding site (). Comparing the binding mode of 9 to that of ADP revealed the overlapping position of the aminopyrimidine and the adenosine rings and suggested to extend 9 toward the small hydrophobic pockets available at the close proximity. Another important feature observed in the binding mode of 9 was that the pyridine ring is twisted around the biaryl axis. Replacing the pyridine ring with an ortho methoxyphenyl substituent preserved this twisted conformation. Furthermore, the methyl group introduced at position 2 of the pyrimidine ring fits well to one of the hydrophobic pockets (). These changes led to compound 10 showing 60 fold improvement in the binding affinity (Kd = 4 μM).
Figure 8. Binding modes of compounds 9 and 10 obtained by X-ray crystallography. Structural waters are indicated by red spots.
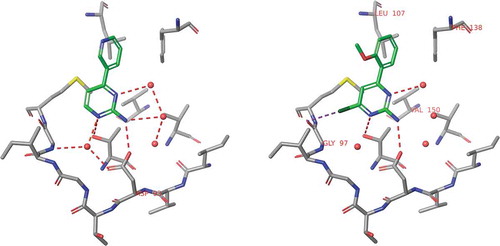
In the next step of the optimization, the 2-methyl substituent at the aminopyperidine core has been replaced by a chlorine (, compound 11) that improved the affinity further 12-fold reaching the submicromolar range (Kd = 0.35 μM).
Figure 9. Thermodynamic profiling of the fragments optimized against HSP90 at Astex.
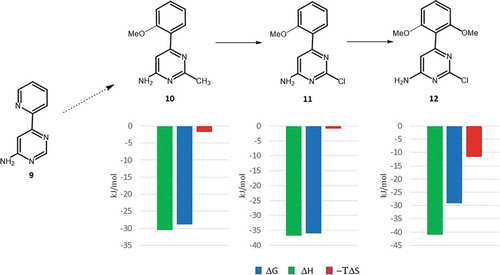
Figure 10. Thermodynamic profiling of protein kinase A ligands obtained by systematic growing of the starting fragment (13).
Comparing the thermodynamic profiles [Citation96] of 10 and 11 there is a significant improvement in the binding enthalpy (ΔΔH = −7.1 kJ/mol) that suggests improved polar interactions to the target. In fact, the X-ray structure of the HSP90-11 complex revealed the formation of a new halogen bond to Gly97 NH that was kept after the introduction of another methoxy group at position 6 of its 2-methoxyphenyl ring (). This step yielded compound 12 with a Kd value of 0.07 μM that represents a 5-fold improvement in the binding affinity relative to that of 11. Analysis of the corresponding thermodynamic profiles suggests this transformation being entropy driven (−ΔTΔS = −10.9 kJ/mol). The entropic character of this step could be readily interpreted by the limited rotational freedom of the 2,6-dimethoxyphenyl ring in solution that results in more beneficial changes in the conformational entropy of 12.
This study demonstrates that careful optimization of the initial fragment hit might provide new polar interactions with the target that improves the binding affinity significantly and the meantime might stabilize the binding mode of the fragment. Structural studies with the parallel evaluation of binding thermodynamics could help identifying the key contacts. This information is specifically useful for late stage optimizations focusing other properties but keeping the beneficial interaction profile to maintain the binding affinity. This optimization also demonstrates the role of restricted ligand flexibility, a well-known optimization strategy applied in many medicinal chemistry programs. Again, structural and thermodynamic analyses help identifying the prioritized conformation of the ligand and the reward realized in the conformational component of the binding entropy.
5.4. Protein kinase A (PKA) ligands
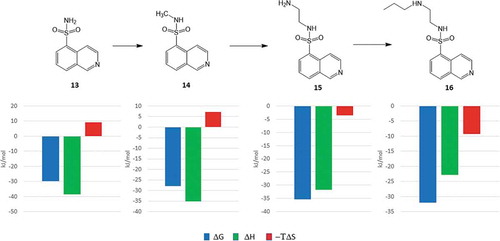
In a recent publication, Klebe and coworkers investigated the thermodynamics of fragment growing using protein kinase A as a model target [Citation40]. Their starting point was isoquinoline-5-sulfonamide (13) that binds to PKA with a Kd of 6.2 μM. Fragment growing was realized in three subsequent steps adding an N-methyl (14), an N-aminoethyl (15), and an N-propylaminoethyl (16) substituent to the sulfonamide group.
