
ABSTRACT
Introduction
American trypanosomiasis, better known as Chagas disease, is a global public health issue. Current treatments targeting the causative parasite, Trypanosoma cruzi, are limited to two old nitroheterocyclic compounds; new, safer drugs are needed. New tools to identify compounds suitable for parasitological cure in humans have emerged through efforts in drug discovery.
Areas covered
Animal disease models are an integral part of the drug discovery process. There are numerous experimental models of Chagas disease described and in use; rather than going through each of these and their specific features, the authors focus on developments in recent years, in particular the imaging technologies that have dramatically changed the Chagas R&D landscape, and provide a critical view on their value and limitations for moving compounds forward into further development.
Expert opinion
The application of new technological advances to the field of drug development for Chagas disease has led to the implementation of new and robust/standardized in vivo models that contributed to a better understanding of host/parasite interactions.
These new models should also build confidence in their translational value for moving compounds forward into clinical development.
1. Chagas disease background
Discovered and characterized more than 100 years ago by Carlos Chagas [Citation1], American trypanosomiasis or Chagas disease (CD) is almost certainly as old as mankind. Indeed, the DNA of Trypanosoma cruzi, the etiological agent of the disease, has been found in tissues of mummies buried in regions between south Peru and northern Chile (the Atacama desert) more than 9,000 years ago [Citation2]. The epidemiology of the disease has changed dramatically over the last few decades. While CD is still endemic in Latin America, with the Gran Chaco region and Bolivia having the highest incidence rate, reaching 20% of the population [Citation3,Citation4], it has become a global public health concern because of its spread into other continents including North America, Europe (especially Spain), Asia (Japan), and Oceania due to migration [Citation5,Citation6,Citation7,Citation8]. Between 6 and 7 million people are estimated to be infected worldwide while around 10,000 deaths occur per year [Citation9,[Citation10,Citation11]. CD makes a significant contribution to cardiovascular disease burden and is the primary cause of infectious cardiomyopathy worldwide [Citation12].
Trypanosoma cruzi spreads primarily through blood-sucking triatomine bugs, but other modes of transmission, such as congenital transmission from mother to child or transmission through contaminated food or drink, are gaining in importance; other potential routes of infection include blood transfusion, organ transplantation, accidental contact in research laboratories, and sexual transmission [Citation13,Citation14,Citation15]. The disease has two clinically defined phases [Citation16,Citation17]:
an acute disease phase that occurs 2–8 weeks post-infection, characterized by elevated parasite load. Symptoms during this period are normally mild, although lethal outcomes may occur in 5% of diagnosed cases, and bloodstream parasites are easily detectable by microscopy.
A strong adaptive immune response brings the parasite load down to low/undetectable levels that last throughout the infection. Infected individuals enter the so-called indeterminate phase of the chronic stage; the majority remain asymptomatic during their entire life, while around 30–40% of those infected eventually develop clinical manifestations such as cardiomyopathy or digestive tract megasyndromes such as megacolon or both. Progressive heart failure and sudden death remain the main causes of death for these patients [Citation18].
Although Chagas pathogenesis is linked to parasite persistence, it has been suggested that the autoimmune response could also play a role; a number of factors probably contribute to the development of Chagas heart disease, including both parasite persistence, altered host immune response and genetics [Citation19,Citation20,Citation21].
While multinational efforts have been undertaken to tackle CD, including vector control and screening of blood banks, challenges such as access to diagnosis and treatment remain. Clinical symptoms of the disease can be dealt with using standard cardiology drugs such as amiodarone, beta-blockers, and others. Treatment options that target the parasite are currently limited to two more than 40-year-old suboptimal nitro-heterocyclic drugs, benznidazole (Abarax/ELEA and Rochagan/LAFEPE) and nifurtimox (Lampit/Bayer). These drugs have been shown to be effective in the acute phase of the disease and evidence is mounting for their efficacy in the chronic phase, especially in children. Their use is limited by poor access and substantial side-effects, not to mention the lack for a test of cure in chronically infected adults [Citation22,Citation23,Citation24,Citation25,Citation26,Citation27]. This highlights a clear priority for better and safer antiparasitic drugs for CD and, importantly, the search for adequate markers or surrogates of parasitological cure.
Parasitological cure following treatment with these drugs has not yet been associated stricto senso with clinical cure, i.e. halting progression of the disease toward cardiac or gastrointestinal pathologies. The recent results of the BENEFIT trial, evaluating whether benznidazole reduces morbidity and mortality in patients with chronic Chagas cardiomyopathy (CCC), showed that this trypanocidal drug had no beneficial effect on these patients, although the debate is still ongoing [Citation28,Citation29]. Nevertheless, there is now enough evidence to suggest that parasite persistence is one of the necessary conditions for the disease to evolve, and that eradication of T. cruzi from the host is a necessary step in the treatment of CD [Citation30]. CD remains a major challenge and all current discovery programs aim to identify ways to eliminate T. cruzi from the human body [Citation31]. Chagas stakeholders developed a target product profile (TPP) for CD (see ), which lists the desirable characteristics for a future product to treat the disease. The target product should be at least as effective as the current standard of care, benznidazole, as assessed using parasitological cure as an endpoint, and better than benznidazole in terms of safety. The target patient population is T. cruzi infected individuals in the asymptomatic phase of CD.
Table 1. Chagas disease target product profile (TPP)
After an absence of significant advances in treatment for the past 50 years, two new potential drugs for CD, posaconazole and a prodrug of ravuconazole, E1224, entered proof-of-concept (PoC) phase 2 clinical trials––but failed dramatically [Citation32,Citation33,Citation34]. Although some investigators attributed the failure of posaconazole and ravuconazole to the suboptimal exposure and/or treatment duration implemented in the clinical trials [Citation35,Citation36] and in very specific cases treatment with posaconazole was shown to be successful [Citation37], these two Phase 2 PoC results were very disappointing, and highlighted major issues around the translation of findings from drug discovery into development phases of Chagas research. These were related to both the types of assays and models used but also, and possibly more importantly, the design and interpretation of the data generated. It was a reminder about the complexity of CD and the fact that a better understanding of its pathology and host/pathogens interactions is essential to the design and development of appropriate assays, and to increasing our confidence in disease models and their translation.
In discovery, there has been substantial progress and improved collaboration over the last eight years, especially for in vitro assays dedicated to the identification of hit compounds with specified properties [Citation38,Citation39,Citation40Citation41]. New assays and a better understanding of some of the features of the T. cruzi parasite have led to an improved screening cascade for moving compounds forward in development. In particular, studies that demonstrated that the current drugs benznidazole and nifurtimox were less potent but more efficacious to eradicate intracellular infection in vitro than ergosterol biosynthesis inhibitors such as posaconazole or ravuconazole led to a completely new vision of drug discovery for CD. It showed the pitfalls of CYP51 inhibitors as potential anti-T. cruzi drugs, and gave a first potential explanation of the clinical failure of this class of compounds [Citation42]. A T. cruzi cytochrome P450 CYP51 (mode of action of azoles) biochemical assay was then developed by the University of Dundee Drug Discovery Unit that allows the triage of phenotypic hit compounds that owe their phenotypic response to inhibition of this enzyme [Citation43].
The failure of azoles in clinical trials also raised the question of the utilization, adequacy, and value of in vivo animal models of CD. Indeed, azoles were considered to have potential for treatment of CD following in vivo assessment in mouse and dog models of the disease [Citation44–46]. Careful analysis of the data published at the time, however, reveals that this class of compounds showed suppressive rather than curative activity in these models. The endpoints used, in particular survival, would not comply with current ethical standards when performing such studies; similarly, considering the current knowledge of the T. cruzi infection and its dynamics, parasitemia, at least alone, would not be judged as an adequate endpoint. This highlights the major issue with the use of CD animal models, and possibly animal models in general: the lack of harmonization in the design of these models.
The various in vivo models and protocols established for CD drug discovery and pre-clinical development for the assessment of new chemical entities (NCEs), natural products and formulation systems such as nanocarriers among others [Citation47,Citation48], and the translational challenges associated with these models have been described in depth elsewhere [Citation49]. Here, we aim to bring a critical view of the advantages and limitations of the new techniques developed over the last 10 years as they relate to drug development, and to assess their relevance and the potential for improvement, as our understanding of CD and the biology of the parasite evolves. Further, we want to demonstrate the translational value, i.e. the transfer of basic in vitro and in vivo research into human applications, of the currently used bioluminescence murine CD model in ongoing drug discovery and development programs.
2. Disease in vivo models: role and translational value in the drug discovery & development process
The use of animals in drug development includes studies dedicated to the demonstration of efficacy and the potential for a specific indication, and assessment of the safety of new drugs before moving forward to clinical trials in humans. Toxicological studies are done in compliance with international regulatory guidance while pharmacodynamics (PD) studies have limitations linked to the so-called translational value of the animal model taken into consideration, i.e. how predictive an animal model can be of a human condition. Technological progress and increased knowledge have led to constant updating and reconsideration of regulatory guidelines as well as to a reduction in animal use, the ‘art’ being to find the right balance between the two [Citation50]. We will focus on the use of animal models for nonclinical PD studies. Basic pharmacokinetic (PK) properties and tolerability are additional parameters that are/should be part of any animal model design. The integration of both parameters into animal models should ideally lead to a good understanding of the PK/PD relationships for a given drug and indication and improve the translational value of the animal model.
2.1. Various applications/roles of disease animal models
One of the first things to consider when developing an animal model or testing compounds in a given model is, ‘is it fit for purpose?’, i.e. is the model under consideration sufficient to answer the research question? There are numerous animal models and they might all be valid for answering specific questions to some extent, if the design and endpoints used are satisfactory.
The animal models used in the R&D process can be broadly categorized into two types, depending on their positioning:
Basic research: The main role at this stage is to answer specific research questions linked to a given pathology of a disease, trying to reproduce as closely as possible the human characteristics in another species. A better understanding of disease pathology (mechanism of action, host/pathogens interactions) is essential to be able to design appropriate assays and better models. It should increase our confidence in the disease models used, their translation and predictive value. Ultimately, such model development can provide new starting points for discovery for a given indication or re-define the properties needed for a compound to fulfil a potential target candidate profile (TCP).
Applied research/drug discovery process: At this stage, a model should be well validated and robust in order to drive development and facilitate the decision-making process, in particular it should:
Provide PoC for a given indication, compound triage during the drug discovery process
Increase confidence in moving forward into clinical trials (linked to increased investment in human and financial resources)
Establish PK/PD relationship and human dose prediction (PD and pharmacokinetic data generated in the animal model of choice)
Basic research data originating from animal models can feed into efficacy assessment models, thus refining them.
Animal models have played a critical role in the exploration and characterization of disease pathophysiology, target identification, and the in vivo evaluation of novel therapeutic agents and treatments, and still do so. However, following clinical trial failures of NCEs that had promising preclinical profiles in animal models irrespective of the therapeutic areas, the predictive value of these models for the efficacy of NCEs in humans has been questioned. However, it is important to be very careful when questioning animal models and evaluating the potential reasons for the lack of translation or predictability, as methodological issues in both preclinical and clinical designs can lead to misconceptions.
Evaluation of PK/PD relationships and the earlier development of validated disease-associated biomarkers are needed to assess target engagement and NCE efficacy when testing disease hypotheses and NCEs in multiple disease models. Additionally, it is essential to transparently integrate efficacy and safety data derived from animal models into the hierarchical data sets generated preclinically in order to derive a level of predictive utility consistent with the degree of validation and inherent limitations of current animal models. The predictive value of an animal model is thus only as useful as the context in which it is interpreted. Finally, rather than dismissing animal models as not being very useful in the drug discovery process, additional resources must be focused on improving existing and developing new animal models [Citation51]. Translational PK/PD modeling to facilitate timely progression of the right compound to clinical studies, simultaneously assuring essential preclinical efficacy and safety knowledge, is essential and should be integrated into the discovery process [Citation52].
2.2. Issues and challenges associated with in vivo disease models
Animal models are still needed to bridge the gap between pre-clinical and clinical research, although the translational value of these models is often questioned. The numerous challenges associated with using animal models, and their translational value, are depicted in . As further developed below, they range from the ethical question of using animals for research purposes to the relevance of the animal species to human physiology, through flaws in study designs, the level of validation and robustness of models, and the definition of endpoints.
Table 2. In vivo animal models’ challenges and potential mitigations
2.2.1. Need
One of the major reservations around the use of animal disease models for drug development is whether they can really predict the outcome of a drug for a specific disease in a human patient. The failure of preclinical animal models to predict clinical efficacy is considered the predominant reason for the poor translation from bench to bedside. Attempts to explain this failure have focused on problems linked to the inability of animal models to mimic the complexity of human conditions, the poor applicability of animal models to clinical settings, and species differences between animals and humans, so the so-called ‘external validity’ of the species used. There are obviously major differences between different animal species and humans with regards to their physiology, genetics, post-translational modifications, immunological response, and other areas, making them an obvious hurdle to translation [Citation53,Citation54].
