
ABSTRACT
Introduction
Our understanding of the mechanism of action of bioactive small molecules contributes to the research and development of new medical drugs, as well as elucidating the pathological mechanisms underlying various diseases. Researchers in this field are committed to a very ambitious goal: the discovery of novel therapeutic compounds along with their molecular targets. To achieve this goal, new methodological developments are indispensable.
Areas covered
This review gives an update on the advancements of phage display (PD) technology in the past decade (2011–2020) for determining the targets of the small molecule therapeutics. In particular, other than providing a brief overview of this field of research, we focus on reporting the research trends and the results solely obtained using this strategy.
Expert opinion
Despite the development of bioinformatics tools and artificial intelligence (AI)-mediated methods, affinity-guided information obtained experimentally are still indispensable to identify drug-protein interactions. By taking advantage of small-molecule-oriented PD methods and their improvements, the extension of the druggable proteome will be further expanded, providing new opportunities to generate small-molecule therapeutics.
KEYWORDS:
1. Introduction
Bioactive small molecules, which have been found in nature or chemically synthesized, are attractive compounds due to their potential to be used as drugs and as diagnostic agents. Due to the diversity of molecular structures and activities, the complete characterization of a small molecule requires multiple steps. These include resolving its molecular structure, identification of the molecular target(s) responsible for its main and adverse effects, determination of the molecule’s biodistribution and pharmacokinetics/dynamics, toxicological test, formulation study, and clinical trials. Even when a drug is approved to be commercialized, sometimes the drug is withdrawn, because of the emergence of unanticipated adverse effects or drug resistance. Thus, developing a new compound for medical applications is a lengthy and costly process, and the establishment of efficient testing methodologies is largely responsible for the success of drug discovery.
The 2018 Nobel Prize for chemistry was awarded to pioneering research on the phage display (PD) technologies and enzymes improving using the same principles used by natural evolution, i.e., genetic change and selection [Citation1]. PD technology has been increasingly adopted as an elegant technology for drug discovery since its original description in 1985 [Citation2]. With this method, it is possible to generate an array of different foreign peptides displayed on a capsid protein of a bacteriophage, a virus that infects bacteria. The foreign peptides displayed on the capsid, called the phage library, are then affinity-selected using the specific target of interest as bait, such as peptides, proteins, antibodies, and other molecules. Once the phage clones displaying the peptides with more affinity to the bait are selected, they can be amplified by infecting the host bacteria to perform other rounds of selection [Citation3,Citation4]. In addition to the general use of peptides or proteins as bait for generating functional peptides or validating protein-protein interactions [Citation5,Citation6], the use of small molecule therapeutics as bait is an essential approach to identify their molecular targets and explaining their bioactivity in the body ().
Figure 1. Schematic representation of the typical biopanning process using a phage-displayed library and a 96 well-formatted microplate coated with the ligand as bait (compounds are generally immobilized onto the surface of solid support with avidin-biotin interaction or organic synthesis etc.). Random DNA or cDNA are inserted into the multi-cloning site on the phage genome where encodes phage capsid protein. The phage library, which displays a population of the diversity (>107) of random peptides or proteins encoded by the DNAs, are allowed to interact with the bait. After extensively washing the well to remove the unbound or nonspecific binding phages, the bait-captured phages are recovered using some appropriate eluate and amplified by host Escherichia coli infection to produce next phage library. By repeating the cycles from 3 to 5 rounds, resulting phages are isolated and part of their DNAs are sequenced to determine the peptides or proteins that specifically recognize the bait molecule. More technical details are summarized in Ref. [7].
![Figure 1. Schematic representation of the typical biopanning process using a phage-displayed library and a 96 well-formatted microplate coated with the ligand as bait (compounds are generally immobilized onto the surface of solid support with avidin-biotin interaction or organic synthesis etc.). Random DNA or cDNA are inserted into the multi-cloning site on the phage genome where encodes phage capsid protein. The phage library, which displays a population of the diversity (>107) of random peptides or proteins encoded by the DNAs, are allowed to interact with the bait. After extensively washing the well to remove the unbound or nonspecific binding phages, the bait-captured phages are recovered using some appropriate eluate and amplified by host Escherichia coli infection to produce next phage library. By repeating the cycles from 3 to 5 rounds, resulting phages are isolated and part of their DNAs are sequenced to determine the peptides or proteins that specifically recognize the bait molecule. More technical details are summarized in Ref. [7].](/cms/asset/72ee52da-e185-4a94-93ea-19c75b9c7ca2/iedc_a_1790523_f0001_oc.jpg)
In a previous review, published in 2010, we summarized the technical basis of PD methods and their use for the determination of small-molecule therapeutic targets since the 1990 s [Citation7]. In that review, we focused on the biochemical engineering, biotechnology, and bioinformatics approaches used in PD to overcome the technical difficulties arising when using small-molecule as baits. During the years 2011–2020, previously unknown interactions between bioactive small-molecules and their molecular target were discovered thanks to PD technology. Some studies reported in this review provided clear examples of the results obtainable by PD technology, which could not have been reached otherwise. The current review summarizes the use of PD for determining the targets of small-molecule therapeutics in the past decade, highlighting the unique findings obtained by PD methods.
2. Distinctive characteristics of the use of PD methods for drug target identification
The molecular structure of bioactive small molecule therapeutics and their cognate interaction properties in living organisms are diverse and depend on the relative molecular target and biological activity. Thus, there are no coherent rules underlying small molecule drug synthesis and the study of drug interaction in the body, differing from drugs based on nucleic acids, peptides, and monoclonal antibodies. To maximize small molecule drug research and development (R&D), the development of efficient and high throughput methods is desirable.
