
1. Introduction
Alzheimer’s disease (AD) is a priority health problem in advanced societies and in many emerging and developing countries, with high cost for public health services, an overload on the economy of families and the consumption of medical and health resources. The cost of dementia exceeds US$800 billion worldwide (>1% of GDP), with an average cost per patient/year that fluctuates between $30,000 and $60,000, depending on the country, social status, quality of medical care and the stage of the disease. The cost of pharmacological treatment for AD and concomitant pathologies represents 20–30% of the overall costs of the disease (approximately €7,000/year in Europe, $15,000/year in the USA, and JPY18 million/year in Japan) [Citation1,Citation2].
Among different forms of dementia, AD is the most frequent (50–60%). Vascular dementia (30–40%), other forms of degenerative dementia (10–15%), and mixed dementia (>70% in patients over 75 years of age) are also relevant. AD is more prevalent in females than in males (mean prevalence: 1–2% at the age of 60; >35% in people older than 80 years). Epidemiological predictions suggest an increase in the prevalence of dementia of approximately 1–2% per year in parallel with the increase in life expectancy of the population.
AD results from the premature death of neurons caused by different factors, including genomic, epigenomic, cerebrovascular and multiple environmental conditions. The clinical picture resulting from the accumulation of pathogenic factors is characterized by progressive cognitive impairment, behavioral alterations (agitation, anxiety, depression, changes in personality and biorhythms) and functional decline (with compromise of psychomotricity and the management of common activities of daily living) [Citation3].
From a nosological point of view, this form of primary degenerative dementia can be conventionally differentiated into (i) early-onset AD (EOAD)(<65 years), commonly associated with Mendelian mutations in high-penetrance pathogenic genes (sometimes, also called familial AD, FAD), and (ii) late-onset AD (LOAD)(>65 years)(also improperly called sporadic AD, sAD), in which multiple genetic defects (susceptibility single nucleotide polymorphisms, SNPs)(>600 genes) and environmental factors may converge with the involvement of epigenetic aberrations to induce progressive brain damage [Citation4,Citation5].
The neuropathological phenotype of AD is characterized by age-related dendritic desarborization, microglial reactivity, astrogliosis, and neuronal loss in critical regions of the CNS, with the presence of extracellular deposits of aggregated β-amyloid (Aβ) in neuritic plaques and vessels, formed due to abnormal processing of APP, together with synergistic intracellular inclusions of neurofibrillary tangles (NFTs), resulting from the hyperphosphorylation of tau protein in microtubules and neurofilaments. Among neurochemical findings, as a consequence of neurodegeneration, AD brains exhibit neurotransmitter deficits (cholinergic, noradrenergic, dopaminergic, serotonergic, glutamatergic, GABAergic, neuropeptidergic), neurotrophic alterations, biochemical markers of neuroinflammatory reactions and disruption of lipid rafts due to oxidative stress-induced lipid peroxidation. All these deleterious events are accompanied by hypoperfusion-related cerebrovascular damage [Citation5–11].
The main challenges facing the scientific community, the medical services and the pharmaceutical industry today are (i) to deepen into a better understanding of the primary causes of the disease and its pathogenic mechanisms, (ii) the characterization and validation of reliable biomarkers that allow for an early diagnosis, (iii) the identification and development of new drugs and therapeutic strategies able to slow-down or halt the course of the disease, and (iv) under optimal conditions, develop new preventive protocols capable of blocking the evolution of the disease in the population at risk at presymptomatic stages, taking into account that brain damage in AD begins several decades before the clinical manifestation of symptoms of dementia [Citation8,Citation10,Citation11].
2. Pathogenic meltdown and cumulative risks
Over the past 30 years, more than 600 genes distributed throughout the human genome have been linked to the risk of AD. Several pathogenic mutations in the amyloid precursor protein (APP) (>50 mutations), PSEN1 (>300 mutations) and PSEN2 genes (>40 mutations), present in less than 10% of AD cases, confer AD the condition of a brain amyloidopathy; MAPT (microtubule-associated protein tau) gene mutations (>100, responsible for diverse tauopathies: frontotemporal dementia, Pick’s disease) link AD to other tauopathies, although MAPT variants are not specific to prototypal forms of AD. Amyloidopathy and tauopathy have been the two dominant hypotheses in the etiopathogenesis of AD for years [Citation12,Citation13].
