
1. Introduction
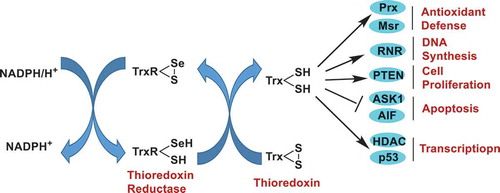
The ubiquitous thioredoxin reductase (TrxR, encoded by TXNRD gene) enzymes are essential to maintain the cellular redox homeostasis. The most outstanding function of TrxR is to keep its native substrate, thioredoxin (Trx, encoded by TXN gene), in a reduced state via taking electrons from NADPH, and the reduced Trx then interacts with diverse downstream proteins, including antioxidant enzymes, DNA repair and synthesis enzymes, apoptosis-regulating kinases, and transcription factors ()[Citation1,Citation2]. To date, three different TrxR isoenzymes, i.e., the cytosolic TrxR1, the mitochondrial TrxR2, and the testis-specific TrxR3, are known. Mammalian TrxRs, quite different from those from low organisms, such as bacteria and fungi, are selenoproteins and contain an indispensable selenocysteine (Sec) residue at their C-terminal active center. Despite localizing in different cell organelles, TrxR1 and TrxR2 share overall similar three-dimensional (3-D) structure and catalytic mechanism. The readers are referred to the recent literature for the detailed structural information of mammalian TrxRs and bacterial TrxRs [Citation1]. The TrxR enzymes are fundamentally important to cells and regulate diverse pathways that control cell growth, differentiation, and death. Compared to normal cells, cancerous cells are more dependent on TrxR functions to maintain tumor phenotypes [Citation1]. Thus, TrxR has been considered as a promising anticancer drug target, and numerous small molecules targeting the enzyme have been disclosed [Citation3–7].
Figure 1. Catalytic mechanism of TrxR and the interaction of Trx with downstream proteins
2. Current progress of TrxR inhibitors
The unique chemical properties of the selenol group in Sec, in particular, its high nucleophilicity and susceptibility to oxidation, are indispensable for its function in selenoenzymes, including TrxR. In addition, as the Sec residue is exposed on the surface of TrxR, it is the target of the majority of the current TrxR inhibitors, which form either covalent or coordinate bond with the Sec residue. Some inhibitors targeting solely the Sec may generate SECTRAP, leading to a shift of TrxR function [Citation8]. We have extensively reviewed TrxR inhibitors published in literatures and claimed in patents [Citation3,Citation5,Citation7]. Readers are recommended to refer to these references for details. Some TrxR/Trx inhibitors, e.g. ethaselen and PX-12, entered clinical trials for cancer treatment. However, due to the presence of much higher concentration of cysteine (Cys) residue and the thiol group in the Cys having similar chemical reactivity to the selenol group in the Sec [Citation9], how to minimize the interference from the Cys residue is a high challenge in developing Sec-targeting TrxR inhibitors. In this editorial, we will present perspectives regarding the improvement of the design of TrxR inhibitors for treating cancer.
3. Expert opinion
3.1. Targeting the Sec residue
Unlike most enzymes that use small molecules as substrates and have well-defined substrate binding pockets, TrxR employs the small protein Trx as the substrate and lacks the small-molecule binding pocket. Thus, the general strategy to construct enzyme inhibitors, e.g., chemical modification of native substrates, is not suitable for the design of TrxR inhibitors. One of the hallmarks of TrxR is that TrxR has a critical and highly reactive Sec residue. Different from other selenoproteins, the Sec residue is on the surface of the enzyme and easily accessible by small molecules. Thus, preparing small molecules with an electrophilic unit is the most popular strategy to construct TrxR inhibitors [Citation3,Citation5,Citation7]. Traditionally, molecules with an electrophilic unit are considered as pan-assay interference compounds (PAINS) and are not welcomed in medicinal chemistry. However, the success of covalent inhibitors, such as ibrutinib, dimethyl fumarate, and KRAS inhibitors [Citation10], in treating diseases has made medicinal chemists reconsider the application of PAINS in drug design. As we discussed earlier, the challenge in design Sec-targeting inhibitors is the presence of much abundant Cys in biological systems. In our previous work, we developed a highly selective probe of selenols over biological thiols via tuning the molecules electrophilicity [Citation11]. This strategy, e.g., tuning the molecules electrophilicity, could be borrowed to design selective TrxR inhibitors. In addition, introducing other groups to facilitate the weak interactions between the molecules and the surrounding residues of Sec may further improve the specificity of inhibitors.
3.2. Targeting protein–protein interaction (PPI)
The reduction of oxidized Trx requires a physical contact between Trx and TrxR. Thus, blocking the TrxR-Trx PPI could be another way to inhibit the function of TrxR. Developing small-molecule inhibitors of PPI is a high challenge as the PPI interfaces are generally large and flat (~1000–2000 A2 per side) [Citation12]. By using the indicated TrxR and Trx mutants (human TrxR1C497S, U498C and human Trx1C35S, C73S), the 3-D crystal structure of TrxR-Trx complex has been resolved by Becker et al., and the complex is stabilized by multiple weak interactions [Citation13]. Notably, the interactions between residues 58–74 in human Trx and residues 103–122 in human TrxR play a significant role in the complex formation [Citation13]. Interestingly, we also observed the formation of different disulfide-linked TrxR-Trx complex by incubating wild-type Trx protein with various single-thiol active site mutants of TrxR [Citation14]. Extracting the peptide sequence at the PPI surface may be a shortcut to find peptide inhibitors of PPI. Peptide inhibitors of PPI may serve as leads for druggable small molecules.
