
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Drug discovery is a laborious process with rising cost per new drug. Peptide macrocycles are promising therapeutics, though conformational flexibility can reduce target affinity and specificity. Recent computational advancements address this problem by enabling rational design of rigidly folded peptide macrocycles.
Areas Covered
This review summarizes currently approved peptide macrocycle therapeutics and discusses advantages of mesoscale drugs over small molecules or protein therapeutics. It describes the history, rationale, and state of the art of computational tools, such as Rosetta, that allow the design of rigidly structured peptide macrocycles. The emerging pipeline for designing peptide macrocycle drugs is described, including current challenges in designing permeable molecules that can emulate the chameleonic behavior of natural macrocycles. Prospects for reducing computational cost and improving accuracy with emerging computational technologies are also discussed.
Expert opinion
To embrace computational design of peptide macrocycle drugs, we must shift current attitudes regarding the role of computation in drug discovery, and move beyond Lipinski’s rules. This technology has the potential to shift failures to earlier in silico stages of the drug discovery process, improving success rates in costly clinical trials. Given the available tools, now is the time for drug developers to incorporate peptide macrocycle design into drug discovery pipelines.
1. Introduction
The development of new drugs is a laborious and costly process. Typically, tens or hundreds of thousands of compounds must be screened for potency in some high-throughput biochemical assay. The hits identified from these screens, representing a small fraction of the initial pool, are examined more closely by low-throughput methods to precisely quantify affinity, determine the structural basis for efficacy, and assess other desirable properties. Cell culture models and animal testing are used to quantify toxicity, identify off-target effects, and determine pharmacokinetic properties, winnowing the number of hits by orders of magnitude to identify a small number of leads to carry forward for refinement and ultimate clinical testing [Citation1]. Even when the top candidates are taken forward to phase 3 human clinical trials, 54% of candidate molecules fail, with 57% of these failures showing poor efficacy and 17% showing unacceptable toxicity or side-effects [Citation2]. Determining how to chemically modify a failed drug to improve efficacy or address toxicity can itself be a long and costly research project. Despite major scientific advancements to the drug development process, and rising investment of person-hours, money, and resources, the number of new molecular entities approved for clinical use each year has remained steady [Citation3,Citation4], meaning that the cost of introducing a new drug is rising [Citation5].
Beginning in the late 1980s, a tantalizing alternative emerged: rather than blindly screening compound libraries for hits, perhaps new drugs could be engineered rationally, based on experimentally determined structures of molecular targets, more efficiently leading to clinic-ready pharmaceuticals [Citation6]. Drug development is, after all, an engineering problem with clear objectives. First, a drug should have high affinity for its target to minimize the lowest dose needed. Second, a drug should have low propensity for off-target interactions to maximize the highest safe dose. Together with the first consideration, this gives rise to the concept of therapeutic index, or the ratio of the maximum safe dose to the minimum effective dose, with the safest and most versatile drugs having the highest therapeutic indices [Citation7]. Beyond these pharmacodynamic considerations, the third consideration is pharmacokinetic: a good drug must persist in the body long enough to produce a desired effect before being degraded or cleared, and must be able to pass through barriers in its way, such as the gut epithelium (for orally administered drugs), the blood–brain barrier (for neuroactive drugs), or the cell membrane (for drugs with intracellular targets) [Citation8–Citation11]. Rational design strategies that try to use prior knowledge, human intuition, or computation to maximize target affinity, minimize off-target effects, and maximize bioavailability have given rise to some successes. Perhaps the most spectacular of these has been the development of the HIV protease inhibitors [Citation6]. First-generation drugs in this class like saquinavir (approved by the FDA in 1996) were designed rationally based on X-ray crystal structures of the HIV-1 protease to mimic the transition state of the reaction catalyzed by this essential viral enzyme. Second-generation inhibitors, such as indinavir, combined features of molecules found from screens with features from the X-ray crystal structure of saquinavir bound to its target. Third-generation inhibitors, such as darunavir, further built on the known structures of first – and second-generation inhibitors, adding chemical groups to maximize favorable interactions with the target [Citation12]. These molecules have transformed the clinical management and worldwide epidemiology of HIV/AIDS [Citation13].
Despite this notable success, most drug development efforts have continued to rely primarily on library screens, using rational structure-based design as a supplementary technique at best [Citation4]. One factor limiting the widespread adoption of rational structure-based design has been the difficulty in synthesizing proposed new molecular entities [Citation14]. If extensive research is required to develop and optimize a synthetic protocol for a computationally designed molecule, rapid computational design methods fail to save drug development time. Another factor is that rational design approaches often strive to maximize the favorable interactions between a drug and its target in a static, bound conformation, and have difficulty considering off-target binding or pharmacokinetic properties. Additionally, since many rational drug design approaches are additive, appending dendritic chemical groups to a starting molecule in order to add favorable interactions with a target, the end product is frequently a large, flexible molecule [Citation6]. Design approaches that fail to take into account the entropic cost of ordering a disordered molecule on binding can easily overestimate the binding affinity, giving rise to one of the major reasons for disconnects between computational predictions of binding affinities and experimentally measured affinities [Citation15].
Many of these problems can be addressed by focusing on peptide and peptide-like molecules. These can be synthesized by standard solid-phase methods, ensuring that new designs require relatively little research or optimization to realize. This also means that peptides are much more mutable than are small molecules, allowing easy experimentation on potentially better variants of a sub-optimal initial hit. Peptide stapling [Citation16,Citation17], cross-linking [Citation18,Citation19], and macrocyclization [Citation20,Citation21] approaches can limit the conformational flexibility of larger molecules, allowing presentation of more chemical groups for favorable interactions with a target without creating an unduly flexible molecule with a prohibitive entropic penalty to binding. However, covalent linkages alone do not guarantee rigidity, let alone rigidity in an active conformation [Citation21,Citation22]. Recently, computational tools have been developed both for rationally designing peptide macrocycles and for reliably stabilizing a peptide in a desired conformation [Citation23–Citation25]. Given the tools now available, the time is ripe for a new wave of rational, structure-based design focusing on peptide macrocycles engineered for both affinity and conformational rigidity. This review summarizes the existing methods for computational design of folded peptide macrocycles, the rationale underlying their development, the successes and current limitations of these methods, and the long-term prospects and challenges for rationally designed peptide macrocycle drugs.
2. Computational design of peptide macrocycles
2.1. Peptide macrocycles: A powerful but underutilized class of drugs
The majority of pharmaceuticals in use today are small molecules: of 3,967 approved drugs indexed in DrugBank [Citation26,Citation27], just over half (2,014 drugs) are molecules obeying the Lipinski criterion of being under 500 Da in mass [Citation28]. Many of the rest are either cellular therapies or protein therapeutics with masses over 10 kDa. Examples of small-molecule drugs are shown in : here, acetaminophen (paracetamol) is a common analgesic, diphenhydramine is a widely used antihistamine, penicillin G is a lactam antibiotic, and darunavir is an HIV protease inhibitor. While small molecules are easier to mass produce, more shelf-stable, easier to administer, frequently orally bioavailable, sometimes cell permeable, and often able to evade the human immune system, their small size limits the surface area that they can present for target recognition. This means that they often suffer from limited target affinity and low specificity, giving rise to low therapeutic indices [Citation29]. Those small molecules with the largest surface areas, such as darunavir, are frequently dendritic and flexible – a factor that can impose a high entropic cost to binding and limit affinity and specificity. Molecules under 45 kDa are also rapidly cleared by the kidneys, limiting serum half-life [Citation30]. Unlike small molecules, protein therapeutics () can present much larger surfaces for very specifically recognizing a target, resulting in high-affinity binding and excellent specificity, and producing high therapeutic indices. Shown are glucarpidase, an enzyme able to remove excess methotrexate from the blood of patients with impaired kidney function, streptokinase, which enzymatically dissolves blood clots, and the Fab fragment of omalizumab, a monoclonal antibody used to treat asthma. Unfortunately, the large size of a protein molecule makes it difficult to administer: typically, protein therapeutics must be injected, and cannot diffuse across the cell membrane, limiting their targets to extracellular molecules [Citation29]. Although the kidneys tend not to remove molecules over 60 kDa [Citation30], the immune system and blood proteases have evolved to recognize and clear foreign proteins, limiting biological half-life while putting patients at risk of an immune response to a therapy [Citation31].
Figure 1. Examples of currently approved molecular therapeutics. (A) Examples of small-molecule drugs, most of which are under 500 Da and less than 1 nm in size, shown as ball-and-stick models with transparent molecular surfaces. Clockwise from top left, Cambridge structural database (CSD) crystal structures COTZAN03, JEMJOA01, BZPENK01, and APUYOA are shown. (B) Examples of peptide macrocycle drugs, of which only 20 are approved for clinical use. Molecules are shown as ball-and-stick models (with apolar hydrogen atoms omitted for clarity) and transparent molecular surfaces. Clockwise from top left, CSD structures YICMUS and DEKSAN, and PDB structures 5L3 F (chain C) and 2VDN (chain C) are shown. (C) Examples of protein drugs, shown as ribbons and molecular surfaces. Clockwise from top, PDB structures 1 CG2 (chains A and B), 1BML (chains C and D), and 5HYS (chains C and D). Insets: small-molecule and peptide macrocycle drugs from panels A and B, respectively, shown to scale.
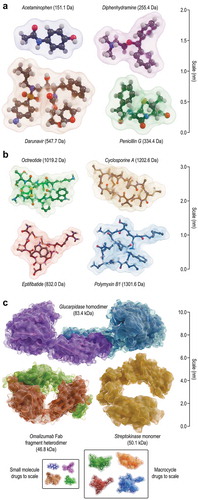
Although they represent a small fraction of known drugs, some of the most potent and versatile therapeutics are intermediate- or mesoscale macrocycles, often built from amino acids or amino acid-like building-blocks. shows octreotide, a synthetic analogue of the natural hormone somatostatin which is used to treat tumors producing growth hormone, cyclosporine A, a potent immunosuppressant, polymyxin B1, an antibiotic that targets Gram-negative bacteria, and eptifibatide, a powerful anticoagulant. Currently, there are 20 peptide macrocycle drugs in use in the clinic (), most of which were discovered or invented prior to 1990 [Citation27,Citation32–Citation59]. These include three molecules (colistin, dactinomycin, and polymyxin B) on the WHO’s Model List of Essential Medicines [Citation60]. In addition, a number of non-peptide macrocycle drugs, such as rifampicin and vancomycin, are in use [Citation61]. Most approved peptide macrocycles are natural products or artificially modified variants. Only five (eptifibatide, octreotide, and three octreotide derivatives) are artificial and rationally designed [Citation42,Citation53].
Table 1. Peptide macrocycle drugs currently approved for clinical use.
