
ABSTRACT
Introduction
Nucleoside analogues represent a cornerstone of achievement in drug discovery, rising to prominence particularly in the fields of antiviral and anticancer discovery over the last 60 years. Traditionally accessed using chemical synthesis, a paradigm shift to include the use of biocatalytic synthesis is now apparent.
Areas covered
Herein, the authors discuss the recent advances using this technology to access nucleoside analogues. Two key aspects are covered, the first surrounding methodology concepts, effectively using enzymes to access diverse nucleoside analogue space and also for producing key building blocks. The second focuses on the use of biocatalytic cascades for de novo syntheses of nucleoside analogue drugs. Finally, recent advances in technologies for effecting enzymatic nucleoside synthesis are considered, chiefly immobilization and flow.
Expert opinion
Enzymatic synthesis of nucleoside analogues is maturing but has yet to usurp chemical synthesis as a first-hand synthesis technology, with scalability and substrate modification primary issues. Moving forward, tandem approaches that harness expertise across molecular microbiology and chemical synthesis will be vital to unlocking the potential of next generation nucleoside analogue drug discovery.
1. Introduction
Since the foundation of nucleoside analogue chemistry in the 1950s, this field has continually evolved to deliver generations of landscape defining therapeutics against viral infections and cancer. Exemplified by the design and licensing of drugs such as gemcitabine, AZT, and sofosbuvir (), chemical synthesis has played a crucial role in enabling atom level structural modifications to canonical nucleoside and nucleotide structures. Most recently, this has paved the way for the approval of Molnupiravir and Remdesivir in tackling the COVID-19 pandemic () [Citation1,Citation2].
Figure 1. Examples of nucleoside analogue drugs.
In addition to the use of purely chemical approaches to synthesize nucleoside analogues, the capability of using enzymes is also not a recent landmark; such syntheses were established more than 50 years ago, particularly for 2’-deoxy systems using thymidine phosphorylase (TP) or pyrimidine nucleoside phosphorylase (PNP) [Citation3]. Typically, simple modifications within the pentose component (e.g. d-arabino vs d-ribo stereochemistry) were effected, alongside changes to the heterocyclic base (e.g. 5-substituted pyrimidines [Citation4]). However, as pharmacophore space surrounding the nucleoside analogue chemotype increased in complexity (compare remdesivir with cytarabine), so thus did the requirements placed upon strategies utilizing enzymes for their synthesis.
Biocatalysis has, however, advanced to a point where enzymes can now be designed to be the perfect catalyst for a particular process [Citation5]. The advent of directed evolution in the early 1990s revolutionized how chemists approached enzymatic transformations, with pioneers such as Arnold demonstrating how artificial evolutionary pressure could transform the function of a protein in extremely short timeframes [Citation6]. Technological advances since then, in particular the power of computation to help understand protein dynamics and the significant reduction in the cost of DNA technologies (e.g., synthesis and sequencing), have delivered a myriad of methods for the efficient evolution of enzymes for synthetic chemistry. A landmark study involved engineering of a transaminase to synthesize the blockbuster diabetes treatment sitagliptin [Citation7]. This enzyme underwent multiple rounds of evolution to deliver a mutant that could operate under conditions akin to that of a synthetic catalyst (200 g L−1 substrate loading, 50% DMSO v/v, 1 M amine donor) and deliver the final amine with perfect stereocontrol. What this study demonstrated was how enzymes could be pushed to function well beyond their natural limits, but still under ambient conditions with perfect selectivity obtained from natural evolution.
Considering these advances in biocatalysis, the challenges surrounding complex enzymatic nucleoside analogue synthesis can begin to be addressed; within this Opinion piece we cover recent highlights, from 2018 onwards, that illustrate these achievements.