Missense APP mutations in EOAD cause AD. Other coding variants (APP A673T) may reduce the risk for AD. AD risk-associated mutations in the APP gene increase Aβ fibrillogenesis and total Aβ levels; in contrast, protective alleles may reduce Aβ levels [Citation12].
Presenilin acts as the catalytic site of γ-secretase and the most dominant mutations associated with familial EOAD occur either in the APP gene encoding the APP substrate or in the PSEN1 and PSEN2 genes encoding the protease (presenilin) responsible for APP cleavage, leading to abnormal Aβ accumulation and deposition in senile plaques and vessels. Apolipoprotein E4 (APOE-4) is the most important risk factor for AD in > 40% of cases. The presence of the APOE-4 allele impairs Aβ clearance from brain tissue. Immunotherapy with different Aβ antibodies (solanezumab, crenezumab, and aducanumab) attempt to reduce Aβ accumulation and slow-down cognitive decline in presymptomatic and/or mild-AD cases, as a novel line of therapeutic intervention [Citation12,Citation13].
In addition to these pathogenic genes, novel mutations have been identified in association with AD in recent NGS (next-generation sequencing) and GWAS (genome-wide association studies) in different ethnic groups, indicating that many pathogenic genes can accumulate in each AD case ().
Figure 1. Frequency and accumulation of pathogenic gene variants in patients with Alzheimer’s disease.
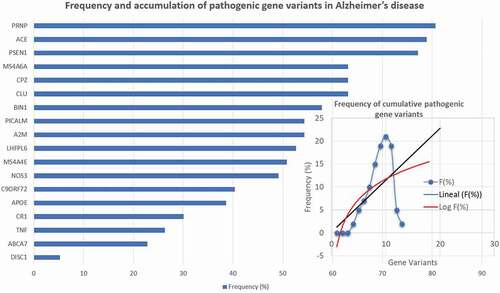
A very important aspect in the analysis of any genetic study in polygenic and complex diseases is to weigh the pathogenic load that each gene has in an individual case. Using a panel with the 18 most influential genes in AD (), it has been found that (i) no patient is a carrier of a single pathogenic gene, (ii) most patients (>60%) are carriers of several pathogenic genes (>10 pathogenic variants per patient), (iii) a considerable number of cerebrovascular risk variants are present in the genotype of AD patients, and (iv) the genes that most frequently (>50%) accumulate pathogenic variants in the same case of AD are A2M (54.38%), ACE (78.94%), BIN1 (57.89%), CLU (63.15%), CPZ (63.15%), LHFPL6 (52.63%), MS4A4E (50.87%), MS4A6A (63.15%), PICALM (54.38%), PRNP (80.70%) and PSEN1 (77.19%) ().
In relation to the pathogenic load that the APOE-4 allele may represent in the clinical expression of AD and in its neuropathological phenotype, the pathogenic influence of the APOE-4 allele, from a quantitative point of view, does not affect more than 35–40% of AD cases. However, the pathogenic role of the genotypes APOE-2/4, APOE-3/4 and especially APOE-4/4 is highly relevant in the age at onset, clinical course, concomitant cardiovascular and cerebrovascular disorders, and therapeutic response to treatment [Citation7–11,Citation14] ().
3. Drug development
Approximately, 20% of papers published during the past decade on AD are related to drug development [Citation15]. The most frequently studied pharmacological categories are the following: natural derivatives with pleiotropic activity (26%), anti-amyloid agents (20%), neurotransmitter enhancers (15%), novel drugs for new targets (12%), reanalyzed old drugs (11%), anti-tau drugs (6%), multitarget drugs (3%), stem cell therapy (1.85%), neuroprotective peptides (1.5%), nanotechnology products (1.45%), anti-inflammatory drugs (1.2%), and other products (1%) [Citation15]. Among the neurotransmitter enhancers, cholinergic drugs predominate (>70%) (), followed by glutamatergic (14.5%), serotonergic (7.5%), dopaminergic (4%), histaminergic (3%), GABAergic (1%), and adrenergic drugs (1%) ().