It is no coincidence that mesoscale natural products can serve as such useful drugs: the evolutionary conditions that select for the biosynthetic pathway that produces a venom, microbicide, other defensive or offensive toxin are similar to the artificial selective criteria used when seeking good drugs. A good toxin must have an effect at low concentration (i.e. bind to its target with high affinity), must not harm the organism that produces it (i.e. must be selective for its target), and must reach the target molecule within the target organism effectively (i.e. must have good pharmacokinetic properties, the ability to evade clearance and degradation, and often, the ability to pass through biological barriers) [Citation62,Citation63]. Nature has repeatedly settled upon mesoscale molecules as an effective solution given these constraints, relying on backbone macrocyclization, disulfide bonds, and other covalent linkages to produce rigid molecules pre-organized in their binding-competent conformations [Citation64,Citation65]. Covalent crosslinking or backbone cyclization can also render these molecules less susceptible to protease degradation [Citation66].
Nature has not exhaustively sampled the chemical space however. Some mesoscale toxins, such as knottins and defensins, are ribosomally synthesized mini-proteins that are heavily cross-linked with disulfide bonds [Citation67,Citation68]. Although loops in these molecules can often be mutated without affecting the fold [Citation69], cross-linked positions cannot; nor can amino acids other than the 20 canonical types be incorporated. Others, such as algal microcystins, are synthesized by dedicated enzymatic pathways that can incorporate nonstandard amino acids [Citation70,Citation71]. The evolution of nonribosomally produced molecules requires successful alteration of a biosynthetic pathway that is unique to each toxin, rather than simple mutations to a single gene, which likely limits the diversity of these molecules. But where this is a limitation in nature, it represents an opportunity for drug discovery: solid-phase peptide synthesis techniques allow diverse sequences to be created in the chemical laboratory using standard methods that allow one amino acid building-block to be swapped for another – natural or non-natural – with ease [Citation72,Citation73]. This means that a vast chemical space, one known to contain molecules with potent drug-like properties but which has not been exhaustively explored by nature, is synthetically accessible to us, provided that we have the means of finding what we seek within that space. Our ability to incorporate nonstandard or non-natural chemical building blocks, such as D-amino acids, also allows the creation of molecules with enhanced resistance to proteases and with reduced immunogenicity [Citation74,Citation75].
Several groups have published methods for synthesizing and screening libraries of peptide macrocycle sequences. These include split-and-pool and teabag syntheses for exploring combinatoric variations on a sequence pattern, microchip-based parallel syntheses, or approaches involving genetic encoding, ribosomal translation, and post-translational cyclization [Citation76–Citation79]. Typically, high-throughput synthetic approaches are hindered by the fact that they can synthesize, at most, thousands of sequences. In comparison, there are approximately one trillion possible sequences for a 10-residue macrocyclic peptide built only from the 20 standard amino acids, a number that balloons when one includes an expanded palette of amino acid building-blocks and choices of chemical cross-linkers, chemically conjugated groups, and exotic cyclization chemistries. This means that libraries can only span an insignificant fraction of the total accessible chemical space. (This is true even if one confines oneself to small-molecule libraries: it has been estimated that there are over 1060 possible small molecules satisfying Lipinski’s rule of five [Citation6]). Peptide libraries are also hindered by the need to purify each peptide away from the harsh chemicals used for synthesis before assaying its effect on a biological system. Given that the number of molecules that may be synthesized for a particular experiment is small, the number that might be assayed in a carefully controlled manner is smaller still, and the number of possible sequences is incredibly vast, there is a clear role for computational design to inform library design so that a library contains sequences worth screening experimentally.
2.2. Discriminating good drug designs from bad: Computational filtering tools
Until recently, most computational techniques were applicable for post hoc filtering, in which large numbers of candidate drugs are considered and those with undesirable computed properties are discarded. In general, filtering methods take a structure as input and produce one or more metrics related to properties of interest as output; these can then be used to discard structures with metric values under or over a chosen threshold. Various groups have published in silico approaches for predicting a drug’s specificity [Citation80,Citation81], solubility and aggregation propensity [Citation82,Citation83], permeability [Citation84,Citation85], and general toxicity [Citation86]. Many of these approaches are based on prior knowledge or machine learning (ML). For example, Li et al. compared the ability of five different ML methods, trained against a database of over 12,000 chemical compounds with experimentally measured LD50 values, to predict acute oral toxicity [Citation87]. Such approaches unfortunately rely on very large databases of chemical compounds with known properties, and have extrapolative power only insofar as similar molecules have similar properties. Unfortunately, a trivially small fraction of the chemical space accessible to peptides has been studied, and small changes in peptide sequence can result in large changes in physical or toxicological properties, limiting the extent to which these approaches are applicable to peptide macrocycle drugs. For this reason, it is often necessary to fall back on unbiased simulations – most often, molecular dynamics (MD) simulations, in which the motions of a molecule and its surrounding solvent are simulated over time – to predict physical properties, but these carry considerable computational expense. Typical MD simulations performed over days on large computing clusters can simulate the behavior of a moderately sized molecular system, such as a peptide macrocycle and its surrounding water, for nanoseconds. The largest MD simulations, carried out on dedicated supercomputing hardware, can simulate the folding of a very small, fast-folding protein on the timescale of a millisecond [Citation88]. Processes involving larger numbers of atoms and longer time-scales, such as the insertion of a peptide macrocycle into a lipid bilayer, tax the abilities of MD, and often require coarse-grained, approximate models [Citation89]. Unfortunately, the expense of MD hinders its applicability for large-scale screening of long lists of candidate drugs, but it can be used to winnow short lists.
2.3. The development of peptide macrocycle design methods that yield rigidly folded molecules
Design methods that propose candidates that have been optimized for some set of desirable properties (affinity, rigidity, solubility, etc.) represent a powerful complement to filtering methods. Although small molecules [Citation6] and proteins [Citation90] have long histories of rational design, the rational design of peptides is a less-explored area, but one in which the successes, despite being guided by human intuition more than by computation, are notable. In 1982, Bauer and colleagues designed the now widely used octreotide by systematically modifying the structure of somatostatin, a natural human hormone [Citation42]. A decade later, using trial-and-error syntheses, Scarborough et al. were able to generate a rigid disulfide-cyclized peptide scaffold (now called eptifibatide) for presenting the RGD sequence that selectively inhibits platelet glycoprotein [Citation53]. Since the late 1990s, using manual design and trial-and-error synthesis, the Gellman group has discovered many peptide ‘foldamers,’ or rigid linear peptide scaffolds that can be functionalized [Citation91–Citation93]. Others, such as the Kritzer group, have pursued linear and cyclic foldamers able to bind to target proteins [Citation94,Citation95]. Despite these successes, extensive exploration of sequence and conformation space has been hindered by the lack of robust computational methods.
Beginning in 2012, my colleagues and I set out to develop computational tools to allow the design of rigidly structured peptide macrocycles, drawing inspiration from the protein design field. The intention was to develop computational methods that take as input all information necessary to define a peptide macrocycle design problem (for example, the structure of a target, a specification of a binding pocket on that target, and a list of desired properties of a molecule designed to bind to that target) and produce as output the sequence and structure of a peptide macrocycle intended to fulfill the requirements of the design problem. When designing proteins or peptides, one faces the challenge of exploring vast conformational and sequence spaces. For example, a 30-residue miniprotein with 2 rotatable bonds per residue and just 3 possible conformations for each rotatable bond has 4.2 × 1028 possible conformations, from which one seeks ‘designable’ conformations that can be stabilized by a suitable choice of sequence and which are also compatible with a desired function. The same 30-residue miniprotein, built from the 20 natural amino acids, has 1.0 × 1039 possible sequences from which one wishes to find one that best stabilizes a desired backbone conformation.
Our first major challenge was developing efficient computational methods for sampling conformations of peptide macrocycles to identify low-energy states when designing or validating designs. MD simulations, which simulate the motions of atoms in large molecular systems with time resolution on the femtosecond scale, producing millions of closely spaced, time-resolved structural snapshots as output, are an inefficient means of doing this, since much of the computation is spent simulating physically plausible transitions from low-energy state to state. Although these transitions are relevant to the kinetic stability of a design (making MD an important complementary validation technique), they are not needed when one is interested only in low-energy states populated at thermodynamic equilibrium. Protein designers draw on the Protein Data Bank (PDB), a repository of more than 157,000 experimentally determined protein structures [Citation96], to simplify the conformational search problem through fragment insertion-based approaches. Like MD, these approaches simulate a molecular system over many trajectory steps to produce a number of static snapshots of plausible low-energy structures. However, fragment insertion-based approaches use contiguous stretches of protein backbone, several amino acids long, taken from PDB structures as templates for large, nonphysical moves performed in the context of Monte Carlo trajectories, allowing rapid exploration of diverse backbone conformations while still limiting sampling to a relatively small, native-like subspace of the overall conformational space [Citation97,Citation98]. The output structural snapshots therefore span more of the low-energy conformation space at lower computational cost than MD. Protein structure-guided sampling approaches have been used in flexible-backbone peptide docking (in which one starts with a peptide sequence and a protein structure as input, and produces plausible docked conformations as output) and design of linear peptide ligands (in which one starts with a target structure as input and produces sequences and bound conformations of newly-designed peptides as output), in, for example, both the PepSpec and FlexPepDock methods [Citation99–Citation101]. An important limitation of this strategy is that it is effective only for peptides built from the 20 canonical amino acids. Incorporation of single D-amino acid residue results in a peptide with conformational preferences unlike those of any natural protein, putting its preferred conformations outside of the subspace sampled by fragment-based methods and rendering this approach ineffective. In addition, heavily covalently constrained molecules are sampled inefficiently by fragment-based approaches, since there is little overlap between the subspace allowed due to the covalent constraints and the subspace spanned by the available protein fragments. As an alternative, the Coutsias and Kortemme groups pioneered the use of kinematic approaches borrowed from the field of robotics to perform unbiased sampling of protein loops [Citation102,Citation103]. These approaches take the atomic-resolution structure of the starting and ending points of a loop, plus the amino acid sequence of the intervening segment, as input, and rapidly produce as output closed conformations in which the intervening chain of amino acids properly connects to the starting and ending points without discontinuities. Building on and generalizing this work, I developed the GeneralizedKIC tool to allow very rapid sampling of closed conformations of macrocycles built from any combination of natural and non-natural building-blocks, limiting sampling to the subspace compatible with the covalent geometry rather than biasing it based on known structures [Citation23,Citation104]. In subsequent years, complementary approaches for sampling macrocycle conformations, such as the combinatorial Prime-MCS method developed by Sindhikara et al., have also been proposed [Citation105,Citation106].