Among all biomolecules, proteins are the most important drug molecular targets, such as G protein-coupled receptors (GPCR), enzymes like protein kinase, transporters, nuclear hormone receptors, transcription factors, and others [Citation8]. Thus, efficient methods allowing the detection of the interaction between small-molecules and these classes of proteins are highly desired. A conventional proteomics approaches is to use cell extracts to obtain the small molecule drug targets by affinity purification [Citation9]. However, there are some technical limitations to this approach. First, due to the small size of the tested compounds, in some cases, the compounds immobilized on a solid support cannot access a potential binding site located internally on the target (). Second, GPCR is often missing in the protein extract because it is a membrane-associated receptor insoluble in water (). Third, cytokines and autacoids form complexes (dimer or tetramer) which are either extracellularly secreted, expressed with low abundance, or have a short lifetime, and thus, these are under-represented in the protein extract (). Therefore, GPCR, cytokines, and autacoids are desirable druggable targets whose identification by conventional proteomics is particularly challenging.
Figure 2. Examples of human druggable proteins and complexes that are potentially challenging to be identified by conventional methods using cell extracts. A, Human DNA topoisomerase I (top I) in nuclei (X-ray diffraction) [PDB ID; 1T8I]. Camptothecin forms a ternary complex with DNA and top I by covalently binding to the active site cavity in the deep part of the complex, thereby inhibiting the action of top I. B, Seven membrane-penetrated rhodopsin receptor, a human G protein-coupled receptor (GPCR) (NMR) [PDB ID: 1LN6]. Because of its poor water solubility, affinity purification often fails to isolate the whole protein. C, Human vascular endothelial growth factor 165 (hVEGF165) (X-ray diffraction) [PDB ID: 1VPF]. The precursor of the active form of hVEGF165 is spliced, forms a stable structure through intramolecular S-S bonds and post-translational modification in the cellular environment. VEGF forms a dimer and complex with the cognate tyrosine kinase receptors by inducing homodimerization.
![Figure 2. Examples of human druggable proteins and complexes that are potentially challenging to be identified by conventional methods using cell extracts. A, Human DNA topoisomerase I (top I) in nuclei (X-ray diffraction) [PDB ID; 1T8I]. Camptothecin forms a ternary complex with DNA and top I by covalently binding to the active site cavity in the deep part of the complex, thereby inhibiting the action of top I. B, Seven membrane-penetrated rhodopsin receptor, a human G protein-coupled receptor (GPCR) (NMR) [PDB ID: 1LN6]. Because of its poor water solubility, affinity purification often fails to isolate the whole protein. C, Human vascular endothelial growth factor 165 (hVEGF165) (X-ray diffraction) [PDB ID: 1VPF]. The precursor of the active form of hVEGF165 is spliced, forms a stable structure through intramolecular S-S bonds and post-translational modification in the cellular environment. VEGF forms a dimer and complex with the cognate tyrosine kinase receptors by inducing homodimerization.](/cms/asset/bca95165-1495-4f11-86ca-eb044be282e4/iedc_a_1790523_f0002_oc.jpg)
The use of PD, especially short peptide-displayed libraries, is an efficient way to overcome these difficulties [Citation10]. The short peptides can sterically contact with any small molecule of interest fixed on a solid support and are capable of providing informative results even for short stretches weakly recognizing the molecular structure of the drug. Another advantage of this technique is that PD allows the identification of different drug-recognizing motifs simultaneously [Citation11]. Subsequent analysis matching the primary amino acid sequence of the drug-recognizing peptides with the proteome enables the identification of the potential drug-binding partners with its binding site. This approach could, in theory, yield the entire drug-protein interactome, which may be associated with the drug’s absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters, which are essential for the next steps in the drug R&D [Citation12]. Thus, with this approach, the physicochemical properties of the protein sample or its interaction properties are irrelevant. Furthermore, this cell-free style approach enables drug and therapeutic target discovery even for infectious pathogens, such as tuberculosis [Citation13,Citation14] and malaria [Citation15,Citation16] that are difficult to handle in normal laboratories.
Conventionally, the lock-and-key docking concept has been considered the main model to explain the bioactivity of a molecule. Unlike protein-protein interactions, a drug binding site in proteins could be located in an unstructured or intrinsically disordered peptide loop [Citation17–Citation19]. The phage-displayed short peptides better mimic the unstructured molecular dynamics during docking and allow the validation of interaction dynamics that cannot be defined by conventional X-ray crystallography or NMR analysis. This advantage of the PD technology contributes to shed light on drug-protein interaction mechanisms such as agonism, antagonism, and protein allostery, which are often linked to the appearance of negative side effects [Citation20,Citation21].
3. Challenges
To maximize the efficiency of affinity selection in PD technology using small molecule bait, various technical developments have been made in the past decade. In general, knowledge of organic or medicinal chemistry is necessary for immobilizing small molecule onto solid support as bait during biopanning. These include direct immobilization by chemical reaction, biotinylation of compounds for avidin-coated solid support, or introduction of self-assembled monolayer unit that assembles on the gold support [Citation7]. The introduction of a biosensor as a platform has extensively contributed to the identification of drug-peptide pairs with relatively weak interactions [Citation22]. Unlike conventional procedure (), this biosensor has also enabled to avoid repeating cycles of multiple rounds of selection, and discarded the elution condition exploration by using the direct recovery of bound phage DNA with host bacterial infection, especially when using the bacteriolytic T7 phage [Citation23]. Furthermore, the combined use of DNA microarray and one-bead-one-compound libraries has increased the diversity of the affinity selection [Citation24,Citation25]. Moreover, the quantitative assessment by real-time PCR and next-generation DNA sequencing accelerated the throughput potential for screening drug-recognizing amino acid sequences [Citation26]. The analysis of the drug-selected peptide population was also supported by the development of ad-hoc bioinformatics tools [Citation27–Citation29]. Finally, the development of all-in-one automatic PD systems has highly contributed to increasing both sample diversity and throughput for PD selection [Citation30,Citation31]. Thus, the advancements of PD performance for the discovery of drug candidates from natural products [Citation32,Citation33] will certainly boost drug R&D capability in the future.