2.2.2. Validation and reporting
Besides the external validity of the preclinical animal models used, the internal validity of the models (e.g. poor study design, lack of measures to control bias, unrepresentative animal samples, etc.) is another key factor that influences translation. Study design is essential and should be scientifically sound; inadequate experimental design may limit the possibility of extrapolating pre-clinical research findings to humans. The main limitations are linked with the level of validation and standardization of disease models. The number of animals per study is often insufficient for powerful statistical analysis and predictability, the reason for this may well be ethical, and the endpoints used might not be appropriate or might be misinterpreted. In the field of osteoarthritis, for example, Smith et al. proposed the Design and Execution of Protocols for Animal Research and Treatment (DEPART) guidelines, with the motto: “DEPART well-prepared and ARRIVE safely” [Citation55]! Denayer et al. reviewed the challenges and limitations associated with animal models and introduced the notion of fit-for-purpose validation of an animal model to answer a specific question [Citation56]. The reporting of such studies can also be very poor. Even if guidelines for the reporting of animal studies exist (e.g. Animals in Research: Reporting In Vivo Experiments – ARRIVE) and are adopted by many, strict implementation of these guidelines needs improvement. Singh et al. bring to light the need for better reporting and propose specific guidelines for animal PK/PD relationship studies; Animal Pharmacokinetic/Pharmacodynamic Studies (APPS) are commonly used to provide meaningful preclinical information that can be used by the scientific community when conducting first-in-human studies [Citation57]. Progress in presenting and interpreting pre-clinical data will certainly improve the reproducibility of studies.
2.2.3. Back-translation
Improving the validity of animal models, so-called back or reverse translation, by feeding clinical data back into the pre-clinical model can have a substantial impact and improve the animal model under consideration. Failure in the clinic is especially important, as it can provide information on the reasons for failure and induce changes in the animal model or redefinition of the endpoints used [Citation58].
Computational modeling of data from humans and animals affected by similar diseases provides an opportunity for parallel drug development in human and veterinary medicine. This “reverse translational” approach needs to be supported by continuing efforts to refine in silico tools that allow extrapolation of results between species [Citation59].
2.2.4. Endpoints, predictive biomarkers
The choice of endpoints and the availability of biomarkers translatable to human disease are critical in animal experimental settings. Biomarkers are defined as any measurable parameters of biological processes. This includes disease pathophysiology and the impact of interventions upon it. Biomarkers are the most important tool of translational science and the development of biomarkers, which are useful and accessible both in animals and in humans, is, therefore, an important focus of translational activities. Imaging (e.g. MRI, PET), for example, is an ideal translational tool as the readouts in disease models and patients are the same or at least very similar. Despite the existence of several approved animal models, most compounds successful in animal tests still fail to be successfully applied to human diseases. To reduce this rate of failure, it is claimed that animal models that better resemble the human context are needed. A new scoring system that should facilitate judgment of the predictability of results from a given animal model for translation to humans for the assessment of translatability has been proposed [Citation60]. It relies on weighed answers about important data features including robust animal data in more than one species, related human data, and accessibility.
2.2.5. Ethical considerations
Animal experimentation or models cannot be discussed without mentioning the ethical considerations associated with testing in animals and animal welfare. The 3 R principles (replacement, reduction, and refinement) were developed over 50 years ago and provide a framework for performing more humane animal research. Since then, they have been embedded in national and international legislation and regulations on the use of animals in scientific procedures, as well as in the policies of organizations that fund or conduct animal research.
Organizations such as NC3R are helping the research community use the latest science and technology to replace animal studies, providing new approaches for biomedical research and avoiding the time and cost associated with in vivo models. Where the use of animals is still required, NC3R can provide help in the design of the best experiments so that their methods and findings are robust and reproducible [Citation61]. Overall, methods that minimize the number of animals used per experiment should be encouraged; in particular, the design and analysis of animal experiments should be appropriately robust and reproducible. But, as described by Taylor, this is still a long and ongoing process [Citation62].
2.2.6. Animal species used in disease models
The choice of animal model and species will depend on many factors, including the type of disease in question and ethical considerations. While it is clear that no model will fully mimic the human pathology under consideration, the choice of animal model for translational research should not only fulfill the needs of the experimental design described above (reproducibility, variability, etc.), but also meet clear external criteria for addressing the hypothesis that targets a clinical endpoint /outcome. The quality of the methodology is of primary importance.
2.2.6.1. Nonhuman primate (NHP) vs other species for disease models
The challenges in selecting an animal model among those that are available to study human disease pathobiology and drug development -translational research- have been well reviewed by Prabhakar [Citation63]. Humans have a close phylogenetic relationship with NHPs and share many physiological parallels, such as highly similar immune systems. Importantly, NHPs can be infected with many human or related simian viruses. In many cases, viruses replicate in the same cell types as in humans, and infections are often associated with the same pathologies. In addition, many reagents that are used to study the human immune response cross-react with NHP molecules. As such, NHPs are often used as models to study viral vaccine efficacy, antiviral therapeutic safety and efficacy, and to understand aspects of viral pathogenesis. With several emerging viral infections becoming epidemic, NHPs are proving to be a very beneficial benchmark for investigating human viral infections [Citation64]. NHPs are the closest animal models to humans regarding genetics, physiology, and behavior. Therefore, NHPs are usually a critical component in translational research projects aimed at developing therapeutics, vaccines, devices, or other interventions aimed at preventing, curing, or ameliorating human disease. NHPs are often used in conjunction with other animal models, such as rodents, and results obtained using NHPs must often be used as the final criterion for establishing the potential efficacy of a pharmaceutical or vaccine before transition to human clinical trials. In some cases, NHPs may be the only relevant animal models for a particular translational study [Citation65].
Rodents may not be relevant models because their physiology and response to infection are not sufficiently close those of humans compared to NHPs; on the other hand the use of NHPs versus other models such as rodents is complicated by availability, cost, and the many human-like characteristics of NHPs leading to ethical questions. While NHP models might be very useful as models of human viral infections, diseases involving the immune system, or for vaccine development programs, if another animal species fulfills key criteria of an ideal animal model for the indication and modality considered then there is no need to use a higher species disease model.
Indeed, the belief that a primate model is an absolute must in order to make sure that a new drug has chances to be effective in human disease remains sometimes a dogma difficult to avoid. One example that illustrates this is the recent history of drug discovery for the parasitological disease, sleeping sickness or human African trypanosomiasis (HAT). Following the development of acute and chronic murine and sheep experimental animal models of HAT, infected by the etiological agent Trypanosoma brucei, that demonstrate CNS involvement with meningitis, Bouteille et al. tested compounds that show efficacy in these models. They went on to state that ‘Further studies of these nitroimidazole compounds, which could be proposed for human use, have to be carried out on a primate model infected by T. b. gambiense’ and subsequently developed a T. b. gambiense primate model of HAT in Cercopithecus aethiops [Citation66]. Other experimental models of the disease using primates (vervet monkeys) were then developed, showing a similar pattern of infection and disease as in human [Citation67]. Some early R&D efforts to develop new drugs for HAT, in particular the Consortium for Parasitic Drug Development that developed novel parenteral diamidines, tested their compounds in primate models before moving forward into clinical trials [Citation68]. As a rationale for selection of one of these compounds for further development, the PK and efficacy of intramuscular (IM) active diamidine (DB829) and oral prodrug (DB868) were compared in the vervet monkey model of second stage HAT [Citation69]. However, recent results have shown that mouse models of the disease are predictive of efficacy in humans and that there is no need for further testing in higher species to increase confidence for moving forward in the development of new compounds. Fexinidazole was shown to cure both acute and chronic HAT disease in mouse models infected with T. brucei species providing evidence that it had the potential to be an effective oral treatment for both the T. b. gambiense and T. b. rhodesiense forms of human sleeping sickness and both stages of the disease [Citation70]. This drug then successfully went through clinical development for the treatment of human sleeping sickness [Citation71] and was found to be as effective and safe for the treatment of T. b. gambiense infection compared with nifurtimox eflornithine combination therapy in late-stage HAT patients [Citation72]. It was then recommended by EMA and is now registered in DRC [Citation73,Citation74,Citation75].
Both acute and chronic mouse models played a critical role in the development of oxaboroles for HAT [Citation76]. SCYX-7158 was identified as an effective, safe, and orally active treatment for HAT. The biological and PK properties of SCYX-7158 (distribution into the cerebrospinal fluid to achieve therapeutically relevant concentrations in this compartment) suggested that this compound should be efficacious and safe to treat stage 2 HAT. SCYX-7158 entered preclinical studies and progressed to phase 1 clinical trials in 2011 [Citation77]. It is now being tested in patients under the name acoziborole and is potentially a single-dose oral cure for both stages of the disease [Citation78].
These examples show that mouse disease models can have sufficient predictability and translational value in specific areas, especially when efficacy data are coupled with other important parameters for further development, such as pharmacokinetics, distribution, and other basic ADME properties.
2.2.6.2. Humanized mice models
While transgenic and knockout techniques have revolutionized the manipulation of rodents and other species in order to gain greater insights into the pathogenesis of human disease, we are far from generating ideal animal models of most human disease states.
To overcome the problem of having to use NHP models, humanized mice, i.e. immunodeficient mice engrafted with functional human cells and tissues, have become increasingly important as small, preclinical animal models for the study of human disease. The development of immunodeficient IL2rgnull mice in the early 2000’s was a major breakthrough, allowing for the study of functional human cells, tissues, and tumors in vivo in a small animal model. Since then, investigators have been able to engraft murine recipients with human hematopoietic stem cells that develop into functional human immune systems. These mice can also be engrafted with human tissues such as islets, liver, skin, and most solid and hematologic cancers. Humanized mice are making possible significant progress in studies of human diseases and are increasingly being used as preclinical models in multiple biological fields including infectious diseases, immunology, cancer, regenerative medicine, hematology, and autoimmunity [Citation79,Citation80].
This rodent model has been used in some parasitological diseases, such as malaria. To address the challenge of the limited range of hosts of malaria-causing Plasmodium species, humanized mice have been constructed in which the tissue compartments relevant to the mammalian stages of plasmodial parasites are humanized. The development of human liver chimeric mice, human erythroid chimeric mice, and dually engrafted mice now allows for faithful replication of the entire Plasmodium falciparum life cycle. Although refinements to these models are still needed, humanized mice provide a promising small-animal model to study development of P. falciparum and, conceivably, other human malarial parasite species, and may also help with drug and vaccine development [Citation81,Citation82]. Obviously, the costs associated with humanized mice make it difficult to apply to NTDs where funding is scarce.
In summary, an appropriate balance between the between the various elements described above -validation level, possible endpoints, used species, guidelines, ethics, PK among others- should be found to have the most predictive fit-for-purpose model that gives the researcher most confidence to answer specific questions and move compounds forward in development.
3. Chagas disease animal models: current status and new developments
Chatelain & Konar have performed an exhaustive review of all the various in vivo models and protocols established for CD drug discovery and pre-clinical development that had been used or considered at that time [Citation49]. Since then, enormous progress has been made in this area, in particular with the application of transgenic T. cruzi parasites and the use of modern imaging techniques. As we will see, this has transformed our understanding of the dynamics of the infection, provided new insights into host/pathogen interactions and allowed members of the Chagas community to develop and standardize a robust in vivo model.
The challenges associated with CD animal models are no different to those for other disease indications.
3.1. Purpose of animal models in Chagas disease: specific questions to be answered
When developing an animal model for CD, it is important to consider what the specific question is that model is intended to answer. It is especially important to define this question, as over the years there has been a lot of controversy over the definition of cure in T. cruzi infected people and the role of the parasite and immune system in the disease etiology.
- Parasitological cure vs. disease progression: One might want a model able to determine if the administration of a new compound to an infected animal will lead to either parasitological cure (i.e. removal of the parasite from the body, both tissues and blood, also termed sterile cure), or halting the progression of the disease (i.e. stopping progression from the asymptomatic to the symptomatic stage of the chronic phase of the disease, looking for example at increase in heart lesions, heart fibrosis or ECG alterations), or possibly both. It is essential to define these questions to allow for proper/adequate experimental design. In the case of a preventive vaccine, design and protocol will have to be adjusted accordingly.
- Acute vs chronic stage: Another important question relates to the stage of the disease to be considered and targeted: Acute vs indeterminate vs chronic organ involvement. Since the TPP for CD refers to the development of an ideal drug that leads to parasitological cure in chronic indeterminate and acute patients, animal models for both stages will be needed. This obviously has an important impact on logistics (keeping animals infected for long periods of time) and the amount of compound needed to be tested. As we will see later, a lot of conclusions/ assumptions about the potential of a compound have relied on data generated in acute stage models.
- Endpoints used: For years, the main endpoints were very often survival and parasitemia in the acute stage of the disease. Considering the 3Rs regulations, as well as the current knowledge of the disease, these endpoints are no longer up-to-date. Azoles, for example, showed suppressive efficacy (e.g. reduction of parasitemia) in a variety of animal models but failed in clinical trials. It is clear that parasite load in tissues is also important and that making assessment of parasites in randomly selected tissues is inadequate.
Another major issue is the lack of biomarkers of parasitological cure that are useful in human clinical trials, which should be introduced early on during the discovery process. PCR for example can be useful in clinical PoC but is not adequate as a pivotal endpoint either in clinical trials or in in vivo animal models, either due to the dynamic of the infection or the lack of correlation between parasitemia and tissue parasitism.
3.2. Issues and challenges
Issues and challenges for in vivo animal models in the field of CD are not that different to those for other animal disease models. The plethora of animal models for CD makes it difficult to select the most adequate [Citation83]. As a consequence, there is a lack of standardization -in particular the use of similar endpoints and design- and acceptance for various models, with the common refrain ‘The model I used is the best model, why should I change?’ In 2010, Romanha et al. proposed some defined steps and protocols for the in vivo assessment of new antiparasitic compounds [Citation84]. This was a good example of an attempt to standardize models within the CD research community, thereby allowing researchers to compare generated data; new developments since then, especially imaging technologies (see below) should lead to an update of the process.
