
1. Introduction
Analyses of cancer landscapes have established multiple activating mutations prompting deliberations on the minimal number required for driving cancer [Citation1]. These are likely to depend on their locations and scenarios. Multiple mutations can co-occur in different genes that encode proteins functioning (i) in the same signaling pathway, (ii) in parallel (redundant) pathways or (iii) in complementary pathways that can combine in cell proliferation [Citation2]. The first strengthens the signal of the specific pathway where the mutations occur. The second drives cancer through an alternative pathway in drug resistance. In the third, the mutations are in complementary pathways. In such a scenario, one can promote cell division and the other cell growth, as in the case of MAPK and PI3K/Akt/mTOR in the G1 to S-phase transition in the cell cycle. MAPK phosphorylation cascade and the role of its downstream ERK activation in proliferation, invasion and metastasis have long been the focus of intense research efforts, and recently increasingly PI3K/Akt/mTOR, one of the most dysregulated pathways in cancer, with over half of the tumors exhibiting aberrant AKT activation [Citation3]. The three groups of multiple mutations are correspondingly illustrated schematically in . Analysis of emerging data now discovers a fourth distinct group (iv) where the double/multiple driver mutations are in cis, that is, within the same gene (allele). These mutations have also been positively selected and are frequent in cancer. The analysis has indicated that they can be derived from an overrepresentation of functionally weak and infrequent mutations that together stimulate more potent oncogenicity, or from a combination of weak and strong mutations [Citation4,Citation5]. Cis mutations act on the same protein, additively or cooperatively more potently activating it [Citation6]. This distinction, same gene versus different genes mutations, is of critical importance since it implicates different functional mechanisms. Especially, it also raises the question of more effective targeted pharmacology. As we discuss below, the combinations of the mutations point to a more powerful oncogenic signaling, but at the same time, higher drug sensitivity and possibly, higher specificity.
Figure 1. Multiple mutations can drive cancer through at least four mechanisms. From left to right: 1) Multiple mutations in different proteins along the same signaling pathway can strengthen oncogenic signals. 2) Multiple mutations in different proteins in parallel pathways. 3) Multiple mutations in different proteins in complementary pathways. 4) Multiple mutations incis, that is, on the same protein, can also activate oncogenic signaling more potently than their single driver components
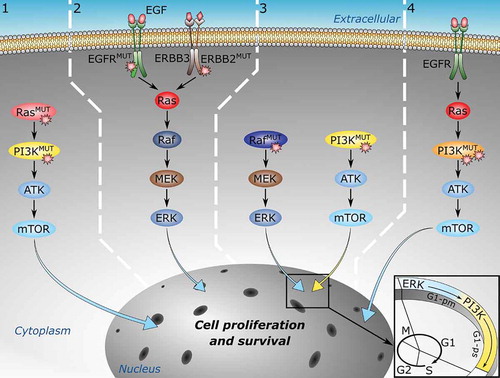
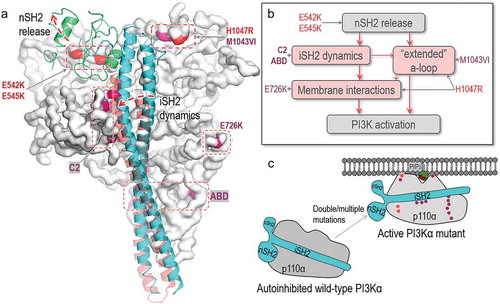
Figure 2. Structural insights into oncogenic mutations in PI3Kα. (A) PI3Kα has hotspot mutations and weak mutations. The hotspot mutations include E542K, E545K in the helical domain and H1047R in the kinase domain. The weak mutations include E726K and M1043VI in the kinase domain, N345K, C420R, E453K/Q in the C2 domain and R38H/C, R88Q, R93Q, R108H, G118D in the ABD domain. The p110α catalytic subunit is shown as the white surface. The hotspot and weak mutations are highlighted in red and pink. The iSH2 and nSH2 domains in p85α regulatory subunit are shown in cyan and green cartoon. (B) The roles of PI3Kα mutations in activation suggest that the hotspot and weak mutations induce the conformational changes for PI3Kα activation. They can be additive or cooperative. “a-loop” denotes activation loop. (C) The schematic diagram shows the activation of PI3Kα by oncogenic mutations
Multiple mutations can drive cancer through at least four mechanisms. From left to right: 1) Multiple mutations in different proteins along the same signaling pathway can strengthen oncogenic signals. 2) Multiple mutations in different proteins in parallel pathways. 3) Multiple mutations in different proteins in complementary pathways. 4) Multiple mutations in cis, that is, on the same protein, can also activate oncogenic signaling more potently than their single driver components.