The thermodynamic profile of this small series of compounds was recorded and each of the ligands was cocrystallized with the target. Since all the ligands of this dataset are derived by systematic construction of the N-substituent of the sulfonamide moiety, it is therefore specifically useful to study the structural and thermodynamic consequences of fragment growing. However, it should be noted that in real life fragment optimizations growing strategies involve multiple often parallel and sometimes dependent growing vectors.
Analyzing the thermodynamic profiles, it is interesting to see that binding enthalpies become less favorable while the entropy rewards are increasing with the increasing size of the ligands (). The increasing number of polar atoms and the increasing number of rotatable bonds from 13 to 16, however, suggest inverse trends for both the enthalpy and the entropy of binding, respectively. Based on the comparative analysis of the corresponding X-ray structures the authors provided reasonable explanations for these observations ().
Figure 11. Superposition of the crystal structures obtained for ligands 13–16. Orientations of sulfonamide oxygens indicate the different binding modes of 13 and 14 vs. 15 and 16. As a consequence, the Gly-rich loop was found in closed and open conformation, respectively.
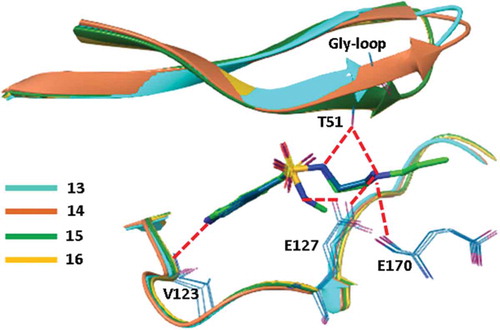
Analyzing the ligand conformations obtained in the binding pocket of PKA, it turned out that the binding modes of 13 and 14 are almost identical. Interestingly, however, the poses of the larger compounds, 15 and 16, obtained by the growing strategy were found to be significantly different. The sulfonamide group was rotated by 80 degrees relative to the conformation found in 13 and 14 that resulted in a completely different interaction pattern for the smaller (13 and 14) and the larger ligands (15 and 16). In the case of 13 and 14, the NH of the sulfonamide forms a H-bond to Glu127. In contrast, the larger N-substituents of 15 and 16 prevent the formation of this H-bond and the sulfonamide NH instead forms interactions with Thr51 of the Gly-rich loop. This H-bond reorganizes the Gly-rich loop that adopts a closed conformation as opposed by the open conformation found in the complexes of 13 and 14. The largely changed interaction patterns might thus explain the decreased enthalpic reward realized in the complex formation of the larger ligands. Entropic effects were rationalized by the analysis of ligand interactions formed with binding site water molecules. Comparative analysis of structural waters identified at the binding pocket revealed that the terminal propyl group of 16 displaced a water molecule present in the crystal structure of 13, 14, and 15 complexes since releasing this water to the bulk improves the binding entropy. Although the analysis of water networks was limited by the quality of the structures molecular dynamics simulations suggested similar mechanisms behind the entropic preference of 15 over 14 binding.
Despite that fragment growing follows different and more rational ways in drug discovery programs, this study shed light to the complexity of structure–thermodynamics relationship. ITC measures the net values of binding thermodynamics that is impacted by the interaction partners, including the protein target, the ligand, and the water molecules at the binding site. In addition to direct protein–ligand contacts, differences in the solvation patterns influence the thermodynamic profile significantly. Together with induced fit effects and conformational rearrangements occurred upon binding, high resolution X-ray crystallography and protein observed NMR measurements might help understanding and interpreting the binding thermodynamics of protein ligands.
6. Conclusion
The analysis of ligand–protein binding thermodynamics revealed links between the thermodynamic profiles and the physicochemical and pharmacokinetic properties of ligands. The opposite trends in available affinity and favorable enthalpy change with ligand size have been recognized and interpreted. They are in line with the demonstrated enthalpy dominated binding of fragment-sized compounds. Moreover, fragments are well suited for enthalpy driven early optimizations using binding thermodynamic and structural data that help understanding ligand binding and support medicinal chemistry optimizations to provide drug candidates with favorable pharmacokinetic profiles.
