
ABSTRACT
Introduction: Drug repurposing provides a cost-effective strategy to re-use approved drugs for new medical indications. Several machine learning (ML) and artificial intelligence (AI) approaches have been developed for systematic identification of drug repurposing leads based on big data resources, hence further accelerating and de-risking the drug development process by computational means.
Areas covered: The authors focus on supervised ML and AI methods that make use of publicly available databases and information resources. While most of the example applications are in the field of anticancer drug therapies, the methods and resources reviewed are widely applicable also to other indications including COVID-19 treatment. A particular emphasis is placed on the use of comprehensive target activity profiles that enable a systematic repurposing process by extending the target profile of drugs to include potent off-targets with therapeutic potential for a new indication.
Expert opinion: The scarcity of clinical patient data and the current focus on genetic aberrations as primary drug targets may limit the performance of anticancer drug repurposing approaches that rely solely on genomics-based information. Functional testing of cancer patient cells exposed to a large number of targeted therapies and their combinations provides an additional source of repurposing information for tissue-aware AI approaches.
1. Introduction
Drug repurposing (also called drug repositioning, reprofiling, redirecting, and drug rediscovery [Citation1]) is a strategy for identifying new therapeutic purposes for approved drugs in medical indications beyond the scope of their original therapeutic use [Citation2]. Drug repurposing offers various advantages over the de-novo development of entirely new drugs, including the possibility to speed-up the discovery process and to reduce failure rates in the clinical development and testing phases [Citation3]. In particular, drug repurposing makes it possible to avoid safety evaluation in preclinical models and humans, hence leading to potentially lower overall development costs, if the safety testing has been completed for the original indication and it displays dose-compatibility with the new indication. Traditionally, drug repurposing success stories have mainly resulted from largely opportunistic and serendipitous findings [Citation4]; for example, sildenafil citrate was originally developed as an antihypertensive drug, but later repurposed by Pfizer and marketed as Viagra for the treatment of erectile dysfunction based on retrospective clinical experience, leading to massive worldwide sales.
Over recent years, a number of computational approaches have been developed for a more systematic drug repurposing process. Popular information sources for in-silico drug repurposing include, for instance, electronic health records, genome-wide association analyses or gene expression response profiles, pathway mappings, compound structures, target-binding assays, and other phenotypic profiling data [Citation4]. Several systematic review articles on the use of computational approaches are available [Citation4], which cover also machine learning (ML) and artificial intelligence (AI) algorithms, such as those based on network propagation, matrix factorization, and completion, as well as recently developed deep learning models [Citation5–8]. Databases and other resources supporting in-silico drug repurposing, such as Drug Repurposing Hub [Citation9] and RepurposeDB [Citation10], have also been recently surveyed [Citation11]. There are also excellent reviews and perspectives on the use of ML and AI approaches in the overall drug discovery and development process [Citation12,Citation13], as well as in the lead optimization or designing of completely new molecules [Citation14].
Our focus here is on supervised ML and AI methods that make use of publicly available databases and information sources. A particular emphasis is placed on the use of comprehensive target activity profiles of drugs as a resource for a systematic repurposing process, in which an existing drug is found to have an off-target effect or a newly recognized on-target effect for a new indication, hence providing sufficient evidence to take it forward for further development and commercial exploitation. Such target-based drug repurposing makes use of the fact that most drugs are not specific for any single target, but rather display a wide spectrum of target activity. In cancer applications, some of the unintended off-targets correspond to known anticancer targets, while others may reveal new cancer vulnerabilities [Citation15]. However, we note that drug repurposing is not by any means limited to anticancer applications alone, but covers various medical indications [Citation16]. For instance, a recent review surveyed how existing drugs may have activity against SARS-CoV-2 to be readily applied to treat COVID-19 patients [Citation17,Citation18]. Similarly, target repositioning [Citation19] can be used in the field of infectious diseases, where a drug is used to inhibit the ortholog target proteins in other species [Citation20,Citation21].
The repurposing process is often initiated after phenotypic observations of adventitious polypharmacological drug activities. For instance, we observed a surprising activity for axitinib, an endothelial growth factor receptor (VEGFR) inhibitor approved for advanced renal cell carcinoma, in primary chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) cells [Citation22]. Since these cancers are driven by the oncogenic BCR-ABL1 fusion protein, we hypothesized that axitinib might bind to BCR-ABL1. This was confirmed by structural and functional analysis, and interestingly, axitinib bound to T315I-mutated BCR-ABL1 with roughly 40 times higher affinity than to the wild-type BCR-ABL1. Currently, axitinib is being investigated in an alternating regimen with bosutinib for CML patients (NCT02782403). Subsequent reports, however, have indicated that axitinib may lose potency when additional compound mutations emerge in BCR-ABL1 [Citation23], and the drug does not seem to be effective against ponatinib-resistant T315I-mutated cells [Citation24]. These observations raise the question whether one could use AI algorithms to predict at least some of the potential drawbacks already before the repurposing process enters the clinical stage.
2. Data resources for in-silico drug repurposing
We start by going through selected data and information resources that we find useful for in-silico drug repurposing. Rather than providing a systematic review of all developed resources, we mainly focus on information sources motivated by the axitinib repurposing study from the previous section, including resources for drug–target activity data, cell-based pharmacogenomic data, and chemical structure information. For more comprehensive surveys of various data resources, the reader is referred to recent reviews [Citation4–6,Citation11]. We will discuss the use of these resources in Section 3.
2.1. Drug–target interaction resources
Comprehensive knowledge about the intended on-targets and non-intended or so-called off-targets of a drug is important for understanding its underlying mechanism of action (MoA), and for modeling its efficacy or toxicity in various tissue and cancer types. As shown in the motivating example of axitinib study, drug–target activity profiles are highly valuable in drug repurposing [Citation22]. In contrast to proprietary resources, which were used e.g. in Drug Repurposing Hub, we promote here the use of publicly available drug–target activity resources and how these can be useful in training supervised ML models for in-silico off-target predictions and drug repurposing. highlights 18 selected compound/target databases, along with various features such as the number of compounds, targets and interactions covered, as well as whether API is provided for programmatic data access for AI-based explorations. For simplicity, we have divided the compound–target activity data types into three categories according the type of activity data they contain: quantitative bioactivity data (e.g. from multi-dose Kd, Ki, or IC50 assays), binary interactions (both active and inactive drug–target pairs), and unary interactions (only active drug–target pairs). These categories determine whether regression or classification algorithms are applicable for the target activity predictions, and whether one has true positive as well as true negative examples for training of the supervised prediction models.