2. Areas covered
We focus here on approaches that deliver nucleoside analogue targets using chemoenzymatic or biocatalytic cascade transformations as the key steps. Enzymatic transformations that have found significant application within other, largely synthetic routes, or for the production of key precursors [Citation8,Citation9], will not be discussed, as they have been reviewed elsewhere [Citation10]. Two key aspects are covered. The first surrounds methodology concepts, effectively using enzymes to access diverse nucleoside analogue space and also for producing key building blocks, for wider use in enzymatic synthesis cascades (e.g., furanose 1-phosphates). The second aspect focuses on the use of biocatalytic enzyme cascades for the synthesis of nucleoside analogue drugs that are either approved (e.g., molnupiravir and didanosine) or in current clinical trials (e.g., islatravir). We also consider important technology improvements for enacting enzymatic nucleoside synthesis, including enzyme immobilization and flow systems.
As mentioned above, demand is increasing for the ‘bio’ synthesis of evermore complex nucleoside analogues. The advent of using promiscuous thermophilic enzymes alongside an ever expanding and rapid capability to perform site-directed mutagenesis to broaden enzyme substrate promiscuity is rising to meet this challenge, and we cover pertinent examples of this within our survey of synthetic developments. This is tensioned with the requirement to balance kinetic and thermodynamic parameters when selecting enzymes for nucleoside synthesis and the consideration of scalability as the transition from analytical to practicable capability is made. The synergy between chemical and enzymatic approaches will ensure future generations of nucleoside analogues can be discovered and synthesized efficiently and this is explored in the Expert Opinion section.
3. Methodology developments for enzymatic nucleoside analogue synthesis
3.1. Enzymatic synthesis of 2-position modified pentose 1-phosphates
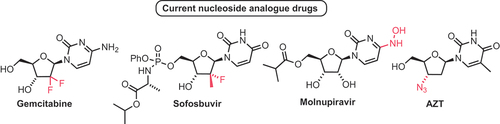
α-d-Pentose 1-phosphates represent key intermediates within canonical nucleoside biosynthesis and metabolic pathways. They are also of significant interest in the design of enzymatic routes to synthesize nucleosides and their analogues. Within this context, approaches to these materials have utilized biocatalytic routes from free pentoses with consecutive ribokinase and phosphopentomutase transformations [Citation11]. An alternative approach based on the nucleoside form was recently developed by Wagner and coworkers [Citation12], utilizing a thermophilic thymidine nucleoside phosphorylase (TNP) to deliver a series of 2-position modified α-d-pentose 1-phosphates. Utilization of this thermostable species demonstrated promiscuity over comparable mesophilic enzymes, and therefore capability in delivering broader spectrum access to 2-position modified α-d-pentose 1-phosphates (). The group demonstrated access to 2-deoxy, 2-deoxy-2-fluoro, 2-arabino, and 2-fluoroarabino systems which, importantly, maps to the prospect of completing biocatalytic synthesis of established nucleoside drugs that contain these modifications, including cytarabine and ribavirin.
Scheme 1. Enzymatic synthesis of α-d-pentose 1-phosphates using a thermostable pyrimidine nucleoside phosphorylase; isolation as Ba2+ salt form facilitates purification by precipitation and gram-scale access; TNP = thymidine nucleoside phosphorylase.
Using a repetitive barium salt precipitation method, gram-scale access to the final materials was demonstrated, and the anomeric integrity of the α-form of the phosphate was confirmed by multinuclear NMR. Stability above pH 9 was confirmed, alongside the materials being stable up to 60 °C, important for wider thermostable enzymatic work.
Interestingly, the final isolated yields of the α-d-ribo and α-d-2-deoxyribo 1-phosphates were highest (25% and 27% respectively) when 200 mM nucleoside substrate concentrations were used (with constant 500 mM phosphate) but the observed enzymatic conversion was lower than when using 50 mM substrate concentrations. The ability to effectively perform phosphorolytic anomeric cleavage was strongly dependent on the 2-position ring substituent. After a reaction time of 6 h, α-d-ribose 1-phosphate had formed in 43% yield whilst α-d-2-deoxy-2-fluoro and α-d-2-fluoroarabino 1-phopshates were only observed in 8% and 6% yields, respectively. This indicates that the electron withdrawing capability of fluorine at C2 within such analogues reduces the rate of cleavage, presumably by disfavoring potential buildup of positive charge at the anomeric carbon during phosphorolysis.