4. Identification of drug-protein interactions using PD technique
Notable examples of target identification using PD have been summarized in this section. Most of the tested compounds were not new, but the discovery of many alternative molecular targets helped to explain bioactivities different from the original effects (i.e., off-targets or drug repurposing), or the appearance of adverse effects, which are problematic in clinical care [Citation34]. The molecular structure of the compounds used in this method are summarized in by considering the importance for drug R&D as well as the usefulness for the experimental design of the viewpoint from organic or medicinal chemistry.
Figure 3. Molecular structure of bioactive small-molecules and their derivatives subjected to PD affinity selection for target identification.
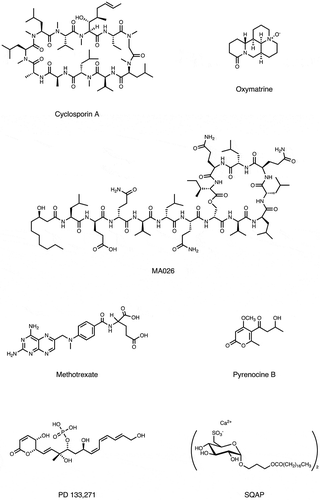
Figure 3. (continued).
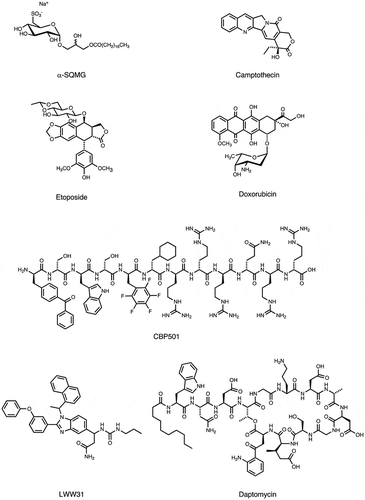
Figure 3. (continued).
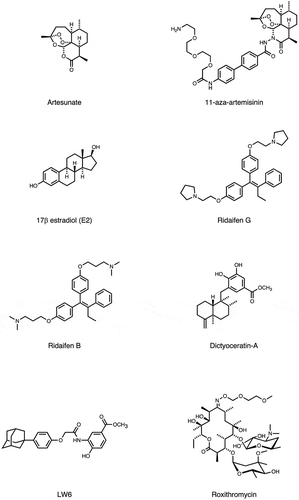
Figure 3. (continued).
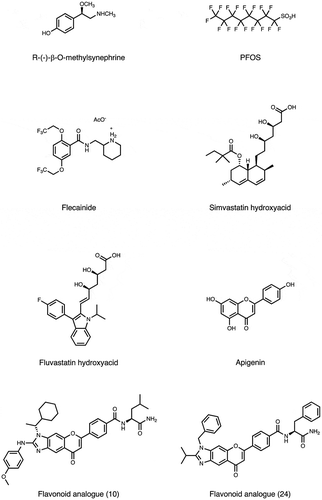
Figure 3. Molecular structure of bioactive small-molecules and their derivatives subjected to PD affinity selection for target identification.
4.1. Cyclosporin A
Cyclosporin A (CsA) is an immunosuppressive agent that complexes with cyclophilin A and inhibits the phosphatase activity of calcineurin, thereby blocking the activation of the nuclear factor of activated T cells transcription factors (NFAT) and its downstream signals for cytokine production [Citation35]. Interestingly, it has been reported that CsA inhibits genome replication of hepatitis C virus (HCV) [Citation36], although the regulation mechanisms are poorly understood. Using a biochemical approach including a T7 PD biopanning, the CsA associated helicase-like protein (CAHL) was identified as a direct binding partner of CsA [Citation37]. This interaction contributes to inhibit the formation of a trimer complex of CAHL with CyPB and HCV-derived RNA polymerase NS5B (CAHL/CyPB/NS5B) that stimulate the HCV replication. Thus, this result might explain the inhibitory activity of CsA against HCV genome replication.
4.2. Methotrexate
Methotrexate (MTX) is a folic acid antagonist that hinders nucleotide biosynthesis by preventing dihydrofolate reductase (DHFR) [Citation38], thereby delaying the growth of cancer cells. A lower dose of MTX is also currently used for the clinical treatment of rheumatoid arthritis (RA) and other inflammatory diseases. A biopannig of T7 phage-displayed protein library found the high mobility group protein B1 (HMGB1) as a molecular target of MTX [Citation39]. The binding site is the receptor for advanced glycation end products (RAGE)-binding region in HMGB1. Upon binding with RAGE, HMGB1 activates downstream cellular events related to inflammation. Indeed, they found that the MTX/HMGB1 interaction (KD: 0.24 ± 0.01 μM) interfered with the HMGB1/RAGE association (KD: 3.71 ± 1.68 μM), inhibiting TNF-α release and mitogenic activity in RAW 264.7 cells [Citation39].
4.3. Pyrenocine B
Pyrenocine B is a natural compound [Citation40] that inhibits the display of endogenous MHC class II-restricted antigens in bone marrow-derived dendritic cells (BMDCs). The Epsin R molecule was identified by T7 PD screening and surface plasmon resonance (SPR) analysis [Citation41]. Epsin R is known to play a role in endosomal trafficking by binding soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) [Citation42]. Downregulation of EpsinR expression in BMDCs by shRNA resulted in a decrease in the responsiveness of CD4+ T cells. Thus, they demonstrated that Epsin R has a previously unknown role in the display of endogenous MHC class II-restricted antigen, which involves SNARE-dependent sorting in the mechanism.