Table 1. Drug–target interaction resources for target activity predictions
Most of the in-silico DTI prediction studies are based on one of the resources listed in [Citation42]. So far, ChEMBL is the most popular target activity resource for regression modeling (i.e. prediction of quantitative drug–target binding affinities). Classification algorithms try to predict whether a drug has sufficient potency against the given target. In addition to the problem formulation (regression vs. classification), we have argued that at least the following factors should be taken into consideration in in-silico target prediction studies to avoid reporting overoptimistic drug–target activity prediction results: (i) multiple evaluation datasets specific to particular drug and target families to evaluate the application domain of the prediction model, (ii) evaluation procedure, where nested cross-validation is preferred over the standard cross-validation, and (iii) prediction problem setting (i.e. whether the training and test sets of compound-target pairs share common drugs and targets, only drugs or targets, or neither, where the latter is often the most challenging case) [Citation43]. Obviously, the more comprehensive is the information present in the databases, e.g. in terms of drug classes and target families, the better coverage the prediction algorithm will have. The predicted target activities should also be experimentally validated before suggesting for drug repurposing [Citation44]. Accordingly, we recently organized an IDG-DREAM Challenge, where the teams used bioactivity data from ChEMBL, DTC, and BindingDB to make quantitative target activity predictions, which were later validated using subsequent experimental assays [Citation42].
2.2. Cell line and patient-derived omics resources
Drug–target bioactivity information offers possibilities to make informed predictions whether the explored compounds have the possibility to modulate a given target or not, and to what extent, but this information is typically cell context independent. However, since the drug MoA is often highly cell context-specific, it is important to actually measure (or predict) the activity of the compound against the cell model or target using cell-based assays. Cell line omics resources contain drug response data along with multi-omics profiles for established cancer cell lines (in vitro models), whereas patient-derived resources include pharmacogenomic information on the patient primary cells tested against various drugs (ex-vivo models). lists a selected set of drug response and omics resources, along with additional features, such as number of drugs, cell lines, patient samples, and whether the resource contains API or drug response visualizations, useful for drug repurposing AI-applications.
Table 2. Cell-based pharmacogenomic resources for drug efficacy predictions
The drawback of most of these resources is that they do not provide programmatic API (except for GDSCtools), and that pharmacogenomic data typically come solely from one lab or study (except for CellMinderCDB and PharmacoDB that integrate data from multiple studies). However, these data are freely available either through GUI (downloadable in many cases) or using batch queries. The patient-derived primary cell data are still limited in these resources, but at least PharmacoDB is currently extending to ex-vivo data as well. We do not consider here more complex preclinical models, such as patient-derived xenografts (PDXs) or other animal models, as the pharmacogenomic data from these models are still rather scarce for AI developments. However, the cell-based omics resources also enable one to predict patient responses to drug treatments, such as those available in The Cancer Genome Atlas (TCGA) resource; see Section 3.3.
2.3. Biological pathway information resources
Biological pathways facilitate the understanding of the inner working of the cells and the cellular responses of the drugs, and can therefore aid the drug repurposing efforts. For instance, mapping of the protein targets of drugs either to the same or orthogonal pathways may help to reveal the MoA of both multi-targeted monotherapies and combination therapies. However, various databases may contain different representations of the same biological pathways, which leads to variable results of statistical target pathway enrichment analysis and predictive models in the context of precision medicine [Citation54]. In this section, we highlight six pathway databases that contain information of compound target pathways, along with their characteristics in terms of the number of proteins, compounds, pathways and interactions (). PathwayCommons [Citation55] and KEGG Pathways [Citation56] are currently the two most comprehensive databases in terms of the number of reactions or interactions. Four out of six pathway databases also provide programmatic access for data using APIs, making them easy for systematic AI model development.
Table 3. Pathway resources for understanding compounds’ mode of action
2.4. Chemical structure and protein property data resources
The chemical structural descriptors and target protein properties provide important information for AI and ML models for drug repurposing. There are various online web-servers and toolkits to calculate chemical descriptors for drugs and target properties of proteins. For instance, ChemCPP calculates kernel functions between the compounds [Citation61]. EDragon software computes more than 1600 topological and geometrical descriptors for the chemicals [Citation62]. The Open Babel toolkit provides several useful features including substructure search and calculation of fingerprints of the chemicals [Citation63]. RDKit provides features including 2D depiction, molecular serialization, fingerprint generation, and similarity analysis for the compounds [Citation64]. Finally, PyDPI is python package that computes molecular descriptors for drugs and structural and physiochemical properties for proteins [Citation65].
There are also web-tools that help to draw chemical structures, compute physiochemical properties and chemical fingerprints. These tools have opened-up various applications for in-silico drug–drug interaction prediction [Citation66] and for drug toxicity prediction [Citation67]. ChemSketch is a package to draw chemical structures including organics, organometallics, polymers, and Markush structures [Citation68]. KNIME comprises features for molecule conversion into various formats, generation of signatures, fingerprints, and molecular properties [Citation69]. PaDEL‐Descriptor is a software for calculating molecular descriptors and 10 different types of fingerprints [Citation70]. BlueDesc is a free tool, which computes 36 different types of fingerprints [Citation71]. However, most of the fingerprint calculation methods are derived from the following five fingerprints: MACCS, PubChem, FP2-based, Atom Pair, and ECFP4.
lists selected open-access databases that contain chemical structural information, such as InchiKeys and SMILES, and that implement options for structure or sub-structural searches either through GUI or API, which we find useful for in-silico drug repurposing.
Table 4. Chemical structure databases using InchiKey searches or structure drawings
3. Supervised ML and AI algorithms for drug repurposing
3.1. Algorithms for drug–target interaction predictions
To accelerate the costly and time-consuming experimental mapping approach to identify DTIs by means of biochemical experiments, various computational approaches have been developed over the past decade, providing a systematic means for prediction of potential DTIs [Citation77–79]. Concomitant with the experimental drug–target discovery efforts that provide either quantitative or qualitative compound–target interactions data (see ), computational tools are being built to predict activities against new molecular targets for drug repurposing. For instance, ML models are using orthogonal drug–target space deconvolution, where the molecular structures of both the drugs and targets help to guide the in-silico predictions [Citation80,Citation81]. Another research line has utilized crowdsourcing-based AI and ML methods to effectively predict target activities for kinase inhibitors [Citation42]. Similarly, Cichonska et al. adopted pairwise multi-kernel learning to predict the compound-kinase target-binding affinities [Citation82]. Extending to other target families, Li et al. predicted compound activity classes for enzyme, ion channel, G protein-coupled receptors (GPCRs), and nuclear receptors using substructure chemical fingerprints and rotation forest classifier [Citation83].
There are excellent review articles that provide a comprehensive overview of AI- and ML-based methods for DTI prediction. For instance, Chen et al. [Citation84] categorized the learning methods into nearest neighbor methods, bipartite local models, matrix factorization methods, and semi-supervised methods, and discussed pros and cons of the method classes. There are also reviews on various classes of DTI prediction methods; for instance, Sachdev et al. provided a review on feature-based chemogenomic methods for DTI prediction [Citation85], and Wu et al. discussed the pros and cons of network-based methods for predicting DTIs [Citation86]. They further sub-divided network-based methods into categories such as network-based inference (NBI) methods, similarity inference methods, random walk-based methods, and other network-based methods. As specific examples of network methods, DTiGEMS+ is a computational approach to predict DTIs using graph mining and similarity-based techniques [Citation87]. Mongia et al. proposed method that is based on multi-graph regularized nuclear norm minimization to identify interactions between drugs and target proteins from three inputs: known DTI network, similarities over drugs, and those over targets [Citation88]. DGraphDTA utilizes graph neural networks to obtain deeper representations for drug–target activity prediction, based on structural information of both molecules and proteins, where the two network graphs of drug molecules and proteins are built up, respectively, [Citation89].