Figure 2. Pharmacological categories in drug development for the treatment of Alzheimer’s disease.
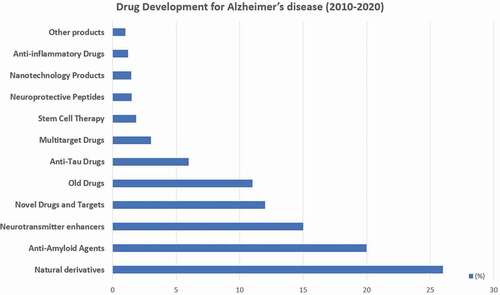
Table 1 Novel cholinesterase inhibitors with potential preclinical effects in AD models (PubMed 2010–2020)
Within the group of cholinergic drugs, acetylcholinesterase inhibitors (AChEIs) represent 76% of the total (many of them derivatives of donepezil>tacrine>rivastigmine>galantamine) (), butyrylcholinesterase inhibitors (BuChEIs) 3.75%, dual inhibitors of AChE and BuChE 3%, dual inhibitors of AChE and monoamine oxidase (MAO) 2.75%, muscarinic receptor agonists 6%, and nicotinic receptor agonists 8%. Other subcategories of pharmaceutical products include dopaminergic (DA D1, D2/3, and D5 receptor agonists; MAO-A (MAOAIs) and MAO-B inhibitors (MAOBIs); serotonergic (5-HT2A, 5-HT4 and 5-HT7 receptor agonists; 5-HT3 and 5-HT6 receptor antagonists); glutamatergic (NMDA receptor antagonists related to memantine, NMDA inhibitors, metabotropic glutamate receptor modulators, AMPA receptor modulators, glutamate modulators, glycine transporter 1 (GlyT1) inhibitors and vesicular glutamate transporter inhibitors); GABAergic (allosteric modulators of GABAA receptors, GABAA and GABAB receptor agonists); histaminergic (HA H3 receptor antagonists, other antihistaminics); and adrenergic drugs (β1 adrenergic receptor (ADRB1) agonists, α2 adrenergic receptor agonists (α2ARA) and selective α2C adrenoceptor antagonists) [Citation15]. In addition, over 150 multi-target drugs, belonging to different pharmacological categories, have been screened in search for anti-dementia properties [Citation15] (). Most multi-target drugs are dual AChEIs and BuChEIs with anti-amyloidogenic activity (). Some of these novel drugs are complex derivatives of classical AChEIs and others belong to the category of bioproducts derived from natural sources. However, many other classes of multi-target drugs, with diverse activity, have been reported, such as dual GSK3β inhibitors and Nrf2 inducers (2,4-Dihydropyrano[2,3-c]pyrazoles; drugs with anti-amyloidogenic and anti-inflammatory properties (CNI-1493); novel dual inhibitors of MAO-A, MAO-B and amyloid-β aggregation (Coumarin-pargyline hybrids); dual-acting AChEI and H3R antagonists (7-(3-(piperidin-1-yl)propoxy)-2,3-dihydropyrrolo[2,1-b]quinazolin-9(1 H)-one)(UW-MD-72); multifunctional iminochromene-2 H-carboxamide derivatives containing aminomethylene triazole with BACE1 inhibitory, neuroprotective and metal chelating properties; multifunctional thioxanthone derivatives with AChE, MAOs and β-amyloid aggregation inhibitory activities; multi-target-directed oxoisoaporphine derivatives with anti-AChE, anti-β-amyloid aggregation and enhanced autophagy activity; and many others ().
Table 2. Selected multi-target drugs in development for the treatment of Alzheimer’s disease
After AChEIs, the most intensively developed anti-dementia products were anti-Aβ agents (>20% of the total), including immunotherapy (>80 new products)(40% of anti-Aβ agents), β-secretase (BACE) inhibitors (>60 new products), ϒ-secretase modulators (≈40 products), and diverse Aβ aggregation inhibitors (>220 new products), together with APP modulators, Aβ scavengers, β-sheet breakers, δ-secretase inhibitors, α-secretase modulators, and Notch inhibitors [Citation15,Citation16].