4.4. PD113271
PD133,271 is a cytotoxic phosphate monoester originally isolated from Streptomyces pulveraceus that shows potent cytotoxic activity against mouse leukemia by targeting topoisomerase II and protein phosphatase 2A (PP2A) [Citation43]. By generating a biotinylated PD133,271 derivative and using a T7 phage library that was constructed from human liver cancer cell line cDNA, it was discovered that PD133,271 binds to the high basic region (HBR) of microtubule-associated protein 1B (MAP1B) (KD: 0.21 μM) [Citation44]. The direct interaction of PD133,271 and MAP1B-HBR resulted in the dissociation of MAP1B from tubulin in MCF cells, thereby inducing an abnormal MAP1B distribution.
4.5. SQAP
Sulfoquinovosylacylpropanediol (SQAP) is a semi-synthetic derivative of a natural sulfoglycolipid [Citation45] that showed anti-tumor activity. A T7 PD screening was performed and identified five binding partners for SQAP: sterol carrier protein 2, multifunctional enzyme type 2, proteasomal ubiquitin receptor, UV excision repair protein, and focal adhesion kinase (FAK) [Citation46]. They further validated the direct binding of SQAP to these proteins by SPR analysis. Next, the authors demonstrated that SQAP inhibits the FAK phosphorylation, which is linked to tumor cell turnover and angiogenesis.
Recently, it has been reported that SQAP has a radiosensitization effect; intravenously administered SQAP increases tumor oxygenation by enhancing oxygen release from the hemoglobin in erythrocytes and tumor perfusion, thereby potentiating the oxygen effect upon ionizing irradiation therapy [Citation47]. Further experiments using the T7 PD and characterizing the target candidates may elucidate the molecular mechanisms underlying the pharmacological effects of SQAP.
4.6. LWW31
One-bead-one-compound (OBOC) combinatorial library is a technique that allows a rapid and cost-effective production of large-scale peptide and small molecule libraries, while simultaneously enabling an efficient screening of such libraries against proteins or other cellular targets to isolate compounds with biological significance [Citation48,Citation49]. Using their own OBOC library screening method in combination with a phage library, a proof of concept (POC) study was conducted for a systematic library-against-library screening approach, to globally identify the bioactive compounds and their interactions and establish a small molecule-protein domain interaction database [Citation25]. The screening using the OBOC benzimidazole small molecule library identified 91 hits binding to the M13 PD cDNA library. Subsequent two-step enzyme-linked colorimetric screening found 4 lead compounds (LWW29, LWW31, LWW32, and LWW55) from 19 randomly selected compounds in the 91 hits. The in vitro assays confirmed that LWW31 directly binds to EIF5B. Moreover, they found that LWW31 is cytotoxic in Jurkat cells, with an IC50 in the tens micromolar range, and is capable of inducing apoptosis. Furthermore, in HepG2 cells, it inhibits protein expression. Thus, this novel strategy might be useful to find compounds and identify new druggable targets, which are the two major tasks required in the pharmaceutical industry for drug discovery.
4.7. Artemisinins
The 2015 Novel Prize in medicine or physiology was awarded for the development of anti-nematode and anti-malaria agents. Among them, artemisinins are the most superior antimalarial agents currently available [Citation50]. In addition to the anti-parasitic activity against Plasmodium falciparum, it was reported that artemisinins have an anti-cancer activity by promoting apoptosis through the mitochondrial pathway in cancer cells, suggesting the presence of a human target for this class of compounds [Citation51]. An unbiased screening using several human cDNA libraries displayed on bacteriophage T7 was performed and found a single target for artesunate; the intrinsically disordered Bcl-2 antagonist of cell death promoter (BAD) [Citation52]. They showed that artesunate averts the phosphorylation of BAD, thereby enhancing the formation of the proapoptotic BAD/Bcl-xL complex and the subsequent apoptotic cascade involving cytochrome c release, PARP cleavage, caspase activation, and finally cell death. These results might explain molecular basis underlying the anti-cancer effect of artesunate, which is independent from the mechanisms related to reactive oxygen species and may lead to information for clinical drug repurposing.
4.8. Dictyoceratin
Due to their chemical and biological diversity, marine organisms are a rich source of drug leads. Dictyoceratin-A and -C were isolated as selective growth inhibitors of cancer cells adapted to hypoxic environments [Citation53]. These compounds, obtained by total synthesis, as well as three derivatives were screened for biological evaluation and target identification using a T7 PD system [Citation54]. After seven rounds of biopanning and subsequent binding assays, RNA polymerase II-associated protein 3 (RPAP3) was identified as a direct binding protein of dictyoceratin. RPAP3 knockdown and overexpression experiments reproduced the effects seen in hypoxic cancer cells treated with dictyoceratin, confirming that dictyoceratin exerts its biological activity by binding to RPAP3. This is a prime example of the use of PD technology for drug discovery, from the isolation of a novel bioactive compound to its target identification.
4.9. Roxithromycin
Macrolides are attractive natural compounds that show a wide variety of bioactivities against different organisms, from bacteria to humans [Citation55]. Among them, a semi-synthetic antibiotic macrolide roxithromycin (RXM) was reported to show anti-angiogenic activity in solid tumors [Citation56]. Multimodal biopanning of T7 phage-displayed peptides was conducted using a 96-well microplate, flow injection-type and cuvette-type quartz-crystal microbalance (QCM) biosensors [Citation57]. The three different biopannings commonly identified angiomotin (Amot) as a RXM receptor. The binding site is the BIG3 extracellular domain, as revealed by the affinity-selected short peptide sequences and a bioinformatics analysis using the Receptor Ligand Contact (RELIC) software suite [Citation29]. Binding assays confirmed that the BIG3 domain on Amot where is the assembling region for angiostatin (Anst), an endogenous anti-angiogenic factor [Citation58], is a binding site for RXM, resulting in the prevention of angiogenesis-dependent tumor growth and metastasis elicited by the RXM treatment.