Recently, several deep learning methods have been developed for predicting DTIs, including convolutional network model that first uses a graph convolutional network to learn the features for each drug–protein pair, and then based on these feature representations as inputs, utilizes deep neural network to classify between positive and negative DTI classes [Citation90]. These in-silico methods provide a deeper understanding of the factors affecting DTI prediction, and have opened novel strategies for computational drug repurposing.
Accurate DTI prediction has the potential to not only complement the experimentally mapped DTI networks but also to provide novel drug repurposing leads by extending the target space of already approved drugs [Citation91]. There are also in-silico methods that make use of DTI mappings or predictions directly in the drug repurposing process. For instance, Mei et al. have proposed a multi-label learning framework to find new uses for approved drugs, and conversely to discover new drugs for known target proteins [Citation92]. In their framework, each drug is treated as a class label and its target proteins as class-specific training data to train l2-regularized logistic regression model. Stratified multi-label cross-validation showed that 84.9% of the known target proteins were correctly predicted at least for one drug, and the proposed framework correctly recognized 86.73% of the independent test DTIs from DrugBank. These results show that the proposed framework could generalize well in the large drug space without requiring the information of drug chemical structures and target protein structures. The recently introduced iDrug method integrates drug repositioning and DTI prediction into one coherent model via cross-network embedding [Citation93]. The embedding approach provides a principled way to transfer knowledge across the drug–target–disease relationships, and in doing so, it enhances the prediction accuracy for both of the prediction tasks (i.e. DTI and drug–disease relationships). The performance of the iDrug method was tested on various real-world datasets, covering multiple disease types, hence making it widely applicable to repurpose drugs for several indications. For more targeted application, Molecule Transformer-Drug Target Interaction (MT-DTI) is a pre-trained deep learning-based drug–target model to identify commercially available drugs that could act on viral proteins for the inhibition of SARS-CoV-2 [Citation94]. Through a detailed analysis, the authors showed that atazanavir, an antiretroviral medication for treatment of HIV, proved to be the most potent drug with an inhibitory potency of Kd = 94.94 nM against the SARS-CoV-2, followed by remdesivir (Kd = 113.13 nM), efavirenz (Kd = 199.17 nM), ritonavir (Kd = 204.05 nM), and dolutegravir (Kd = 336.91 nM).
3.2. Algorithms for molecular docking and molecular dynamic simulations
Molecular docking is a widely used in-silico method in structure-based drug design, due to its ability to predict the binding-conformation of small molecule ligands to the appropriate target-binding site [Citation95–97]. The drawback of molecular docking is that the 3D structures of many target proteins have not yet been resolved, which is required for running the docking simulations. Furthermore, the accuracy of docking-based methods decreases in cases where the number of known ligands for a protein is not sufficient [Citation98]. Regardless of these limitations, there are several examples of successful docking-based drug off-target activity predictions [Citation99]. For instance, antipsychotic agent thioridazine was found among 1500 FDA-approved compounds to possess anti-inflammatory activity by binding and inhibiting IκB kinase, which is critical for the NF-ΚB pathway [Citation100]. Similarly, virtual docking accurately predicted inhibitory activity of five compounds from a collection of more than 1400 FDA-approved drugs against Pseudomonas aeruginosa quorum-sensing (population-wide virulence) mechanisms, with antipsychotic agent pimozide displaying potent in vitro activity in inhibiting bacterial virulence gene expression [Citation101]. Moreover, AI is also emerging as an increasingly accurate approach for predicting the 3D structures of proteins from their amino-acid sequences [Citation102,Citation103].
We recently implemented VirtualKinomeProfiler, an efficient computational platform that captures distinct representations of the chemical similarity space of the druggable kinome for speeding-up drug discovery and repurposing process for highly promiscuous kinase inhibitors [Citation104]. An ensemble support vector machine (eSVM) algorithm enabled activity classification for >30 M compound-kinase pairs, using which we carried out in-silico activity predictions for >151 K compounds in terms of their drug repositioning and lead molecule potential. Experimental testing with biochemical assays validated 19 of the 51 of the predicted interactions, leading to a 1.5-fold increase in precision and 2.8-fold decrease in false-discovery rate, which demonstrated its potential to expedite the kinome-specific drug discovery process. There are also several other case studies, where structural information of chemicals has been directly utilized for drug repurposing applications in various target classes. For instance, CATNIP is ML model for drug repurposing that requires only similarity information of the molecules based on their structural, target, or pathway information [Citation105]. Another model utilized chemical fingerprint information to predict that 22 FDA-approved drugs have potential activities on heart failure, and confirmed experimentally 8 of the 22 of the cardioprotective activities in vitro [Citation106].
3.3. Algorithms for cell and tissue-based drug response predictions
Once the target activity potential of a drug has been predicted or established, either by using DTI prediction algorithms or molecular docking methods, the next important prediction task involves the investigation whether the drug has efficacy in a relevant cell context. This is critical because biochemical compound affinity and structure-based modeling provide only hypotheses of compound activity against a particular disease target, and these predictions need to be further investigated using a relevant disease model. In anticancer applications, cancer cell line models and patient-derived primary cells are widely used for such predictive purposes (see ).
As an early community effort and an example for other in-silico precision oncology studies, NCI-DREAM Drug Sensitivity Prediction Challenge benchmarked in 2013 a number of supervised ML algorithms based on genome-wide omics and drug response profiles of 53 human breast cancer cell lines [Citation107]. Notable, the predictive models that made use of multiple omics profiles of the cancer cell lines had the best performance, suggesting that the genomic, transcriptomic, epigenomic, and proteomic profiles each provides complementary predictive signal for the cell-based drug response modeling. The best-performing approach was based on the Bayesian efficient multiple kernel learning (BEMKL) model [Citation108], a kernelized regression model that makes use of multi-task and multi-omics learning, where the pairwise similarities of cell lines in terms of the multiple omics profiles are first represented as separate profile kernels, and a multiple kernel learning algorithm then calculates a combined kernel as the weighted sum of all profile-specific kernels. Finally, multi-task learning allows one to estimate the BEMKL model simultaneously for all the drugs as related prediction tasks.
After the DREAM Drug Sensitivity Prediction Challenge, hundreds of prediction algorithms have been developed for matching cancer cell omics features to the cell-based drug efficacies. Some common features of the best-performing methods can be inferred from two recent systematic analyses in cancer cell lines datasets [Citation109,Citation110]. Both of these comparative analyses focused on multi-omics and multi-target learning approaches, and concluded that matrix-factorization and kernel-based methods performed best in drug response prediction across various cancer cell lines. More specifically, similarity-regularized matrix factorization (SRMF) approximates the drug response matrix by the product of two low-rank similarity matrices; one that uses the cell line omics profiles, and the other that is based on drug structural similarities [Citation111]. Similarly, pairwise multi-kernel learning (pairwiseMKL) method integrates heterogeneous cell line and chemical structure information into a single model, enabling the joint analysis of the kernel mixture weights for the different information sources [Citation82]. Importantly, SRMF and pairwiseMKL methods showed robust and improved performance in various cell line datasets and in terms of different evaluation metrics [Citation109,Citation110].