2. Driver mutations can be strong or weak; frequent or rare
Recent reports pointed to double/multiple cis mutations commonly consisting of individually weak mutations that collectively promote oncogenesis [Citation5] or combinations where one mutation is a strong driver, the other weak [Citation4]. Combinations of strong drivers have not been observed in the smaller scale analysis [Citation4] but were observed in large-scale studies [Citation5]. The observation that weak drivers can additively combine to drive cancer signals was expected and can be understood [Citation7].
Driver mutations are largely discovered through their statistics, that is, their frequencies of occurrence in cancer genomes [Citation8]. Rare drivers have extremely low frequencies. With low statistics, at the tail of the distribution, rare driver mutations are challenging to detect. Unlike high-frequency drivers, low-frequency, or weak drivers can be tissue specific [Citation9]. This raises the question – if driver mutations are under positive selection pressure for potent oncogenic signaling, why are they often statistically infrequent or rare [Citation7]? To explain this conundrum, it was proposed that it could be a matter of the statistical calculation to detect drivers. In order to switch the protein functional state from an inactive conformation to an active one, two or more mutations need to act together, and this may hold for hot spot drivers and weak drivers. Whereas sequence analyses of the cancer genome identify the high frequency hot spots, they may not identify the low-frequency ones. These could be discovered if the analyses were performed for pairs or more residues; however, the small sample sizes could prove prohibitive for such analyses.
Driver mutations can occur directly at functional sites, in which case they are orthosteric drivers; however, they are frequently allosteric [Citation10]. Allosteric drivers commonly work through a conformational change, by shifting the conformational ensemble of the protein. Thus, a binary classification into Yes (driver, observable functional change) or No (passenger, no observable cancer hallmarks) is inaccurate since there can be a functional change – except that it may not be sufficiently strong to have an observable effect. This argues for an additional driver category. A ‘latent driver’ is an allosteric mutation that alone cannot execute oncogenic activation; however, in combination with another mutation it can [Citation7].
3. Why double (multiple) driver mutations can activate more potently than the single drivers
Oncogenic mutations are commonly observed to be differentially expressed in tumors [Citation11,Citation12]. Although mostly unidentified (thus appearing rare), weak drivers are expected to be common. Rare and weak activating mutations can promote proliferation in vivo and in vitro, however, they can collaborate with hotspot (strong, common) mutations additively leading to more potent activation. A combination of strong and weak drivers can enhance activity and proliferation at a level that can be sustained. A pair of weak mutations may also additively promote proliferation. Now we know that double/multiple mutations are indeed common and promote proliferation.
With the available crystal structures [Citation13,Citation14], detailed activation mechanism at the atomic scale [Citation15,Citation16], and experimental and clinical data [Citation4,Citation5], PI3Kα lipid kinase provides a good example that can help in understanding how a combination of strong and weak, or other mutational pairs, can work. PI3Kα is a lipid kinase in the PI3K/Akt/mTOR signaling pathway feeding into the cell cycle to promote cell growth via phosphorylation of signaling lipid PIP2 to PIP3 at the membrane [Citation17]. Aberrant signaling by the PI3K pathway is associated with multiple diseases related to PIP3 production and regulation of downstream effectors. Analyses of available data of its cis double mutations suggest that as expected [Citation6,Citation7], a combination of its weak mutations can drive cancer as well. Phosphorylation events play a key role in PI3Kα autoinhibition and activation [Citation18].