4.10. Flecainide
Flecainide acetate is an anti-arrhythmic agent that inhibits a cardiomyocyte plasma membrane sodium-channel, which is used in the prevention and treatment of ventricular tachycardia and other cardiac arrhythmias [Citation59]. Previously, the development of the Magic Tag technique, a method for immobilizing a small molecule onto solid support through photo cross-linkage, was reported [Citation60]. Given the fixed flecainide with this technique, a T7 PD library constructed from cDNAs of Drosophilia melanogaster was screened [Citation61]. After three rounds of selection and ELISA validation, myosin regulatory light chain 2 (Mlc2), which corresponds to the human gene MYL2, has been identified as a potential off-target of flecainide, which might be linked to sudden cardiac death reported when using this compound [Citation62].
4.11. Apigenin
Flavonoids are dietary phytochemicals whose cellular targets remain largely unknown [Citation63,Citation64]. The identification of human cellular targets of apigenin, a flavonoid abundantly present in fruits and vegetables, was attempted using a high throughput T7 PD in combination with second-generation sequencing [Citation65]. This strategy identified 160 high-confidence apigenin target candidates. Most of them are associated with GTPase activation, membrane transport, and mRNA metabolism/alternative splicing. Among the candidates, the authors focused on the validation of hnRNPA2, which was identified by both this strategy and a conventional method. Binding analysis and amplified luminescent proximity assay confirmed the direct interaction between apigenin and the glycine-rich domain (GRD) of hnRNPA2 located in the C-terminal. The binding of apigenin to hnRNPA2 inhibits hnRNPA2 dimerization and mRNA splicing. Thus, this innovative method comprehensively identified the binding proteins, which might explain how a dietary flavonoid interacts with multiple targets and show the broad biological effects usually associated with dietary phytochemicals.
4.12. Other molecules
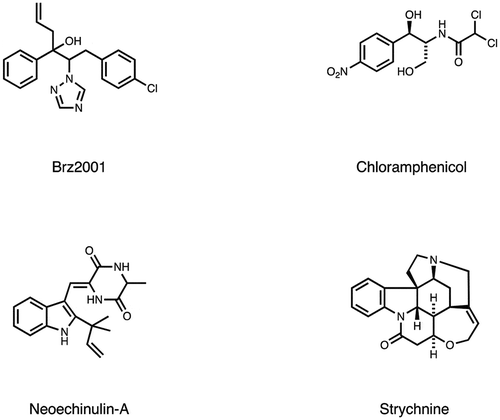
Other small molecules were screened to identify their interaction with endogenous peptides. Examples include a plant-derived natural product oxymatrine to treat chronic hepatitis B (CHB) [Citation66], a lipocyclodepsipeptide MA026 that shows anti-hepatitis C virus (HCV) activity [Citation67]. Off-target for conventional anti-cancer compounds, such as α-SQMG, camptothecin (CPT), etoposide (Etp), or doxorubicin [Citation68–Citation71], as well as for anti-tumor compounds with unique mechanism (CBP501, daptomycin, 11-aza-artemisinin, estradiol or Ridaifens) were identified [Citation72–Citation77]. Furthermore, following bioactive compounds were used for phage display biopanning as bait; tumor hypoxia-targeting LW6 [Citation78], anti-angiogenic methylsynephrine or PFOS [Citation79,Citation80], simvastatin and fluvastatin used to treat cardiovascular disease [Citation81], flavonoid analogues [Citation82], an inhibitor for plant hormone biosynthesis (Brz2001) [Citation83], or the rice blast fungus differentiation (roxithromycin) or appressorium formation (chloramphenicol) [Citation84,Citation85], neuroprotective agents (neoechinulin A) [Citation86], and central nervous system stimulants [Citation87]. A comprehensive list of these compounds is shown in and .
Table 1. List of compounds whose molecular targets were identified by PD (2011–20).
5. Conclusion
In this review, we summarized the use of PD technology for determining therapeutic small molecule targets. Over the past decade, this technique has mainly been used for off-target identification of known drugs, rather than discovering novel compounds. In addition to further corroborating the mechanism of action underlying the primary effect, the discovery of off-targets will contribute toward clarifying the pharmacodynamics of the administered drugs, and this could also lead to drug repurposing for other therapies. PD allows a comprehensive identification of molecular target candidates within the proteome, which depends on the identification of drug-recognizing amino acid sequences and the comparison of the primary sequence with similar proteins. PD technology is capable of transcending the limitations of conventional approaches by providing distinctive technical advantages. We are confident that this technology will be crucial for the next-generation R&D of novel drugs and their targets.
6. Expert opinion
Drug and its target discovery are still difficult processes, despite the recent technological advancements. After drugs are approved for clinical use, information about their safety should be continuously updated. Unanticipated positive outcomes during clinical treatment might lead to drug repurposing. Indeed, target validation by PD led to repurposing in some cases, as discussed in this review. Furthermore, many drugs bind to multiple proteins in the body. A comprehensive profile of a drug interactome could be used as training data by AI-based approaches to generate superior drugs.
To improve next-generation drug R&D methods, there are some key points to be considered. Drug behavior in vivo (pharmacodynamics) should be considered during drug screening. This enables the early identification of modulators for specific physiological conditions, such as blood perfusion and oxygen dependence (e.g., hypoxia in tumors) that are responsible for malignant transformation or therapeutic resistance. In many cases, conventional drug screening has been conducted using a static endpoint method, such as the inhibition of the enzyme activity, signal transduction, cell proliferation, or cytotoxicity. This approach often results in the drug’s failure in the preclinical or clinical phase, due to drug inactivation effects, insufficient blood concentration, and the presence of adverse effects in patients. Alternatively, some compounds, such as hormones or autacoids, may provide a medical benefit only when tested in the living body. Indeed, SQAP, a semi-synthetic derivative of a natural product isolated from sea algae, enhanced the radiosensitivity of tumors only in vivo [Citation47]. Experiments using inducible pluripotent stem cells (iPSCs) and immune checkpoint inhibitors showed that the drug developed by testing their effect on reprogramming and immunomodulation resulted in a better prognosis than the agents developed with conventional testing (selective enzyme inhibition or cell killing). Moreover, the identification of drugs targeting proteins involved in cell metabolism could normalize cells, vessels, and tissues affected in inflammatory and other pathological states. The use of PD technology in vivo might be a strategy to investigate pharmacodynamics and contribute to the development of new drugs.