Another part of our challenge was adapting protein-centric energy functions for peptide macrocycle design. Energy functions take as input a structural model and produce as output an estimate of conformational energy, or some other score sensitive to covalent connectivity and conformation. These are used repeatedly to guide sampling when exploring conformations or sequences during structure prediction, docking, or design, and these also provide a means of comparing final designs. Although MD-inspired potentials have been applied to non-canonical heteropolymer design in the past [Citation107], protein structures have proven invaluable for calibrating the more reliable scoring functions used for protein design. The Rosetta energy function, for example, is composed of both physics-based terms and statistical terms based on distributions of features observed in PDB structures [Citation108,Citation109]. The observed distribution across PDB structures of backbone and side-chain conformations for each canonical amino acid is used both to construct knowledge-based potentials for amino acid conformation and to guide conformational sampling [Citation97,Citation110,Citation111]. An important limitation of this approach is that it cannot easily be extended to non-canonical amino acids that are poorly represented in PDB structures, though we realized that the D-amino acids that are the mirror images of canonical L-amino acids are an exception: in this special case, statistical potentials can be used, provided that all dihedral values are inverted [Citation23]. For the more general case, physics-based methods of precomputing torsional potentials, using either MD or quantum mechanics (QM) computations, are necessary [Citation112,Citation113]. Through all of this, we strove to preserve and augment the protein-centric energy function rather than replacing it. Mindful of the goal of accurately modeling peptide macrocycles binding to proteins, which requires an energy function that is as accurate as possible for proteins as well as for peptide macrocycles, my colleagues and I incorporated all enhancements into the protein-optimized Rosetta ref2015 energy function [Citation108,Citation109].
Sequence design itself was another challenge. Given a fixed peptide backbone (produced beforehand by a backbone-sampling algorithm) and a set of candidate side-chain identities and conformations (collectively called rotamers) for each backbone position as input, a sequence design algorithm produces as output a selection of one rotamer per position that optimizes some objective function (often an energy function) that corresponds to the quality of a design for a given design goal. The combinatorial problems associated with sequence design have long been recognized as a major factor limiting computational design of peptide therapeutics [Citation114]. While ML-based approaches have been applied for canonical protein design [Citation115], these methods are also limited to the 20 canonical amino acids present in the training data. The most general approaches, compatible with non-natural building blocks, are unbiased algorithms such as the dead-end elimination approaches featured in Osprey or Toulbar2 [Citation116,Citation117], or heuristics such as the Rosetta packer [Citation107,Citation118]. An energy-based scoring function may be augmented with additional terms intended to penalize undesirable or promote desirable physical features in the designed peptide, a strategy frequently needed to ensure solubility, or to encourage incorporation of conformationally constrained amino acid residues such as D- or L-proline that promote rigid structure [Citation119]. Using Rosetta’s design tools (the Rosetta packer) in combination with the sampling and scoring approaches described above, my colleagues and I have designed, computationally validated, synthesized, and experimentally verified dozens of peptide macrocycles in recent years, representative examples of which are shown in . The smallest of these (panels A and B) are 7 to 10 residue macrocycles comparable in size to existing peptide macrocycle drugs like octreotide (8 residues) or cyclosporine A (11 residues) [Citation24,Citation42,Citation120,Citation121]. At intermediate scale (panel C) it is possible to design exotic secondary and tertiary structures that cannot be built from natural amino acids alone, such as the closely-packing helices of opposite handedness shown [Citation23]. The largest of these (panel D) are mini-proteins of up to 60 residues which can incorporate artificial cross-linking agents to produce exotic folds unlike anything found in nature [Citation25]. The robust design process producing these molecules is made available through the Rosetta software suite, and may be controlled through the RosettaScripts or PyRosetta scripting languages [Citation122,Citation123].
Figure 2. Nuclear magnetic resonance (NMR) spectroscopy structures of designed synthetic peptide and polypeptide macrocycles with rigid folds. One representative structure from the NMR ensemble is shown in each case. Structures are shown as ball-and-stick models with apolar hydrogen atoms omitted, and as transparent molecular surfaces; ribbons represent backbone secondary structure (helices) in the larger polypeptides. Peptide names are italicized, and amino acid sequences are displayed with lower- and upper-case letters representing D- and L-amino acid residues, respectively. (A) and (B) Designed 7- and 11-residue peptide macrocycles with rigid structures (PDB IDs 6BF3 and 6BES) [Citation24]. (C) Designed 26-residue polypeptide macrocycle NC_cHh_DL_D1, containing helices of opposite handedness packing against one another (PDB ID 5KX0) [Citation23]. (D) Designed threefold-symmetric 60-residue macrocyclic mini-protein CovCore 3H1, with a hydrophobic core consisting of a central, artificial, covalent 1,3,5-tris(bromomethyl)benzene cross-linker (PDB ID 5V2 G) [Citation25].
![Figure 2. Nuclear magnetic resonance (NMR) spectroscopy structures of designed synthetic peptide and polypeptide macrocycles with rigid folds. One representative structure from the NMR ensemble is shown in each case. Structures are shown as ball-and-stick models with apolar hydrogen atoms omitted, and as transparent molecular surfaces; ribbons represent backbone secondary structure (helices) in the larger polypeptides. Peptide names are italicized, and amino acid sequences are displayed with lower- and upper-case letters representing D- and L-amino acid residues, respectively. (A) and (B) Designed 7- and 11-residue peptide macrocycles with rigid structures (PDB IDs 6BF3 and 6BES) [Citation24]. (C) Designed 26-residue polypeptide macrocycle NC_cHh_DL_D1, containing helices of opposite handedness packing against one another (PDB ID 5KX0) [Citation23]. (D) Designed threefold-symmetric 60-residue macrocyclic mini-protein CovCore 3H1, with a hydrophobic core consisting of a central, artificial, covalent 1,3,5-tris(bromomethyl)benzene cross-linker (PDB ID 5V2 G) [Citation25].](/cms/asset/4abf014b-bc3f-4070-9f65-0ef43258e25f/iedc_a_1751117_f0002_oc.jpg)
2.4. Computational validation of conformational rigidity
Although many relatively low-cost computational validation approaches exist to winnow pools of candidate designs, when we began development of our peptide design pipeline, no tool was available to predict whether a particular design uniquely favors the designed conformation for final selection of designs worth testing experimentally; nor was there a standard metric for quantifying or reporting the extent to which a design is predicted to favor its designed conformation. Since conformational flexibility, and the entropic cost of ordering a disordered molecule on binding, is a major reason that rational design fails for larger drug molecules [Citation15], Rosetta’s simple_cycpep_predict application was developed. This application takes a designed peptide macrocycle sequence and structure as input, and uses GeneralizedKIC to sample conformations, carrying out side-chain conformational optimization and energy minimization on each conformation sampled to report a minimized energy and root-mean-squared deviation (RMSD) to the designed conformation [Citation124]. With the output energy and RMSD values for a representative set of sampled conformations, the application quantifies the extent to which the design uniquely favors the desired conformation, providing the PNear metric as final output. PNear is a general metric applicable to any large-scale sampling approach for validating designs, shown in EquationEquation 1(1)
(1) [Citation23,Citation24]:
In the above, the sums are over all sampled conformations, RMSDi is the root mean squared deviation of the ith sample from the desired conformation, Ei and Ej are the energies of the ith and jth samples, kBT is the Boltzmann temperature, and λ is a tuneable parameter (typically set around 1.5 Å) governing the tolerance to small deviations from the desired conformation. The output value (PNear) ranges from 0 to 1, where 0 represents no propensity to favor the desired conformation and 1 represents full preference for the desired conformation. Intuitively, this expression may be thought of as an approximation of the Boltzmann probability that a molecule sampled from an equilibrium distribution is within a fuzzily defined region of conformation space near a desired conformation; replacing the Gaussian term with a hat function makes the definition of state precise. The use of a soft boundary (the Gaussian) instead of a hard threshold is practically advantageous for numerical stability given stochastic sampling variation. In our experience, macrocycles with computed PNear values over 0.9 nearly always adopt the predicted conformations in real-world experiments, and those with values over 0.8 are often worth testing experimentally. In addition to being used to validate the macrocycles shown in -C [Citation23,Citation24], the simple_cycpep_predict application has proven useful for selecting candidate drug-like molecules. Est et al. were recently able to show improvements to the amyloid inhibitory activity of a transthyretin-derived cyclic peptide using mutations and disulfide placements screened virtually with the simple_cycpep_predict application. They found experimentally that conformational rigidity in a slightly twisted active β-sheet conformation was a key determinant of anti-amyloid activity, and were able to predict experimental success of peptide designs using the application [Citation22].
2.5. The emerging pipeline for computational design of rigid peptide macrocycle drugs
shows the typical drug discovery process. Since later steps in this process, particularly clinical trials, involve enormous investment of resources, computational predictions can play an invaluable role to improve the probability of success in these later steps by informing earlier steps, both as filters and as design tools. The emerging process for de novo design of peptide macrocycles for therapeutic development is examined in more detail in . This process takes as input the known structure of a target protein and begins by sampling candidate peptide conformations and binding modes in the context of a binding site in the target. For each candidate conformation, an amino acid sequence is built, attempting both to maximize favorable interactions with the target and to maximally stabilize the bound conformation of the peptide macrocycle in the absence of the target. This produces an initial large pool of candidate designs, which can be pruned by low-cost filters that discard poor designs based on prior knowledge (for example, those unlikely to be soluble, those with too few conformationally constrained amino acids to be likely to fold, or those with undesirable features such as buried unsatisfied hydrogen bond donors or acceptors). As a final computational step, the conformational flexibility of the macrocycle in the absence of the target is assessed. These steps can be performed using tools within the Rosetta software suite (GeneralizedKIC, FastDesign, and simple_cycpep_predict [Citation23,Citation24]), or with other software tools. and D show typical output from simple_cycpep_predict for a macrocycle predicted to favor its binding-competent conformation uniquely, and for one predicted to have a high entropic cost to binding, illustrating how poor designs (those with a high entropic cost of ordering the macrocycle when it binds its target) are distinguished from good designs. As shown, the quantitative metric PNear (EquationEquation 1(1)
(1) ) clearly distinguishes designs with a unique low-energy state matching the designed conformation from designs with no unique low-energy state or with an alternative low-energy state, allowing automated discrimination. The final output of the design pipeline is a set of designed sequences and structures, with metrics such as interaction energy to the target and peptide fold propensity in the absence of target (PNear) that can be used to rank designs to prioritize experiments.