PI3Kα activation requires relieving the autoinhibition exerted by the nSH2 domain of the p85α subunit and having the kinase domain favorably associated with the membrane. Its driver mutations act to fulfill both requirements. Under physiologically regulated conditions, the high-affinity interaction of the nSH2 with a phosphorylated tyrosine motif at the C-terminal of an activated receptor tyrosine kinase (RTK) triggers conformational changes that result in exposure to the active site. Hotspot mutations E542K and E545K at the surface of the helical domain of p110α, mimic and compete with the RTK–nSH2 interactions, reorganizing the active site. With opposite charges, the lysine residues disrupt the interfacial salt bridges. In turn, hotspot mutation H1047R, on the surface of the kinase domain strengthens the membrane interaction, substituting for the role exerted by active Ras under physiological conditions. PI3Kα also has rare and weak activating mutations [Citation19]. Clinically, in breast cancer, weak mutations (E453 K/Q, E726K, and M1043V/I) are often coupled with the hotspots (E542K, E545K, and H1047R) [Citation4]. Their strong additive effects can be understood by their location in PI3Kα and the mechanism through which they can contribute to activation ().
E726K is on the surface of the N-lobe of the kinase domain, and M1043V/I is in the interior of the C-lobe. Similar to H1047R on the surface of the C-lobe, E726K charge reversal enhances the interaction with the membrane. E726K is weaker than H1047R since it is close to other positive residues. However, together, the mutations accomplish a stronger outcome. The M1043V/I mutations are in the regulatory arch, strengthening the hydrophobic core interactions. Its mutation to valine or isoleucine with shorter hydrophobic side chains may alter the local hydrophobic interactions with the activation loop in the kinase domain, promoting activation. Its weak effect may imply that it is a latent driver. The weak mutations in the C2 domain of p110α (N345K, C420R, and E453 K/Q) and ABD (R38H/C, R88Q, R93Q, R108H, and G118/D) can also enhance transformation through the iSH2 domain which acts in the release of the autoinhibition.
Above, we focused on PI3Kα. PI3Kα has abundant experimental and clinical data [Citation4,Citation5], and its detailed, atomistic-scale mechanism of activation [Citation6], can explain the double/multiple observed driver mutations. Multiple driver mutations were also observed in 10% of the EGFR samples and in at least 5% of the samples from 9 other oncogenes that were explored. Overall, significant enrichments of multiple driver mutations were identified in 14 oncogenes that act in the PI3K/AKT/mTOR pathway, including PPP2R1A and mTOR, members of the RAS family of GTPases (KRAS and NRAS), and receptor tyrosine kinases (e.g. EGFR, ERBB2). The collaborative contributions of the mutations in these proteins still await detailed mechanistic analysis. The recent availability of the cryo-electron microscopy structures of mTOR1 [Citation20,Citation21] could facilitate mTOR exploration. There too, we expect additive contributions which would enhance their oncogenicity. Minor somatic mutations were observed to be common in cis in oncogenes that contained major hotspots, including EGFR A289, CTNNB1 S33 and S37, and KRAS Q61, and near them. Combinations of major hotspots, such as NRAS/KRAS G12–G13 mutation pairs, were observed on different alleles. The mechanisms of these are different and complex and are not discussed here.
4. Identification of weak and rare driver mutations
Double/multiple mutations can consist of a combination of weak and/or rare drivers, weak and strong or only strong, as observed by Kataoka and his colleagues [Citation5], although these are more challenging to understand considering that strong activation can lead to senescence or to apoptosis, restricting proliferation thus acting as tumor suppression mechanisms. Oncogene-induced senescence (OIS) can be driven by excessive RAS/ERK signaling, and by strong constitutive PI3K/AKT/mTORC1 pathway activation which promotes an OIS-like phenotype. For example, the so-called AKT-induced senescence (AIS) can be promoted by constitutively active AKT, and by PIK3CA driver mutations, along with inactivation, or deletion of the PTEN tumor repressor gene [Citation22].