Using PD technology will also be invaluable to identify druggable targets among intrinsically disordered proteins and regions (IDPs and IDPRs) [Citation19], which cannot be elucidated by conventional approaches. Moreover, PD technology provides a better understanding of the molecular docking dynamics between a protein and small organic compound, similar to the allosteric regulation of proteins. PD technology will be involved in the identification and characterization of a new category of drugs such as iPSCs, anti-aging agents, oxygen modulators for radiosensitizing tumors, immunomodulators, and regulators of molecular metabolism. Together with the advancements in the understanding of biological aspects (e.g., evolution, development, and differentiation) and the identification of drug candidates in nature (inspired by traditional medicine), the use of PD will enable the next generation drug R&D.
Article highlights
The use of phage display (PD) technology in the years 2011-2020 for target determination of small molecule therapeutics has been summarized.
Approximately 30 organic molecules were used as bait for target discovery using PD methods.
Most of the drug-like compounds used in these studies are not new but PD provided valuable information about the drug’s off-target, which will lead to a better understanding of the drug’s absorption, distribution, metabolism, and toxicity (ADMET), as well as expanding the research on drug repurposing.
Independent of the physicochemical properties of proteins or the pathological state, affinity-guided selection of phage-displayed peptides can further expand the druggable proteomes, which would otherwise be almost undetectable.
This box summarizes key points contained in the article.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors would like to thank Editage (www.editage.com) for English language editing.
Additional information
Funding
References
- Smith GP. Phage display: simple evolution in a Petri Dish (Nobel Lecture). Angew Chem Int Ed Engl. 2019;58:14428–14437.
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317.
- Sioud M. Phage Display Libraries: from Binders to Targeted Drug Delivery and Human Therapeutics. Mol Biotechnol. 2019;61:286–303.
- Sidhu SS, Geyer CR, editors. Phage Display in Biotechnology and Drug Discovery. 2nd ed. Abingdon: CRC Press; 2017.
- Iaccino E, Mimmi S, Dattilo V, et al. Monitoring multiple myeloma by idiotype-specific peptide binders of tumor-derived exosomes. Mol Cancer. 2017;16:159.
- Rimbault C, Maruthi K, Breillat C, et al. Engineering selective competitors for the discrimination of highly conserved protein-protein interaction modules. Nat Commun. 2019;10:4521.
- Takakusagi Y, Takakusagi K, Sugawara F, et al. Use of phage display technology for the determination of the targets for small-molecule therapeutics. Expert Opin Drug Discov. 2010;5:361–389.
- Santos R, Ursu O, Gaulton A, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19–34.
- Ziegler S, Pries V, Hedberg C, et al. Target identification for small bioactive molecules: finding the needle in the haystack. Angew Chem Int Ed Engl. 2013;52:2744–2792.
- Molek P, Strukelj B, Bratkovic T. Peptide phage display as a tool for drug discovery: targeting membrane receptors. Molecules. 2011;16:857–887.
- Yang M, Sunderland K, Mao C. Virus-Derived Peptides for Clinical Applications. Chem Rev. 2017;117:10377–10402.
- Makowski L. Quantitative analysis of peptide libraries. In: Petrenko VA, Smith GP, editors. Phage Nanobiotechnology. Cambridge, United Kingdom: RSC Publishing; 2011. p. p 33–54.
- Luo D, Wang L, Liu H, et al. Ribokinase screened from T7 phage displayed Mycobacterium tuberculosis genomic DNA library had good potential for the serodiagnosis of tuberculosis. Appl Microbiol Biotechnol. 2019;103:5259–5267.
- Talwar H, Rosati R, Li J, et al. Development of a T7 Phage Display Library to Detect Sarcoidosis and Tuberculosis by a Panel of Novel Antigens. EBioMedicine. 2015;2:341–350.
- Leow CH, Fischer K, Leow CY, et al. Isolation and characterization of malaria PfHRP2 specific VNAR antibody fragments from immunized shark phage display library. Malar J. 2018;17:383.
- Lanzillotti R, Coetzer TL. Phage display: a useful tool for malaria research? Trends Parasitol. 2008;24:18–23.
- Gibbs EB, Cook EC, Showalter SA. Application of NMR to studies of intrinsically disordered proteins. Arch Biochem Biophys. 2017;628:57–70.
- Frederick KK, Michaelis VK, Corzilius B, et al. Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus. Cell. 2015;163:620–628.
- Uversky VN. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin Drug Discov. 2012;7:475–488.
- Liu J, Nussinov R. Allostery: An Overview of Its History, Concepts, Methods, and Applications. PLoS Comput Biol. 2016;12:e1004966.
- Nussinov R, Tsai CJ. Allostery in disease and in drug discovery. Cell. 2013;153:293–305.
- Pavan S, Berti F. Short peptides as biosensor transducers. Anal Bioanal Chem. 2012;402:3055–3070.
- Takakusagi Y, Takakusagi K, Sugawara F, et al. Using the QCM Biosensor-Based T7 Phage Display Combined with Bioinformatics Analysis for Target Identification of Bioactive Small Molecule. Methods Mol Biol. 2018;1795:159–172.
- Yang Y, Adelstein SJ, Kassis AI. Target discovery from data mining approaches. Drug Discov Today. 2012;17(Suppl):S16–23.