7. Expert opinion
Ligand–protein binding is associated with free energy change whose enthalpic and entropic components reflect the type and quality of interactions. As a general principle, the formation of polar, directional interactions between the ligand and the protein contributes to favorable enthalpy, while the desolvation of apolar surfaces of the interacting partners is the major source of favorable entropy. This implies that ligands with appropriate polar groups have the chance of enthalpy driven binding. However, optimal polar interactions with substantial enthalpy gain are restricted to a small range of the relative positions of the interacting partners. In contrast, the entropy gain achieved with apolar interactions are less sensitive to the geometrical parameters and the introduction of apolar groups in the ligand offers a more facile way to improve the binding free energy. This has the consequence that affinity increase is easier to achieve via entropic, rather than enthalpic optimizations [Citation5–Citation7]. This, however, has an unfavorable impact on the drug like properties of ligands, since large and apolar compounds tend to have suboptimal pharmacokinetic parameters [Citation45–Citation47]. The recognized relationship between the binding thermodynamic profile and the physicochemical and pharmacokinetic parameters suggests that monitoring and controlling binding thermodynamics may help improving the quality of drug candidates.
Analysis of a large amount of binding thermodynamic data revealed that maximal ligand affinity increases with ligand size, however, the favorable enthalpy tends to decrease with ligand size. Moreover, it has been shown that compounds with optimal affinity tend to have lower than maximal favorable binding enthalpy [Citation1]. These findings point out the limitation of the simultaneous optimization of affinity and enthalpy and suggest that moderate, rather than very high affinities can be conciliated with favorable physicochemical and pharmacokinetic profiles. This is also reflected by the observation that historical oral drugs tend to have affinity in the double digit nanomolar range [Citation53,Citation54].
The thermodynamic profiling of ligand binding in medicinal chemistry optimizations is most often used together with X-ray crystallography based atomic resolution structural analysis. Studies of congeneric series reveal important details like the role of water molecules [Citation33–Citation36] and the interplay between hydrogen-bonding and hydrophobic contacts [Citation37]. They highlight the complexity of ligand binding thermodynamics, and point out that thermodynamic quantities cannot be used as direct endpoints, but they are useful in analyzing and supporting the optimization of specific systems [Citation42,Citation43].
Theoretical considerations suggest and experimental data confirm that fragment-sized compounds exhibit enthalpy driven binding to proteins [Citation59,Citation61,Citation62]. Moreover, fragments are well suited for the optimization of polar interactions when supported by thermodynamic profiling and structural studies [Citation61]. These considerations represent the thermodynamic rationale for FBDD. Monitoring the thermodynamic profile is therefore beneficial in fragment-based optimization programs. Published case studies confirmed that thermodynamic characterization is useful analyzing the key interactions and rationalizing the binding mode. These pieces of information were found to be crucial in successful fragment optimization programs.
Article Highlights
Ligand–protein binding thermodynamics is related to the type and quality of interactions
Optimal geometry polar interactions provide favorable binding enthalpy
The desolvation of apolar groups is the major source of favorable binding entropy
Enthalpy, rather than entropy driven optimizations tend to provide compounds with favorable pharmacokinetic profiles
Enthalpic optimizations are more challenging than entropic optimizations
Binding thermodynamic profiling with structural studies supports medicinal chemistry optimizations
Available ligand affinity shows increasing, and favorable enthalpy shows decreasing trend with ligand size
Enthalpy dominated binding is a general feature of fragment-sized compounds
Fragments are well suited for enthalpy driven early optimizations
This box summarizes key points contained in the article.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
Related Research Data
References
- Ferenczy GG, Keserű GM. Enthalpic efficiency of ligand binding. J Chem Inf Model. 2010;50:1536–1541.
- Todd MJ, Luque I, Velázquez-Campoy A, et al. Thermodynamic basis of resistance to HIV-1 protease inhibition: calorimetric analysis of the V82F/I84V active site resistant mutant. Biochemistry. 2000;39:11876–11883.
- Velazquez-Campoy A, Todd MJ, Freire E. HIV-1 protease inhibitors: enthalpic versus entropic optimization of the binding affinity. Biochemistry. 2000;39:2201–2207.