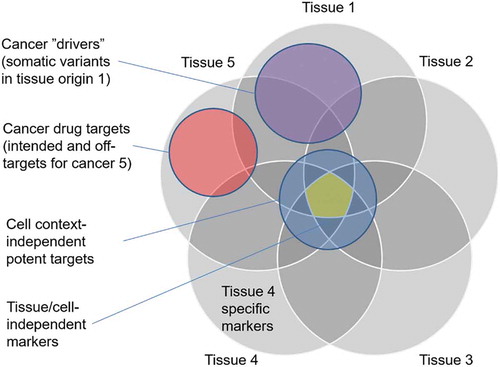
However, there are still some critical missing pieces that need to be addressed in these drug efficacy prediction methods when used for drug repurposing. The first challenge is how to identify panels of multi-omics features that are predictive of the drug efficacy in the target cancer type. While matrix factorization and kernel-based methods often provide high predictive accuracy, they cannot directly identify clinically actionable biomarkers among the genome-wide omics profiles [Citation112]. Toward feature selection, the use of drug–target activity information has been shown to improve the predictive performance and interpretability of drug efficacies [Citation113]. Recent systematic analysis demonstrated how rather simple feature selection methods enabled identifying relative small feature panels using prior information on targets and pathways of molecularly targeted drugs, whereas wider feature sets were required for drugs affecting general cellular mechanisms (i.e. standard chemotherapies) [Citation114]. These results indicate that there are both target-based and non-target-based features that can be predictive of specific drug efficacies in various cancer types (see ).
Figure 1. Schematic illustration of overlaps between cancer-related gene sets. There are both target-based and non-target-based features that can be predictive of specific drug efficacies in various cancer types. The cancer genes and protein targets should be studies separately for each tissue type (e.g. breast cancer) and inhibitor class (e.g. HER2 inhibitors). Selective efficacies are preferred in the repurposing predictions, as tissue of origin-independent targets may lead to toxic side effects
The next challenge is how to best predict treatment outcomes in cancer patients (e.g. clinical in vivo responses to treatments), rather than merely drug efficacies in established cell lines (in vitro responses), as the former enables straightforward translational precision oncology applications and drug repurposing opportunities. A recent systematic analysis investigated the importance of a number of modeling components for the clinical treatment response prediction of cancer patients [Citation115]. As expected, the sample size of the patient response data was found as an important determinant for the predictive modeling, along with experimental noise within the data that can easily deteriorate the models’ robustness. Rather surprisingly, the in vitro drug treatment profile was not among the most predictive feature when predicting the clinical response of the same drug in actual cancer patients. These results indicate that even cell line models of high accuracy do not necessarily translate to accurate predictions of drug response processes in cancer patients in vivo [Citation115].
For drug repurposing, there is an added need for accurate tissue-specific drug efficacy predictions to study the efficacy of a drug in a relevant tissue-of-origin. Recent models, such as tissue-guided LASSO, make use of information on samples’ tissue-of-origin to improve in vivo prediction performance [Citation116]. It was shown that tissue-guided LASSO improves the clinical predictions and was able to distinguish resistant and sensitive patients for selected drugs. Furthermore, the method identified genes associated with the drug response, including known targets and pathways involved in the drugs’ MoA. Surprisingly, the use of information on the tissue-of-origin did not improve the prediction results, suggesting that there is still room for improvement for tissue-aware drug efficacy predictions. We further argue that one needs to consider several drug response-informative gene sets when predicting the potential efficacy and toxicity of specific drugs, some of which are illustrated in . Finally, one needs to avoid inhibiting so-called anti-targets, i.e. proteins that are involved in normal cellular processes, which may lead to severe toxic side effects if modulated.
4. Conclusion
This review described the use of supervised ML and AI models, with accompanying data resources, for three levels of prediction tasks related to drug repurposing process. First, biochemical bioactivity predictions for new DTIs; second, cell-based compound response predictions for drug–cell line/patient interactions; and third, drug repurposing predictions by means of novel drug–disease relationships. Each of these levels is important for understanding the MoA of the repurposed drugs in terms of their on/off target potencies and tissue-based response profiles. In addition to the identified protein targets, repurposed drugs may reveal additional molecular targets and pathways that can be further exploited therapeutically using other drugs or their combinations. Polypharmacological effects originating either from combination therapies or multi-targeted drugs are important for treating complex diseases, including many cancers and viral infections, but the potential toxicity of polytherapies needs to be carefully predicted using computational and experimental models. We also note that the entire field of drug repurposing is at risk of publication bias in the sense that much of the content of the various data and information sources is derived from published research; this introduces biases, e.g. well-known drugs tend to have more publications, and therefore weighting evidence more heavily than for lesser-studied drugs.
5. Expert opinion
In this section, we highlight our opinion on drug repurposing specifically in cancer research, where large-scale cancer sequencing efforts are being carried out to identify genomic aberrations specific to each tumor type. These genomic data are invaluable to match drug therapies targeting specific aberrations, either using the drug’s intended medical indications or repurposed drugs. However, even though the extent of genomic testing and the diversity of our pharmacological portfolio are constantly increasing, we argue that genomics alone is currently insufficient to identify therapeutic options for the majority of patients, especially for those with advanced disease or cases without known cancer drivers and rare cancer types. The scarcity of clinical patient data and focus on genetic aberrations as the primary drug targets may further limit the accuracy of those drug repurposing approaches that rely solely on genomics-based information. We and others believe that this limitation can be partly addressed by functional testing of cancer patient cells exposed to large number of both targeted and conventional therapies using drug testing assays in patient-derived cell models ex vivo, and later verified in patient-derived organoids (PDO) or xenograft (PDX) models in vivo [Citation117–119]. Cell-based drug testing enables identification of patient-selective target activities, rather than broadly toxic effects that often lead to severe toxic side-effects. Compared to the genomics-only approach, predictions from drug testing are often pharmaceutically actionable. However, we believe that integration of mutation profiling and drug sensitivity testing leads to improved, and sometimes unexpected drug repurposing options (e.g. axitinib for CML and ALL [Citation17]).
In addition to the data from in vitro or ex vivo model systems (), there is also a need for flexible computational models that can speed-up the early investigation of both the therapeutic and toxic effects of small molecules before entering into lengthy and costly animal or clinical studies. Rather than using single outcomes to rank the in-silico predictions, we argue that it is important to carefully dissect various readouts, such as those quantifying efficacy, toxicity, or synergy of multi-targeting mono- and combinatorial therapies in the pre-clinical model systems, when developing safe and effective therapeutic regimens for cancers and other diseases [Citation120]. The use of both in-silico and preclinical pharmacogenomic predictions can greatly reduce the extensive cost, time and risks associated with drug discovery process, before entering clinical trials. While a large number of in-silico drug repurposing approaches have been developed, including AI and ML models, what is unclear, however, is how useful these methods are in producing clinically efficacious repositioning hypotheses. Most computational studies perform analytic validation, where the prediction results are compared to existing biomedical knowledge. When examining the repositioning literature, however, there appeared no consistent practices for validation of the methods [Citation121]. To address this unmet need, Brown and Patel reviewed the computational repositioning literature, focusing on the studies in which authors claimed to have validated their work. Their analysis revealed a widespread variation in the types of strategies, predictions made, and databases used as ‘gold standards’ [Citation121]. This suggests that further developments are needed to make the in-silico drug repurposing predictions more actionable.