Other drugs and/or therapeutic strategies in development, with minor quantitative impact so far, include anti-tau agents (with a 50% increase over the past 10 years), APOE modulators, anti-inflammatory drugs, polyunsaturated fatty acids, neurotrophic factors, neuroprotective peptides, nootropic-like cognitive enhancers, epigenetic drugs, gene therapy, stem cell therapy, nanotherapeutics, and some different combination drug regimes, the most frequent of which are donepezil + memantine or AChEIs in combination with other neuroprotectants [Citation7–11,Citation15,Citation17–19].
Strikingly, natural products and derivatives with pleiotropic activity plus novel drugs for new targets represented >40% of contributions to drug development in AD for the past decade. Only in the 2013–2017 interval, over 1,500 new natural products and 500 novel drugs were discovered and characterized [Citation15].
Of the 121 drugs studied in 2020 for the treatment of AD, 22% were in phase-I trials, 54% in phase-II trials and 24% in phase-III trials. About 10% of these drugs were cognitive enhancers, another 10% were tested to treat neuropsychiatric symptoms of dementia, and 80% were medications aimed at slowing-down the course of the disease [Citation20].
The only winner of this intense race to identify a useful drug for the treatment of AD was Aducanumab, formally approved by the FDA on 7 June 2021 as the first drug with a putative disease-modifying mechanism for AD, although not devoid of controversy in the scientific community [Citation21–27]. Aducanumab is a human monoclonal antibody with selective activity on aggregated Aβ. Aducanumab penetrates into the brain, binds parenchymal Aβ, and dose-dependently reduces soluble and insoluble Aβ. One year of monthly intravenous infusions of aducanumab in patients with incipient cognitive disorder or mild-AD reduces brain Aβ in a time- and dose-dependent manner, with parallel slowing of cognitive decline [Citation22–25].
Aducanumab and other injectable antibodies (gantenerumab, BAN2401), and a small molecule oral agent, ALZ-801, are amyloid-targeting drugs with variable efficacy and safety, and differential effects in terms of brain penetration, time-to-peak brain exposure, plasma half-life, and selectivity for Aβ oligomers. Recent studies show that the degree of selectivity for Aβ oligomers and brain exposure drive the magnitude and onset of clinical efficacy; in contrast, the clearance of plaques with the highest doses of aducanumab and BAN2401 induces vasogenic brain edema, especially in APOE4 carriers [Citation26,Citation27]. ALZ-801, an optimized oral prodrug of tramiprosate with selective anti-oligomer activity shows efficacy in homozygous APOE4/4 AD subjects, without evidence of vasogenic edema [Citation26].
4. Expert opinion
Although from a theoretical perspective and 100 years of history, AD appears to be pathogenically defined, the accumulation of multiple genetic defects in AD patients suggest that the current pathogenic conception of AD does not fully explain its more intimate pathogenic mechanisms. This would partly explain the therapeutic failure derived from concentrating all pharmacological research efforts on the cholinergic hypothesis in the years 1980–2000 and the amyloidogenic-tauopathic hypothesis from 2000 to the present, using biased animal models with results which could not be replicated in humans [Citation28]. There are still many pathogenic unknowns to be clarified. More than fifty genes encoding transporter proteins are altered in a certain number of patients with AD. The role of glia in the process of premature neuronal death remains an enigma. Recent studies indicate that astrocytes actively kill neurons in neurodegenerative disorders. APOE- and APOJ-contained saturated lipids may mediate astrocyte-induced neurotoxicity and these effects can be neutralized by eliminating the formation of long-chain saturated lipids in astrocytes through inhibition of the elongation of very long chain fatty acids-like 1 enzyme encoded by the ELOVL1 gene (1p34.2) [Citation29].
We still need to dig much deeper into the molecular mechanisms that induce premature neuronal death when the brain stops maturing and neurodegenerative mechanisms are activated decades before the onset of dementia symptoms. We also need presymptomatic biomarkers that allow us to identify the risk to intervene in asymptomatic periods of the disease.