Figure 3. Uses of computation to inform design of peptide macrocycle drugs. (A) Steps of the drug discovery process (blue), and means by which computation may inform early steps of this process (lilac ovals) to increase the probability of success in later steps requiring greater investment of resources. Computation may be used for filtering (e.g. by predicting physical, pharmacokinetic, or toxicological properties, and discarding candidates with undesirable properties), or for generating new possibilities to test. The most aggressive use of computation lies in de novo design to produce initial compound pools to screen for hits (boldface). (B) The emerging pipeline for designing peptide macrocycle drugs. The steps involve diminishing numbers of possibilities, and invest more and more computational effort per possibility for design and validation. The largest investment of computing resources is necessary for assessing the conformational rigidity of a design, through large-scale conformational sampling (boldface). (C) and (D) Example output from a method of assessing conformational rigidity of designed peptide macrocycles (in this case, the Rosetta simple_cycpep_predict application). Each point represents the output of a separate conformational sampling trajectory, with the final energy and RMSD to a target conformation plotted. Sequences are shown above the plots, with X representing 2-aminoisobutyric acid, B representing 2,3-diaminopropionic acid, and upper- and lower-case letters representing L- and D-amino acids, respectively. Panel C shows a design predicted to have a unique low-energy state closely matching the desired conformation (blue arrow), with a PNear value near 1. Panel D shows a design with many low-energy states and no unique-folded state (red arrows), with a PNear value near 0. Computations were performed on the Mira and Theta supercomputers (Argonne Leadership Computing Facility).
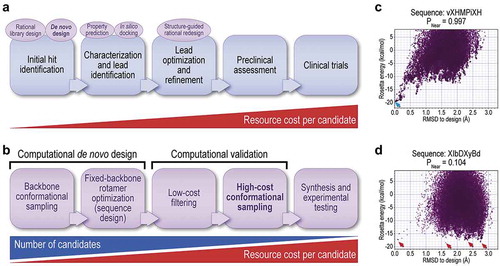
As a practical consideration, the design pipeline progresses from considering very large numbers of possibilities, at low computational cost per possibility, to settling on a few final designs, validated at high computational cost per design [Citation25]. By discarding designs first by the lowest-cost method available (albeit with high false-positive rates), and applying high-cost computations (with low false-positive rates) only to those candidates that remain, one minimizes the total computational cost to find a hit. The final number of computational designs produced by this process is ultimately determined by the capacity of the experimental screen that this process informs. Since the resource cost of experiments invariably exceeds the resource cost of computation, accurate computations that are able to winnow designs that are unlikely to succeed experimentally, and which can produce designs that are likely to succeed, can greatly reduce the overall resource cost of producing a successful molecule.
2.6. Ongoing challenges: Designing cell-permeable peptide macrocycles
Although currently approved peptide macrocycle therapeutics generally exhibit good bioavailability, indicating the potential of this class of molecule, artificial peptide macrocycles are frequently limited in their ability to cross biological barriers – particularly cell membranes. Adding permeability to the twin goals of creating high-affinity binders with high specificity represents a challenge that ongoing research aims to address. In the past, the Lipinski rule of five was often dogmatically treated as the key to drug permeability [Citation28]; however, there is mounting recognition that many useful drugs lie ‘beyond the rule of five,’ and yet can still potentially possess important properties like permeability. Although there is some potential even for non-permeable peptide macrocycles as injected therapeutics targeting extracellular proteins, learning to design peptide macrocycles that retain barrier permeability despite violating the Lipinski rules will greatly expand the range of targets and applicability of peptide macrocycle drugs.
One strategy is to try to design molecules with dynamic behavior similar to cyclosporine A. Cyclosporine has fairly high oral bioavailability, ranging from 10% to 60% depending on formulation [Citation10], and is able to reach intracellular targets despite violating Lipinski’s rules for mass (well over 500 Da) and hydrogen bond acceptors (over 10) [Citation28]. Structures of cyclosporine in apolar and aqueous solvent reveal a large conformational change between the two environments, as shown in . In apolar environments (like the lipid bilayer), this macrocycle forms internal hydrogen bonds satisfying all of its backbone amide protons. Other amide groups, which would have protons pointing outward in this conformation, are N-methylated [Citation120]. In aqueous solvent, the conformation shifts radically to allow all polar groups to form hydrogen bonds with water and to bury three of the N-methyl groups [Citation125,Citation126]. This ‘chameleonic’ behavior – converting from a water-favoring to a lipid-favoring state – is thought to be what allows cyclosporine to partition from aqueous to apolar environments, and to diffuse passively across the lipid bilayer into cells [Citation121,Citation127–Citation130]. A recent nuclear magnetic resonance study of four non-peptidic macrocycle drugs that are also cell-permeable despite violating Lipinski’s rules revealed similar chameleonic behavior, suggesting that this may be a key to membrane permeability in the size range beyond 500 Da [Citation131]. Several studies have suggested that polar chemical group content or exposed polar surface area are some of the better predictors of poor membrane permeability [Citation129,Citation132–Citation134]. This is intuitive: the more polar groups that are exposed in the membrane-penetrant state, the greater the favorability of remaining in the aqueous phase, and the more difficulty one would expect a molecule to have in passing through a membrane. The Lipinski rules, which impose hard and arbitrary maximum values on the size of a drug molecule (500 Da) and the number of hydrogen bond donors (5) and acceptors (10) [Citation28], may be a too-strict approximation of a simpler, more permissive rule: that exposed polar surface area must be minimized to allow solubility in an apolar environment in order to penetrate a membrane. Unfortunately, as molecules grow larger and more flexible, it is difficult to satisfy this simpler rule without explicitly considering conformational flexibility during design. Indeed, the most precise correlations have been found between the conformationally averaged exposed polar surface area and membrane permeability [Citation129,Citation131]. Recent enhancements to conformational sampling software, such as Rosetta’s simple_cycpep_predict application, allow predictions to be made about a peptide macrocycle’s ensemble-averaged dynamically exposed polar surface area [Citation124], making after-the-fact filtering of designs possible.
Figure 4. Conformational dynamics of cyclosporine A. Apolar hydrogen atoms are omitted in ball-and-stick renderings. (A) X-ray crystal structure of cyclosporine A in an apolar environment (CSD crystal structure DEKSAN) [Citation120]. All backbone amide protons, labeled H1, H6, H9, and H11 in blue, form internal hydrogen bonds to backbone carbonyl oxygen atoms. Backbone amides on residues 2–5, 7–8, and 10, which would otherwise be unsatisfied in apolar solvent, are N-methylated (green labels), allowing solubility in apolar solvent. (B) NMR ensemble of cyclosporine A in aqueous solvent (PDB ID 1CYB) [Citation125]. Backbone amide protons (blue labels) point outward, forming hydrogen bonds with water. N-methyl groups on residues 3, 5, and 10, which were exposed to organic solvent in panel A, now point inward, forming a small hydrophobic core. (C) Cyclosporine A, shown as a ball-and-stick model with a transparent blue molecular surface, bound to human cyclophilin G, shown as an opaque tan molecular surface (PDB ID 2WFJ) [Citation126]. The binding conformation is similar to the aqueous conformation shown in panel B, with most backbone amides exposed to water, and most N-methyl groups forming a small hydrophobic core (residues 3, 5, and 10) and/or making hydrophobic contacts with the target (residues 2, 3, 4, and 7).
![Figure 4. Conformational dynamics of cyclosporine A. Apolar hydrogen atoms are omitted in ball-and-stick renderings. (A) X-ray crystal structure of cyclosporine A in an apolar environment (CSD crystal structure DEKSAN) [Citation120]. All backbone amide protons, labeled H1, H6, H9, and H11 in blue, form internal hydrogen bonds to backbone carbonyl oxygen atoms. Backbone amides on residues 2–5, 7–8, and 10, which would otherwise be unsatisfied in apolar solvent, are N-methylated (green labels), allowing solubility in apolar solvent. (B) NMR ensemble of cyclosporine A in aqueous solvent (PDB ID 1CYB) [Citation125]. Backbone amide protons (blue labels) point outward, forming hydrogen bonds with water. N-methyl groups on residues 3, 5, and 10, which were exposed to organic solvent in panel A, now point inward, forming a small hydrophobic core. (C) Cyclosporine A, shown as a ball-and-stick model with a transparent blue molecular surface, bound to human cyclophilin G, shown as an opaque tan molecular surface (PDB ID 2WFJ) [Citation126]. The binding conformation is similar to the aqueous conformation shown in panel B, with most backbone amides exposed to water, and most N-methyl groups forming a small hydrophobic core (residues 3, 5, and 10) and/or making hydrophobic contacts with the target (residues 2, 3, 4, and 7).](/cms/asset/97496411-8b37-4045-9609-8e42c4e62a86/iedc_a_1751117_f0004_oc.jpg)
Advances in correctly modeling the energetics of folding in a membrane environment have already permitted the design of multi-pass transmembrane proteins [Citation135,Citation136]. The same energy functions could also be used to develop biconformational peptide macrocycles with different low-energy conformations in aqueous and apolar environments. Fully satisfying hydrogen bond donors and acceptors in apolar environments to minimize exposed polar surface area will be a challenge, but some donors can be removed by incorporating N-methylated amino acids or peptoid residues (in which the side chain extends from the backbone nitrogen). Both N-methylation and peptoid incorporation are associated with greater passive permeability [Citation128]. However, N-methylation greatly alters the conformational preferences of amino acids, so any addition of N-methyl groups must consider the effects on the molecule’s folding propensity. Peptoids are functionalized glycine, possessing the expanded conformational flexibility of an achiral building-block, so they too have an enormous impact on the folding behavior of a molecule [Citation137,Citation138]. Recently, support has been added for N-methylated amino acids, with QM-based precomputed mainchain potentials, to the Rosetta software suite [Citation139], and limited support for peptoids is also offered [Citation140], opening up the possibility of designing cyclosporine-like, membrane-permeable macrocycles – an ongoing area of research. Multistate design strategies, which consider many backbone conformations simultaneously rather than a single backbone conformation during the sequence design steps [Citation141], will ultimately be necessary to create molecules that are both able to undergo conformational transitions to pass through membranes, and which have high propensity to favor a binding-competent conformation in aqueous solvent, and which have high shape- and charge-complementarity to a target.