Another major issue is the quality of reporting of in vivo studies performed in CD and compliance with the ARRIVE guidelines. Gulin et al. in their systematic review on the subject reported that there is an urgent need for the scientific community to improve the description of animal use and the animal models employed, and for transparent reporting and experimental design to facilitate transfer and application of results to the affected human population [Citation85]. This would improve the reproducibility and assessment of the design of such studies.
When comparing all data generated with benznidazole published in the literature, Molina et al. showed the extent of the heterogeneity of experimental design and the problem of generating an overall conclusion relating to the standard of care [Citation86]. Standardization is further weakened by the use of different animal species (mice, NHPs, dogs, etc.) with different genetic and immune backgrounds as hosts of the infection, and the use of different T. cruzi parasite strains.
3.3. Brief overview of models used in the Chagas field
For an exhaustive description of the variety of animal models used in the CD field, we refer the reader to the description in Chatelain and Konar’s extensive review [Citation49]. Here we highlight the main species used so far and consider a few more exotic models that are rarely described.
3.3.1. From zebrafish to nonhuman primates
A model of infection in zebrafish was developed by Akle et al. in an attempt to evaluate T. cruzi motility in the vascular system in a live vertebrate model [Citation87]. Indeed, and until recently, it has been a challenge to understand and visualize parasite behavior during the initial stages of infection in humans. Using light sheet fluorescence microscopy (LSFM), circulating parasites could be visualized in fluorescent-T. cruzi-infected transparent zebrafish larvae, and their tropism, migration patterns, and motility in the dynamic environment of the cardiovascular system of a live animal could be evaluated. T. cruzi parasites were observed traveling in the circulatory system of live zebrafish in different-sized blood vessels and the yolk, and to the atrioventricular valve, despite the strong forces associated with heart contractions. However, the relevance to humans is yet to be established.
Dogs represent an important reservoir of the parasite and it therefore makes sense to assess this species as potential model of CD. Bahia’s group has been active in developing a canine model of the disease using mainly the Beagle dog [Citation88]. They suggested it was a suitable model because it reproduces the clinical and immunological findings described in chagasic patients, and because dogs can develop cardiomyopathy [Citation89]. The canine model of CCC was, indeed, shown to mimic human disease, as it could reproduce the percentage of individuals that develop heart failure during the chronic infection. A robust echocardiographic evaluation of left ventricular size and function in dogs chronically infected by T. cruzi showed a significant left ventricular ejection fraction (EF) and fractional shortening reduction in infected animals compared to controls, with no significant variation in volumes probably due the limited period of observation. Using the cutoff value of EF ≤ 40%, established for dilated cardiomyopathy in dogs, only 28% of the infected dogs were affected by the chronic infection [Citation90]. A recent study in dogs showed no correlation between parasite load in tissues/myocardium and fibrosis at either the acute or chronic stage. This led the authors to conclude that the symptomatic progression of the disease cannot be explained solely by parasite persistence [Citation91]. The dog model was also used to assess ravuconazole, and results were the basis, rightly or wrongly, for moving this compound into clinical trials in human. The investigators concluded that ravuconazole had potent suppressive but not curative activity in the canine model of acute Chagas’ disease, and postulated that the unfavorable PK properties (half-life, 8.8 h) could have been responsible for the lack of parasitological cure observed. Since this compound had a longer half-life in humans (4 to 8 days), it was postulated that it could be a promising drug for use as chemotherapy in human CD [Citation45]. A prodrug of ravuconazole, E1224, went through to a PoC Phase 2 clinical trial, but failed to show lack of relapse of parasitemia as assessed by quantitative PCR (qPCR) [Citation34].
The long lifespan of dogs, together with both ethical and cost considerations, makes it difficult to carry out routine efficacy assessments of compounds; this is reflected in the low number of studies using this species that can be found in the literature [Citation92].
Another species often mentioned for CD, and sometimes considered as the obligatory way forward, is the nonhuman primate (NHP). Like dogs, monkeys serve as a reservoir for the parasite T. cruzi. Old studies have looked at experimental infection of rhesus monkeys with T. cruzi and shown a similar pattern of disease progression as in humans [Citation93,Citation94]. The results from nonhuman primate models for CD are controversial. For example, following infection with various strains of T. cruzi, Cebus apella sp. monkeys were found to present normal ECG despite having high parasitemia and positive serology, and did not develop the disease [Citation95]. Studies with naturally infected baboons in the chronic phase of the disease found that posaconazole treatment at similar drug blood levels as in humans failed to completely clear the parasite from any of the animals. The authors concluded that the NHP model should be used more widely, as the same compound was found to have curative activity in mouse models [Citation96]. However, the authors forgot to compare the endpoints used in both cases (survival, parasitemia) and the definition of parasitological cure in the mouse model at the time [Citation46,Citation97]. At the time of writing, there is no validated NHP model for CD. Moreover, the notion of cure and comparison of endpoints and methodologies for studies that were performed with at least a 10-year interval may be questionable.
In addition, if we consider costs (even if a small number of animals were sufficient to give results with significant statistical power), ethics, and the lack of validated models, NHP are not currently a suitable model for assessment of efficacy of new compounds.
Murine models of CD are, for various reasons (costs, logistics, handling, application of new technologies), the most favored and widely used models to study T. cruzi infection and assess potential anti-parasitic drugs. They recapitulate many aspects of the human disease, are easy to manipulate genetically and are amenable to study by small animal imaging technologies, as described in 3.4.
3.3.2. Other less well-known Chagas models
3.3.2.1. Investigation of T. cruzi sexual transmission in animals
Trypanosoma cruzi infection is transmitted congenitally from a chagasic mother to her offspring, but the male partner’s contribution to in utero contamination is unknown. Although the possibility of sexual transmission of T. cruzi has been suggested since its discovery, few studies have been published on this subject. Experimentally infected mice have been used to evaluate the potential sexual transmission of T. cruzi. Male and female mice in the chronic phase of CD were mated with naive partners. Parasitological, serological, and molecular tests demonstrated the presence of parasites in tissues and blood of partners and confirmed the potential for sexual transmission of T. cruzi in mice [Citation98]. These results have been corroborated in acutely infected mice; following mating with naïve partners, infection was detected in the majority of mice following serology assays detecting anti-T. cruzi antibodies. The possibility of sexual transmission was also confirmed by visualization of amastigotes in the testes [Citation99,Citation100].
These results demonstrate that sexual transmission of T. cruzi is possible and may contribute to maintenance of the parasite’s enzootic cycle, and that this is a potential additional route of transmission for the parasite.
3.3.2.2. Models to study the chronic gastrointestinal manifestations in T. cruzi-infected species
There are very few descriptions of the chronic gastrointestinal manifestations of CD in animal models. They are mainly a result of impairment of the enteric nervous system caused by T. cruzi infection and associated inflammation and fibrosis of tissues. The anatomical locations most commonly described to be affected by CD are the salivary glands, esophagus, lower esophageal sphincter, stomach, small intestine, colon, gallbladder, and biliary tree. Megacolon is caused by myenteric plexular denervation in the intestinal mucosa, causing motility disorders associated with colonic constipation and dilatation [Citation101,Citation102].
Experimental animal models of the disease looking at these pathologies and abnormalities are few and far between. Although difficult, it has been possible to establish alterations in gastrointestinal tract (GI) function in T. cruzi infected mice [Citation103]. X-rays and magnetic resonance imaging (MRI) techniques allow monitoring of alterations in the GI tract such as intestinal dilation and megasyndromes in the bladder of T. cruzi chronically infected mice [Citation104]. Histological examination of the digestive tract of all infected mice showed extensive changes of the intestinal muscle layer, such as the diminution of the muscular and mucous layers and the loss of colonic folds and myoenteric plexus. There is clear evidence that infection with T. cruzi causes megasyndromes of the GI tract [Citation105,Citation106]. More recently, attempts were made to develop a murine model to elucidate megacolon pathogenesis and associated adaptive and neuromuscular intestinal disorders [Citation107]. In the acute phase, all animals presented inflammatory lesions associated with intense and diffuse parasitism of the muscular and submucosa layers, which were enlarged when compared with the controls. The occurrence of intense degenerative inflammatory changes and increased reticular fibers suggests inflammatory-induced necrosis of muscle cells. In the chronic phase, parasitism was insignificant; however, there was a focus on the architecture of the Aüerbach plexus which became inflamed, and a significant decrease was detected in the number of neurons and in the density of intramuscular nerve bundles. Other changes that were observed included increased thickness of the colon wall, diffuse muscle cell hypertrophy, and increased collagen deposition, indicating early fibrosis in the damaged areas. Mast cell count significantly increased in the muscular layers. The authors hypothesize that the long-term inflammatory process mediates neuronal damage and intramuscular and intramural denervation, leading to phenotypic changes in smooth muscle cells associated with fibrosis. These long-term structural changes may represent the basic mechanism for the formation of Chagasic megacolon. A similar attempt was performed using male Wistar rats infected with the Y strain of T. cruzi; the changes manifested in the colon are not directly proportional to the size of the inoculum to which the animals were submitted, but to the duration of infection [Citation108].
3.3.2.3. Application of Chagas models to the study of the immune response and vaccine development
A therapeutic vaccine could bolster the protective Th1-mediated immune response, thereby slowing or halting the progression of CCC. Prior work in mice has demonstrated the therapeutic efficacy of a Tc24 recombinant protein vaccine -formulated with an emulsion containing the Toll-like receptor 4 agonist E6020 as an immunomodulatory adjuvant- in the acute phase of CD. Since most CD patients are in the chronic phase of the disease, it is important to reproduce the same study in a mouse model of chronic T. cruzi infection; one study has looked at the therapeutic efficacy of a vaccine prototype containing recombinant protein Tc24 [Citation109]. The models used to test this are similar to those used to assess NCEs but the readouts are different, concentrating on the immune response markers and disease progression. The results were very encouraging, as the therapeutic vaccination significantly reduced cardiac fibrosis in chronically infected mice. This is the first study demonstrating therapeutic efficacy of a prototype vaccine against cardiac fibrosis in a mouse model of chronic T. cruzi infection.
Chronically T. cruzi-infected individuals exhibit a deterioration of T cell function, a state of immune exhaustion characterized by poor cytokine production and increased inhibitory receptor co-expression, suggesting that these changes are potentially related to the progression of CD. Moreover, an effective anti-parasitic treatment appears to reverse this state and improve the T cell response. Taking these findings into account, the state of functionality of T cells might provide a potential correlate of protection to detect individuals who will or will not develop the severe forms of CD. T cell response was analyzed by flow cytometry to assess cytokines/cytotoxic molecules and the expression of inhibitory receptors, in a murine model of acute (10 and 30 days) and chronic (100 and 260 days) CD, characterized by parasite persistence for up to 260 days post-infection and moderate inflammation of the colon and liver of T. cruzi-infected mice. Acute CD induced a high antigen-specific multifunctional T cell response by producing IFN-γ, TNF-α, IL-2, granzyme B, and perforin; and a high proportion of T cells co-expressed 2B4, CD160, CTLA-4, and PD-1. In contrast, chronically infected mice with moderate inflammatory infiltrate in liver tissue exhibited monofunctional antigen-specific cells, high cytotoxic activity (granzyme B and perforin), and elevated levels of inhibitory receptors (predominantly CTLA-4 and PD-1) co-expressed on T cells. Taken together, these data support the idea that, similar to in humans, T. cruzi persistence in mice promotes the dysfunctionality of T cells, and that these changes might correlate with CD progression. They also show that an in-depth search for immune markers and correlates of protection, as well as long-term studies of new immunotherapy strategies for CD using these models, is possible [Citation110].
3.4. Impact of imaging technologies on the development of new animal models
The recent development of imaging technologies (fluorescence, bioluminescence) together with genetic editing techniques and transgenic pathogens has facilitated animal studies in a number of diseases.
Traditional methods for localization and quantification of the presence of pathogenic microorganisms in living experimental animal models of infection have mostly relied on sacrificing the animals, dissociating the tissue, and counting the number of colony forming units. However, the discovery of several varieties of the light producing enzyme luciferase and the genetic engineering of bacteria, fungi, parasites, and mice to make them emit light, either after administration of the luciferase substrate or, in the case of the bacterial lux operon, without any exogenous substrate, has opened up new alternatives. Dedicated bioluminescence imaging (BLI) cameras can record the light emitted from living animals in real time allowing noninvasive, longitudinal monitoring of the anatomical location and growth of infectious microorganisms as measured by strength of the BLI signal, which is proving useful for the dynamic monitoring of a variety of cellular functions. BLI technology has been used to follow bacterial infections in a wide range of tissues and conditions such as traumatic skin wounds and burns, osteomyelitis, infections in intestines, mycobacterial infections, otitis media, lung infections, biofilm and endodontic infections, and meningitis. Fungi that have been engineered to be bioluminescent have been used to study infections caused by yeasts (Candida) and by filamentous fungi. Parasitic infections caused by malaria, Leishmania, trypanosomes and toxoplasma have all been monitored by BLI. Light producing transgenic rodents are emerging as key tools in the study of host response to infection, shedding light on the host-pathogen relationship. Viruses such as vaccinia, herpes simplex, hepatitis B and C, and influenza, have also been studied using BLI. This enables longitudinal studies in which the spectrum of the disease process and its response to therapies can be monitored. This rapidly growing technology is expected to continue to provide much useful information, while drastically reducing the numbers of animals needed in experimental studies [Citation111,Citation112,Citation113].
3.4.1. Application to kinetoplastids
The introduction of transgenic parasite strains that allow real-time monitoring of parasite infection in mice using image techniques based on the emission of fluorescence or bioluminescence has been a major advance in the field of in vivo models for the associated diseases. It has allowed parasite burdens to be tracked throughout the chronic stage of infection and the investigation of specific tropism and dynamics.