- Carbonell T, Freire E. Binding thermodynamics of statins to HMG-CoA reductase. Biochemistry. 2005;44:11741–11748.
- Freire E. Do enthalpy and entropy distinguish first in class from best in class? Drug Discov Today. 2008;13:869–874.
- Ladbury JE, Klebe G, Freire E. Adding calorimetric data to decision making in lead discovery: a hot tip. Nat Rev Drug Discov. 2010;9:23–27.
- Ferenczy GG, Keserű GM. Thermodynamics guided lead discovery and optimization. Drug Discov Today. 2010;15:919–932.
- Freyer MW, Lewis EA. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 2008;84:79–113.
- Falconer RJ. Applications of isothermal titration calorimetry - the research and technical developments from 2011 to 2015. J Mol Recognit. 2016;29:504–515.
- Tellinghuisen J. Van’t Hoff analysis of K° (T): how good … or bad? Biophys Chem. 2006;120:114–120.
- Torres FE, Kuhn P, De Bruyker D, et al. Enthalpy arrays. Proc Natl Acad Sci. 2004;101:9517–9522.
- Recht MI, Sridhar V, Badger J, et al. Identification and optimization of PDE10A inhibitors using fragment-based screening by nanocalorimetry and X-ray crystallography. J Biomol Screen. 2014;19:497–507.
- Rajarathnam K, Rösgen J. Isothermal titration calorimetry of membrane proteins — progress and challenges. Biochim Biophys Acta - Biomembr. 2014;1838:69–77.
- Paketurytė V, Zubrienė A, Ladbury JE, et al. Intrinsic thermodynamics of protein-ligand binding by isothermal titration calorimetry as aid to drug design. In: Ennifar E, editor. Microcalorim. Biol. Mol. Methods Mol Biol. Humana Press, New York, NY. 2019;1964:61–74.
- Chodera JD, Mobley DL. Entropy-enthalpy compensation: role and ramifications in biomolecular ligand recognition and design. Annu Rev Biophys. 2013;42:121–142.
- Cabani S, Gianni P, Mollica V, et al. Group contributions to the thermodynamic properties of non-ionic organic solutes in dilute aqueous solution. J Solution Chem. 1981;10:563–595.
- Olsson TSG, Williams MA, Pitt WR, et al. The thermodynamics of protein-ligand interaction and solvation: insights for ligand design. J Mol Biol. 2008;384:1002–1017.
- Eisenhaber F. Hydrophobic regions on protein surfaces. Derivation of the solvation energy from their area distribution in crystallographic protein structures. Protein Sci. 1996;5:1676–1686.
- Walther KA, Gräter F, Dougan F, et al. Signatures of hydrophobic collapse in extended proteins captured with force spectroscopy. Proc Natl Acad Sci. 2007;104:7916–7921.
- Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct Funct Bioinforma. 1991;11:281–296.
- Robertson AD, Murphy KP. Protein structure and the energetics of protein stability. Chem Rev. 2002;97:1251–1268.
- Searle MS, Williams DH. The cost of conformational order: entropy changes in molecular associations. J Am Chem Soc. 1992;114:10690–10697.
- Murray CW, Verdonk ML. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J Comput Aided Mol Des. 2002;16:741–753.
- Mobley DL, Gilson MK. Predicting Binding Free Energies: frontiers and Benchmarks. Annu Rev Biophys. 2017;46:531–558.
- Abel R, Wang L, Mobley DL, et al. A critical review of validation, blind testing, and real- world use of alchemical protein-ligand binding free energy calculations. Curr Top Med Chem. 2017;17:2577–2585.
- Wang L, Berne BJ, Friesner RA. Ligand binding to protein-binding pockets with wet and dry regions. Proc Natl Acad Sci U S A. 2011;108:1326–1330.
- Lazaridis T. Inhomogeneous fluid approach to solvation thermodynamics. 1. Theory. J Phys Chem B. 1998;102:3531–3541.
- Baroni M, Cruciani G, Sciabola S, et al. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J Chem Inf Model. 2007;47:279–294.
- Goodford PJ. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem. 1985;28:849–857.
- Mason JS, Bortolato A, Weiss DR, et al. High end GPCR design: crafted ligand design and druggability analysis using protein structure, lipophilic hotspots and explicit water networks. Silico Pharmacol. 2013;1:23.