However, the heterogeneous preclinical data are currently housed in various locations. Drug–target bioactivity profiles are being collected in drug/target databases (), which provide insights into the potential use of small-molecule compounds to modulate various on- and off-targets, including mutant targets and wild-type proteins. Cell-based drug response phenotypic data () provide further evidence that the compound is actually effective in a given cell context or patient-derived sample (and not broadly effective in many cell types, which may be a sign of toxic effects). Finally, drug–target potencies and gene–drug associations can be linked to tumor genomic profiles and associated lifestyle and clinical data to make informed decisions about therapeutic efficacies, hence leading to translationally actionable drug repurposing opportunities. The scattered location of the preclinical pharmacogenomic data means that these information sources are currently available in formats that are not interoperable with each other, greatly limiting our ability to use these data in a systematic manner in AI-based predictive models. In the past, the lack of common standards for cancer models and chemical compounds, as well as meta-data for quantitative drug response profiles, further prevented the wider translational re-use of such data. Recent data harmonization efforts, such as DrugTargetCommons [Citation122] for compound-target activities, PharmacoDB [Citation123] for cell-based drug response profiles, as well as Cell Model Passports [Citation124] and Xeva [Citation125] for in vitro, ex vivo and in vivo models, are likely make their integrated use more straightforward in the AI models.
Although genomic sequencing and cell-based drug testing technologies continue to improve, wider adoption of genomics-based precision oncology and functional drug repurposing in the clinics has been held back by several logistic, regulatory and financial issues. For instance, even though the off-target potencies of approved drugs should lead to rather straightforward drug repurposing opportunities, it is often unclear for the academic researcher how to deal with approvals of off-label use of drugs or investigational molecules that show potency in patient-derived samples ex vivo, perhaps in combination with agents from other pharma companies. At the regulatory level, new types of clinical trials may be needed to get molecules approved sometimes for very narrow and specific indications, e.g. basket trials for molecularly targeted patient subgroups, or umbrella trials for rare cancer types. Furthermore, sharing and re-use of the pharmacogenomic data for new research or translational purposes is often complicated by uncertainties at the legal or ethical level, as different countries adopt divergent legislations. For translational applications, working with early phase diagnostic patients, rather than with the late stage relapsed cases, should lead to improved and sometimes also more durable outcomes. For routine cancer diagnosis and prognosis, cell-based drug sensitivity testing ex vivo cannot be implemented for each cancer patient, which calls for accurate response predictive biomarkers inferred, for instance, by computational AI models. This requires collaborative and multidisciplinary effort between experimental scientists, computational biologists and clinicians or translational researchers to solve these and other future challenges.
A recent comprehensive review of the time and cost expenditures of drug repurposing clinical trials in acute myeloid leukemia (AML) debunked the common dogmas associated with drug repurposing, namely (1) drug repurposing saves time, (2) phase I clinical trials can be skipped, and (3) repurposed drugs are safe as their toxicity profile is known [Citation126]. However, the realities are much more complex, and in particular the toxicities of drug combinations can be unexpected, and should not be underestimated. For example, combination with cholesterol medication pravastatin with idarubicin and cytarabine resulted in multi-organ failure in AML patients [Citation126]. Thus, it remains vital to develop better AI and ML models to predict combinatorial toxicities. Furthermore, there is a need to further improve our capacity to understand the effects of tumor subclonality and adaptive responses to drug responses, repurposed or otherwise. Notably, a recent report featuring single-cell DNA sequencing of 123 primary AML samples revealed simultaneous co-evolution of several independent but leukemogenic tumor subclones in each patient sample [Citation127], implying a requirement for multi-targeting treatments for a lasting tumor control using either drug combinations or promiscuous drugs [Citation128]. Fortunately, computational tools are being developed to help us decipher the multiple cellular drug targets and their associated pathways, with the aim to better predicting toxicities and targeting multiple subdiseases in the patient. Open-access, crowdsourced web-based resources to complement missing drug activity annotations [Citation122], combined with AI-based predictive models and analytic visualizations should facilitate manual efforts by automated data mining approaches toward more systematic and accurate drug repurposing leads.
We also note that many computational repurposing predictions are mechanistic or statistical only, and will require separate evaluation for specific medical indications and patient populations. It is well known, for instance, that drug metabolism and pharmacodynamics are influenced by gender, age, concomitant medications and food intake, as well as underlying physiological states, and thus drug repurposing from one indication to another still necessitates a thorough understanding of the individual and disease-specific clinical safety parameters [Citation129,Citation130]. FDA maintains both the ‘passive’ postmarketing pharmacovigilance database FAERS (FDA Adverse Event Reporting System) and the ‘active’ sentinel system, which collect information on adverse events that may occur in patients outside the clinical trials in the long term. In cancer treatment, for instance, genetic alterations that may negatively or positively influence drug efficacy in the malignant tissue are being collected in databases such as OncoPDSS [Citation131], but the germline changes, and epigenetic and non-genetic physiological states that impact efficacy and safety outside the tumor context have not been similarly annotated. Yet, single nucleotide variation and other genetic alterations can combine with physiological states to deviate drug responses. For instance, individual nucleotide variances in drug metabolizing CYP450 cytochrome family enzymes that alter drug metabolism, such as CYP2C19, drastically influence both efficacy and safety of several drugs, such as antiplatelet agent clopidogrel. Taken together, while the process of drug repurposing can be initiated through drug–target or pathway interactions, the actual clinical translation will depend on several additional biological and physiological checkpoints.
Article highlights
AI-guided drug repurposing benefits from large drug–target binding affinity resources for compound off-target activity predictions
Repurposing leads needs to be further explored in cell-based pharmacogenomic resources for drug efficacy and toxicity predictions
A wide variety of supervised machine learning algorithms have been developed for drug–target activity and drug response predictions
There is critical need for context-specific modeling of tissue-specific drug mode of action for more actionable drug repurposing applications
Scattered location of heterogeneous preclinical pharmacogenomic data limits our ability to use these data in AI-based drug repurposing
This box summarizes key points contained in the article.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, et al. Drug repositioning and repurposing: terminology and definitions in literature. Drug Discov Today. 2015;20: 1027–1034. Elsevier Ltd.
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673–683.
- Pantziarka P, Bouche G, Meheus L, et al. The repurposing drugs in oncology (ReDO) project. Ecancermedicalscience. 2014;8.
- Pushpakom S, Iorio F, Eyers PA, et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov. 2019;18:41–58.
- Luo H, Li M, Yang M, et al. Biomedical data and computational models for drug repositioning: a comprehensive review. Brief Bioinform. 2020. Online ahead of print. DOI:https://doi.org/10.1093/bib/bbz176.