Bioluminescence imaging of pathogens expressing firefly luciferase (emission maximum 562 nm) has been adopted in a number of in vivo models of disease to monitor dissemination, drug-treatment, and the role of immune responses. The original lack of sensitivity for detecting deep tissue bioluminescence at wavelengths below 600 nm has prevented the widespread application of in vivo imaging to investigating infections with T. brucei and other trypanosomatids. McLatchie et al. developed an improved system that allows the detection of fewer than 100 bioluminescent T. brucei parasites in a murine mode using a red-shifted firefly luciferase (PpyRE9H) that has a peak emission of 617 nm. Parasite dissemination and drug efficacy could be monitored in real time, and brain infections were readily detectable. The level of sensitivity in vivo was significantly greater than was achievable with a yellow firefly luciferase reporter. The optimized bioluminescent reporter line significantly enhanced the application of in vivo imaging to study stage II African trypanosomiasis in murine models [Citation114,Citation115]. Using bioluminescent T. b. brucei GVR35 VSL-2 and an IVIS® Lumina II imaging system to detect parasites in mice and evaluate parasite localization and disease progression, Burrell-Saward et al. confirmed the efficacy of drugs such as diamidines (DB829) and fexinidazole that were previously evaluated using the standard murine models. They showed complete elimination of bioluminescent parasites by treatment with melarsoprol and DB829, and could monitor trypanosome infection in different areas of the brain and assess the dose and rate of kill effect of fexinidazole in infected mice. It was shown that drug dose-response can be evaluated using bioluminescence imaging, and confirmed quantification of tissue parasite load using qPCR. Most importantly, the model was also able to detect drug relapse earlier than traditional blood film detection, even in the absence of any detectable peripheral parasites [Citation116,Citation117].
Similarly, bioluminescent imaging was recently successfully applied to both BALB/c murine and golden Syrian hamster VL experimental models with Leishmania donovani and infantum species, genetically modified to express the firefly luciferase. These models are now fairly well validated (validation with known reference drugs amphotericin B and miltefosine, with a robust bioluminescent signal in target organs, such as the liver and the spleen). It allows infections to be followed in real-time longitudinally (same mouse assessed over time, i.e. reduction of the number of laboratory animals used) and treatment efficacy is assessed using whole-mouse bioluminescence imaging without the need to wait several weeks for spleen infections to be detectable, as is the case for invasive methods, thereby opening up new perspectives for testing of compounds for visceral leishmaniasis [Citation118,Citation119].
3.4.2. Application to the field of Chagas disease
As early as 2008, luminescent T. cruzi trypomastigotes and amastigotes were imaged in infections of rat myoblast cultures in vitro and a clear correlation of photon emission signal strength to the number of parasites used was observed [Citation120]. Similar pictures were observed in mice infected with different numbers of luminescent parasites, where a clear correlation was identified early on between photon emission and parasite number at the site of inoculation, followed by dissemination of parasites to different sites over the course of a 25-day infection, as seen by whole animal imaging from ventral, dorsal, and lateral perspectives. Tissue distribution of T. cruzi was further determined by imaging heart, spleen, skeletal muscle, lungs, kidneys, liver, and intestines ex vivo. These results illustrated the natural dissemination of T. cruzi during infection and provided a new tool for studying a number of aspects of CD, including rapid screening of potential therapeutic agents, roles of parasite and host factors in the outcome of infection, and analysis of differential tissue tropism in various parasite-host strain combinations. Further, developments in noninvasive imaging technologies have permitted the study of the same animal over an extended period of time by multiple imaging modalities, thus permitting the study of the transition from acute infection through the chronic stage and during administration of therapeutic regimens [Citation103].
Canavaci et al. described the development and validation of improved methods to test anti-T. cruzi compounds in vitro and in vivo using parasite lines expressing the firefly luciferase (luc) or the tandem tomato fluorescent protein (tdTomato) [Citation121]. In vivo, signal intensity was measured as a surrogate for parasite load at the site of infection before and after initiation of drug treatment in mice infected in the footpads with fluorescent or bioluminescent parasites. Importantly, the efficacy of various drugs as determined in this short-term (<2 weeks) assay mirrored that of a 40-day standard treatment course used by the investigators. It was then argued that the broader and higher-throughput screening of compounds needed to identify potential new drugs for the treatment of T. cruzi infection could become feasible using these methods, allowing their subsequent rapid validation in vivo [Citation121,Citation122].
From a technical point of view, the imaging of animals infected with parasites expressing luciferase opened up new possibilities for following the fate of parasites in infected mammals. To produce bioluminescence, infected and control mice receive a luciferin solution through intraperitoneal injection. Mice are then immediately anesthetized with 2% isofluorane, and imaged 10 minutes later. Infected tissues and organs are evaluated ex vivo in a 24-well plate following incubation with D-luciferin diluted in PBS. Images are captured using an IVIS Lumina image system. Dissected organs can also be evaluated by microscopy of hematoxylin-eosin stained sections.
Results obtained using a genetically modified Dm28c strain of T. cruzi, expressing firefly luciferase to keep track of infection by bioluminescence imaging, showed a progressive infection in vivo in BALB/c mice at various intervals after infection. The bioluminescent signal was immediately observed at the site of T. cruzi inoculation, and it was disseminated in the peritoneal cavity one day post infection (dpi). A similar pattern in the cavity was observed on 7 dpi, but the bioluminescence was more intense in the terminal region of the large intestine, rectum, and gonads. On 14 and 21 dpi, bioluminescent parasites were also observed in the heart, snout, paws, hind limbs, and forelimbs. From 28 dpi to 180 dpi in chronically infected mice, bioluminescence declined in regions of the body but was concentrated in the gonad region. Ex vivo evaluation of dissected organs and tissues by bioluminescent imaging confirmed the in vivo bioluminescent foci. Histopathological analysis of dissected organs demonstrated parasite nests at the rectum and snout, in muscle fibers of mice infected with Dm28c-WT and with Dm28c-luc, corroborating the results of bioluminescent imaging [Citation123].
These results showed that bioluminescence imaging is accurate for tracking parasites in vivo, and that this methodology could become important for gaining a better understanding of the infection, tissue inflammation, and parasite biology in terms of host cell interaction, proliferation, and parasite clearance to subpatent levels. In 2015, Rogers subsequently suggested that bioluminescent parasites could be of major help in R&D for Chagas [Citation124].
Since then, this new technology has boomed, leading to the validation of a very robust and reproducible model applicable for assessment of compounds for CD.
A novel red-shifted variant of luciferase able to emit a tissue penetrating red-orange light (as opposed to the light emission in the blue-green region of the spectrum) has enabled the standardization and validation of an in vivo image-based technique that allows high sensitivity real-time tracking of parasite infection in mice during chronic stage infections. A robust correlation between parasite numbers and whole animal bioluminescence was observed, with a limit of detection of close to 100 parasites [Citation125].
3.4.3. A specific example of the development and translational value of the BLI mouse model using red-shifted luciferase CL Brener strain (TcVI)
The use of BLI with a red-shifted luciferase transgenic T. cruzi parasite of the CL Brener strain (TcVI) has allowed John Kelly’s group to develop a very robust and informative murine model of T. cruzi infection [Citation125]. It has generated a lot of new information and knowledge about parasite infection that could be incorporated into a new model that would be very useful for assessing the potential of new compounds to provide parasitological cure, potentially increasing its translational value to human.
3.4.3.1. New knowledge about T. cruzi infection acquired from the BLI mouse model
The major issue with in vivo models in the past was the lack of a method that would allow the tracking of parasite burdens from the acute stage to throughout the chronic stage of infection. The development of a highly sensitive in vivo imaging system based on bioluminescent T. cruzi, which expresses a red-shifted luciferase that emits light in the tissue-penetrating orange-red region of the spectrum, addressed this issue. This system enabled long-term serial evaluation of parasite burdens in individual mice in real time with an in vivo limit of detection of significantly less than 1000 parasites.
Two major pieces of information emerged from these developments [Citation125]:
First, one can follow T. cruzi infection in real-time and reproduce the different stages, from the acute stage, characterized by very high levels of bioluminescence in all organs, to entry into the chronic stage where parasite burden was very low, characterized by much lower bioluminescence (logarithmic scale) and focal distribution. Chronically infected mice developed myocarditis and cardiac fibrosis, despite the absence of locally persistent parasites in the heart.
Second, infection in the chronic stage showed a very dynamic spatiotemporal and focal distribution of parasites, not localized specifically to the heart and possibly other organs as often previously speculated. T. cruzi parasites were rapidly moving from one site to another. The only sites where T. cruzi infection was consistently observed were reservoir sites in the gastro-intestinal tract, specifically the colon and stomach. The primary reservoir sites in humans are not definitively known, and it remains to be seen whether they will be the same as those identified in mice.
In short, BLI has allowed a link to be established between parasite persistence and the pathogenesis of Chagas heart disease. Parasite burden in the chronic phase is controlled by effective, but nonsterilizing immune responses. BLI data would suggest that long-term persistence of T. cruzi is likely to involve episodic reinvasion as well as continuous infection, to an extent that varies between tissues. It is a dynamic, not a static process, localized in specific organs. Infected cells are difficult to detect because they are scarce and focally distributed in multiple sites. These new insights into T. cruzi dynamics and reservoir sites are key to a better understanding of the association between persistence, pathogenesis, and immunity, and to optimizing treatment [Citation126,Citation127].
3.4.3.2. Model validation for use in R&D and compound efficacy assessment
The knowledge generated by these preliminary studies using luciferase transgenic T. cruzi and BLI has helped to design a better model for compound efficacy assessment.
A lot of work has been done to ensure the robustness and reproducibility of the model over time. Important parameters that were tackled in John Kelly’s lab included transgene expression throughout the different stages of the T. cruzi parasite, the stability of the expression of luciferase over time, a reproducible pattern of infection in mice, assessment of the impact of the number of parasites used for infection, as well the routes used for infection. It is critical that a model is reproducible over time. This was achieved in BALB/c mice infected i.p. with specific amounts of blood trypomastigotes from luminescent CL Brener T. cruzi issued from infected SCID mice. End-point ex vivo bioluminescence imaging allowed tissue-specific quantification of parasite loads with minimal sampling bias. During chronic infections, qPCR-inferred parasite loads correlated with ex vivo bioluminescence confirmed the gut as the parasite reservoir. Benznidazole was used as a control drug in this model and showed good efficacy in eliminating the parasite [Citation128].
Once validated and the robustness shown, attempts were made to assess the value of this new experimental model for testing compounds for their efficacy against both acute and chronic stages of T. cruzi infections, and to define whether this new tool should be included into the screening cascade of drug development programs for CD (see for the current CD screening cascade). As discussed earlier, back-translation is a good way to feed clinical data back into a preclinical model in order to assess and improve its validity. We therefore decided to try to reproduce the outcome of clinical trials in T. cruzi infected people when treated with benznidazole and posaconazole using this new BLI model and protocol (see ). In two recent clinical trials involving patients in the chronic indeterminate stage, posaconazole failed to show efficacy when compared with benznidazole, i.e. patients treated with posaconazole did not achieve sustained negative T. cruzi PCR in blood after one year follow-up but rather showed parasite relapse in blood [Citation32,Citation33]. Mice inoculated with bioluminescent T. cruzi were treated with either benznidazole or posaconazole, and the efficacy of these drugs was assessed by in vivo and ex vivo imaging. Immunosuppression obtained following three rounds of cyclophosphamide treatment was used to rapidly detect relapse. Posaconazole was found to be significantly inferior to benznidazole as a treatment for both acute and chronic T. cruzi infections. Whereas 20 days treatment with benznidazole was 100% successful in achieving sterile cure, posaconazole failed in almost all cases [Citation129]. These data showed that the efficacy of different compounds in the various stages of CD could be distinguished using highly sensitive bioluminescence imaging of the murine infection model. To gather further data on the potential of this model for determining the efficacy of compounds, three nitroheterocycles were evaluated in both acute and chronic stages of the CD model – the two current standard of care treatments, benznidazole and nifurtimox, and a promising new candidate, fexinidazole. The results were to some extent unexpected as benznidazole and nifurtimox were found more effective at curing chronic than acute stages of the infection, judged by treatment duration and therapeutic dose; in addition, this was not associated with factors that differentially influence plasma drug concentrations in the two disease stages [Citation130].