Even before we find a preventive treatment for AD, we cannot forget that in over 90% of cases of dementia, multifactorial treatment is needed, which involves the simultaneous administration of several pharmaceutical categories (>10 drugs/day) with the consequent risk of adverse drug reactions (ADRs) and drug-drug interactions (DDIs) [Citation11,Citation14,Citation19]. According to the phenotypic profile of patients with mild-to-moderate dementia, the most frequent concomitant diseases are the following: behavioral disorders (95%), functional decline (90%), cerebrovascular disorder (90%), obesity (70%), depression (65%), anxiety (60%), cardiovascular disease (40%), hypercholesterolemia (40%), hypertension (28%), diabetes (26%), hypertriglyceridemia (20%), metabolic syndrome (20%), hyperbilirubinemia (15%), metabolic deficits (15%), transaminitis (11%), cancer (10%), anemia (7%), and endocrine disorders (5%). Cardiovascular disorders and blood pressure changes in AD are currently reported in association with increased risk of brain damage and increased cognitive deterioration. Furthermore, APOE variants are associated with AD, cardiovascular disorders, atherosclerosis, and cerebrovascular damage in dementia [Citation7,Citation8,Citation17]. Lipid metabolism disorder and the cerebrovascular component of AD have been extensively studied, and alterations in cholesterol, changes in cell membrane lipids and arteriosclerosis lead to ischemia and cerebral hypoperfusion that contributes to accelerating the premature death of neurons in patients with predisposition to AD [Citation8].
The only manner to reduce ADRs and DDIs in AD patients treated with antidementia agents plus drugs for the management of concomitant diseases, is the incorporation of pharmacogenetics into the daily clinic to personalize pharmacological treatments [Citation30].
The pharmacoepigenetic machinery responsible for the safety and efficacy of any type of drug is composed of a set of at least 400 genes that encode proteins, enzymes and receptors related to the target that each drug has to reach and the process of deactivation and elimination of metabolites and residual products. More than 90% of the drugs act as substrates, inducers or inhibitors of the products of these pharmagenes. The expression or repression of these genes is under the strict control of the epigenetic apparatus, which acts promiscuously and redundantly for safety reasons, and whose functional failure can lead to pharmacological ineffectiveness and/or the appearance of ADRs [Citation31,Citation32].
From a didactic point of view, there are at least five categories of genes related to the genomic device that modulates the pharmacogenetic complex: pathogenic genes associated with AD (); the mechanistic genes that encode components of the biochemical pathways that explain the mechanism of action of each drug; metabolic genes encoding phase-I and II enzymes responsible for hepatic and tissue metabolism, as well as the elimination of drugs; genes encoding xenobiotic agent transport proteins that facilitate the flux and efflux of drugs in the brain, allowing their access to the target or preventing their penetration through the blood-brain barrier; and hundreds of pleiotropic genes that participate in a multitude of metabolomic pathways since a xenobiotic agent penetrates an organism, circulates in blood, reaches the target and disintegrates in the form of metabolites that have to be processed in the tissues and liver prior to their subsequent elimination by urine, feces, bile or sweat [Citation30,Citation31] ().
Figure 3. Frequency and accumulation of defective pharmagene variants in patients with Alzheimer’s disease.
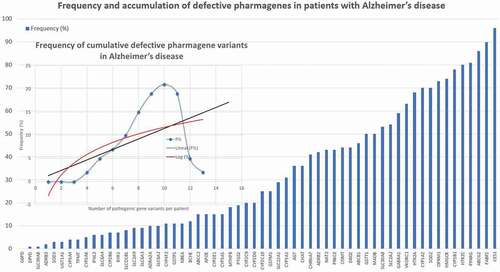
Furthermore, in the general population and among AD cases only 20–30% are normal metabolizers for major enzymes of the CYP gene family [Citation8,Citation10,Citation14,Citation30].
Of a cluster of 60 genes potentially involved in the pharmacogenetics of common drugs, the most dysfunctional genes frequently found (>20%) in AD are the following: CYP1A1 (31.47%), CYP1B1 (82.23%), MAOB (47.72%), CES1 (96.45%), CHAT (36.04%), COMT (30.96%), GSTM1 (54.82%), GSTT1 (25.38%), NAT2 (43.65%), SOD2 (67.51%), ABCB1 (44.16%), ABCG2 (90.36%), FABP2 (90.86%), SLCA2 (59.90%), SLC22A1 (34.52%), SLC30A8 (48.22%), ADRB2 (41.62%), AGT (42.13%), APOE (30.96%), CHRNA7 (42.13%), DRD2 (45.18%), GABRA1 (55.33%), HMGCR (73.60%), HTR1A (65.48%), HTR2C (79.19%), OPRM1 (69.54%), PPARG (81.73%), PRKCE (41.62%), and VKORC1 (65.99%) (). There is an accumulation of defective pharmagenes in approximately 85% of AD patients (). AD-related pathogenic genes and pharmagenes are under the redundant and promiscuous control of the epigenetic machinery (DNA methylation/demethylation, histone/chromatin remodeling, miRNA regulation) which regulates their expression and/or repression in the brain and other tissues [Citation31].