2.7. Ongoing research: Enhancing peptide macrocycle design with emerging computational approaches
Ongoing research aims to tackle the problems of increasing the accuracy and decreasing the computational cost of designing peptide macrocycle therapeutics. These problems are closely related: more accurate methods would allow fewer designs to be created and screened virtually, saving computational time, and lower-cost methods would allow more computational rigor (e.g. more virtual replicates, more samples, etc.) to be applied, improving accuracy. Much effort is being devoted into improved methods of computing conformational energies, a task for which there is a trade-off between speed and accuracy. Newtonian force fields, such as CHARMM [Citation142], AMBER [Citation143,Citation144], or the Rosetta ref2015 energy function [Citation108,Citation109], can be evaluated in milliseconds for large structures, allowing large-scale conformational sampling, but this speed comes at the cost of poorly capturing complex many-body effects such as induced dipoles or electrostatic screening. QM molecular modeling packages, such as GAMESS [Citation145], Psi4 [Citation146], or NWChem [Citation147], allow far greater precision and accuracy at the cost of much longer computation times (minutes to hours for systems of dozens of atoms) that hinder large-scale sampling. Where Newtonian force field have computing times that scale, at worst, with the square of the number of atoms in the system, QM methods scale exponentially, so that mesoscale molecules like peptide macrocycles are only tractable by breaking the systems into fragments [Citation148]. Additionally, QM packages allow very accurate and precise calculations of enthalpies, but much of the conformational and binding energy for a drug molecule comes from the hydrophobic effect, which is entropic in origin and often necessitates either large-scale sampling of water, or limited-accuracy implicit solvation models [Citation149,Citation150]. Strategies that use one-time, computationally expensive simulations or computations to train lower-cost approaches could give drug designers more accurate results without repeatedly applying more expensive calculations. For example, QM calculations have been used to pre-compute potentials that can be incorporated into Newtonian force fields [Citation112]. The idea of training ML models against accurate-but-expensive simulations is gaining traction in other fields as well: recently, astrophysicists trained neural networks to predict outputs of expensive cosmological simulations, yielding surprisingly accurate predictions of emergent properties of the simulated systems at much lower computational cost [Citation151]. Ongoing peptide macrocycle design research aims to train neural networks against the output of more expensive classical or QM simulations, allowing the output of these simulations to be approximated in milliseconds for large-scale sampling.
Modern and emerging hardware technologies can also make more accurate drug design computationally tractible. Graphics processing units (GPUs) use thousands of relatively slow processing cores that can simultaneously evaluate functions that can be broken into thousands of simpler calculations. These have proven invaluable for accelerating Newtonian [Citation152,Citation153] and QM [Citation154,Citation155] calculations. Field-programmable gate arrays (FPGAs), which can be reconfigured on the fly for highly efficient evaluation of a given function, could also accelerate such computations [Citation156], but to date, they have proven less popular, in part because the programming model is more complex and the hardware is available to fewer researchers. The massive parallelism of large supercomputing clusters can also be leveraged to accelerate minutes-long QM calculations [Citation112,Citation145] or to accelerate folding simulations by carrying out separate conformational sampling trajectories on separate computing cores [Citation23,Citation24], though the speed of inter-node communication limits the extent to which a large supercomputer can accelerate already-fast Newtonian energy calculations for a single conformation of a molecular system.
These hardware strategies typically accelerate computations by a constant factor, but cannot alter the scaling of the computation in question. Two of the most difficult problems in peptide macrocycle drug design remain the conformational sampling problem and the sequence sampling problem, and both involve search spaces that scale exponentially with the number of amino acid residues. Although the strategy of limiting sampling to a small subregion of the space is applied for proteins (using biased fragment-based methods), the same strategy cannot be used for peptides built from non-natural building-blocks, for which there are not large databases of known structures to use to bias the sampling. For this reason, perhaps the most intriguing emerging hardware technology is the quantum computer. Quantum computers offer the enticing prospect of modeling astronomical numbers of possible solutions to difficult combinatorial search problems simultaneously, and of sampling efficiently from the optimal solutions; as such, they could one day revolutionize peptide macrocycle structure prediction and design. Moreover, their parallel modeling capacity scales exponentially with the number of quantum bits, or qubits [Citation157]. Although today’s quantum computers are hindered by thermal noise, limited inter-qubit connectivity, small qubit counts [Citation158], early and ongoing work on the D-Wave quantum annealer has produced quantum algorithms for conformational sampling in a simplified peptide-like system [Citation159] and for designing real peptide sequences without simplification [Citation160]. Applying this technology practically to drug design, and demonstrating an advantage over classical approaches, is the subject of ongoing research.
3. Conclusion
Although drug development is a costly process offering diminishing returns on the time and resources invested, peptide macrocycles represent an underexplored class of drugs with enormous potential for improving success rates. Since they lie between large proteins and small molecules in size, they have the potential to combine the advantages of both, providing larger interaction surfaces for more specific target recognition than is possible for a small molecule, while offering potentially better permeability and immune system evasion than is possible for a large protein. Since diverse peptide sequences can be synthesized using standard solid-phase methods, research and development time need not be spent finding a route to synthesis. Computational design methods promise to accelerate drug development and drop the costs associated with bringing a new peptide macrocycle therapeutic to the clinic.
Recently developed computational methods provide means both for creating pools of candidate peptide macrocycle designs with desired properties, and for predicting properties that affect usefulness as a drug, such as rigidity, conformational preferences, specificity and toxicity, solubility, and permeability. The emerging tools use both ML approaches, trained on the limited databases of drugs and druglike macrocycles, and physics-based simulations, relying on molecular dynamics or more rapid conformational sampling techniques. These methods have lately begun to address one of the deficiencies of structure-based design methods for mesoscale therapeutics: the decrease in target affinity and specificity that arises as a drug grows in size, becomes more flexible and gives rise to an entropic penalty associated with ordering a flexible molecule on binding. By designing for conformational rigidity of the macrocycle while simultaneously optimizing target affinity, the emerging tools provide a solution to this problem. Software suites such as Rosetta offer an end-to-end pipeline for peptide macrocycle design and computational validation, but other complementary design tools exist, such as Osprey and Toulbar2 for design; CHARMM and AMBER for MD-based validation; and GAMESS, Psi4, and NWChem for quantum mechanics-based validation. Many of these tools are publicly available either as open-source software or as free software for academic and nonprofit research [Citation161–Citation168], permitting researchers to experiment with new strategies for incorporating computational design and validation into existing drug development pipelines.
Although the existing peptide design and validation pipeline is powerful, it has its limitations. The robust design of membrane-permeable peptides remains a challenge. Approaches that seek to satisfy hydrogen bond donors and acceptors, minimize conformation-averaged solvent-accessible polar surface area, and permit emulation of the chameleonic behavior of cyclosporine A and other permeable macrocycle drugs will ultimately be necessary to address this. Improving the accuracy of computations and better integrating high-cost, high-accuracy methods, such as QM approaches, with large-scale sampling tools are ongoing challenges. ML-based approximations of high-cost calculations may improve accuracy. Computational cost also remains an obstacle, with much of the cost per design incurred during the validation step, where expensive MD simulations or conformational sampling trajectories must be carried out. Here, too, ML-based filtering methods trained against high-cost validation computations may help to bring down the cost of screening large numbers of computational designs to find high-quality designs for experimental study.
Despite these challenges, a robust pipeline is emerging for peptide macrocycle drug design, providing an enticing alternative to blind screening to identify hits, one which could give rise to more promising hits and to leads with better probabilities of success in clinical trials. As this technology continues to mature, it has the potential to begin to address the inefficiencies in the current drug discovery pipeline that make it such a laborious and costly process today.
4. Expert Opinion
The cost of drug development and the high failure rate of clinical trials create a clear need for better drug design methods. The recent advancements in computational design and structure prediction of peptide macrocycles that I have reviewed here provide a path to a future in which peptide macrocycle drugs are rationally designed on the computer, shifting failures to earlier, in silico stages of the drug development pipeline, prior to expensive investment of laboratory or clinical resources, and increasing the probability of success in laboratory and clinical phases. Designed peptide macrocycles also provide a means of avoiding costly late-stage failure modes for a drug development project. Peptides can be synthesized by standard methods, eliminating the research and development that would be needed to synthesize a computationally designed small molecule, and reducing the likelihood of failure due to inability to synthesize a compound. Meso-scale macrocycles present larger surface areas for target recognition than do small molecules, permitting higher-affinity, higher-specificity interactions and reducing the likelihood of a lead compound failing due to off-target effects during costly clinical trials. At the same time, mesoscale macrocycles have greater potential to cross biological barriers, evade the immune system, and resist proteolytic degradation than do large protein therapeutics, addressing factors that might prevent a protein therapeutic from reaching the clinic. With the advent of robust computational tools for designing and identifying rigidly folded macrocycles, one of the major disadvantages of mesoscale molecules – the flexibility that they typically possess, which creates entropic barriers to high-affinity binding – has now been addressed. Since the possibility of eliminating the current barriers to rapid and robust low-cost drug development is in sight, and since many experimentally validated examples now exist of designed, rigidly folded peptide macrocycles with precisely controlled structure, it is my view that the time is now ripe for a new wave of rational design of peptide macrocycle therapeutics.
To embrace computational design of these molecules, I believe that we must challenge certain drug discovery paradigms that are broadly accepted today. First, attitudes regarding the appropriate use of computation in drug discovery tend to oscillate between extremes. Computational approaches allow a researcher to generate informed predictions or hypotheses that have reasonable chances, but which are not guaranteed, to be correct. This leads to two unfortunate beliefs. The first is the naïve notion that since computers are mathematically precise, their predictions are always reliable. This leads to rapid disillusionment: given the simplifying assumptions made when modeling complex phenomena and the limited data available with which to train or develop modeling algorithms, computational approaches inevitably have limits to their accuracy. The second notion is the other extreme: that, because any given computational prediction may be incorrect, computation should be shunned in favor of experiment alone. Some oppose computational drug design in part because it is wrongly perceived to be a competitor, rather than a complement, to high-throughput experimental methods, and, as such, is viewed as lacking sufficient accuracy to be the better approach. The ability to screen hundreds of thousands or millions of chemical compounds experimentally can lead to false confidence in one’s ability to explore chemical space: in actuality, even the largest high-throughput screens can only cover a trivially small fraction of the accessible space. A more balanced viewpoint is most reasonable: computation is an important tool, not for replacing large-scale experimental screens, but for informing them. A prime example is the generation of peptide macrocycle sequences for experimental validation. No experiment can exhaustively screen all possible sequences, or even a significant fraction of possible sequences, so it is essential to have a means of enriching the pool that will be screened for likely hits. By thinking of the computer as an hypothesis generator, and not as a result generator, we can obtain useful hypotheses that can inform experiments – experiments that could not have been performed in the absence of those hypotheses or the means of generating them.
Second, I would claim that we must reevaluate our preconceived notions of what a ‘good drug’ must look like. Drug development has long focused primarily on small molecules (influenced heavily in the twenty-first century by the dogmatic application of the Lipinski rule of five), with a minority of efforts directed at large proteins. Efforts to develop mesoscale therapeutics such as peptide macrocycles have lagged. Inspired by the examples of highly successful mesoscale drugs like cyclosporine A, however, more and more researchers are beginning to question whether useful drugs must strictly conform to the conservative and rather arbitrary cutoffs that describe only about half of our existing pharmacopoeia. I would add my voice to those claiming that, rather than limiting ourselves by clinging to too-strict rules that force us to focus on smaller and more hydrophobic molecules, we should be actively moving into the ‘beyond rule-of-five’ space that is currently so sparsely explored, embracing rational design of peptide macrocycles as a means of finding handholds within this space.