- Wu CY, Wang DH, Wang X, et al. Rapid Discovery of Functional Small Molecule Ligands against Proteomic Targets through Library-Against-Library Screening. ACS Comb Sci. 2016;18:320–329.
- Monjezi R, Tan SW, Tey BT, et al. Detection of hepatitis B virus core antigen by phage display mediated TaqMan real-time immuno-PCR. J Virol Methods. 2013;187:121–126.
- Halperin RF, Stafford P, Emery JS, et al. GuiTope: an application for mapping random-sequence peptides to protein sequences. BMC Bioinformatics. 2012;13:1.
- Huang J, Ru B, Dai P. Bioinformatics resources and tools for phage display. Molecules. 2011;16:694–709.
- Mandava S, Makowski L, Devarapalli S, et al. RELIC–a bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics. 2004;4:1439–1460.
- Miersch S, Li Z, Hanna R, et al. Scalable high throughput selection from phage-displayed synthetic antibody libraries. J Vis Exp. 2015;95:51492.
- ACW C, Ahmad A, Konthur Z, et al. Nanoparticle-Based Semi-Automated Antibody Phage Display Biopanning. Methods Mol Biol. 2019;1904:377–400.
- Piggott AM, Karuso P. Identifying the cellular targets of natural products using T7 phage display. Nat Prod Rep. 2016;33:626–636.
- Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661.
- Keiser MJ, Setola V, Irwin JJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181.
- Fruman DA, Klee CB, Bierer BE, et al. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci U S A. 1992;89:3686–3690.
- Watashi K, Hijikata M, Hosaka M, et al. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38:1282–1288.
- Morohashi K, Sahara H, Watashi K, et al. Cyclosporin A associated helicase-like protein facilitates the association of hepatitis C virus RNA polymerase with its cellular cyclophilin B. PLoS One. 2011;6:e18285.
- Gargaro AR, Soteriou A, Frenkiel TA, et al. The solution structure of the complex of Lactobacillus casei dihydrofolate reductase with methotrexate. J Mol Biol. 1998;277:119–134.
- Kuroiwa Y, Takakusagi Y, Kusayanagi T, et al. Identification and characterization of the direct interaction between methotrexate (MTX) and high-mobility group box 1 (HMGB1) protein. PLoS One. 2013;8:e63073.
- Sparace SA, Mudd JB. The Biosynthesis of the Pyrenocines in Cultures of Pyrenochaeta terrestris. Plant Physiol. 1985;79:308–310.
- Shishido T, Hachisuka M, Ryuzaki K, et al. EpsinR, a target for pyrenocine B, role in endogenous MHC-II-restricted antigen presentation. Eur J Immunol. 2014;44:3220–3231.
- Miller SE, Collins BM, McCoy AJ, et al. E-adaptor interaction is a new mode of cargo recognition in clathrin-coated vesicles. Nature. 2007;450:570–574.
- Stampwala SS, Bunge RH, Hurley TR, et al. Novel antitumor agents CI-920, PD 113,270 and PD 113,271. II. Isolation and characterization. J Antibiot (Tokyo). 1983;36:1601–1605.
- Takeuchi T, Imai T, Ishi K, et al. The anti-tumor antibiotic PD 113,271 binds to microtubule-associated protein 1B (MAP1B). Medchemcomm. 2011;2:1174–1180.
- Benson AA, Daniel H, Wiser R. A Sulfolipid in Plants. Proc Natl Acad Sci U S A. 1959;45:1582–1587.
- Izaguirre-Carbonell J, Kawakubo H, Murata H, et al. Novel anticancer agent, SQAP, binds to focal adhesion kinase and modulates its activity. Sci Rep. 2015;5:15136.
- Takakusagi Y, Naz S, Takakusagi K, et al. A Multimodal Molecular Imaging Study Evaluates Pharmacological Alteration of the Tumor Microenvironment to Improve Radiation Response. Cancer Res. 2018;78:6828–6837.
- Gray BP, Brown KC. Combinatorial peptide libraries: mining for cell-binding peptides. Chem Rev. 2014;114:1020–1081.
- Liu R, Li X, Lam KS. Combinatorial chemistry in drug discovery. Curr Opin Chem Biol. 2017;38:117–126.
- Wang J, Zhang CJ, Chia WN, et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun. 2015;6:10111.
- Efferth T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr Drug Targets. 2006;7:407–421.
- Gotsbacher MP, Cho SM, Kim NH, et al. Reverse Chemical Proteomics Identifies an Unanticipated Human Target of the Antimalarial Artesunate. ACS Chem Biol. 2019;14:636–643.
- Arai M, Kawachi T, Sato H, et al. Marine spongian sesquiterpene phenols, dictyoceratin-C and smenospondiol, display hypoxia-selective growth inhibition against cancer cells. Bioorg Med Chem Lett. 2014;24:3155–3157.
- Kawachi T, Tanaka S, Fukuda A, et al. Target Identification of the Marine Natural Products Dictyoceratin-A and -C as Selective Growth Inhibitors in Cancer Cells Adapted to Hypoxic Environments. Mar Drugs. 2019;17:163.
- Omura S. Macrolide Antibiotics: Chemistry Biology, and Practice. Cambridge: Academic Press; 2002.
- Fernandes P, Martens E, Pereira D. Nature nurtures the design of new semi-synthetic macrolide antibiotics. J Antibiot (Tokyo). 2017;70:527–533.
- Takakusagi K, Takakusagi Y, Suzuki T, et al. Multimodal biopanning of T7 phage-displayed peptides reveals angiomotin as a potential receptor of the anti-angiogenic macrolide Roxithromycin. Eur J Med Chem. 2015;90:809–821.
- O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328.
- Watanabe H, Chopra N, Laver D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009;15:380–383.