Whatever new drugs we develop in the future to effectively treat AD, drug development procedures should not ignore pharmacogenetics to optimize resources, time, and money. Aducanumab is a good example: potentially useful in APOE-3 carriers, but harmful in patients homozygous for the APOE-4 allele (27). In most pharmacogenetic studies, APOE-3 carriers tend to be good responders and APOE-4 carriers usually respond poorly in different studies [Citation7–11,Citation17–19,Citation30].
Various regulatory hurdles and rigidity of criteria by regulatory agencies in dominant countries have contributed to hindering the development of new treatments for AD. For more than 30 years, much of the outcome measures for validating an anti-dementia drug focused on a simple improvement in cognitive and/or behavioral functions, with little reference to an anti-neurodegenerative effect, in part due to the lack of efficient biomarkers. Costly long-term clinical trials, laxity in patient selection – mainly based on neuropsychological criteria – inspecificity of the few existing biomarkers and concomitant diseases – which required additional treatments – have also been limiting factors for the development of new drugs. But perhaps the most damaging factor has been to focus attention on symptomatic treatments in impaired patients, when the disease has already killed billions of neurons, without ignoring that symptomatic treatments are also necessary once the disease has manifested itself phenotypically at the cognitive, behavioral and functional levels. The present and future development of new anti-dementia treatments forces us to modify this regulatory and protocol paradigm in pre-symptomatic and symptomatic clinical trials in which new outcome measures must be incorporated and the regulatory rigidity prevailing until now must be made more flexible.
In conclusion, in light of current knowledge, the potential causes of therapeutic failure in AD may be related to the following: misconceptions in the pathogenic cascade of deleterious events leading to AD-specific neurodegeneration; erratic procedures in drug development, inadequate animal models and drug targets; delay in the implementation of the treatment, which should be in early periods of the disease and, optimally, in asymptomatic phases identified with highly reliable biomarkers; inertial obsession to look for a single drug instead of a multitarget product to treat a multifactorial neurodegenerative process; absence of pharmacogenetic procedures to personalize and optimize the development of new drugs and redefine the utility old drugs with neuroprotective properties; and inappropriate regulations.
Declaration of interest
The author is President and a stockholder of EuroEspes SA and the International Agency for Brain Research and Aging SL (IABRA). The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The author would like to thank his collaborators Natalia Cacabelos, Vinogran Naidoo and Juan C. Carril, at the International Center of Neuroscience and Genomic Medicine, for technical assistance.
Additional information
Funding
References
- Cantarero-Prieto D, Leon PL, Blazquez-Fernandez C, et al. The economic cost of dementia: a systematic review. Dementia. 2020;19(8):2637–2657.
- Sado M, Ninomiya A, Shikimoto R, et al. The estimated cost of dementia in Japan, the most aged society in the world. PLoS One. 2018;13(11):e0206508.
- Arvanitakis Z, Shah RC, Bennett DA. Diagnosis and management of dementia: review. Jama. 2019;322:1589–1599.
- Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12(3):292–323.
- Masters CL, Bateman R, Blennow K, et al. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056.
- Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci. 2020;23(10):1183–1193.
- Cacabelos R, Cacabelos P, Torrellas C, et al. Pharmacogenomics of Alzheimer’s disease: novel therapeutic strategies for drug development. Methods Mol Biol. 2014;1175:323–556.
- Cacabelos R, Carril JC, Cacabelos P, et al. Pharmacogenomics of Alzheimer’s Disease: genetic determinants of phenotypic variation and therapeutic outcome. J Genomic Med Pharmacogenomics. 2016;1:151–209.