This is not to say that entering the ‘beyond rule-of-five’ space is not difficult, or that the peptide macrocycle design field is not without its challenges. The Lipinski rules, which exclude almost all peptide macrocycles, have historically provided a convenient means of confining oneself to a space containing many examples of possible drugs. To move beyond this space, we must examine the true physical principles underlying permeability and other desired drug properties, and learn to recognize and emulate the way in which nature has been able to achieve these properties in toxins and defensive molecules without being confined to the small molecular sizes and narrow range of properties suggested by the Lipinski rules. Evidence is mounting from many groups that minimizing solvent-accessible polar surface area across the ensemble of conformations accessible to a macrocycle, maximizing internal satisfaction of hydrogen bond donors and acceptors, and enabling chameleonic conformational switching from polar to apolar environments are necessary for membrane permeability, but we have too few examples of permeable mesoscale molecules to yet know whether this is sufficient. Computational peptide macrocycle design permits us to pose these questions, turning an observational field that was largely limited to making discoveries about sparse natural products into an experimental one in which we can construct hypotheses about the needed dynamic properties of druglike peptides and make molecules with and without these properties to test these hypotheses. Methods are now emerging to estimate properties such as exposed surface area across the entire conformational ensemble of a molecule, including those that have been added to software tools like the Rosetta simple_cycpep_predict application. In the near future, it is my hope that such methods will allow hypothesis-testing to yield a fuller understanding of the precise determinants of permeability and other desirable drug-like properties, particularly in the ‘beyond rule-of-five’ space.
The high computational cost of current peptide design methods, which puts such methods beyond the reach of some researchers, is another challenge. In this area, some of the most exciting ongoing research is using high-cost simulations to train low-cost neural networks or other ML models that are then able to approximate the output of the higher-cost approaches. This again follows the pattern of permitting failure at lower cost – in this case, promising to allow many poor designs to be discarded with a computation that occurs in milliseconds on a single CPU rather than in hundreds of CPU-hours on a computing cluster – which again lowers the overall cost of finding a successful design.
The field of computational peptide design is still in its early years, and the available tools are rapidly evolving. Although the Rosetta software suite currently offers a toolset for all steps of the process, new approaches are being introduced continuously, and we will hopefully see complementary and competitive software approaches developed in the near future. In the coming years, we can expect to see greater integration of predictive methods for the diverse properties that are desirable in a drug, permitting better multi-objective design and validation. Simultaneously, I anticipate that the computational cost of these methods will drop as the field shifts from reliance on brute-force simulations to ML-based approximate methods for processing large numbers of designs. In the slightly longer term, I am excited by the possibility that the next generation of quantum computers will eliminate barriers to more complex design problems, and permit the full palette of thousands of chemical building-blocks that are accessible to chemists to be used to solve design problems. However, regardless the potential for using current and future computational technologies to improve design methodologies, I would encourage drug developers to explore ways to incorporate computational peptide macrocycle design into their drug discovery pipelines today. The prospects for peptide macrocycle therapeutics are bright, and the emerging design pipeline could soon ease the developmental bottlenecks that currently limit the rate at which new drugs reach the clinic. A near future in which the dominant drug development paradigm involves rationally designing and eliminating most dead ends in silico, and reserving laboratory and clinical trials for compounds with a high probability of success, is foreseeable.
Article Highlights
Drug discovery typically involves costly library screens, yielding diminishing returns on time and resources invested.
Peptide macrocycles represent an underexplored class of drugs with properties that could address current deficiencies of small-molecule drugs.
Recently developed computational peptide macrocycle design tools, and validation tools able to predict a peptide’s rigidity in a binding-competent conformation, are described.
Ongoing challenges include design for membrane permeability and computational cost of validation, but current research aims to address these.
Now is an excellent time for drug developers to incorporate computational peptide macrocycle design into existing drug discovery pipelines, to shift failures to earlier, less-costly in silico steps and increase success in later laboratory and clinical steps.
Declaration of interest
V Mulligan is a co-founder of Menten AI, in which he owns equity. He has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Illustrations were rendered with Blender 2.81 (https://www.blender.org/). An award of computer time provided to the author by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program was used to produce the output shown in . This research used resources of the Argonne Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357.
Additional information
Funding
References
- Lombardino JG, Lowe JA. The role of the medicinal chemist in drug discovery – then and now. Nat Rev Drug Discov. 2004;3:853–862.
- Hwang TJ, Carpenter D, Lauffenburger JC, et al. Failure of investigational drugs in late-stage clinical development and publication of trial results. JAMA Intern Med. 2016;176:1826–1833.
- Mullard A. 2015 FDA drug approvals. Nat Rev Drug Discov. 2016;15:73–76.
- Chai CL, Mátyus P. One size does not fit all: challenging some dogmas and taboos in drug discovery. Future Med Chem. 2016;8:29–38.
- Scannell JW, Blanckley A, Boldon H, et al. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200.
- Bohacek RS, McMartin C, Guida WC. The art and practice of structure-based drug design: a molecular modeling perspective. Med Res Rev. 1996;16:3–50.
- Muller PY, Milton MN. The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov. 2012;11:751–761.
- Dahlgren D, Lennernäs H. Intestinal permeability and drug absorption: predictive experimental, computational and in vivo approaches. Pharmaceutics. 2019;11.
- Patel MM, Patel BM. Crossing the blood-brain barrier: recent advances in drug delivery to the brain. CNS Drugs. 2017;31:109–133.
- Wang CK, Craik DJ. Cyclic peptide oral bioavailability: lessons from the past. Biopolymers. 2016;106(6):901–909.
- Troeira Henriques S, Craik DJ. Cyclotide structure and function: the role of membrane binding and permeation. Biochemistry. 2017;56(5):669–682.
- Ghosh AK, Gemma S. HIV-1 Protease Inhibitors for the Treatment of HIV Infection and AIDS: design of Saquinavir, Indinavir, and Darunavir. In: Ghosh AK and Gemma S, editors. Structure-Based Design of Drugs and Other Bioactive Molecules. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA; 2015. p. 237–270.
- Fettig J, Swaminathan M, Murrill CS, et al. Global epidemiology of HIV. Infectious Disease Clinics of North America. 2014;28(3):323–337.
- Anderson AC. The process of structure-based drug design. Chem Biol. 2003;10(9):787–797.
- Hassan Baig M, Ahmad K, Roy S, et al. Computer aided drug design: success and limitations. Current Pharmaceutical Design. 2016;22(5):572–581.
- Watkins AM, Arora PS. Structure-based inhibition of protein–protein interactions. Eur J Med Chem. 2015;94:480–488.
- Moiola M, Memeo MG, Quadrelli P. Stapled peptides-a useful improvement for peptide-based drugs. Molecules. 2019;24.
- Rentero Rebollo I, Heinis C. Phage selection of bicyclic peptides. Methods. 2013;60(1):46–54.
- Peraro L, Siegert TR, Kritzer JA. Conformational restriction of peptides using dithiol bis-alkylation. Meth Enzymol. 2016;580:303–332.
- White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nat Chem. 2011;3(7):509–524.
- Wang CK, Swedberg JE, Northfield SE, et al. Effects of cyclization on peptide backbone dynamics. J Phys Chem B. 2015;119(52):15821–15830.
- Est CB, Mangrolia P, Murphy RM. ROSETTA-informed design of structurally stabilized cyclic anti-amyloid peptides. Protein Eng Des Sel. 2019;gzz016. DOI:10.1093/protein/gzz016.
- Bhardwaj G, Mulligan VK, Bahl CD, et al., Accurate de novo design of hyperstable constrained peptides. Nature. 2016;538(7625):329–335.
- Hosseinzadeh P, Bhardwaj G, Mulligan VK, et al., Comprehensive computational design of ordered peptide macrocycles. Science. 2017;358(6369):1461–1466.
- Dang B, Wu H, Mulligan VK, et al. De novo design of covalently constrained mesosize protein scaffolds with unique tertiary structures. Proceedings of the National Academy of Sciences. 2017;114:10852–10857.
- Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(90001):D668–672.
- Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced Drug Delivery Reviews 23 (1997) 3–25. 1. Advanced Drug Delivery Reviews. 2001;46(1–3):3–26.
- Craik DJ, Fairlie DP, Liras S, et al. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–147.
- Jia L, Zhang L, Shao C, et al. An attempt to understand kidney’s protein handling function by comparing plasma and urine proteomes. PLoS ONE. 2009;4(4):e5146.
- Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. Aaps J. 2012;14(2):296–302.
- Hollstein U, Breitmaier E, Jung G. Carbon-13 nuclear magnetic resonance study of actinomycin D. J Am Chem Soc. 1974;96(26):8036–8040.
- Hotchkiss RD, Dubos RJ. Bactericidal fractions from an aerobic sporulating bacillus. J Biol Chem. 1940;136:803–804.
- Johnson BA, Anker H, Meleney FL. Bacitracin: A new antibiotic produced by a member of the B. subtilis group. Science. 1945;102:376–377.
- Benedict RG, Langlykke AF. Antibiotic activity of Bacillus polymyxa. J Bacteriol. 1947;54:24.
- Koyama Y. A new antibiotic “colistin” produced by spore-forming soil bacteria. J Antibiot. 1950;3:457–458.
- Ehrlich J, Smith RM, Penner MA, et al. Antimicrobial activity of streptomyces floridae and of viomycin. Am Rev Tuberc. 1951;63(1):7–16.
- Finlay AC, Hobby GL, Hoschstein F, et al. Viomycin a new antibiotic active against mycobacteria. Am Rev Tuberc. 1951;63(1):1–3.
- Herr EB Jr, Haney ME, Pittenger GE. Capreomycin: A new peptide antibiotic. Am Chem Soc. 1961;Abstract 49C. 140th Meeting.
- Jolles G, Terlain B, Thomas JP. Metabolic investigations on pristinamycin. Nature. 1965;207(4993):199–200.
- Heusler K, Pletscher A. The controversial early history of cyclosporin.. Swiss Med Wkly. 2001;131(21–22):299–302.
- Bauer W, Briner U, Doepfner W, et al., SMS 201–995: A very potent and selective octapeptide analogue of somatostatin with prolonged action. Life Sci. 1982;31(11):1133–1140.
- Eliopoulos GM, Willey S, Reiszner E, et al. In vitro and in vivo activity of LY 146032, a new cyclic lipopeptide antibiotic. Antimicrobial Agents and Chemotherapy. 1986;30(4):532–535.