Overall, this work demonstrated a scalable access to important building blocks for the wider synthesis of nucleosides, offering advantages over traditional chemical synthesis (number of steps, chromatographic purification, and final anomeric purity), especially for α-d-2-fluoroarabino 1-phopshates.
Within a context of accessing nucleoside phosphate derivatives, human deoxycytidine kinase (HdCK) was recently evaluated for its potential to effect the synthesis of native and non-native ribonucleotides (5’-monophosphates) from the corresponding nucleosides [Citation13], demonstrating important potential as a capability to convert nucleoside analogues toward their nucleotide forms for nucleic acid synthesis.
3.2. A biocatalytic approach to access 4’-modified nucleosides
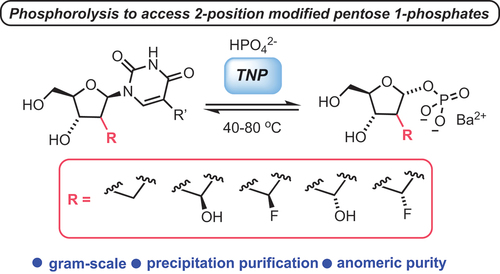
The chemical synthesis of 4’-modified nucleosides is challenging and lengthy, requiring multiple steps and protecting group manipulations and is often molecule specific, presenting little opportunity for scaffold diversification. In 2021, the Kurreck group demonstrated a biocatalytic approach, using a pyrimidine nucleoside phosphorylase (PyNP) from Thermus thermophilus (TtPyNP), to divergently access 4’-methylated nucleosides () [Citation14]. This approach compliments a scalable chemical synthesis of nucleosides (which included 4’-modifications) recently reported by Britton and colleagues [Citation15]. Nucleoside phosphorylases catalyze the reversible phosphorolysis of nucleosides to their corresponding pentose 1-phosphate and purine/pyrimidine components. A subsequent reverse phosphorolysis can then incorporate an alternative nucleobase [Citation16]. This transglycosylation effectively interconverts the nucleobase components across a pentose scaffold and was used effectively for 4’-alkynyl installation within the Merck biocatalytic synthesis of Islatravir (vide infra) [Citation17].
Scheme 2. Diversification of 4’-Me substituted nucleosides using the nucleoside phosphorylase TtDyNP; transglycosylation under thermodynamic reaction control enables the insertion of pyrimidines or purines; R = Me, F, Br, I, CF3; R1 = NH2 or OH; R2 = NH2, H or Cl.
Having identified TtPyNP as a suitably promiscuous PyNP, kinetic studies of phosphorolysis showed that 1 was converted to the corresponding glycosyl 1-phosphate 2, albeit with a lower substrate affinity than for native uridine (KM = 3.37 mM for 1 versus KM = <1 mM for U). Complimentary molecular docking studies suggested that a subtle, space-creating mutation of threonine to serine within the TtPyNP active site might contribute to the observed conversion of 1, in comparison to other established PyNPs [Citation18].
Following this, the thermodynamic parameters for the action of TtPyNP with 1 were shown to closely represent those of uridine, opening a prospect to perform transglycosylation and diversify the scaffold. Accordingly, the group demonstrated access to a range of analogues () using minimal phosphate and excess base to deliver 4’-Me transglycosylated products; 5-substituted pyrimidines from 2 and 2-/6-purine modifications using a promiscuous purine nucleoside phosphorylase from G. thermoglucosidasius.