Strong driver mutations are frequent and statistics-based strategies can discover them in the cancer genome as, for example, shown by the TCGA initiative [Citation23]. A strong driver possesses a potent activation mechanism. The potency of a weak driver is lower. That is, it may not populate cancers where the protein is a major operator. The case of K-RasG12D versus K-RasA146T provides an example. K-RasA146T is a weak driver. It is mostly in colorectal and hematopoietic cancers, but not in pancreatic cancer [Citation24] where K-RasG12D is extremely highly populated [Citation25]. K-RasA146T acts by promoting nucleotide exchange rather than blocking GTP hydrolysis, which is the common mechanism for strong K-Ras mutations, such as K-RasG12D. Cell-specific analysis can detect K-RasA146T in the tissues it populates.
Identification of rare drivers is challenging. Under such circumstances, in addition to crystallography, NMR and long-timescale molecular dynamic simulations can pinpoint their conformation (e.g. state 1 or state 2 for Ras) and capture the changes in the atomic interactions as shown for K-RasD33E [Citation26]. Computational strategies can also include functional impact score and three-dimensional mutational clustering analyses [Citation7]. The mechanism adopted by the mutation, which can be inferred from the structure and its conformational dynamics, is also indicative. One tell-tale clue is autoinhibition; if the mutation relieves it – it is a driver no matter its frequency.
5. Conclusions
Single driver mutations can be insufficient for oncogenic cell survival and proliferation, leading to efforts to estimate the minimal number of mutations needed for cancers to develop [Citation1], which may depend on the cancer type [Citation27]. Cancer landscapes provide a wealth of information on positive selection of the mutations in the cancer cell. These data couple with detailed structural studies of single driver mutations and the mechanisms through which they activate the respective proteins. However, recently, large-scale experimental mapping observed that the double/multiple mutations are also in-cis, that is, in the same gene. These observations are significant because of their implications in drug discovery. They raise the question of whether, or to what extent, drugs targeting a protein harboring multiple mutations in cis should differ from those initially designed against single drivers and argue for an extension of the drug discovery effort to these mutants as well.
The mechanistic descriptions provided here for single and multiple collaborating mutations point to complementary and additivity in their activation of the protein, which attests to their enhanced mutational strength, but not to new mechanistic scenarios. The stronger signaling outcome also points to the expected more potent drug action, but not to a new drug paradigm since the mechanisms of activation in the wild type, in single mutants, and in double/multiple mutants, are essentially the same. Thus, drugs designed for one of these are likely to work for the others as well. However, as we discuss below, the double/multiple co-occurring mutations in cis scenarios also indicate a likelihood of conformational and dynamic alterations which can be exploited in drug optimization. At the same time, the problem of drug resistance through alterations in the expression and mutations in other proteins – downstream in the same pathway or in parallel pathways – remains imminent [Citation28].
6. Expert opinion
The cancer genome landscape of multiple strong driver mutations coexisting in the same allele is still unclear. The lack of their observation in an earlier analysis [Citation4] can be attributed to the statistical sample size and low frequencies. They may also be inherently disfavored under certain conditions due to a possible senescence (or apoptosis) outcome. A similar argument has been advanced for activation mutations in PI3Kα which are not favored to co-exist with deactivating mutations in PTEN since both result in high levels of signaling lipid PIP3. Multiple drivers commonly co-exist in different proteins within the same or parallel pathways in drug resistance (TCGA [Citation1]). Recently, multiple co-occurring drivers were documented in cis on a large scale in PIK3CA, NOTCH1 and EGFR. The recorded combinations of strong and weak or rare drivers, or only weak/rare drivers, demonstrate the abundance of preexisting, weak mutations before cancer emerges. Alone, their functional change is subtle; but when combined with another mutation, the signal is strengthened leading to stronger activation. The more potent oncogenic signal is also more strongly inhibited even with the same drugs [Citation29]. The fact that nearly a quarter of all tumors have two different mutations in the same gene [Citation30] emphasizes the significance of the observation of the multiple mutations in-cis. Considering that they act additively, the function of the segment where they appear, and its environment may be the more relevant features than their locations along the sequence.