- Fass RJ, Helsel VL. In vitro activity of LY146032 against staphylococci, streptococci, and enterococci.. Antimicrobial Agents and Chemotherapy. 1986;30(5):781–784.
- Knapp CC, Washington JA. Antistaphylococcal activity of a cyclic peptide, LY146032, and vancomycin. Antimicrobial Agents and Chemotherapy. 1986;30(6):938–939.
- Adrian HJ, Dörr U, Bach D, et al. Biodistribution of 111In-pentetreotide and dosimetric considerations with respect to somatostatin receptor expressing tumor burden. Hormone and Metabolic Research. Supplement Series. 1993;27:18–23.
- Joseph K, Stapp J, Reinecke J, et al. Receptor scintigraphy with 111In-pentetreotide for endocrine gastroenteropancreatic tumors. Hormone and Metabolic Research. Supplement Series. 1993;27:28–35.
- Dörr U, Räth U, Sautter-Bihl M-L, et al. Improved visualization of carcinoid liver metastases by indium-111 pentetreotide scintigraphy following treatment with cold somatostatin analogue. Eur J Nucl Med. 1993;20(5):431–433.
- Ernst ME, Klepser ME, Wolfe EJ, et al. Antifungal dynamics of LY 303366, an investigational echinocandin B analog, against Candida ssp. Diagnostic Microbiology and Infectious Disease. 1996;26(3–4):125–131.
- Bartlett MS, Current WL, Goheen MP, et al. Semisynthetic echinocandins affect cell wall deposition of Pneumocystis carinii in vitro and in vivo. Antimicrobial Agents and Chemotherapy. 1996;40(8):1811–1816.
- Del Poeta M, Schell WA, Perfect JR. In vitro antifungal activity of pneumocandin L-743,872 against a variety of clinically important molds. Antimicrobial Agents and Chemotherapy. 1997;41(8):1835–1836.
- Vazquez JA, Lynch M, Boikov D, et al. In vitro activity of a new pneumocandin antifungal, L-743,872, against azole-susceptible and -resistant Candida species. Antimicrobial Agents and Chemotherapy. 1997;41(7):1612–1614.
- Scarborough RM, Naughton MA, Teng W, et al. Design of potent and specific integrin antagonists. peptide antagonists with high specificity for glycoprotein IIb-IIIa. The Journal of Biological Chemistry. 1993;268(2):1066–1073.
- Ueda H, Nakajima H, Hori Y, et al. FR901228, a novel antitumor bicyclic depsipeptide produced by chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. The Journal of Antibiotics. 1994;47(3):301–310.
- Tawara S, Ikeda F, Maki K, et al. In vitro activities of a new lipopeptide antifungal agent, FK463, against a variety of clinically important fungi. Antimicrob Agents Chemother. 2000;44(1):57–62.
- Kwekkeboom DJ, Teunissen JJ, Bakker WH, et al. Radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr 3]Octreotate in patients with endocrine gastroenteropancreatic Tumors. JCO. 2005;23(12):2754–2762.
- Decristoforo C, Knopp R, von Guggenberg E, et al. A fully automated synthesis for the preparation of 68Ga-labelled peptides. Nucl Med Commun. 2007;28(11):870–875.
- Win Z, Rahman L, Murrell J, et al. The possible role of 68Ga-DOTATATE PET in malignant abdominal paraganglioma. European Journal of Nuclear Medicine and Molecular Imaging. 2006;33(4):506.
- Win Z, Al-Nahhas A, Towey D, et al. 68Ga-DOTATATE PET in neuroectodermal tumours: first experience. Nucl Med Commun. 2007;28(5):359–363.
- World health organization model list of essential medicines, 21st list [Internet]. World Health Organization; 2019 [ cited 2019 Dec 16]. Available from: https://apps.who.int/iris/bitstream/handle/10665/325771/WHO-MVP-EMP-IAU-2019.06-eng.pdf.
- Yu X, Sun D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules. 2013;18(6):6230–6268.
- Kumar R, Feltrup TM, Kukreja RV, et al. Evolutionary features in the structure and function of bacterial toxins. Toxins (Basel). 2019;11(1):15.
- Peigneur S, Tytgat J. Toxins in drug discovery and pharmacology. Toxins (Basel). 2018;10.
- May JP, Perrin DM. Tryptathionine bridges in peptide synthesis. Biopolymers. 2007;88(5):714–724.
- Mir R, Karim S, Kamal MA, et al. Conotoxins: structure, therapeutic potential and pharmacological applications. Current Pharmaceutical Design. 2016;22(5):582–589.
- Clark RJ, Fischer H, Dempster L, et al. Engineering stable peptide toxins by means of backbone cyclization: stabilization of the -conotoxin MII. Proceedings of the National Academy of Sciences 2005;102(39):13767–13772.
- Conibear AC, Craik DJ. The chemistry and biology of theta defensins. Angewandte Chemie International Edition. 2014;53(40):10612–10623.
- Akondi KB, Muttenthaler M, Dutertre S, et al. Discovery, synthesis, and structure–activity relationships of conotoxins. Chemical Reviews. 2014;114(11):5815–5847.
- Ireland DC, Colgrave ML, Craik DJ. A novel suite of cyclotides from viola odorata: sequence variation and the implications for structure, function and stability. Biochem J. 2006;400(1):1–12.
- Kohli RM, Walsh CT. Enzymology of acyl chain macrocyclization in natural product biosynthesis. Chemical Communications. 2003;3:297–307. DOI:10.1039/b208333g
- Bouaïcha N, Miles CO, Beach DG, et al. Structural diversity, characterization and toxicology of microcystins. Toxins (Basel). 2019;11(12):714.
- Behrendt R, White P, Offer J. Advances in Fmoc solid-phase peptide synthesis. Journal of Peptide Science. 2016;22(1):4–27.
- Mäde V, Els-Heindl S, Beck-Sickinger AG. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J Org Chem. 2014;10:1197–1212.
- Weinstock MT, Francis JN, Redman JS, et al. Protease-resistant peptide design-empowering nature’s fragile warriors against HIV. Biopolymers. 2012;98(5):431–442.
- Dintzis HM, Symer DE, Dintzis RZ, et al. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins: Structure, Function, and Genetics. 1993;16(3):306–308.
- Ashby M, Petkova A, Gani J, et al. Use of peptide libraries for identification and optimization of novel antimicrobial peptides. Curr Top Med Chem. 2016;17(5):537–553.
- Passioura T, Katoh T, Goto Y, et al. Selection-based discovery of druglike macrocyclic peptides. Annual Review of Biochemistry. 2014;83(1):727–752.
- Zaretsky S, Tan J, Hickey JL, et al. Macrocyclic templates for library synthesis of peptido-conjugates. Methods Mol Biol. 2015;1248:67–80.
- Pellois JP, Zhou X, Srivannavit O, et al. Individually addressable parallel peptide synthesis on microchips. Nat Biotechnol. 2002;20(9):922–926.
- Playe B, Azencott C-A, Stoven V. Efficient multi-task chemogenomics for drug specificity prediction. Plos One. 2018;13(10):e0204999.
- Yan Z, Wang J. Specificity quantification of biomolecular recognition and its implication for drug discovery. Sci Rep. 2012;2(1):309.
- Boothroyd S, Kerridge A, Broo A, et al. Solubility prediction from first principles: a density of states approach. Phys Chem Chem Phys. 2018;20(32):20981–20987.
- Bergström CAS, Larsson P. Computational prediction of drug solubility in water-based systems: qualitative and quantitative approaches used in the current drug discovery and development setting. Int J Pharm. 2018;540(1–2):185–193.
- Egan WJ, Lauri G. Prediction of intestinal permeability. Advanced Drug Delivery Reviews. 2002;54(3):273–289.
- Dickson CJ, Hornak V, Pearlstein RA, et al. Structure–kinetic relationships of passive membrane permeation from multiscale modeling. Journal of the American Chemical Society. 2017;139(1):442–452.
- Zhang L, Zhang H, Ai H, et al. Applications of machine learning methods in drug toxicity prediction. Curr Top Med Chem. 2018;18(12):987–997.
- Li X, Chen L, Cheng F, et al., In silico prediction of chemical acute oral toxicity using multi-classification methods. J Chem Inf Model. 2014;54(4):1061–1069.
- Shaw DE, Maragakis P, Lindorff-Larsen K, et al., Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330(6002):341–346.
- Hwang H, Schatz G, Ratner M. coarse-grained molecular dynamics study of cyclic peptide nanotube insertion into a lipid bilayer†. J Phys Chem A. 2009;113(16):4780–4787.
- Kuhlman B, Dantas G, Ireton GC, et al., Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302(5649):1364–1368.
- Appella DH, Christianson LA, Klein DA, et al. Residue-based control of helix shape in β-peptide oligomers. Nature. 1997;387(6631):381–384.
- Giuliano MW, Horne WS, Gellman SH. An α/β-Peptide helix bundle with a pure β3 -amino acid core and a distinctive quaternary structure. Journal of the American Chemical Society. 2009;131(29):9860–9861.
- Horne WS, Gellman SH. Foldamers with Heterogeneous Backbones. Acc Chem Res. 2008;41(10):1399–1408.
- Slough DP, McHugh SM, Cummings AE, et al. Designing well-structured cyclic pentapeptides based on sequence–structure relationships. J Phys Chem B. 2018;122(14):3908–3919.
- Kritzer JA, Hodsdon ME, Schepartz A. Solution structure of a β-Peptide ligand for hDM2. Journal of the American Chemical Society. 2005;127(12):4118–4119.
- Burley SK, Berman HM, Bhikadiya C, et al. RCSB protein data bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019;47(D1):D464–D474.
- Handl J, Knowles J, Vernon R, et al. The dual role of fragments in fragment-assembly methods for de novo protein structure prediction. Proteins: Structure, Function, and Bioinformatics. 2012;80(2):490–504.
- Simons KT, Bonneau R, Ruczinski I, et al. Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins. 1999;Suppl 3:171–176.
- King CA, Bradley P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins: Structure, Function, and Bioinformatics. 2010;78(16):3437–3449.
- Raveh B, London N, Schueler-Furman O. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins. 2010;78(9):2029–2040.
- London N, Raveh B, Schueler-Furman O. Peptide docking and structure-based characterization of peptide binding: from knowledge to know-how. Current Opinion in Structural Biology. 2013;23(6):894–902.
- Coutsias EA, Seok C, Jacobson MP, et al. A kinematic view of loop closure. J Comput Chem. 2004;25(4):510–528.
- Mandell DJ, Coutsias EA, Kortemme T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nature Methods. 2009;6(8):551–552.