- Kamps JJAG, Huang J, Poater J, et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat Commun. 2015;6:8911.
- Christopher JA, Orgován Z, Congreve M, et al. Structure-based optimization strategies for G Protein-Coupled Receptor (GPCR) allosteric modulators: a case study from analyses of new metabotropic glutamate receptor 5 (mGlu 5) X-ray structures. J Med Chem. 2019;62:207–222.
- Biela A, Sielaff F, Terwesten F, et al. Ligand binding stepwise disrupts water network in thrombin: enthalpic and entropic changes reveal classical hydrophobic effect. J Med Chem. 2012;55:6094–6110.
- Shimokhina N, Bronowska A, Homans SW. Contribution of ligand desolvation to binding thermodynamics in a ligand-protein interaction. Angew Chemie Int Ed. 2006;45:6374–6376.
- Biela A, Khayat M, Tan H, et al. Impact of ligand and protein desolvation on ligand binding to the S1 pocket of thrombin. J Mol Biol. 2012;418:350–366.
- Snyder PW, Mecinovic J, Moustakas DT, et al. Mechanism of the hydrophobic effect in the biomolecular recognition of arylsulfonamides by carbonic anhydrase. Proc Natl Acad Sci. 2011;108:17889–17894.
- Baum B, Muley L, Smolinski M, et al. Non-additivity of functional group contributions in protein-ligand binding: A comprehensive study by crystallography and isothermal titration calorimetry. J Mol Biol. 2010;397:1042–1054.
- Kazlauskas E, Petrikaitė V, Michailovienė V, et al. Thermodynamics of aryl-dihydroxyphenyl-thiadiazole binding to human Hsp90. Vertessy BG, editor. PLoS One. 2012;7:e36899.
- Kišonaitė M, Zubrienė A, Čapkauskaitė E, et al. Intrinsic thermodynamics and structure correlation of benzenesulfonamides with a pyrimidine moiety binding to carbonic anhydrases I, II, VII, XII, and XIII. Khodarahmi R, editor. PLoS One. 2014;9:e114106.
- Wienen-Schmidt B, Wulsdorf T, Jonker HRA, et al. On the implication of water on fragment-to-ligand growth in kinase binding thermodynamics. Chem Med Chem. 2018;13:1988–1996.
- Biela A, Nasief NN, Betz M, et al. Dissecting the hydrophobic effect on the molecular level: the role of water, enthalpy, and entropy in ligand binding to thermolysin. Angew Chemie Int Ed. 2013;52:1822–1828.
- Scott AD, Phillips C, Alex A, et al. Thermodynamic optimisation in drug discovery: A case study using carbonic anhydrase inhibitors. Chem Med Chem. 2009;4:1985–1989.
- Geschwindner S, Ulander J, Johansson P. Ligand binding thermodynamics in drug discovery: still a hot tip? J Med Chem. 2015;58:6321–6335.
- Klebe G. Applying thermodynamic profiling in lead finding and optimization. Nat Rev Drug Discov. 2015;14:95–110.
- Teague SJ, Davis AM, Leeson PD, et al. The Design of Leadlike Combinatorial Libraries. Angew Chemie Int Ed. 1999;38:3743–3748.
- Morphy R. The influence of target family and functional activity on the physicochemical properties of pre-clinical compounds. J Med Chem. 2006;49:2969–2978.
- Hann MM, Leach AR, Harper G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J Chem Inf Comput Sci. 2001;41:856–864.
- Tyrchan C, Blomberg N, Engkvist O, et al. Physicochemical property profiles of marketed drugs, clinical candidates and bioactive compounds. Bioorg Med Chem Lett. 2009;19:6943–6947.
- Oprea TI, Davis AM, Teague SJ, et al. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci. 2001;41:1308–1315.
- Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881–890.
- Ferenczy GG, Keser GM. The impact of binding thermodynamics on medicinal chemistry optimizations. Future Med Chem. 2015;7:1285–1303.
- Kuntz ID, Chen K, Sharp KA, et al. The maximal affinity of ligands. Proc Natl Acad Sci. 1999;96:9997–10002.
- Gleeson MP, Hersey A, Montanari D, et al. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov. 2011;10:197–208.
- Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov. 2012;11:355–365.
- Erlanson DA, Fesik SW, Hubbard RE, et al. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016;15:605–619.
- Lamoree B, Hubbard RE. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017;61:453–464.
- Jacquemard C, Kellenberger E. A bright future for fragment-based drug discovery: what does it hold? Expert Opin Drug Discov. 2019;14:413–416.
- Leach AR, Hann MM. Molecular complexity and fragment-based drug discovery: ten years on. Curr Opin Chem Biol. 2011;15:489–496.
- Ferenczy GGG, Keserű GM, Keseru GM. Thermodynamics of fragment binding. J Chem Inf Model. 2012;52:1039–1045.
- Giordanetto F, Jin C, Willmore L, et al. Fragment hits: what do they look like and how do they bind? J Med Chem. 2019;62:3381–3394.
- Ferenczy GG, Keserű GM. On the enthalpic preference of fragment binding. Medchemcomm. 2016;7:332–337.
- Williams G, Ferenczy GG, Ulander J, et al. Binding thermodynamics discriminates fragments from druglike compounds: a thermodynamic description of fragment-based drug discovery. Drug Discov Today. 2017;22:681–689.
- Murray CW, Rees DC. Opportunity knocks: organic chemistry for fragment-based drug discovery (FBDD). Angew Chemie Int Ed. 2016;55:488–492.
- O’Reilly M, Cleasby A, Davies TG, et al. Crystallographic screening using ultra-low-molecular-weight ligands to guide drug design. Drug Discov Today. 2019;24:1081–1086.
- Ferenczy GG, Keserű GM. How are fragments optimized? A retrospective analysis of 145 fragment optimizations. J Med Chem. 2013;56:2478–2486.
- Steuber H, Heine A, Podjarny A, et al. Merging the binding sites of aldose and aldehyde reductase for detection of inhibitor selectivity-determining features. J Mol Biol. 2008;379:991–1016.
- Gloster TM, Macdonald JM, Tarling CA, et al. Structural, thermodynamic, and kinetic analyses of tetrahydrooxazine- derived inhibitors bound to β-glucosidases. J Biol Chem. 2004;279:49236–49242.
- Mecinović J, Snyder PW, Mirica KA, et al. Fluoroalkyl and alkyl chains have similar hydrophobicities in binding to the “Hydrophobic Wall” of carbonic anhydrase. J Am Chem Soc. 2011;133:14017–14026.
- Krishnamurthy VM, Kaufman GK, Urbach AR, et al. Carbonic anhydrase as a model for biophysical and physical-organic studies of proteins and protein-ligand binding. Chem Rev. 2008;108:946–1051.
- Binford JS, Lindskog S, Wadsö I. A calorimetric study of the binding of sulfonamides and cyanate to carbonic anhydrase. BBA - Enzymol. 1974;341:345–356.
- Protasevich II, Brouillette CG, Snow ME, et al. Role of inhibitor aliphatic chain in the thermodynamics of inhibitor binding to Escherichia coli enoyl-ACP reductase and the Phe203Leu mutant: A proposed mechanism for drug resistance. Biochemistry. 2004;43:13380–13389.
- Dunford JE, Kwaasi AA, Rogers MJ, et al. Structure-activity relationships among the nitrogen containing bisphosphonates in clinical use and other analogues: time-dependent inhibition of human farnesyl pyrophosphate synthase. J Med Chem. 2008;51:2187–2195.
- Balendiran GK, Schnütgen F, Scapin G, et al. Crystal structure and thermodynamic analysis of human brain fatty acid-binding protein. J Biol Chem. 2000;275:27045–27054.
- Bingham RJ, Findlay JBC, Hsieh SY, et al. Thermodynamics of Binding of 2-Methoxy-3-isopropylpyrazine and 2-Methoxy-3-isobutylpyrazine to the major urinary protein. J Am Chem Soc. 2004;126:1675–1681.
- Malham R, Johnstone S, Bingham RJ, et al. Strong solute-solute dispersive interactions in a protein-ligand complex. J Am Chem Soc. 2005;127:17061–17067.
- Barratt E, Bronowska A, Vondrášek J, et al. Thermodynamic penalty arising from burial of a ligand polar group within a hydrophobic pocket of a protein receptor. J Mol Biol. 2006;362:994–1003.