At the same time, mutations where detailed mechanistic structural data are available suggest that their mechanisms mimic those of their single mutation components in the same protein, no matter their specific driver combinations. This is reasonable: the activation mechanism of driver mutations has been established to follow that of the wild-type protein – except that it is unregulated. We expect that this trend will persist. However, minor conformational changes can take place, exposing surfaces, deepening pockets, or for PI3Kα altering the ATP pocket enabling a more isoform-specific drug design. To uncover these, conformational dynamics from detailed studies is expected to be helpful. Such discoveries may foretell a new generation of more potent higher affinity drugs. Combinatorial pharmacology can then target the mutant protein and protein(s) in a parallel signaling pathway likely to emerge in drug resistance.
Data suggest that drugs inhibit more effectively double mutations than single mutations [Citation4]. This is expected considering the more potent outcome of double/multiple mutations [Citation6]. Exploiting oncogenic proteins with double/multiple mutations in drug discovery may prove a productive route even for single mutants where no specific effective drugs are available, as in the case of PI3Kα [Citation29]. Even though the activation mechanisms of proteins with single mutations and double mutations are similar, with both mimicking the activation mechanism of the wild type, the conformational details will vary given the residue substitutions, thus altering chemistry, volume, and interactions. Double/multiple mutations can uncover hidden pockets existing in only a low population of the single mutants. However, the conformation containing them can become stabilized by the double mutations, and thus more populated. Their discovery will make not only the double mutant but also the single hotspot mutant targetable by drugs fitting the now exposed cavity. This rationale holds since every protein exists in an ensemble of conformations. Covalent (e.g. mutations, post-translational modifications) and noncovalent bonds (interactions), or changes in the environment will not alter the conformations in the ensemble. They will however alter their relative abundance. That is, conformations which are abundant in the single mutants may not be as abundant for the double/multiple mutants, and those which have a high fraction in the double mutants may exist in only a low fraction in the single mutant protein. Detailed conformational analysis by crystallography, NMR and molecular dynamics simulations may discover these. Combined with clinical data on the relative occurrences of certain combinations of mutations in patients may yield clues to emerging targetable cavities. Active, ongoing research proceeds within this framework. At the same time, innovative, non-mechanistic approaches for single or multiple mutations are being conceived as, for example, an alternative strategy for targeting AKT, involving a pan-AKT degrader consisting of an ATP-competitive AKT inhibitor conjugated to lenalidomide, a recruiter of the E3 ubiquitin ligase substrate adaptor Cereblon [Citation3].
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations that imply endorsement by the U.S. Government.
Additional information
Funding
References
- Martincorena I, Raine KM, Gerstung M, et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell. 2017 Nov 16;171(5):1029–1041 e21.
- Nussinov R, Tsai CJ, Jang H. Are Parallel Proliferation Pathways Redundant? Trends Biochem Sci. 2020 Jul;45(7):554–563.
- You I, Erickson EC, Donovan KA, et al. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem Biol. 2020 Jan 16;27(1):66–73 e7.
- Vasan N, Razavi P, Johnson JL, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3K alpha inhibitors. Science. 2019 Nov 8;366(6466):714–723.
- Saito Y, Koya J, Araki M, et al. Landscape and function of multiple mutations within individual oncogenes. Nature. 2020 Jun;582(7810):95–99.
- Zhang M, Jang H, Nussinov R. PI3K driver mutations: a biophysical membrane-centric perspective. Cancer Res. 2020. 10.1158/0008-5472.CAN-20-0911
- Nussinov R, Tsai CJ, Jang H. Why are some driver mutations rare? Trends Pharmacol Sci. 2019 Dec;40(12):919–929.
- Brown AL, Li M, Goncearenco A, et al. Finding driver mutations in cancer: elucidating the role of background mutational processes. PLoS Comput Biol. 2019 Apr;15(4):e1006981.