- Cacabelos R, Carril JC, Cacabelos N, et al. Sirtuins in Alzheimer’s disease: SIRT2-related genophenotypes and implications for PharmacoEpiGenetics. Int J Mol Sci. 2019;20:E1249.
- Cacabelos R. Population-level pharmacogenomics for precision drug development in dementia. Exp Rev Precis Med Drug Dev. 2018;3(3):163–188.
- Cacabelos R. Pharmacogenomics of cognitive dysfunction and neuropsychiatric disorders in dementia. Int J Mol Sci. 2020;21(9):3059.
- Tcw J, Goate AM. Genetics of β-amyloid precursor protein in Alzheimer’s disease. Cold Spring Harb Perspect Med. 2017;7(6):a024539.
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608.
- Cacabelos R, Cacabelos N, Carril JC. The role of pharmacogenomics in adverse drug reactions. Exp Rev Clin Pharmacol. 2019;12(5):407–442.
- Cacabelos R. Have there been improvement in Alzheimer’s disease drug discovery over the past 5 years? Exp Opin Drug Discovery. 2018;13(6):523–538.
- Cacabelos R. How plausible is an Alzheimer’s disease vaccine? Expert Opin Drug Discov. 2020;15(1):1–6.
- Cacabelos R, Goldgaber D, Vostrov A, et al. APOE-TOMM40 in the Pharmacogenomics of demetia. J Pharmacogen Pharmacoprot. 2014;5:135.
- Cacabelos R. Pharmacogenomic of drugs to treat brain disorders. Exp Rev Precis Med Drug Dev. 2020;5(3):181–234.
- Cacabelos R, Carril JC, Corzo L, et al. Influence of pathogenic and metabolic genes on the pharmacogenetics of mood disorders in Alzheimer’s disease. Pharmaceuticals. 2021;14:366.
- Cummings J, Lee G, Ritter A, et al. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement. 2020;6(1):e12050. *Updating of drug development and clinical trials in AD.
- Lalli G, Schott JM, Hardy J, et al. Aducanumab: a new phase in therapeutic development for Alzheimer’s disease? EMBO Mol Med. 2021;13(8):e14781.
- Mukhopadhyay S, Banerjee D. A primer on the evolution of aducanumab: the first antibody approved for treatment of Alzheimer’s disease. J Alzheimers Dis. 2021;83(4):1537–1552.
- Knopman DS, Jones DT, Greicius MD. Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021;17(4):696–701.
- Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50–56.
- Ferrero J, Williams L, Stella H, et al. First-in-human, double-blind, placebo-controlled, single-dose escalation study of aducanumab (BIIB037) in mild-to-moderate Alzheimer’s disease. Alzheimers Dement. 2016;2(3):169–176.
- Tolar M, Abushakra S, Hey JA, et al. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther. 2020;12(1):95.
- VandeVrede L, Gibbs DM, Koestler M, et al. Symptomatic amyloid-related imaging abnormalities in an APOE ε4/ε4 patient treated with aducanumab. Alzheimers Dement. 2020;12(1):e12101.
- Cacabelos R, Carrera I, Martínez-Iglesias O, et al. What is the gold standard model for Alzheimer’s disease drug discovery and development? Expert Opin Drug Discov. 2021;25:1–26.
- Guttenplan KA, Weigel MK, Prakash P, et al. Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature. 2021;599(7883):102–107.
- Cacabelos R. Pharmacogenetic considerations when prescribing cholinesterase inhibitors for the treatment of Alzheimer’s disease. Exp Opin Drug Metab Toxicol. 2020;16(8):673–701.
- Cacabelos R, Carril JC, Sanmartín A, et al. Pharmacoepigenetic processors: epigenetic drugs, Drug resistance, Toxicoepigenetics, and nutriepigenetics. In: Cacabelos R, editor. Pharmacoepigenetics. Oxford: Academic Press/Elsevier; 2019. p. 191–424.
- Cacabelos R, Cacabelos P, Carril JC. Epigenetics and pharmacoepigenetics of age-related neurodegenerative disorders. In: Cacabelos R, editor. Pharmacoepigenetics. Oxford: Academic Press/Elsevier; 2019. p. 903–950.