- Sharrow SD, Novotny MV, Stone MJ. Thermodynamic analysis of binding between mouse major urinary protein-I and the pheromone 2-sec-butyl-4,5-dihydrothiazole. Biochemistry. 2003;42:6302–6309.
- Neumann L, Von Knig K, Ullmann D. HTS Reporter displacement assay for fragment screening and fragment evolution toward leads with optimized binding kinetics, binding selectivity, and thermodynamic signature. In: Kuo LC, editor. Methods Enzymol. Academic Press. 2011. vol. 493, p. 299–320.
- Papalia GA, Giannetti AM, Arora N, et al. Thermodynamic characterization of pyrazole and azaindole derivatives binding to p38 mitogen-activated protein kinase using Biacore T100 technology and van’t Hoff analysis. Anal Biochem. 2008;383:255–264.
- Silvestre HL, Blundell TL, Abell C, et al. Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery. Proc Natl Acad Sci U S A. 2013;110:12984–12989.
- Scott DE, Dawes GJ, Ando M, et al. A fragment-based approach to probing adenosine recognition sites by using dynamic combinatorial chemistry. ChemBioChem. 2009;10:2772–2779.
- Bullock AN, Debreczeni JÉ, Fedorov OY, et al. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM-1) kinase. J Med Chem. 2005;48:7604–7614.
- Dai R, Geders TW, Liu F, et al. Fragment-based exploration of binding site flexibility in mycobacterium tuberculosis BioA. J Med Chem. 2015;58:5208–5217.
- Poget SF, Legge GB, Proctor MR, et al. The structure of a tunicate C-type lectin from Polyandrocarpa misakiensis complexed with D-galactose. J Mol Biol. 1999;290:867–879.
- Calamini B, Santarsiero BD, Boutin JA, et al. Kinetic, thermodynamic and X-ray structural insights into the interaction of melatonin and analogues with quinone reductase 2. Biochem J. 2008;413:81–91.
- Fisher BM. Coulombic effects of remote subsites on the active site of ribonuclease A. Biochemistry. 1998;37:17386–17401.
- Weber PC, Pantoliano MW, Simons DM, et al. Structure-based design of synthetic azobenzene ligands for streptavidin. J Am Chem Soc. 1994;116:2717–2724.
- Rühmann E, Betz M, Fricke M, et al. Thermodynamic signatures of fragment binding: validation of direct versus displacement ITC titrations. Biochim Biophys Acta Gen Subj. 2015;1850:647–656.
- Perozzo R, Jelesarov I, Bosshard HR, et al. Compulsory order of substrate binding to herpes simplex virus type 1 thymidine kinase: A calorimetric study. J Biol Chem. 2000;275:16139–16145.
- Terán W, Krell T, Ramos JL, et al. Effector-repressor interactions, binding of a single effector molecule to the operator-bound TtgR homodimer mediates derepression. J Biol Chem. 2006;281:7102–7109.
- Dolezal O, Doughty L, Hattarki MK, et al. Fragment Screening for the Modelling Community: SPR, ITC, and Crystallography. Aust J Chem. 2013;66:1507.
- Talhout R, Engberts JBFN. Thermodynamic analysis of binding of p-substituted benzamidines to trypsin. Eur J Biochem. 2001;268:1554–1560.
- Bertini I, Calderone V, Fragai M, et al. Exploring the subtleties of drug-receptor interactions: the case of matrix metalloproteinases. J Am Chem Soc. 2007;129:2466–2475.
- Ferenczy GG, Keserű GM. Thermodynamics of Ligand Binding. Luque J, Barril X, editors. Physico-Chemical Comput. Approaches to drug discov. RSC Drug Discov. Ser. Cambridge: Royal Society of Chemistry; 2012. p. 23–79.
- Murray CW, Carr MG, Callaghan O, et al. Fragment-based drug discovery applied to hsp90. discovery of two lead series with high ligand efficiency. J Med Chem. 2010;53:5942–5955.
- Torres FE, Recht MI, Coyle JE, et al. Higher throughput calorimetry: opportunities, approaches and challenges. Curr Opin Struct Biol. 2010;20:598–605.