Figure 1. Chagas Disease screening cascade: From a confirmed hit toward pre-clinical candidate selection – Positioning of the in vivo animal models
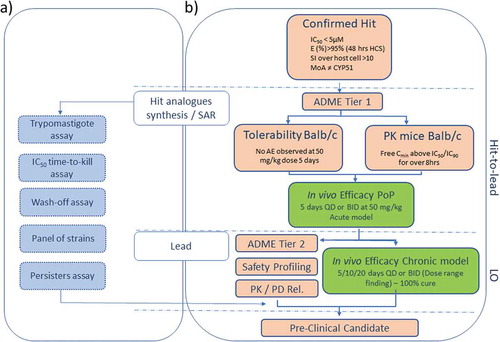
Figure 2. Chagas disease in vivo bioluminescence imaging model procedure
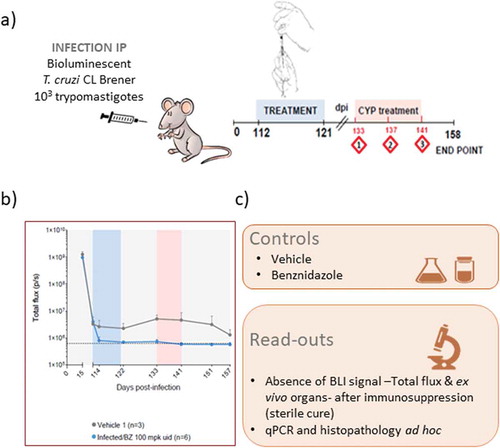
In order to remove any specificity of the results obtained linked to the use of a specific strain of T. cruzi (in this case CL Brener belonging to Tc VI DTU) or a specific mouse background, cardiomyopathy caused by highly divergent parasite strains (TcVI CL Brener and TcI JR) was investigated in BALB/c, C3H/HeN, and C57BL/6 mice. As in preliminary studies with CL Brener strain and BALB/c mice, the GI tract was found to be the primary site of chronic infection in all models. Immunosuppression induced expansion of parasite loads in the gut and was followed by widespread dissemination. These data indicate that immune control of T. cruzi differs between tissues and shows that the large intestine and stomach provide permissive niches for active infection. The end-point frequency of heart-specific infections ranged from 0% in TcVI-CLBR-infected C57BL/6 to 88% in TcI-JR-infected C3H/HeN mice. Nevertheless, infection led to fibrotic cardiac pathology in all models [Citation131]. Another strain, T. cruzi Colombiana expressing nanoluciferase, was used to infect Swiss Webster mice, and bioluminescent monitoring was undertaken for 126 days, a point at which total animal luminescence could still be measured while parasites remained undetectable in blood by microscopy in most animals. Ex vivo imaging of specific tissues and organs dissected postmortem at 126 dpi revealed widespread parasite distribution in the skeletal muscle, heart, GI tract, and mesenteric fat [Citation132].
Overall, these results show that although differences are observed depending on the genetic background of mice and the T. cruzi strain, there are common features such as development of cardiomyopathy and the GI tract as a reservoir.
3.4.3.3. Future applications for further understanding the disease and host /pathogen interactions
Our understanding of parasite biology and disease pathogenesis is still limited, and the complexity of the disease, the long-term nature of the infection, and the fact that parasites are barely detectable during the chronic stage are complicating factors. Further, the limited genetic manipulation technology applicable to the T. cruzi parasite (no RNAi, for example) restricts functional dissection of T. cruzi biology. The advent of new technologies such as gene editing using CRISPR-Cas9 and imaging technologies are opening new avenues never thought possible before.
Two technical innovations that will allow better assessment of the role of the parasite in disease progression were described by Costa et al. [Citation133].
First, a double transgene T. cruzi reporter strain that expresses a fusion protein comprising red-shifted luciferase and green fluorescent protein domains allows both the kinetics of infection within a single animal to be followed using bioluminescence (as described above), and specific foci of infection in excised tissues to be pinpointed; additionally, individual parasites can be visualized using fluorescence in tissue sections, allowing the study of host-parasite interactions at a cellular level. Using this strategy, finding individual parasites within chronically infected murine tissues became feasible.
Second, the incorporation of a streamlined CRISPR/Cas9 functionality into this reporter strain facilitates genome editing using a PCR-based approach that does not require DNA cloning. This system allows the rapid generation of null mutants and fluorescently tagged parasites in a context where the in vivo phenotype can be rapidly assessed. These techniques will no doubt have multiple applications for studying various aspects of T. cruzi biology and CD pathogenesis, previously inaccessible to conventional approaches.
The oral route is increasingly important for transmission of CD and several outbreaks associated with consumption of contaminated food or drink have occurred in Latin America in recent years. Although the associated long-term pathogenesis is not yet completely understood, it leads to severe acute infection. BLI models could be helpful in following infection dynamics post oral contamination. Early studies demonstrated that mice displayed only mild acute symptoms but later developed significantly increased myocardial collagen content, indicative of fibrosis. Similar to i.p. infection, oral infection resulted in gastrointestinal tissues and skin being principal chronic infection reservoirs, and in myocardial fibrosis [Citation134]. In another study, Silva-Dos-Santos et al. studied the presence of T. cruzi Dm28c luciferase (Dm28c-luc) parasites in orally infected mice using bioluminescence and real-time qPCR. During the acute phase, following nasomaxillary invasion, the parasite spread into different tissues such as the mandibular lymph nodes, pituitary gland, heart, liver, small intestine, and spleen at 7 dpi, and further disseminated to other tissues, such as the brain, stomach, esophagus, and large intestine at 21 dpi [Citation135].
3.5. Translational value of animal models in CD: Toward a standardized mouse BLI CD model that translates?
The very often misguided belief that NHPs are the best models and should be used as a final gate point in CD needs to be addressed. So far, there are no validated NHP models for CD. Some exploratory studies were performed with posaconazole and ravuconazole, apparently reproducing the outcome of the clinical trials, but these have not been published. Considering the cost of such models and the fact that they might not represent any advantage over mice models, it seems judicious at that stage to concentrate instead on a simpler, cheaper, quicker, reproducible, and validated model in another species.
Indeed, in-depth analysis of the BLI murine model for CD shows that this model actually complies to a large extent to the often cited characteristics of an ideal animal model of human disease- see , namely: pathogenesis similar to human disease, similar phenotypic and histologic characteristics, similar demonstrated biomarkers of disease, reliable toxicity prediction, and similar response to proven therapies in human [Citation64].
Table 3. Current translational value of the murine BLI model of CD: Characteristics and further improvements
3.5.1. Pathogenesis similar to human disease
A thorough review of the literature on the subject reveals that murine, dog, and NHP models of CD (either experimental or natural infection) can show a pathogenesis of the disease that is similar to that found in human. Indeed, in all three most studied species, cardiomyopathy is observed following long-term infection and ECG abnormalities can be monitored. Mouse models have shown that it is also possible to observe GI complications, such as megacolon, following experimental T. cruzi infection. In a study using bioluminescence looking at the effect of benznidazole on the progression of disease and development of pathology, an assessment of inflammation and fibrosis (markers of cardiac pathology) of the hearts of the mice (5 to 6 months postinfection) showed myocardial collagen content at a group level (i.e. not all the mice developed fibrosis and/or cardiac inflammation), indicative of fibrotic pathology. This showed similarity to human where chagasic heart disease develops in 30–40% of those infected with the protozoan parasite T. cruzi, but which can take decades to become symptomatic [Citation136]. Moreover, the study showed that when mice were treated with benznidazole and cured in the acute stage, the development of pathology was completely blocked. These experiments therefore demonstrate that curative benznidazole treatment early in murine T. cruzi infections can prevent the development of cardiac fibrosis. They also show that treatment during the chronic stage can block the pathology, but the effectiveness varies between infection models. If these findings are extendable to humans, it implies that widespread chemotherapeutic intervention targeted at early-stage infections could play a crucial role in reducing CD morbidity at a population level, as suggested by Pecoul et al. following the BENEFIT trial outcome [Citation137].
3.5.2. Provide similar response to proven therapies in human
Indeed, mouse models of CD disease can predict the outcome of experimental drugs in human. The advent of new technologies such as BLI is adding new parameters that should strengthen our confidence in their translational value. Looking at the back-translation exercise of the clinical trials with azoles and comparison with benznidazole, the BLI model would have predicted the outcome of the clinical trials performed [Citation129]. Some researchers had reservations about these assumptions, questioning the need for parasite elimination in CD, in addition these studies were performed with one specific strain of the parasite, and what is more, a transgenic parasite [Citation138]; however, the bioluminescent CL clone of the parasite was shown to have similar characteristics to the wild type [Citation139]. The results obtained using the BLI technique were confirmed at almost the same time by Khare et al. in another mouse background and using other strains of T. cruzi (C57Bl/6 mice and T. cruzi CL and Y strains) [Citation140]. In their studies, since they did not use BLI, parasitological cure was defined by the absence of parasitemia recrudescence after immunosuppression as assessed by qPCR. Twenty-day therapy with benznidazole (100 mg/kg regimen) resulted in parasitological cure in all treated mice. In contrast, all mice remained infected after a 25-day treatment with posaconazole at all tested doses (10 to 100 mg/kg/day) and further extension of posaconazole therapy to 40 days resulted in only a marginal improvement of treatment outcome.
In a systematic study with the two front-line drugs benznidazole and nifurtimox and using BLI, it was shown that both drugs were efficacious and led to parasitological cure, but that the acute stage of the infection was more difficult to cure than the chronic stage. In general, there is a consensus that the front-line drugs benznidazole and nifurtimox are more effective against the acute stage in both clinical and experimental settings. However, it is very difficult to confirm or demonstrate parasitological cure in human due to the lack of surrogate markers. Seroconversion is the only available marker of parasitological cure: in mouse models, seroreversion of anti-T. cruzi IgG upon antiparasitic treatment is faster than in other models of infection more closely related phylogenetically to man such as dog and NHPs, and is therefore an additional advantage of the murine model [Citation141]. If these findings are translatable to human patients, they will have important implications for treatment strategies. Studies with benznidazole also suggest that dose reduction of the drug is possible and still leads to parasitological cure in the experimental model if one extends the treatment duration [Citation130]. Back-translation of the results of the BENDITA clinical trial in human will be helpful to further assess the translational value of the mouse BLI models [Citation142,Citation143]. In addition, rounds of immunosuppression are very often used in experimental chronic CD models to induce the release of any remaining parasites, or ensuring that a given compound has eradicated all the parasites in the animal. In cases of relapse, i.e. release or detection of parasites as observed by various means (BLI, parasitemia by PCR), the use of immunosuppression confirms the very important role of the strong immune response to control T. cruzi infection, and to a certain extent reproduces what is happening Chagas patients when co-infected with HIV or immunosuppressed during specific treatments.
Given this, why is there still a belief that animal models of CD might not very useful and that the results of these models do not translate?
One of the reasons is the lack of standardization of the models used in the Chagas research community. There were attempts to standardize the testing of compounds in in vivo animal models ten years ago [Citation84], since then new technologies and the knowledge acquired should allow for an updated, harmonized standard protocol for testing, and a consensus to use BLI as a standard. The review of all benznidazole data obtained in animal models of CD is compelling and showed that it is difficult to compare data and make a clear statement of the efficacy of this compound, contrary to the authors’ statement [Citation86]. One cannot compare data from acute stage with those generated in the chronic stage of infectious, using different endpoints with different design, and so on.
Issues with study design and endpoints are other important reasons for conflicting results or interpretations. These are critical to the usefulness of an animal model; bad design and endpoint choices can result in flawed results from an otherwise good animal model. One of the main issues is the definition of the question a researcher wants to answer with the model. Because of the complexity of CD and the different theories and dogmas that have surrounded the Chagas community over the years, there has always been and possibly still are questions around what a clinician wants to achieve, e.g. reduction in parasitemia versus parasitological cure. There is general agreement that parasite persistence is one of the drivers of the development of Chagas pathology and, therefore, until proven contrary, the consensus should be that drugs are needed that eradicate the parasite from the human body. In this case, measurement of parasitemia is possibly interesting but not a relevant endpoint in an animal model. This is significant since careful analysis of the data related to posaconazole or ravuconazole in the dog model, for example, would have already predicted the outcome of the PoC clinical trials, since only suppressive activity was observed and not parasitological cure as defined by the current standards. Until a few years ago it was difficult to assess parasitism in tissues (both in the clinic and in animal models) and thus parasitemia was a standard endpoint. The imaging system brought important information related to the dynamics of infection with T. cruzi. Before BLI, we had no idea of the extent of the dynamics of the parasite over time; this might explain the variability of results when assessing tissue parasitism; indeed, even negative tissue PCR could misclassify treatment failures as cures because T. cruzi moves around and is not in a static state in a tissue. On the contrary, a PCR positive result, as is the case when used in clinical PoC trials, would be a clear indication of failure.
Another endpoint used in the past – in principle no longer adequate, considering ethical guidelines and the 3Rs concept – was linked to survival. A lot of data generated with CD animal models used death/mortality as an endpoint in the acute stage, although not a direct endpoint in human (even if there is an approximate death rate of 5% in the acute stage in infected individuals). Most of the studies in animal models were performed in the acute stage of the disease for the sake of cost and experimental ease.
One remaining issue with current animal models for CD is the lack of biomarkers of cure that could be used in human. Indeed, assessment of parasitological cure in human is still a major challenge. This has possibly led to the belief, and often repeated statement, that the current drugs benznidazole and nifurtimox are more efficacious in acutely infected patients than in the chronic indeterminate stage, although there is no way to assess treatment efficacy and/or make these assumptions in the latter category, and the last PoC clinical trials showed that that for 80% or more of patients treated, no relapse of parasitemia could be observed after one year of follow-up (although obviously this is not a proof of efficacy, but it is at least a lack of failure, which is a caveat of the endpoint used).
4. Conclusions
Experimental animal models have played an important role in the Chagas research community. This is illustrated by the impressive number of models using various species described in the literature as well as the number of studies published dealing with the testing of compounds [Citation49,Citation83]. Challenges and issues associated with disease animal models common to any disease have been identified. One of the challenges relates to the complexity of the disease, the need to understand host/pathogen interactions, and the evolution over recent decades of the various theories to explain the pathology and progression of the disease. Another issue is linked to the number of models used that have produced a huge variability of results when testing the same compound. This highlights a clear lack of harmonization even if there have been efforts toward this. Thirdly, the very small number of compounds that have gone into development makes it hard to determine the real translational value of these models in the R&D process. On the other hand, it is interesting to notice that a selection of models available do indeed reproduce the human disease and pathologies fairly well, including cardiomyopathy and pathologies associated with the GI tract.