- Sam E, Athri P. Web-based drug repurposing tools: a survey. Brief Bioinform. 2019;20:299–316.
- Lotfi Shahreza M, Ghadiri N, Mousavi SR, et al. A review of network-based approaches to drug repositioning. Brief Bioinform. 2018;19:878–892.
- Zhao K, So HC. Using Drug Expression Profiles and Machine Learning Approach for Drug Repurposing. In: Vanhaelen Q, editor. Computational Methods for Drug Repurposing. Methods in Molecular Biology, vol 1903. Humana Press, New York, NY; 2019. https://doi.org/10.1007/978-1-4939-8955-3_13
- Corsello SM, Bittker JA, Liu Z, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23:405–408.
- Shameer K, Glicksberg BS, Hodos R, et al. Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Brief Bioinform. 2017;19:656–678.
- Tanoli Z, Seemab U, Scherer A, et al. Exploration of databases and methods supporting drug repurposing: a comprehensive survey. Brief Bioinform. 2020. Online ahead of print. DOI:https://doi.org/10.1093/bib/bbaa003.
- Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18:463–477.
- Koromina M, Pandi M-T, Patrinos GP. Rethinking drug repositioning and development with artificial intelligence, machine learning, and omics. Omics A J Integr Biol. 2019;23:539–548.
- Duca J, Jansen H, Schneider P, et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov. 2019. DOI:https://doi.org/10.1038/s41573-019-0050-3.
- Palve V, Liao Y, Rix LLR, et al. Turning liabilities into opportunities: off-target based drug repurposing in cancer. Semin Cancer Biol. 2020. Elsevier. DOI:https://doi.org/10.1016/j.semcancer.2020.02.003.
- Rice C, Colon BL, Chen E, et al. Discovery of repurposing drug candidates for the treatment of diseases caused by pathogenic free-living amoebae. PLoS Negl Trop Dis. 2020 Sep 24;14(9):e0008353. doi: https://doi.org/10.1371/journal.pntd.0008353. PMID: 32970675.
- Mohanty S, Rashid MHA, Mridul M, et al. Application of Artificial Intelligence in COVID-19 drug repurposing. Diabetes Metab Syndr Clin Res Rev. 2020. DOI:https://doi.org/10.1016/j.dsx.2020.06.068.
- Pantziarka P, Vandeborne L, Meheus L, et al. Covid19db–An online database of trials of medicinal products to prevent or treat COVID-19, with a specific focus on drug repurposing. medRxiv. 2020. https://doi.org/10.1101/2020.05.27.20114371
- Parisi D, Adasme MF, Sveshnikova A, et al. Drug repositioning or target repositioning: a structural perspective of drug-target-indication relationship for available repurposed drugs. Comput Struct Biotechnol J. 2020;18:1043.
- Bélgamo JA, Alberca LN, Pórfido JL, et al. Application of target repositioning and in silico screening to exploit fatty acid binding proteins (FABPs) from Echinococcus multilocularis as possible drug targets. J Comput Aided Mol Des. 2020;34:1275–1288.
- Klug DM, Gelb MH, Pollastri MP. Repurposing strategies for tropical disease drug discovery. Bioorg Med Chem Lett. 2016;26:2569–2576.
- Pemovska T, Johnson E, Kontro M, et al. Axitinib effectively inhibits BCR-ABL1 (T315I) with a distinct binding conformation. Nature. 2015;519:102–105.
- Zabriskie MS, Eide CA, Yan D, et al. Extreme mutational selectivity of axitinib limits its potential use as a targeted therapeutic for BCR-ABL1-positive leukemia. Leukemia. 2016;30:1418–1421.
- Okabe S, Tauchi T, Tanaka Y, et al. Anti-leukemic activity of axitinib against cells harboring the BCR-ABL T315I point mutation. J Hematol Oncol. 2015;8:97.
- Gilson MK, Liu T, Baitaluk M, et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016;44:D1045–D1053.
- Tamborero D, Rubio Pérez C, Déu Pons J, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018 Dec;10:25. 10 25.
- Gaulton A, Hersey A, Nowotka M, et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016;45:D945–D954.
- Duran-Frigola M, Pauls E, Guitart-Pla O, et al. Extending the small-molecule similarity principle to all levels of biology with the Chemical Checker. Nat Biotechnol. 2020;38:1–10.
- Ursu O, Holmes J, Bologa CG, et al. DrugCentral 2018: an update. Nucleic Acids Res. 2019 [cited 2019 Apr 23];47:D963–D970. Available from: https://academic.oup.com/nar/article/47/D1/D963/5146206
- Tanoli Z, Alam Z, Vähä-Koskela M, et al. Drug Target Commons 2.0: a community platform for systematic analysis of drug–target interaction profiles. Database. 2018;2018:1–13.
- Tanoli Z, Alam Z, Ianevski A, et al. Interactive visual analysis of drug–target interaction networks using Drug Target Profiler, with applications to precision medicine and drug repurposing. Brief Bioinform. 2018 [cited 2019 Mar 26]; Available from: https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bby119/5232987
- Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672.
- Wagner AH, Coffman AC, Ainscough BJ, et al. DGIdb 2.0: mining clinically relevant drug–gene interactions. Nucleic Acids Res. 2015;44:gkv1165.
- Alexander SPH, Kelly E, Marrion NV, et al. The Concise Guide to PHARMACOLOGY 2017/18: overview. Br J Pharmacol. 2017;174.
- Okuno Y, Tamon A, Yabuuchi H, et al. GLIDA: GPCR–ligand database for chemical genomics drug discovery–database and tools update. Nucleic Acids Res. 2008 [cited 2019 Apr 18];36:D907–12. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17986454
- Wang Y, Bryant SH, Cheng T, et al. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2016;45:D955–D963.
- Roth BL, Lopez E, Patel S, et al. The multiplicity of serotonin receptors: uselessly diverse molecules or an embarrassment of riches? Neurosci. 2000 [cited 2019 Apr 23];6:252–262. Available from: http://journals.sagepub.com/doi/10.1177/107385840000600408
- Skuta C, Popr M, Muller T, et al. Probes & Drugs portal: an interactive, open data resource for chemical biology. Nat Methods. 2017 [cited 2019 Aug 28];14:759–760. Available from: http://www.nature.com/articles/nmeth.4365
- Hewett M, Oliver DE, Rubin DL, et al. PharmGKB: the pharmacogenetics knowledge base. Nucleic Acids Res. 2002;30:163–165.
- Hecker N, Ahmed J, von Eichborn J, et al. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012 [cited 2019 Apr 23];40:D1113–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22067455
- Szklarczyk D, Santos A, von Mering C, et al. STITCH 5: augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44:D380–D384.
- Cichonska A, Ravikumar B, Allaway RJ, et al. Crowdsourced mapping of unexplored target space of kinase inhibitors. BioRxiv. 2020:2012–2019. https://doi.org/10.1101/2019.12.31.891812
- Pahikkala T, Airola A, Pietilä S, et al. Toward more realistic drug–target interaction predictions. Brief Bioinform. 2015;16:325–337.