3.3. Prodrug strategy
Prodrugs are molecules that are activated and converted into pharmacologically active drugs after administration. As TrxR is generally overexpressed in different types of tumors, the development of TrxR-dependent prodrugs is also a strategy to treat tumors. In our efforts to develop the TRFS series fluorescent probes of TrxR, we disclosed that the 1,2-dithiolane (five-membered cyclic disulfide) scaffold could be reduced by TrxR exclusively [Citation15,Citation16]. Based on this discovery, we prepared a first TrxR-dependent prodrug S-Gem and demonstrated that S-Gem shows TrxR-dependent cytotoxicity [Citation17]. Compared to the inhibition of TrxR for cancer treatment, the success of S-Gem provides a completely new strategy to develop therapeutic molecules that target TrxR. We expect that the 1,2-dithiolane unit may be a general scaffold to construct TrxR-dependent prodrugs.
3.4. High-throughput screening (HTS)
HTS is a key process used in modern drug discovery to identify hits from compound libraries that may become leads for further medicinal chemistry optimization. HTS has become increasingly important in medicinal chemistry and chemical biology. Due to the availability and high price of TrxR, the application of HTS to discover TrxR inhibitors has not been widely adopted [Citation18]. We have developed an HTS model for discovering TrxR inhibitors using crude tissue extracts as the source of TrxR and identified dozens of natural product inhibitors of TrxR [Citation16]. This HTS model does not require the expensive TrxR enzyme, and may work in any common labs. Many compound libraries are commercially available, and thus HTS could accelerate the discovery of TrxR inhibitors, especially those inhibitors with new chemical scaffolds.
3.5. Concluding remarks
TrxR has increasingly recognized as a promising target for the development of anticancer drugs. Although numerous inhibitors have been disclosed, the number of highly selective or specific inhibitors is limited. This is largely due to the inhibition mechanism, e.g., targeting the Sec residue, employed in designing TrxR inhibitors. Currently, targeting Sec is still a mainstream in developing TrxR inhibitors. We present our opinion to discuss the strategies in design of TrxR inhibitors in this editorial, i.e., targeting the Sec residue, targeting TrxR-Trx PPI, prodrug strategy, and HTS. We expect these strategies would promote the discovery of TrxR inhibitors for cancer treatment.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Zhang J, Li X, Han X, et al. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol Sci. 2017;38(9):794–808.
- Dagnell M, Schmidt EE, Arner ESJ. The A to Z of modulated cell patterning by mammalian thioredoxin reductases. Free Radic Biol Med. 2018;115:484–496.
- Cai W, Zhang L, Song Y, et al. Small molecule inhibitors of mammalian thioredoxin reductase. Free Radic Biol Med. 2012;52(2):257–265.
- Arnér ESJ. Targeting the selenoprotein thioredoxin reductase 1 for anticancer therapy. Adv Cancer Res. 2017;136:139–151.
- Zhang B, Zhang J, Peng S, et al. Thioredoxin reductase inhibitors: a patent review. Expert Opin Ther Pat. 2017;27(5):547–556.
- Bian M, Fan R, Zhao S, et al. Targeting the thioredoxin system as a strategy for cancer therapy. J Med Chem. 2019;62(16):7309–7321.
- Zhang J, Zhang B, Li X, et al. Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: an update. Med Res Rev. 2019;39(1):5–39.
- Anestal K, Prast-Nielsen S, Cenas N, et al. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PloS One. 2008;3(4):e1846.
- Xu J, Cheng Q, Arner ES. Details in the catalytic mechanism of mammalian thioredoxin reductase 1 revealed using point mutations and juglone-coupled enzyme activities. Free Radic Biol Med. 2016;94:110–120.
- Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. .
- Zhang B, Ge C, Yao J. et al. Selective selenol fluorescent probes: design, synthesis, structural determinants, and biological applications. J Am Chen Soc. 2015;137(2):757–769.
- Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21(9):1102–1114.
- Fritz-Wolf K, Kehr S, Stumpf M, et al. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat Commun. 2011;2:383.
- Xu J, Eriksson SE, Cebula M, et al. The conserved Trp114 residue of thioredoxin reductase 1 has a redox sensor-like function triggering oligomerization and crosslinking upon oxidative stress related to cell death. Cell Death Dis. 2015;6:e1616.
- Zhang L, Duan D, Liu Y, et al. Highly selective off-on fluorescent probe for imaging thioredoxin reductase in living cells. J Am Chem Soc. 2014;136(1):226–233.
- Li X, Zhang B, Yan C, et al. A fast and specific fluorescent probe for thioredoxin reductase that works via disulphide bond cleavage. Nat Commun. 2019;10:2745.
- Li X, Hou Y, Meng X, et al. Selective activation of a prodrug by thioredoxin reductase providing a strategy to target cancer cells. Angew Chem Int Ed Engl. 2018;57(21):6141–6145.
- Prast-Nielsen S, Dexheimer TS, Schultz L, et al. Inhibition of thioredoxin reductase 1 by porphyrins and other small molecules identified by a high-throughput screening assay. Free Radic Biol Med. 2011;50(9):1114–1123.