The development of new technologies, both genetic tools allowing the production of specific transgenic parasites and new imaging systems, has led to both a new understanding of T. cruzi infection dynamics during the chronic stage of the disease and has introduced new endpoints (e.g. bioluminescence flux, images in vivo and ex vivo). These developments brought together a new experimental mouse model – the so-called Chagas mouse BioLuminescence Imaging (BLI) model – that has showed real promise so far. Not only does it contribute significantly to the 3Rs concept since it allows the same mouse to be followed in real-time for a long period (thereby reducing the number of animals to be used), but it has also been shown to be very robust and allows a very reproducible pattern of T. cruzi infection in the animal. Most importantly, the first datasets are alluding to the potentially very good translational value of this model for PoC phase 2 in adults and efficacy assessment in children, as back-translation of the few clinical trial data in the field have indicated that the outcome could be predicted using this animal model. Overall, the improved predictability of the currently available murine BLI model has increased confidence about moving compounds forward into the next stage of development.
Notwithstanding these improvements and the involvement of various institutions from both industry and academia, further developments and assessments are still needed to give a definitive answer and some of the requirements and approaches to future improvements will be discussed in the next section.
5. Expert opinion
Major progress has been made during the last decade in the field of experimental models for CD. However, there are opposing views about their translational value to drug discovery and how important or essential they are as a gate keeper in the R&D process for moving compounds forward into further development. Following careful review of the literature and considering the general attributes of an optimal disease model, we believe, possibly in contrast to the general consensus, that the major challenge for CD animal models may not be their limited reproduction of the human pathology. Indeed, it appears that some experimental animal models in different species can reproduce the pattern of infection and pathologies associated with CD such as cardiomyopathies and complication of the GI tract, such as megacolon, fairly well. Rather, we believe that the major issue is linked with assay design and the use of inadequate endpoints to answer specific questions.
The complexity of CD, our incomplete understanding of the mechanism of disease progression and host/pathogen interactions, and the very small number of drugs that have been tested in clinical trials in human are other major hurdles for the proper assessment of the translational value of CD animal models. Further, the lack of appropriate markers of parasitological cure in infected adult individuals (seroreversion, that can take decades to occur in adults, is currently the only marker of cure) makes it very difficult, if not impossible, to assess efficacy of a given treatment in adults without decades of follow-up, thereby further complicating the assessment of the translational value of preclinical animal models.
With current knowledge of the parasite biology and the technologies available today, we believe that the BioLuminescent Imaging mouse model should be the model of reference to be used in R&D. There are, in our opinion, no reasons to favor a potential NHP model and, as described earlier, the BLI model is bringing a new perspective and the endpoints are outperforming the former classical murine model. Although NHP are, for various reasons, often considered to be the most adequate models most closely reproducing human disease, there are no validated models in CD so far and the associated costs are not attractive. Taking into consideration the current standards (the 3Rs concept, ethical considerations) and since there does not seem to be any advantage to focusing on a NHP model compared to other species for the assessment of NCEs, the use of lower species with similar or better translational value should be preferred. NHPs could, however, still have a place in the development of vaccine. While larger animals have been employed, mice remain the most favored animal model, as they recapitulate many aspects of the human disease, are easy to manipulate genetically, and are amenable to study by small animal imaging technologies.
Indeed, the BLI mouse model for CD is a very valuable new tool that allows the T. cruzi infection dynamic to be followed and parasite loads in specific organs to be quantified in the same animal during a complete study. Using this new imaging technique, it has been possible to gain a better understanding and interpretation of previously generated results; in particular, it has shown the pantropic nature of acute stage infections, and the restriction of parasites to specific and small focal sites, predominantly the GI tract during the chronic stage. Other tissues and organs, including the heart and skeletal muscle, are infected sporadically. The survival of the small parasite foci within apparently tolerant sites or reservoirs is crucial for long-term infection. Another contributor to the long-term nature of T. cruzi infections could be the recently postulated phenomenon of parasite dormancy; individual intracellular amastigotes can enter an apparently quiescent state in which they cease to replicate and exhibit reduced drug sensitivity [Citation144]. A better understanding of host/parasite interactions and the disease itself through the use of these new technologies will enable the design of better in vivo models. As such, the BLI mouse model has real value for assessing the efficacy of compounds and determining their potential to meet the TPP, and for taking these new insights into infection/parasite dynamics, in particular the role of reservoirs, through the imaging ability and associated endpoints. These new developments also help to explain the variability observed in earlier models, when parasites in tissues were assessed by qPCR; the choice of organs was random and the dynamics of the infection were not taken into consideration. This model also has the potential to answer two different questions, depending on the endpoints selected: efficacy of compounds for parasitological cure and efficacy of compounds to stop or avoid progression of the disease, as described by Francisco et al. [Citation136]. Similar to the outcome in human, not all mice develop heart fibrosis and inflammation.
All these points make the BioLuminescent Imaging mouse model an ideal system to be the model of reference to be used in R&D for CD. Its translational value has been preliminarily assessed using benznidazole and posaconazole clinical data; it has been shown that this model would have predicted the outcome of these trials in human and the failure of azoles in PoC Phase 2. To further strengthen its translational value, data from current completed clinical trials (BENDITA and fexinidazole) should be translated back into this model. There is also a need for NCEs to move into clinical trials to further validate – or otherwise – the value of this predictive mouse model, further understand its predictability and bring more confidence into the R&D decision-making process.
Standardization of experimental animal models in the CD R&D community has been identified as a major drawback [Citation78]. There has been a continuous debate within the Chagas drug discovery community about whether the experimental conditions of assay protocols should be standardized. Our current opinion is that, in addition to complying with the best practices of assay validation and reporting, validation with known drugs, alignment of the protocol and endpoints used to assess new compounds, and for the sake of harmonization and comparison of data, the mouse BLI model should be the model of reference to be used by the research community in the future. Even if progress is being made and more laboratories are now using the BLI model, there should be a broader consensus and acceptance by the Chagas community for this new state of the art in vivo model and a protocol for testing efficacy of compounds (see Supplementary Information for a proposed standardized BLI model draft protocol for compound efficacy assessment in chronically infected mice). The science has evolved since the first harmonization proposal in 2010 [Citation79], thus there is a need for adaptation /adjustment of new protocols taking into account new data; this is the basis and philosophy of scientific progress. The sharing of biological material such as parasite strains, reagents, and cell lines should be fairly easy, as these reagents have been generated as a community resource and are freely available on request; this should facilitate the path toward harmonization.
Obviously, research is a not a static activity. There is still room for improvement and adjustment of the new model. There is no doubt that in the near future new reagents will become available through the use of the CRISPR-cas9 gene editing system that could allow the development of assays using T. cruzi parasites deficient in specific genes, for example [Citation145]. The role of the immune system/markers could be further assessed or integrated into the current models. There is no doubt that image informatics and other technological developments will allow the development of even more sophisticated and varied assays that might allow, for example, the further lowering the limit of detection of parasites, currently set at around 100 T. cruzi parasites in vivo. Indeed, and this is still a potential limitation of the imaging power, the ‘purists’ might state that in the model we should talk about ‘lack of parasite relapse following immunosuppression’ rather than parasitological cure. One way to deal with this issue would be to use PCR in all organs to increase sensitivity.
Finally, a clear gap in all current experimental models of CD is the absence of biomarkers at that stage that could be used and would translate to the clinic. There is therefore an urgent need to integrate at the discovery stage the identification of biomarkers of parasitological cure that are useful in the animal models and would translate to human. Indeed, until markers or surrogates for treatment efficacy -either parasitological cure or clinical benefit, i.e. halt progression of the disease to the symptomatic stage- in adults patients are identified, validated and accepted by the regulatory authorities, it will remain difficult to fully prove or disprove the translational value of CD animal models.
In conclusion, to improve their current translational value, we believe that animal models of efficacy should not be considered in isolation and only look at PD endpoints. On the contrary, data generated from several complementary assessments or endpoints should be considered, thereby ensuring better predictability and more confidence when moving forward. We should move to a more integrated and multiparametric platform, using multimodality imaging approaches (integrated technology with BLI and PET, serial imaging, imaging of the heart and GI), combining, for example, monitoring dilatation of the right ventricular chamber in mice using MRI [Citation146] and BLI, and, when possible, associated blood sampling during the in life-experiment to allow direct PK/PD analysis in infected animals (as opposed to assessing drug exposure in satellite non-infected groups).
This should lead to the design of a fit-for purpose animal model for CD with the potential to answer specific questions that will certainly improve the translation and predictability of in vitro results into in vivo conditions and further downstream development. This should also lead to the design of a more efficient screening cascade for CD R&D and give researchers more confidence to move compounds forward in development.
At the end of the day, a model is, by definition, a simplified representation of a phenomenon, and the very process of simplification means that some of the complex reality is lost [Citation147]. As the statistician George Box once stated: ‘essentially all models are wrong, but some are useful’ [Citation148]. However, they can help explain and interpret data and possibly predict efficacy of compounds in humans or build drug developers’ confidence in moving compounds forward into clinical trials. In the long run, only human clinical trial data will validate a standardized animal model.
The chances of success will obviously depend not only on the approaches taken and developed to identify and move drug candidates and biomarkers forward but also, and more critically, on the funding available for these efforts and on a collaborative spirit within the Chagas research community.
Article highlights
CD is a global public health problem; there is a need for new, better, and safer drugs and markers of treatment efficacy but also a better understanding of the disease
Animal models are still essential for both exploratory and applied research to increase our knowledge of the disease and host /T. cruzi interaction but also bridge the translational gap in Chagas R&D
Harmonization of models used in the Chagas scientific community is still needed (in particular, alignment of the protocol and endpoints used to assess new compounds)
Animal models using BLI and associated endpoints should become the new standard as they represent a major improvement from both an ethical and a scientific point of view; whenever possible, associated blood sampling during in life-experiment should be considered to allow direct PK/PD analysis in infected animals, and research on biomarkers of cure should be integrated earlier in the discovery process for potential translation to human and use in the clinic
The role of the immune system should be further assessed in current models -integration of immune markers, or through the development of specific models
Further increased confidence in CD in vivo animal models and their translational value will be achieved through back-translation of clinical data for new compounds that will move into proof of concept phase 2 clinical trials and beyond (pending feasibility of treatment efficacy assessment)
This box summarizes key points contained in the article.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Chronic_Chagas_protocol_review_Suppl_Info_Revised.docx
Download MS Word (34.2 KB)Acknowledgments
The authors wish to thank Dr Louise Burrows for the editing of this manuscript and Fanny Escudié for critical reading and help with the figures and supplementary data. DNDi is grateful to its donors, public and private, who have provided funding to DNDi since its inception in 2003. A full list of DNDi’s donors can be found at http://www.dndi.org/donors/donors/
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Chagas C. Nova tripanozomiase humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem Inst Oswaldo Cruz. 1909;1:159–218.
- Aufderheide AC, Salo W, Madden M, et al. A 9,000-year record of Chagas’ disease. Proc Natl Acad Sci U S A. 2004 Feb 17;101(7):2034–2039.
- Sartor P, Colaianni I, Cardinal MV, et al. Improving access to Chagas disease diagnosis and etiologic treatment in remote rural communities of the Argentine Chaco through strengthened primary health care and broad social participation. PLoS Negl Trop Dis. 2017;11(2):e0005336.
- Pinazo MJ, Pinto J, Ortiz L, et al. A strategy for scaling up access to comprehensive care in adults with Chagas disease in endemic countries: the Bolivian Chagas Platform. PLoS Negl Trop Dis. 2017;11(8):e0005770.
- Schmunis GA, Yadon ZE. Chagas disease: a Latin American health problem becoming a world health problem. PLoS Negl Trop Dis. 2015;9(2):e0003540.
- Meymandi SK, Hernandez S, Forsyth CJ. A community-based screening program for Chagas disease in the USA. Trends Parasitol. 2017;S1471–4922(17):30166.
- Requena-Méndez A, Aldasoro E, de Lazzari E, et al. Prevalence of Chagas disease in Latin-American migrants living in Europe: a systematic review and meta-analysis. PLoS Negl Trop Dis. 2015;9(2):e0003540.
- Jackson Y, Pinto A, Pett S. Chagas disease in Australia and New Zealand: risks and needs for public health interventions. Trop Med Int Health. 2014;19:212–218.
- WHO Fact Sheet No 340. [ cited 2020 Feb 29]. Available from: http://www.who.int/mediacentre/factsheets/fs340/en/
- Cucunubá ZM, Okuwoga O, Basáñez MG, et al. Increased mortality attributed to Chagas disease: a systematic review and meta-analysis. Parasit Vectors. 2016;9:42.
- Martins-Melo FR, Ramos AN Jr, Alencar CH, et al. Mortality due to Chagas disease in Brazil from 1979 to 2009: trends and regional differences. J Infect Dev Ctries. 2012;6:817–824.
- Moolani Y, Bukhman G, Hotez PJ. Neglected tropical diseases as hidden causes of cardiovascular disease. PLoS Negl Trop Dis. 2012;6(6):e1499.
- Coura JR. The main sceneries of Chagas disease transmission. The vectors, blood and oral transmissions–a comprehensive review. Mem Inst Oswaldo Cruz. 2015;110:277–282.
- Filigheddu MT, Górgolas M, Ramos JM. Orally-transmitted Chagas disease. Med Clin (Barc). 2017 Feb 9;148(3):125–131.
- Araujo PF, Almeida AB, Pimentel CF, et al. Sexual transmission of American trypanosomiasis in humans: a new potential pandemic route for Chagas parasites. Mem Inst Oswaldo Cruz. 2017 June;112(6):437–446.