- Dilly SJ, Bell MJ, Clark AJ, et al. A photoimmobilisation strategy that maximises exploration of chemical space in small molecule affinity selection and target discovery. Chem Commun (Camb). 2007;2808–2810.
- Taylor PC, Clark AJ, Marsh A, et al. A chemical genomics approach to identification of interactions between bioactive molecules and alternative reading frame proteins. Chem Commun (Camb). 2013;49:9588–9590.
- Cardiac Arrhythmia Suppression Trial I. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–412.
- Panche AN, Diwan AD, Chandra SR. Flavonoids: an overview. J Nutr Sci. 2016;5:e47.
- Nijveldt RJ, van Nood E, van Hoorn DE, et al. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr. 2001;74:418–425.
- Arango D, Morohashi K, Yilmaz A, et al. Molecular basis for the action of a dietary flavonoid revealed by the comprehensive identification of apigenin human targets. Proc Natl Acad Sci U S A. 2013;110:E2153–62.
- Sun YH, Zhang XY, Xie WQ, et al. Identification of UQCRB as an oxymatrine recognizing protein using a T7 phage display screen. J Ethnopharmacol. 2016;193:133–139.
- Shimura S, Ishima M, Nakajima S, et al. Total synthesis and anti-hepatitis C virus activity of MA026. J Am Chem Soc. 2013;135:18949–18956.
- Kusayanagi T, Tsukuda S, Shimura S, et al. The antitumor agent doxorubicin binds to Fanconi anemia group F protein. Bioorg Med Chem. 2012;20:6248–6255.
- Takami M, Takakusagi Y, Kuramochi K, et al. A screening of a library of T7 phage-displayed peptide identifies E2F-4 as an etoposide-binding protein. Molecules. 2011;16:4278–4294.
- Takakusagi Y, Takakusagi K, Ida N, et al. Binding region and interaction properties of sulfoquinovosylacylglycerol (SQAG) with human vascular endothelial growth factor 165 revealed by biosensor-based assays. Medchemcomm. 2011;2:1188–1193.
- Manita D, Toba Y, Takakusagi Y, et al. Camptothecin (CPT) directly binds to human heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) and inhibits the hnRNP A1/topoisomerase I interaction. Bioorg Med Chem. 2011;19:7690–7697.
- Matsumoto Y, Shindo Y, Takakusagi Y, et al. Screening of a library of T7 phage-displayed peptides identifies alphaC helix in 14-3-3 protein as a CBP501-binding site. Bioorg Med Chem. 2011;19:7049–7056.
- Gotsbacher MP, Cho S, Kwon HJ, et al. Daptomycin, a last-resort antibiotic, binds ribosomal protein S19 in humans. Proteome Sci. 2016;15:16.
- Chen XY, Yin Y, Xi J, et al. 11‐Aza‐artemisinin Derivatives Exhibit Anticancer Activities by Targeting the Fatty Acid Binding Protein 6 (FABP6). Chin J Chem. 2018;36:1197–1201.
- Van Dorst B, Mehta J, Rouah-Martin E, et al. The identification of cellular targets of 17beta estradiol using a lytic (T7) cDNA phage display approach. Toxicol In Vitro. 2011;25:388–393.
- Ikeda K, Kamisuki S, Uetake S, et al. Ridaifen G, tamoxifen analog, is a potent anticancer drug working through a combinatorial association with multiple cellular factors. Bioorg Med Chem. 2015;23:6118–6124.
- Tsukuda S, Kusayanagi T, Umeda E, et al. Ridaifen B, a tamoxifen derivative, directly binds to Grb10 interacting GYF protein 2. Bioorg Med Chem. 2013;21:311–320.
- Kim BS, Lee K, Jung HJ, et al. HIF-1alpha suppressing small molecule, LW6, inhibits cancer cell growth by binding to calcineurin b homologous protein 1. Biochem Biophys Res Commun. 2015;458:14–20.
- Kim NH, Pham NB, Quinn RJ, et al. The Small Molecule R-(-)-beta-O-Methylsynephrine Binds to Nucleoporin 153 kDa and Inhibits Angiogenesis. Int J Biol Sci. 2015;11:1088–1099.
- Miyano Y, Tsukuda S, Sakimoto I, et al. Exploration of the binding proteins of perfluorooctane sulfonate by a T7 phage display screen. Bioorg Med Chem. 2012;20:3985–3990.
- Marsh A, Casey-Green K, Probert F, et al. Simvastatin Sodium Salt and Fluvastatin Interact with Human Gap Junction Gamma-3 Protein. PLoS One. 2016;11:e0148266.
- Yao N, Chen CY, Wu CY, et al. Novel flavonoids with antiproliferative activities against breast cancer cells. J Med Chem. 2011;54:4339–4349.
- Takakusagi Y, Manita D, Kusayanagi T, et al. Mapping a disordered portion of the Brz2001-binding site on a plant monooxygenase, DWARF4, using a quartz-crystal microbalance biosensor-based T7 phage display. Assay Drug Dev Technol. 2013;11:206–215.
- Nozaka A, Nishiwaki A, Nagashima Y, et al. Chloramphenicol inhibits eukaryotic Ser/Thr phosphatase and infection-specific cell differentiation in the rice blast fungus. Sci Rep. 2019;9:9283.
- Ishii A, Kumasaka M, Nagashima Y, et al. A eukaryotic molecular target candidate of roxithromycin: fungal differentiation as a sensitive drug target analysis system. Biosci Biotechnol Biochem. 2013;77:1539–1547.
- Kamisuki S, Himeno N, Tsurukawa Y, et al. Identification of proteins that bind to the neuroprotective agent neoechinulin A. Biosci Biotechnol Biochem. 2018;82:442–448.
- Zhang F, Wang M, Qiu Z, et al. Identification and Characterization of Strychnine-Binding Peptides Using Phage-Display Screening. Protein Pept Lett. 2017;24:626–632.