- Poulin EJ, Bera AK, Lu J, et al. Tissue-specific oncogenic activity of KRAS A146T. Cancer Discov. 2019 Jun;9(6):738–755.
- Tan ZW, Guarnera E, Tee WV, et al. AlloSigMA 2: paving the way to designing allosteric effectors and to exploring allosteric effects of mutations. Nucleic Acids Res. 2020 Jul 2;48(W1):W116–W124.
- Xue JM, Liu Y, Wan LH, et al. Comprehensive analysis of differential gene expression to identify common gene signatures in multiple cancers. Med Sci Monit. 2020 Feb;8(26):e919953.
- Souza-Santos PT, Soares Lima SC, Nicolau-Neto P, et al. Mutations, differential gene expression, and chimeric transcripts in esophageal squamous cell carcinoma show high heterogeneity. Transl Oncol. 2018 Dec;11(6):1283–1291.
- Burke JE, Perisic O, Masson GR, et al. Oncogenic mutations mimic and enhance dynamic events in the natural activation of phosphoinositide 3-kinase p110alpha (PIK3CA). Proc Natl Acad Sci U S A. 2012 Sep 18;109(38):15259–15264.
- Huang CH, Mandelker D, Schmidt-Kittler O, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007 Dec 14;318(5857):1744–1748.
- Zhang M, Jang H, Nussinov R. The mechanism of PI3Kalpha activation at the atomic level. Chem Sci. 2019Mar28;10(12):3671–3680.
- Chakrabarti M, Gabelli SB, Amzel LM. Allosteric activation of PI3Kalpha results in dynamic access to catalytically competent conformations. Structure. 2020 Apr 7;28(4):465–474 e5.
- Tangye SG. Defects in human B-cell development and differentiation due to disease-causing mutations in the Pi3k pathway. J Clin Immunol. 2016 Apr;36(3):298–299.
- Nussinov R, Zhang M, Tsai CJ, et al. Phosphorylation and driver mutations in PI3Kalpha and PTEN autoinhibition. Mol Cancer Res. 2020 Dec;7. DOI:10.1158/1541-7786.MCR-20-0818
- Sun M, Hillmann P, Hofmann BT, et al. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc Natl Acad Sci U S A. 2010 Aug 31;107(35):15547–15552.
- Anandapadamanaban M, Masson GR, Perisic O, et al. Architecture of human Rag GTPase heterodimers and their complex with mTORC1. Science. 2019 Oct 11;366(6462):203–210.
- Rogala KB, Gu X, Kedir JF, et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science. 2019 Oct 25;366(6464):468–475.
- Chan KT, Blake S, Zhu H, et al. A functional genetic screen defines the AKT-induced senescence signaling network. Cell Death Differ. 2020 Feb;27(2):725–741.
- Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018 Apr 5;173(2):371–385 e18.
- Bera AK, Lu J, Wales TE, et al. Structural basis of the atypical activation mechanism of KRAS(V14I). J Biol Chem. 2019 Sep 20;294(38):13964–13972.
- Jiang W, Li H, Liu X, et al. Precise and efficient silencing of mutant Kras(G12D) by CRISPR-CasRx controls pancreatic cancer progression. Theranostics. 2020;10(25):11507–11519.
- Lu J, Bera AK, Gondi S, et al. KRAS Switch Mutants D33E and A59G Crystallize in the State 1 Conformation. Biochemistry. 2018 Jan 23;57(3):324–333.
- Martinez-Jimenez F, Muinos F, Sentis I, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020 Oct;20(10):555–572.
- Li K, Du Y, Li L, et al. Bioinformatics approaches for anti-cancer drug discovery. Curr Drug Targets. 2020;21(1):3–17.
- Zhang M, Jang H, Nussinov R. PI3K inhibitors: review and new strategies. Chem Sci. 2020 Jun 21;11(23):5855–5865.
- Gorelick AN, Sanchez-Rivera FJ, Cai Y, et al. Phase and context shape the function of composite oncogenic mutations. Nature. 2020 Jun;582(7810):100–103.