- Mulligan VK Generalized kinematic closure (GeneralizedKIC) tutorial series [Internet]. 2017 [ cited 2019 Nov 14]. Available from: https://www.rosettacommons.org/demos/latest/tutorials/GeneralizedKIC/generalized_kinematic_closure_1.
- Sindhikara D, Spronk SA, Day T, et al. Improving accuracy, diversity, and speed with prime macrocycle conformational sampling. Journal of Chemical Information and Modeling. 2017;57(8):1881–1894.
- Sindhikara D, Borrelli K. High throughput evaluation of macrocyclization strategies for conformer stabilization. Sci Rep. 2018;8(1):6585.
- Renfrew PD, Choi EJ, Bonneau R, et al., Incorporation of noncanonical amino acids into rosetta and use in computational protein-peptide interface design. Uversky VN, editor. PLoS One. 2012;7(3):e32637.
- Alford RF, Leaver-Fay A, Jeliazkov JR, et al. The rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput. 2017;13(6):3031–3048.
- Park H, Bradley P, Greisen P, et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J Chem Theory Comput. 2016;12(12):6201–6212.
- Dunbrack RL, Karplus M. Backbone-dependent rotamer library for proteins application to side-chain prediction. Journal of Molecular Biology. 1993;230(2):543–574.
- Shapovalov MV, Dunbrack RL. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19(6):844–858.
- Mironov V, Alexeev Y, Mulligan VK, et al. A systematic study of minima in alanine dipeptide. J Comput Chem. 2019;40(2):297–309.
- Renfrew PD, Butterfoss GL, Kuhlman B. Using quantum mechanics to improve estimates of amino acid side chain rotamer energies. Proteins: Structure, Function, and Bioinformatics. 2007;71(4):1637–1646.
- Vanhee P, van der Sloot AM, Verschueren E, et al. Computational design of peptide ligands. Trends Biotechnol. 2011;29(5):231–239.
- Wang J, Cao H, Zhang JZH, et al. computational protein design with deep learning neural networks. Sci Rep. 2018;8(1):6349.
- Traoré S, Allouche D, André I, et al. A new framework for computational protein design through cost function network optimization. Bioinformatics. 2013;29(17):2129–2136.
- Hallen MA, Martin JW, Ojewole A, et al., OSPREY 3.0: open-source protein redesign for you, with powerful new features. J Comput Chem. 2018;39(30):2494–2507.
- Leaver-Fay A, Tyka M, Lewis SM, et al. Rosetta3. In: Johnson ML and Brand L, editors. Methods in Enzymology. San Deigo (CA): Elsevier; 2011. p. 545–574.
- Mulligan VK Design-centric guidance terms [Internet]. 2017 [ cited 2019 Dec 12]. Available from: https://www.rosettacommons.org/docs/latest/rosetta_basics/scoring/design-guidance-terms.
- Loosli H-R, Kessler H, Oschkinat H, et al. Peptide conformations. Part 31. The conformation of cyclosporin A in the crystal and in solution. Helvetica Chimica Acta. 1985;68(3):682–704.
- Witek J, Keller BG, Blatter M, et al. Kinetic models of cyclosporin a in polar and apolar environments reveal multiple congruent conformational states. J Chem Inf Model. 2016;56:1547–1562.
- Fleishman SJ, Leaver-Fay A, Corn JE, et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE. 2011;6:e20161.
- Chaudhury S, Lyskov S, Gray JJ. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691.
- Mulligan VK Simple cyclic peptide prediction (simple_cycpep_predict) application [internet]. 2015 [ cited 2019 Nov 18]. Available from: https://www.rosettacommons.org/docs/latest/structure_prediction/simple_cycpep_predict.
- Fesik SW, Gampe RT, Eaton HL, et al. NMR studies of [U-13C]cyclosporin A bound to cyclophilin: bound conformation and portions of cyclosporin involved in binding. Biochemistry. 1991;30:6574–6583.
- Stegmann CM, Seeliger D, Sheldrick GM, et al. The thermodynamic influence of trapped water molecules on a protein-ligand interaction. Angew Chem Int Ed Engl. 2009;48:5207–5210.
- El Tayar N, Mark AE, Vallat P, et al. Solvent-dependent conformation and hydrogen-bonding capacity of cyclosporin A: evidence from partition coefficients and molecular dynamics simulations. J Med Chem. 1993;36:3757–3764.
- Peraro L, Kritzer JA. Emerging methods and design principles for cell-penetrant peptides. Angew Chem Int Ed Engl. 2018;57:11868–11881.
- Rossi Sebastiano M, Doak BC, Backlund M, et al. Impact of dynamically exposed polarity on permeability and solubility of chameleonic drugs beyond the rule of 5. J Med Chem. 2018;61:4189–4202.
- Wang CK, Swedberg JE, Harvey PJ, et al. Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. J Phys Chem B. 2018;122:2261–2276.
- Danelius E, Poongavanam V, Peintner S, et al. Solution conformations explain the chameleonic behaviour of macrocyclic drugs. Chem Eur J. 2020;chem.201905599.
- Matsson P, Kihlberg J. How big is too big for cell permeability? J. Med Chem. 2017;60:1662–1664.
- Pye CR, Hewitt WM, Schwochert J, et al. nonclassical size dependence of permeation defines bounds for passive adsorption of large drug molecules. J Med Chem. 2017;60:1665–1672.
- Over B, Matsson P, Tyrchan C, et al. Structural and conformational determinants of macrocycle cell permeability. Nat Chem Biol. 2016;12:1065–1074.
- Alford RF, Koehler Leman J, Weitzner BD, et al. An integrated framework advancing membrane protein modeling and design. PLoS Comput Biol. 2015;11:e1004398.
- Lu P, Min D, DiMaio F, et al. Accurate computational design of multipass transmembrane proteins. Science. 2018;359:1042–1046.
- Butterfoss GL, Renfrew PD, Kuhlman B, et al. a preliminary survey of the peptoid folding landscape. J Am Chem Soc. 2009;131:16798–16807.
- Butterfoss GL, Drew K, Renfrew PD, et al. Conformational preferences of peptide-peptoid hybrid oligomers: conformational preferences of peptide-peptoid oligomers. Biopolymers. 2014;102:369–378.
- Lewis SM Release Notes – rosetta 3.11 [Internet]. Rosetta Commons; 2019 [ cited 2019 Dec 15]. Available from: https://www.rosettacommons.org/docs/latest/Release-Notes#rosetta-3-11.
- Drew K, Renfrew PD, Craven TW, et al. Adding diverse noncanonical backbones to rosetta: enabling peptidomimetic design. Uversky VN, editor. PLoS One. 2013;8:e67051.
- Leaver-Fay A, Jacak R, Stranges PB, et al. a generic program for multistate protein design. Plos One. 2011;6:e20937.
- Huang J, Rauscher S, Nawrocki G, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14:71–73.
- Maier JA, Martinez C, Kasavajhala K, et al. FF14SB: improving the accuracy of protein side chain and backbone parameters from FF99SB. J Chem Theory Comput. 2015;11:3696–3713.
- Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688.
- Alexeev Y, Mazanetz MP, Ichihara O, et al. GAMESS as a free quantum-mechanical platform for drug research. Curr Top Med Chem. 2012;12:2013–2033.
- Parrish RM, Burns LA, Smith DGA, et al. Psi4 1.1: an open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J Chem Theory Comput. 2017;13:3185–3197.
- Valiev M, Bylaska EJ, Govind N, et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun. 2010;181:1477–1489.
- Fedorov DG, Brekhov A, Mironov V, et al. molecular electrostatic potential and electron density of large systems in solution computed with the fragment molecular orbital method. J Phys Chem A. 2019;123:6281–6290.
- Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10.
- Lazaridis T, Karplus M. Discrimination of the native from misfolded protein models with an energy function including implicit solvation. J Mol Biol. 1999;288 Fersht AR 11Edited by. 477–487.
- He S, Li Y, Feng Y, et al. Learning to predict the cosmological structure formation. PNAS. 2019;116:13825–13832.
- Bauer BA, Davis JE, Taufer M, et al. Molecular dynamics simulations of aqueous ions at the liquid-vapor interface accelerated using graphics processors. J Comput Chem. 2011;32:375–385.
- Mermelstein DJ, Lin C, Nelson G, et al. Fast and flexible GPU accelerated binding free energy calculations within the amber molecular dynamics package. J Comput Chem. 2018;39:1354–1358.
- Fernandes KD, Renison CA, Naidoo KJ. Quantum supercharger library: hyper-parallelism of the Hartree-Fock method. J Comput Chem. 2015;36:1399–1409.
- Wilkinson KA, Sherwood P, Guest MF, et al. Acceleration of the GAMESS-UK electronic structure package on graphical processing units. J Comput Chem. 2011;32:2313–2318.
- Chiu M, Herbordt MC. molecular dynamics simulations on high-performance reconfigurable computing systems. 2010;3. ACM Trans Reconfigurable Technol Syst.
- Feynman RP. Simulating physics with computers. Int J Theor Phys. 1982;21:467–488.
- Preskill J. Quantum Computing in the NISQ era and beyond. Quantum. 2018;2:79.
- Babej T, Ing C,Fingerhuth M. Coarse-grained lattice protein folding on a quantum annealer. arXiv:1811.00713 [quant-ph] [ Internet]. 2018 [ cited 2019 Dec 12]; Available from: http://arxiv.org/abs/1811.00713.
- Mulligan VK, Melo H, Merritt HI, et al. Designing peptides on a quantum computer. bioRxiv. 2019;752485.
- RosettaCommons website [Internet]. [ cited 2019 Dec 12]. Available from: https://www.rosettacommons.org/.
- Donald B. Osprey 3.0, donald lab at duke university [internet]. [cited 2019 Dec 13]. Available from: https://www2.cs.duke.edu/donaldlab/osprey.php.
- de Givry S Toulbar2: an exact solver for cost function networks [Internet]. [cited 2019 Dec 13]. Available from: https://miat.inrae.fr/toulbar2/.
- CHARMM Registration [Internet]. [ cited 2020 Mar 21]. Available from: http://charmm.chemistry.harvard.edu/.
- Download Amber MD [Internet]. [ cited 2020 Mar 21]. Available from: https://ambermd.org/GetAmber.php.
- Gordon Group/GAMESS Homepage [Internet]. [ cited 2020 Mar 21]. Available from: https://www.msg.chem.iastate.edu/gamess/index.html.
- Psi4: Open-source quantum chemistry [internet]. [ cited 2020 Mar 21]. Available from: http://www.psicode.org/.
- NWChem: open source high-performance computational chemistry [internet]. [ cited 2020 Mar 21]. Available from: http://www.nwchem-sw.org/index.php/Main_Page.