- Cichonska A, Ravikumar B, Parri E, et al. Computational-experimental approach to drug-target interaction mapping: a case study on kinase inhibitors. PLoS Comput Biol. 2017;13:13.
- Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017 [cited 2019 Apr 17];171:1437–1452.e17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29195078
- Seashore-Ludlow B, Rees MG, Cheah JH, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;5:1210–1223.
- Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603.
- Rajapakse VN, Luna A, Yamade M, et al. CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience. 2018 [cited 2019 May 29];10:247–264. Available from: https://www.sciencedirect.com/science/article/pii/S2589004218302190?via%3Dihub
- Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell. 2017 [cited 2019 Aug 28];170:564–576.e16. Available from: https://www.sciencedirect.com/science/article/pii/S0092867417306517
- Cokelaer T, Chen E, Iorio F, et al. GDSCTools for mining pharmacogenomic interactions in cancer. Bioinformatics. 2018;34:1226–1228.
- Klijn C, Durinck S, Stawiski EW, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015;33:306–312.
- Brimacombe KR, Zhao T, Eastman RT, et al. An OpenData portal to share COVID-19 drug repurposing data in real time. bioRxiv. 2020. DOI:https://doi.org/10.1101/2020.06.04.135046.
- Yu C, Mannan AM, Yvone GM, et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol. 2016;34:419–423.
- Mubeen S, Hoyt CT, Gemünd A, et al. The impact of pathway database choice on statistical enrichment analysis and predictive modeling. Front Genet. 2019;10:1203.
- Cerami EG, Gross BE, Demir E, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690.
- Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361.
- Fabregat A, Jupe S, Matthews L, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649–D655.
- Caspi R, Billington R, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018;46:D633–D639.
- Licata L, Lo Surdo P, Iannuccelli M, et al. SIGNOR 2.0, the SIGnaling network open resource 2.0: 2019 update. Nucleic Acids Res. 2020;48:D504–D510.
- Wishart DS, Li C, Marcu A, et al. PathBank: a comprehensive pathway database for model organisms. Nucleic Acids Res. 2020;48:D470–D478.
- Perret J-L, Mahe P, Vert J-P Chemcpp: an open source c++ toolbox for kernel functions on chemical compounds. 2007. Software. Available from: http://chemcpp.sourceforge.net.
- Mauri A, Consonni V, Pavan M, et al. Dragon software: an easy approach to molecular descriptor calculations. Match. 2006;56:237–248.
- O’Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33.
- Landrum G, Penzotti JE, Putta S. Feature-map vectors: a new class of informative descriptors for computational drug discovery. Journal of computer-aided molecular design. 2006;20:751-762. DOI:https://doi.org/10.1007/s10822-006-9085-8
- Cao D-S, Liang Y-Z, Yan J, et al. PyDPI: freely available python package for chemoinformatics, bioinformatics, and chemogenomics studies. J Chem Inf Model. 2013 Nov 25;53(11):3086–3096. doi:https://doi.org/10.1021/ci400127q
- Ekins S, Wrighton SA. Application of in silico approaches to predicting drug-drug interactions. J Pharmacol Toxicol Methods. 2001;45:65–69.
- Lagunin AA, Dubovskaja VI, Rudik AV, et al. CLC-Pred: a freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS One. 2018;13:e0191838.
- Hunter AD, ACD/ChemSketch 1.0 (freeware); ACD/ChemSketch 2.0 and its tautomers, dictionary, and 3D plug-ins; ACD/HNMR 2.0; ACD/CNMR 2.0. J Chem Educ. 1997 [cited 2020 Aug 21];74:905. Available from: http://www.msi.com
- Beisken S, Meinl T, Wiswedel B, et al. KNIME-CDK: workflow-driven cheminformatics. BMC Bioinformatics. 2013 [cited 2020 Aug 21];14:257. Available from: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471–2105–14–257
- Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011 [cited 2020 Aug 21];32:1466–1474. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/jcc.21707
- University of Tübingen: BlueDesc . [cited 2020 Aug 21]. Available from: http://www.ra.cs.uni-tuebingen.de/software/bluedesc/welcome_e.html.
- Pence HE, Williams A. ChemSpider: An Online Chemical Information Resource. J Chem Educ. 2010;87(11):1123–1124. https://doi.org/10.1021/ed100697w
- Chen JH, Linstead E, Swamidass SJ, et al. ChemDB update full-text search and virtual chemical space. Bioinformatics. 2007 [cited 2019 Mar 27];23:2348–2351. Available from: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btm341
- Moghadam PZ, Li A, Wiggin SB, et al. Development of a Cambridge Structural Database Subset: A Collection of Metal–Organic Frameworks for Past, Present, and Future. Chem Mater. 2017;29(7):2618–2625. https://doi.org/10.1021/acs.chemmater.7b0044
- Tomasulo P, ChemIDplus–super source for chemical and drug information. Med Ref Serv Q. 2002 [cited 2020 Aug 21];21:53–59. Available from: https://pubmed.ncbi.nlm.nih.gov/11989279/
- Sterling T, Irwin JJ, ZINC 15 - Ligand Discovery for Everyone. J Chem Inf Model. 2015 [cited 2020 Aug 21];55:2324–2337. Available from: https://clinicaltrials.gov
- Lavecchia A, Cerchia C. In silico methods to address polypharmacology: current status, applications and future perspectives. Drug Discov Today. 2016;21:288–298. Elsevier Ltd.
- Zheng M, Liu X, Xu Y, et al. Computational methods for drug design and discovery: focus on China. Trends Pharmacol Sci. 2013 [cited 2020 Aug 21];34:549–559. Available from: https://pubmed.ncbi.nlm.nih.gov/24035675/
- Chen X, Yan CC, Zhang X, et al. Drug–target interaction prediction: databases, web servers and computational models. Brief Bioinform. 2015;17:696–712.
- Daina A, Michielin O, Zoete V, SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019 [cited 2019 Aug 9];47:W357–W364. Available from: https://academic.oup.com/nar/article/47/W1/W357/5491750
- Mervin LH, Bulusu KC, Kalash L, et al. Orthologue chemical space and its influence on target prediction. Bioinformatics. 2018 [cited 2019 Aug 9];34:72. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5870859/
- Cichonska A, Pahikkala T, Szedmak S, et al. Learning with multiple pairwise kernels for drug bioactivity prediction. Bioinformatics. 2018;34:i509–i518.
- Li Y, Huang Y-A, You Z-H, et al. Drug-target interaction prediction based on drug fingerprint information and protein sequence. Molecules. 2019;24:2999.
- Chen R, Liu X, Jin S, et al. Machine learning for drug-target interaction prediction. Molecules. 2018;23:2208.
- Sachdev K, Gupta MK. A comprehensive review of feature based methods for drug target interaction prediction. J Biomed Inform. 2019;93:103159.
- Wu Z, Li W, Liu G, et al. Network-based methods for prediction of drug-target interactions. Front Pharmacol. 2018 [cited 2019 May 29];9:1134. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30356768
- Thafar MA, Olayan RS, Ashoor H, et al. DTiGEMS+: drug–target interaction prediction using graph embedding, graph mining, and similarity-based techniques. J Cheminform. 2020;12:1–17.