- Pérez-Molina JA, Molina I. Chagas disease. Lancet. 2018Jan6;391(10115):82–94.
- Lidani KCF, Bavia L, Ambrosio AR, et al. The complement system: a prey of Trypanosoma cruzi. Front Microbiol. 2017 Apr 20;8:607.
- Ayub-Ferreira SM, Mangini S, Issa VS, et al. Mode of death on Chagas heart disease: comparison with other etiologies. a subanalysis of the REMADHE prospective trial. PLoS Negl Trop Dis. 2013;7:e2176.
- Tarleton RL. Parasite persistence in the aetiology of Chagas disease. Int J Parasitol. 2001May1;31(5–6):550–554.
- Gironès N, Fresno M. Etiology of Chagas disease myocarditis: autoimmunity, parasite persistence, or both? Trends Parasitol. 2003 Jan;19(1):19–22.
- Teixeira AR, Hecht MM, Guimaro MC, et al. Pathogenesis of chagas’ disease: parasite persistence and autoimmunity. Clin Microbiol Rev. 2011 July;24(3):592–630.
- Guedes PM, Silva GK, Gutierrez FR, et al. Current status of Chagas disease. Expert Rev Anti Infect Ther. 2011;9:609–620.
- Brum-Soares L, Cubides JC, Burgos I, et al. High seroconversion rates in Trypanosoma cruzi chronic infection treated with benznidazole in people under 16 years in Guatemala. Rev Soc Bras Med Trop. 2016;49(6):721–727.
- Crespillo-Andújar C, Venanzi-Rullo E, López-Vélez R, et al. Safety profile of benznidazole in the treatment of Chronic chagas disease: experience of a referral centre and systematic literature review with meta-analysis. Drug Saf. 2018 Nov;41(11):1035–1048.
- Crespillo-Andújar C, Chamorro-Tojeiro S, Norman F, et al. Toxicity of nifurtimox as second-line treatment after benznidazole intolerance in patients with chronic Chagas disease: when available options fail. Clin Microbiol Infect. 2018 Dec;24(12):1344.e1–1344.e4.
- Olivera MJ, Cucunubá ZM, Álvarez CA, et al. Safety profile of nifurtimox and treatment interruption for chronic Chagas disease in colombian adults. Am J Trop Med Hyg. 2015;93:1224–1230.
- de Lana M, Martins-Filho OA. Revisiting the posttherapeutic cure criterion in chagas disease: time for new methods, more questions, doubts, and polemics or time to change old concepts? Biomed Res Int. 2015;2015:65298516–65298520.
- Morillo CA, Marin-Neto JA, Avezum A, et al. Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. N Engl J Med. 2015;373(14):1295–1306.
- Rassi A Jr, Neto MJA, Rassi A. Chronic Chagas cardiomyopathy: a review of the main pathogenic mechanisms and the efficacy of aetiological treatment following the BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) trial. Mem Inst Oswaldo Cruz. 2017;112(3):224–235.
- Gutierrez FR, Guedes PM, Gazzinelli RT, et al. The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol. 2009 Nov;31(11):673–685.
- Chatelain E. Chagas disease research and development: is there light at the end of the tunnel? Comput Struct Biotechnol J. 2016;15:98–103.
- Morillo CA, Waskin H, Sosa-Estani S, et al. Benznidazole and posaconazole in eliminating parasites in asymptomatic T. Cruzi Carriers: the STOP-CHAGAS Trial. J Am Coll Cardiol. 2017;69(8):939–947.
- Molina I, Gómez I Prat J, Salvador F, et al. Randomized trial of posaconazole and benznidazole for Chronic chagas’ disease. N Engl J Med. 2014;370:1899–1908.
- Torrico F, Gascon J, Ortiz L, et al. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: a proof-of-concept, randomised, placebo-controlled trial. Lancet Infect Dis. 2018 Apr;18(4):419–430.
- Urbina JA. Recent clinical trials for the etiological treatment of chronic Chagas disease: advances, challenges and perspectives. J Eukaryot Microbiol. 2015;62(1):149–156.
- Urbina JA. The long road towards a safe and effective treatment of chronic Chagas disease. Lancet Infect Dis. 2018;18(4):363–365.
- Pinazo MJ, Espinosa G, Gállego M, et al. Successful treatment with Posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am J Trop Med Hyg. 2010;82(4):583–587.
- Chatelain E. Chagas disease drug discovery: toward a new era. J Biomol Screen. 2015 Jan;20(1):22–35.
- Chatelain E, Ioset JR. Phenotypic screening approaches for Chagas disease drug discovery. Expert Opin Drug Discov. 2018 Feb;13(2):141–153.
- Villalta F, Rachakonda G. Advances in preclinical approaches to Chagas disease drug discovery. Expert Opin Drug Discov. 2019 Nov;14(11):1161–1174.
- MacLean LM, Thomas J, Lewis MD, et al. Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl Trop Dis. 2018 July 12;12(7):e0006612.
- Moraes CB, Giardini MA, Kim H, et al. Nitroheterocyclic compounds are more efficacious than CYP51 inhibitors against Trypanosoma cruzi: implications for Chagas disease drug discovery and development. Sci Rep. 2014 Apr 16;4:4703.
- Riley J, Brand S, Voice M, et al. Development of a fluorescence-based Trypanosoma cruzi CYP51 inhibition assay for effective compound triaging in drug discovery programmes for Chagas disease. PLoS Negl Trop Dis. 2015 Sept 22;9(9):e0004014.
- Urbina JA, Payares G, Sanoja C, et al. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int J Antimicrob Agents. 2003 Jan;21(1):27–38.
- Diniz Lde F, Caldas IS, Guedes PM, et al. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob Agents Chemother. 2010 July;54(7):2979–2986.
- Urbina JA, Payares G, Contreras LM, et al. Antiproliferative effects and mechanism of action of SCH 56592 against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies. Antimicrob Agents Chemother. 1998 July;42(7):1771–1777.
- Pereira RM, Greco GMZ, Moreira AM, et al. Applicability of plant-based products in the treatment of Trypanosoma cruzi and Trypanosoma brucei infections: a Systematic review of preclinical in vivo evidence. Parasitology. 2017;144(10):1275–1287.
- Arrua EC, Seremeta KP, Bedogni GR, et al. Nanocarriers for effective delivery of benznidazole and nifurtimox in the treatment of Chagas disease: a review. Acta Trop. 2019;198:105080.
- Chatelain E, Konar N. Translational challenges of animal models in Chagas disease drug development: a review. Drug Des Devel Ther. 2015 Aug 19;9:4807–4823.
- van Meer PJ, Graham ML, Schuurman HJ. The safety, efficacy and regulatory triangle in drug development: impact for animal models and the use of animals. Eur J Pharmacol. 2015 July;15(759):3–13.
- McGonigle P, Ruggeri B. Animal models of human disease: challenges in enabling translation. Biochem Pharmacol. 2014 Jan 1;87(1):162–171.
- Bueters T, Ploeger BA, Visser SA. The virtue of translational PKPD modeling in drug discovery: selecting the right clinical candidate while sparing animal lives. Drug Discov Today. 2013 Sept;18(17–18):853–862.
- Pound P, Ritskes-Hoitinga M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J Transl Med. 2018 Nov 7;16(1):304.
- Martić-Kehl MI, Schibli R, Schubiger PA. Can animal data predict human outcome? Problems and pitfalls of translational animal research. Eur J Nucl Med Mol Imaging. 2012 Sept;39(9):1492–1496.
- Smith MM, Clarke EC, Little CB. Considerations for the design and execution of protocols for animal research and treatment to improve reproducibility and standardization: “DEPART well-prepared and ARRIVE safely”. Osteoarthritis Cartilage. 2017 Mar;25(3):354–363.
- Denayer T, Stöhr T, Van Roy M. Animal models in translational medicine: validation and prediction. New Horiz Transl Med. 2014;2:5–11.
- Singh J, Elbarbry F, Lan K, et al. Animal pharmacokinetic/pharmacodynamic studies (APPS) reporting guidelines. Eur J Drug Metab Pharmacokinet. 2018 Oct;43(5):483–494.
- ‘T Hart BA. Reverse translation of failed treatments can help improving the validity of preclinical animal models. Eur J Pharmacol. 2015 July 15;759:14–18.
- Schneider B, Balbas-Martinez V, Jergens AE, et al. Model-based reverse translation between veterinary and human medicine: the one health initiative. CPT Pharmacometrics Syst Pharmacol. 2018 Feb;7(2):65–68.
- Wendler A, Wehling M. The translatability of animal models for clinical development: biomarkers and disease models. Curr Opin Pharmacol. 2010 Oct;10(5):601–606.
- [ cited 2020 Feb 20]. Available from: https://www.nc3rs.org.uk/
- Taylor K. Reporting the implementation of the Three Rs in European primate and mouse research papers: are we making progress? Altern Lab Anim. 2010 Dec;38(6):495–517.
- Prabhakar S. Translational research challenges: finding the right animal models. J Investig Med. 2012 Dec;60(8):1141–1146.
- Estes JD, Wong SW, Brenchley JM. Nonhuman primate models of human viral infections. Nat Rev Immunol. 2018 June;18(6):390–404.
- Harding JD. Nonhuman primates and translational research: progress, opportunities, and challenges. Ilar J. 2017 Dec 1;58(2):141–150.
- Bouteille B, Millet P, Enanga B, et al. Human African trypanosomiasis, contributions of experimental models. Bull Soc Pathol Exot. 1998;91(2):127–132.
- Ouwe-Missi-Oukem-Boyer O, Mezui-Me-Ndong J, Boda C, et al. The vervet monkey (Chlorocebus aethiops) as an experimental model for Trypanosoma brucei gambiense human African trypanosomiasis: a clinical, biological and pathological study. Trans R Soc Trop Med Hyg. 2006 May;100(5):427–436.
- Thuita JK, Wang MZ, Kagira JM, et al. Pharmacology of DB844, an orally active aza analogue of pafuramidine, in a monkey model of second stage human African trypanosomiasis. PLoS Negl Trop Dis. 2012;6(7):e1734.
- Thuita JK, Wolf KK, Murilla GA, et al. Chemotherapy of second stage human African trypanosomiasis: comparison between the parenteral diamidine DB829 and its oral prodrug DB868 in vervet monkeys. PLoS Negl Trop Dis. 2015 Feb 5;9(2):e0003409.
- Kaiser M, Bray MA, Cal M, et al. Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob Agents Chemother. 2011 Dec;55(12):5602–5608.
- Torreele E, Bourdin Trunz B, Tweats D, et al. Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis. 2010 Dec 21;4(12):e923.
- Mesu VKBK, Kalonji WM, Bardonneau C, et al. Oral fexinidazole for late-stage African Trypanosoma brucei gambiense trypanosomiasis: a pivotal multicentre, randomised, non-inferiority trial. Lancet. 2018 Jan 13;391(10116):144–154.
- Pelfrene E, Harvey Allchurch M, Ntamabyaliro N, et al. The European Medicines Agency’s scientific opinion on oral fexinidazole for human African trypanosomiasis. PLoS Negl Trop Dis. 2019 June 27;13(6):e0007381.
- Deeks ED. Fexinidazole: first Global Approval. Drugs. 2019 Feb;79(2):215–220.
- Neau P, Hänel H, Lameyre V, et al. Innovative partnerships for the elimination of human African trypanosomiasis and the development of fexinidazole. Trop Med Infect Dis. 2020 Jan 27;5(1):pii: E17.
- Nare B, Wring S, Bacchi C, et al. Discovery of novel orally bioavailable oxaborole 6-carboxamides that demonstrate cure in a murine model of late-stage central nervous system african trypanosomiasis. Antimicrob Agents Chemother. 2010 Oct;54(10):4379–4388.
- Jacobs RT, Nare B, Wring SA, et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl Trop Dis. 2011 June;5(6):e1151.
- Dickie EA, Giordani F, Gould MK, et al. New drugs for human African trypanosomiasis: a twenty first century success story. Trop Med Infect Dis. 2020 Feb 19;5(1):pii: E29.
- Walsh NC, Kenney LL, Jangalwe S, et al. Humanized mouse models of clinical disease. Annu Rev Pathol. 2017 Jan 24;12:187–215.
- Pearson T, Greiner DL, Shultz LD. Humanized SCID mouse models for biomedical research. Curr Top Microbiol Immunol. 2008;324:25–51.
- Siu E, Ploss A. Modeling malaria in humanized mice: opportunities and challenges. Ann N Y Acad Sci. 2015 Apr;1342:29–36.
- Minkah NK, Schafer C, Kappe SHI. Humanized mouse models for the study of human malaria parasite biology, pathogenesis, and immunity. Front Immunol. 2018 Apr 19;9:807.
- Gulin JEN. In vivo drug testing for experimental trypanosoma cruzi infection. In: Altcheh J, Freilij H, editors. Chagas disease. Cham: Springer; 2019. p. 313–321.
- Romanha AJ, Castro SL, Soeiro MN, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz. 2010 Mar;105(2):233–238.
- Gulin JE, Rocco DM, García-Bournissen F. Quality of reporting and adherence to ARRIVE guidelines in animal studies for chagas disease preclinical drug research: a systematic review. PLoS Negl Trop Dis. 2015Nov20;9(11):e0004194.
- Molina I, Perin L, Aviles AS, et al. The effect of benznidazole dose among the efficacy outcome in the murine animal model. A quantitative integration of the literature. Acta Trop. 2020 Jan;201:105218.
- Akle V, Agudelo-Dueñas N, Molina-Rodriguez MA, et al. Establishment of larval zebrafish as an animal model to investigate trypanosoma cruzi motility in vivo. J Vis Exp. 2017 Sept;30(127):e56238.