- Mongia A, Majumdar A, Olier I. Drug-target interaction prediction using multi graph regularized nuclear norm minimization. PLoS One. 2020;15:e0226484.
- Jiang M, Li Z, Zhang S, et al. Drug–target affinity prediction using graph neural network and contact maps. RSC Adv. 2020;10:20701–20712.
- Zhao T, Hu Y, Valsdottir LR, et al. Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief Bioinform. 2020. DOI: https://doi.org/10.1093/bib/bbaa044.
- Cichonska A, Rousu J, Aittokallio T. Identification of drug candidates and repurposing opportunities through compound–target interaction networks. Expert Opin Drug Discov. 2015;10:1333–1345.
- Mei S, Zhang K. A multi-label learning framework for drug repurposing. Pharmaceutics. 2019;11:466.
- Chen H, Cheng F, Li J. IDrug: integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput Biol. 2020 [cited 2020 Oct 20];16. Available from: https://pubmed.ncbi.nlm.nih.gov/32667925/
- Beck BR, Shin B, Choi Y, et al. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J. 2020 [cited 2020 Oct 20];18:784–790. Available from: https://pubmed.ncbi.nlm.nih.gov/32280433/
- Pujadas G, Vaque M, Ardevol A, et al. Protein-ligand docking: a review of recent advances and future perspectives. Curr Pharm Anal. 2008;4:1–19.
- Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 2007;25:71–75.
- Li H, Gao Z, Kang L, et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006;34:W219–W224.
- Jacob L, Vert J-P. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics. 2008;24:2149–2156.
- Pinzi L, Rastelli G. Molecular docking: shifting paradigms in drug discovery. Int J Mol Sci MDPI AG. 2019 [cited 2020 Aug 21];20:4331. Available from: /pmc/articles/PMC6769923/?report=abstract
- Baig MS, Roy A, Saqib U, et al. Repurposing Thioridazine (TDZ) as an anti-inflammatory agent. Sci Rep. 2018;8:8.
- Mellini M, Di Muzio E, D’Angelo F, et al. In silico selection and experimental validation of FDA-approved drugs as anti-quorum sensing agents. Front Microbiol. 2019 [cited 2019 Nov 8];10:2355. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31649658
- Callaway E. “It will change everything”: deepMind’s AI makes gigantic leap in solving protein structures. Nature. 2020;588:203–204.
- Shi Y, Zhang X, Mu K, et al. D3Targets-2019-nCoV: a webserver for predicting drug targets and for multi-target and multi-site based virtual screening against COVID-19. Acta Pharm Sin B. 2020 [cited 2020 Aug 21];10:1239. Available from: /pmc/articles/PMC7169934/?report=abstract
- Ravikumar B, Timonen S, Alam Z, et al. Chemogenomic analysis of the druggable kinome and its application to repositioning and lead identification studies. Cell Chem Biol. 2019;26:1608–1622.
- Gilvary C, Elkhader J, Madhukar N, et al. A machine learning and network framework to discover new indications for small molecules. PLoS Comput Biol. 2020 [cited 2020 Oct 20];16:16. Available from: https://pubmed.ncbi.nlm.nih.gov/32764756/
- Peng Y, Wang M, Xu Y, et al. Drug repositioning by prediction of drug’s anatomical therapeutic chemical code via network-based inference approaches. Brief Bioinform. 2020 [cited 2020 Oct 20]; Available from: https://pubmed.ncbi.nlm.nih.gov/32221552/
- Costello JC, Heiser LM, Georgii E, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 2014;32:1202–1212.
- Gönen M, Khan S, Kaski S. Kernelized Bayesian matrix factorization. Int Conf Mach Learn. 2013;864–872.
- Güvenç Paltun B, Mamitsuka H, Kaski S. Improving drug response prediction by integrating multiple data sources: matrix factorization, kernel and network-based approaches. Brief Bioinform. 2021;22:232-246.
- Chen J, Zhang L. A survey and systematic assessment of computational methods for drug response prediction. Brief Bioinform. 2021;22:232–246.
- Wang L, Li X, Zhang L, et al. Improved anticancer drug response prediction in cell lines using matrix factorization with similarity regularization. BMC Cancer. 2017;17:1–12.
- Ali M, Aittokallio T. Machine learning and feature selection for drug response prediction in precision oncology applications. Biophys Rev. 2019;11:31–39.
- Knowles DA, Bouchard G, Plevritis S. Sparse discriminative latent characteristics for predicting cancer drug sensitivity from genomic features. PLOS Comput Biol. 2019;15:e1006743.
- Koras K, Juraeva D, Kreis J, et al. Feature selection strategies for drug sensitivity prediction. Sci Rep. 2020;10:1–12.
- Schätzle L-K, Esfahani AH, Schuppert A. Methodological challenges in translational drug response modeling in cancer: a systematic analysis with FORESEE. PLOS Comput Biol. 2020;16:e1007803.
- Huang EW, Bhope A, Lim J, et al. Tissue-guided LASSO for prediction of clinical drug response using preclinical samples. PLoS Comput Biol. 2020;16:e1007607.
- Friedman AA, Letai A, Fisher DE, et al. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15:747–756.
- Dienstmann R, Tabernero J. Cancer: a precision approach to tumour treatment. Nature. 2017;548:40–41.
- Letai A. Functional precision cancer medicine—moving beyond pure genomics. Nat Med. 2017;23:1028.
- Giri AK, Ianevski A, Aittokallio T. Genome-wide off-targets of drugs: risks and opportunities. Cell Biol Toxicol. 2019;35:485–487.
- Brown AS, Patel CJ. A review of validation strategies for computational drug repositioning. Brief Bioinform. 2018;19:174–177.
- Tang J, Tanoli Z-R, Ravikumar B, et al. Drug Target Commons: a Community Effort to Build a Consensus Knowledge Base for Drug-Target Interactions. Cell Chem Biol. 2018;25:224–229.
- Smirnov P, Kofia V, Maru A, et al. PharmacoDB: an integrative database for mining in vitro anticancer drug screening studies. Nucleic Acids Res. 2018;46:D994–D1002.
- van der Meer D, Barthorpe S, Yang W, et al. Cell model passports—a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res. 2019;47:D923–D929.
- Mer AS, Ba-Alawi W, Smirnov P, et al. Integrative pharmacogenomics analysis of patient-derived xenografts. Cancer Res. 2019;79:4539–4550.
- Valli D, Gruszka AM, Alcalay M. Has drug repurposing fulfilled its promise in acute myeloid leukaemia? J Clin Med. 2020;9:1892.
- Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nature Communications. 2020;11. Article number: 5327.
- Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25:744–750.
- Gandhi A, Moorthy B, Ghose R. Drug disposition in pathophysiological conditions. Curr Drug Metab. 2012;13:1327–1344.
- Tyson RJ, Park CC, Powell JR, et al. Precision dosing priority criteria: drug, disease, and patient population variables. Front Pharmacol. 2020;11:420.
- Xu Q, Zhai J-C, Huo C-Q, et al. OncoPDSS: an evidence-based clinical decision support system for oncology pharmacotherapy at the individual level. BMC Cancer. 2020;20:1–10.