- Guedes PM, Veloso VM, Tafuri WL, et al. The dog as model for chemotherapy of the Chagas’ disease. Acta Trop. 2002 Oct;84(1):9–17.
- Guedes PM, Veloso VM, Afonso LC, et al. Development of chronic cardiomyopathy in canine Chagas disease correlates with high IFN-gamma, TNF-alpha, and low IL-10 production during the acute infection phase. Vet Immunol Immunopathol. 2009 July 15;130(1–2):43–52.
- Carvalho EB, Ramos IPR, Nascimento AFS, et al. Echocardiographic measurements in a preclinical model of chronic chagasic cardiomyopathy in dogs: validation and reproducibility. Front Cell Infect Microbiol. 2019 Sept;24(9):332.
- Caldas IS, Menezes APJ, Diniz LF, et al. Parasitaemia and parasitic load are limited targets of the aetiological treatment to control the progression of cardiac fibrosis and chronic cardiomyopathy in Trypanosoma cruzi-infected dogs. Acta Trop. 2019 Jan;189:30–38.
- Fonseca-Berzal C, Arán VJ, Escario JA, et al. Experimental models in Chagas disease: a review of the methodologies applied for screening compounds against Trypanosoma cruzi. Parasitol Res. 2018 Nov;117(11):3367–3380.
- Seah SK, Marsden PD, Voller A, et al. Experimental Trypanosoma cruzi infection in rhesus monkeys–the acute phase. Trans R Soc Trop Med Hyg. 1974;68(1):63–69.
- Carvalho CM, Andrade MC, Xavier SS, et al. Chronic Chagas’ disease in rhesus monkeys (Macaca mulatta): evaluation of parasitemia, serology, electrocardiography, echocardiography, and radiology. Am J Trop Med Hyg. 2003 June;68(6):683–691.
- de Almeida EA, Navarro MR, Guariento ME, et al. [The experimental infection of Cebus apella sp. monkeys with Trypanosoma cruzi. Its clinical, electrocardiographic and anatomicopathological assessment]. [Article in Portuguese]. Rev Soc Bras Med Trop. 1992 Jan–Mar;25(1):7–12.
- Cox LA, Olivier M, Spradling-Reeves K, et al. Nonhuman primates and translational research-Cardiovascular disease. Ilar J. 2017 Dec 1;58(2):235–250.
- Molina J, Martins-Filho O, Brener Z, et al. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob Agents Chemother. 2000 Jan;44(1):150–155.
- Ribeiro M, Nitz N, Santana C, et al. Sexual transmission of Trypanosoma cruzi in murine model. Exp Parasitol. 2016 Mar;162:1–6.
- Rios A, Ribeiro M, Sousa A, et al. Can sexual transmission support the enzootic cycle of Trypanosoma cruzi? Mem Inst Oswaldo Cruz. 2018 Jan;113(1):3–8.
- Almeida AB, Araújo PF, Bernal FM, et al. Sexual transmission of American trypanosomes from males and females to naive mates. J Vis Exp. 2019 Jan 27;(143). DOI:10.3791/57985
- Matsuda NM, Miller SM, Evora PR. The chronic gastrointestinal manifestations of Chagas disease. Clinics (Sao Paulo). 2009;64(12):1219–1224.
- Coura JR, Dias JC. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem Inst Oswaldo Cruz. 2009 July;104(Suppl 1):31–40.
- Jelicks LA, Tanowitz HB. Advances in imaging of animal models of Chagas disease. Adv Parasitol. 2011;75:193–208.
- Boczko J, Tar M, Melman A, et al. Trypanosoma cruzi infection induced changes in the innervation, structure and function of the murine bladder. J Urol. 2005 May;173(5):1784–1788.
- Guillén-Pernía B, Lugo-Yarbuh A, Moreno E. Digestive tract dilation in mice infected with Trypanosoma cruzi. [Article in Spanish]. Invest Clin. 2001 Sept;42(3):195–209.
- Ny L, Li H, Mukherjee S, et al. A magnetic resonance imaging study of intestinal dilation in Trypanosoma cruzi-infected mice deficient in nitric oxide synthase. Am J Trop Med Hyg. 2008 Nov;79(5):760–767.
- Campos CF, Cangussú SD, Duz ALC, et al. Enteric neuronal damage, intramuscular denervation and smooth muscle phenotype changes as mechanisms of chagasic megacolon: evidence from a long-term murine model of Trypanosoma cruzi infection. PLoS ONE. 2016;11(4):e0153038.
- Fontes CER, Abreu AP, Gasparim AZ. Radiological study of megacolon in Trypanosoma cruzi infected rats. Arq Bras Cir Dig. 2018 Mar 1;31(1):e1341.
- Barry MA, Versteeg L, Wang Q, et al. A therapeutic vaccine prototype induces protective immunity and reduces cardiac fibrosis in a mouse model of chronic Trypanosoma cruzi infection. PLoS Negl Trop Dis. 2019 May 30;13(5):e0007413.
- Mateus J, Guerrero P, Lasso P, et al. An animal model of acute and chronic chagas disease with the reticulotropic Y strain of Trypanosoma cruzi that depicts the multifunctionality and dysfunctionality of T cells. Front Immunol. 2019 Apr;26(10):918.
- Kelkar M, De A. Bioluminescence based in vivo screening technologies. Curr Opin Pharmacol. 2012 Oct;12(5):592–600.
- Suff N, Waddington SN. The power of bioluminescence imaging in understanding host-pathogen interactions. Methods. 2017 Aug 15;127:69–78.
- Avci P, Karimi M, Sadasivam M, et al. In-vivo monitoring of infectious diseases in living animals using bioluminescence imaging. Virulence. 2018 Jan 1;9(1):28–63.
- Myburgh E, Coles JA, Ritchie R, et al. In vivo imaging of trypanosome-brain interactions and development of a rapid screening test for drugs against CNS stage trypanosomiasis. PLoS Negl Trop Dis. 2013 Aug 22;7(8):e2384.
- McLatchie AP, Burrell-Saward H, Myburgh E, et al. Highly sensitive in vivo imaging of Trypanosoma brucei expressing “red-shifted” luciferase. PLoS Negl Trop Dis. 2013 Nov 21;7(11):e2571.
- Burrell-Saward H, Rodgers J, Bradley B, et al. A sensitive and reproducible in vivo imaging mouse model for evaluation of drugs against late-stage human African trypanosomiasis. J Antimicrob Chemother. 2015 Feb;70(2):510–517.
- Burrell-Saward H, Harris AJ, de LaFlor R, et al. Dose-dependent effect and pharmacokinetics of fexinidazole and its metabolites in a mouse model of human African trypanosomiasis. Int J Antimicrob Agents. 2017 Aug;50(2):203–209.
- Melo GD, Goyard S, Lecoeur H, et al. New insights into experimental visceral leishmaniasis: real-time in vivo imaging of Leishmania donovani virulence. PLoS Negl Trop Dis. 2017 Sept 25;11(9):e0005924.
- Mendes Costa D, Cecílio P, Santarém N, et al. Murine infection with bioluminescent Leishmania infantum axenic amastigotes applied to drug discovery. Sci Rep. 2019 Dec 12;9(1):18989.
- Hyland KV, Asfaw SH, Olson CL, et al. Bioluminescent imaging of Trypanosoma cruzi infection. Int J Parasitol. 2008 Oct;38(12):1391–1400.
- Canavaci AM, Bustamante JM, Padilla AM, et al. In vitro and in vivo high-throughput assays for the testing of anti-Trypanosoma cruzi compounds. PLoS Negl Trop Dis. 2010 July 13;4(7):e740.
- Bustamante JM, Tarleton RL. Methodological advances in drug discovery for Chagas disease. Expert Opin Drug Discov. 2011 June;6(6):653–661.
- Henriques C, Henriques-Pons A, Meuser-Batista M, et al. In vivo imaging of mice infected with bioluminescent Trypanosoma cruzi unveils novel sites of infection. Parasit Vectors. 2014 Mar 3;7:89.
- Rogers N. Bugging out over Chagas: bioluminescent protozoans and old drugs might help unravel kissing-bug disease. Nat Med. 2015 Oct;21(10):1108–1110.
- Lewis MD, Fortes Francisco A, Taylor MC, et al. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell Microbiol. 2014 Sept;16(9):1285–1300.
- Lewis MD, Kelly JM. Putting infection dynamics at the heart of Chagas disease. Trends Parasitol. 2016 Nov;32(11):899–911.
- Francisco AF, Jayawardhana S, Lewis MD, et al. Biological factors that impinge on Chagas disease drug development. Parasitology. 2017 Dec;144(14):1871–1880.
- Lewis MD, Francisco AF, Taylor MC, et al. A new experimental model for assessing drug efficacy against Trypanosoma cruzi infection based on highly sensitive in vivo imaging. J Biomol Screen. 2015 Jan;20(1):36–43.
- Francisco AF, Lewis MD, Jayawardhana S, et al. Limited ability of posaconazole to cure both acute and chronic trypanosoma cruzi infections revealed by highly sensitive in vivo imaging. Antimicrob Agents Chemother. 2015 Aug;59(8):4653–4661.
- Francisco AF, Jayawardhana S, Lewis MD, et al. Nitroheterocyclic drugs cure experimental Trypanosoma cruzi infections more effectively in the chronic stage than in the acute stage. Sci Rep. 2016 Oct;17(6):35351.
- Lewis MD, Francisco AF, Taylor MC, et al. Host and parasite genetics shape a link between Trypanosoma cruzi infection dynamics and chronic cardiomyopathy. Cell Microbiol. 2016 Oct;18(10):1429–1443.
- Silberstein E, Serna C, Fragoso SP, et al. A novel nanoluciferase-based system to monitor Trypanosoma cruzi infection in mice by bioluminescence imaging. PLoS One. 2018 Apr 19;13(4):e0195879.
- Costa FC, Francisco AF, Jayawardhana S, et al. Expanding the toolbox for Trypanosoma cruzi: a parasite line incorporating a bioluminescence-fluorescence dual reporter and streamlined CRISPR/Cas9 functionality for rapid in vivo localisation and phenotyping. PLoS Negl Trop Dis. 2018 Apr 2;12(4):e0006388.
- Lewis MD, Francisco AF, Jayawardhana S, et al. Imaging the development of chronic Chagas disease after oral transmission. Sci Rep. 2018 July 26;8(1):11292.
- Silva-Dos-Santos D, Barreto-de-Albuquerque J, Guerra B, et al. Unraveling Chagas disease transmission through the oral route: gateways to Trypanosoma cruzi infection and target tissues. PLoS Negl Trop Dis. 2017 Apr 5;11(4):e0005507.
- Francisco AF, Jayawardhana S, Taylor MC, et al. Assessing the effectiveness of curative benznidazole treatment in preventing chronic cardiac pathology in experimental models of Chagas disease. Antimicrob Agents Chemother. 2018 Sept 24;62(10):pii: e00832–18.
- Pecoul B, Batista C, Stobbaerts E, et al. The BENEFIT Trial: where Do We Go from Here? PLoS Negl Trop Dis. 2016 Feb 25;10(2):e0004343.
- Urbina JA, McKerrow JH. Drug susceptibility of genetically engineered Trypanosoma cruzi strains and sterile cure in animal models as a criterion for potential clinical efficacy of Anti-T. cruzi drugs. Antimicrob Agents Chemother. 2015 Dec;59(12):7923–7924.
- Francisco AF, Lewis MD, Jayawardhana S, et al. Reply to “drug susceptibility of genetically engineered Trypanosoma cruzi strains and sterile cure in animal models as a criterion for potential clinical efficacy of anti-T. cruzi drugs”. Antimicrob Agents Chemother. 2015 Dec;59(12):7925.
- Khare S, Liu X, Stinson M, et al. Antitrypanosomal Treatment with benznidazole is superior to posaconazole regimens in mouse models of chagas disease. Antimicrob Agents Chemother. 2015 Oct;59(10):6385–6394.
- de Lana M, Martins-Filho OA. Revisiting the post-therapeutic cure criterion in Chagas disease: time for new methods, more questions, doubts, and polemics or time to change old concepts? Biomed Res Int. 2015;2015:652985.
- Drugs for Neglected Diseases. BENDITA BEnznidazole New Doses Improved Treatment and Associations (BENDITA). In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000 [cited 2020 Aug 03]. Available from: https://clinicaltrials.gov/ct2/show/NCT033786611 NLM Identifier: NCT033786611
- The BENDITA study. Chagas BENDITA Study Briefing Document. [ cited 2020 Feb 29]. Available from: https://www.dndi.org/wp-content/uploads/2019/03/2page_BenditatStudyOverview_ENG.pdf
- Sánchez-Valdéz FJ, Padilla A, Wang W, et al. Spontaneous dormancy protects Trypanosoma cruzi during extended drug exposure. Elife. 2018 Mar 26;7:pii: e34039.
- Cui Y, Yu L. Application of the CRISPR/Cas9 gene editing technique research on functional genomes of parasites. Parasitol Int. 2016;65(6PtA):641–644.
- de Souza AP, Tang B, Tanowitz HB, et al. Magnetic resonance imaging in experimental Chagas disease: a brief review of the utility of the method for monitoring right ventricular chamber dilatation. Parasitol Res. 2005 Sept;97(2):87–90.
- Capitanio JP. Naturally occurring nonhuman primate models of psychosocial processes. Ilar J. 2017 Dec 1;58(2):226–234.
- Box GEP, Draper NR. Empirical Model-building and response surfaces. New York (NY): John Wiley & Sons; 1987. p. 424.