Relatedly, a PyNP from Thermus thermophilus displayed a broad activity profile (pH 4–10, operational temperatures of up to 100 °C), but its application was limited by nucleobase inhibition at low concentrations [Citation19]. Transglycosylation has also been used recently by the same group to effect the synthesis of 2-selenopyrimidines [Citation20], the inclusion of which within wider nucleic acid chemistries has an established capability to deliver structure-to-function tools [Citation21]. Enzymatic synthesis of 2-selenouridine and 2-selenothymidine was effected using uridine and thymidine as donors and a 2-selenyl heterobase. Significantly larger equilibrium constants were observed for these non-native transglycosylation processes, requiring an excess of the nucleoside donor to access reaction yields of 40–48%.
3.3. Thermodynamic control of nucleoside phosphorolysis
Nucleoside phosphorylases (NPases) catalyze a phosphorolytic cleavage of canonical nucleosides to the corresponding 1-phosphate and heterobase. Two general classifications exist, one for purine systems and one for pyrimidines. Much of the work surrounding their use in a biocatalysis setting has focused on establishing and optimizing kinetic parameters for specific enzymes for a given phosphorolysis or transglycosylation [Citation22]. Nonetheless, such reactions tend not to reach full completion, and whilst specific catalyst modifications through enzyme immobilization [Citation23,Citation24] or engineering [Citation2,Citation25] can improve the outcome, the lower yields observed have been suggested to be influenced by the thermodynamic properties of the nucleosides involved in the given reaction.
The groups of Alexeev and Wagner have both recently shown that nucleoside phosphorolysis and transglycosylation are equilibrium reactions under thermodynamic control, adhering to the law of mass action [Citation18,Citation26]. Ultimately, this enables the concentrations of the reaction components to dictate the outcome, irrespective of the identity and amount of biocatalyst used. Substrate-specificapparent equilibrium constants are thus accessible and can be used to predict maximum yields for NPase-catalyzed reactions. Given this move away from kinetically controlled biocatalytic reactions for nucleoside analogue synthesis, efforts from this work direct toward the development of enzyme systems that can reach equilibrium faster.
Noteworthy herefrom is a recent enzymatic synthesis of a series of dihalogenated purines using a thermostable phosphorylase and uridine or thymidine as the sugar donor [Citation27]. Following analytical-scale reactions, maximum product yields were calculated from the derived equilibrium constants, and an approximate five-fold excess of the donor was required to deliver a 90% reaction yield. This represents a viable platform for the synthesis of new libraries or established nucleoside analogue drugs.
4. Full biocatalytic synthesis of nucleoside analogue drugs
The recognized efficiency of biocatalysis to deliver perfect stereo- and chemoselectivity has seen it rise to prominence for nucleoside analogue synthesis in recent years [Citation28,Citation29]. A number of enzymes have been identified within biosynthetic pathways for the natural production and deconstruction of nucleosides. These have provided inspiration for the redesign of these pathways to afford de novo biocatalytic routes to non-natural nucleoside analogues, offering an alternative to their synthetic counterparts.
4.1. Didanosine
A pioneering example of a fully engineered biosynthetic pathway was reported by Birmingham et al. [Citation30]. The authors described an engineering campaign of a suite of biocatalysts, to deliver a five-enzyme cascade toward the nucleoside analogue didanosine 5, which is used as a combination antiretroviral treatment for HIV/AIDS [Citation31]. A bioretrosynthetic approach was used to design the pathway to 5. Inspired by a nucleoside salvage pathway (), the group focussed on the pathway for inosine 3, closely related to their target. By running this degradative pathway in reverse, it was theorized that the enzymes could be engineered for activity toward non-natural components and deliver a biocatalytic cascade to synthesize 5.
Scheme 3. A) Nucleoside salvage pathway enzymes for the degradation of inosine. B) An engineered pathway for didanosine production. C) Biocatalytic synthesis of islatravir. Engineered enzymes are shown in blue, accessory enzymes in red. RK = ribokinase, PPM = phosphopentomutase, PNP = purine nucleoside phosphorylase, PanK = pantothenate kinase, DERA = deoxyribose 5-phosphate aldolase, PK = pyruvate kinase, AK = adenylate kinase, GOase = galactose oxidase, HRP = horseradish peroxidase.
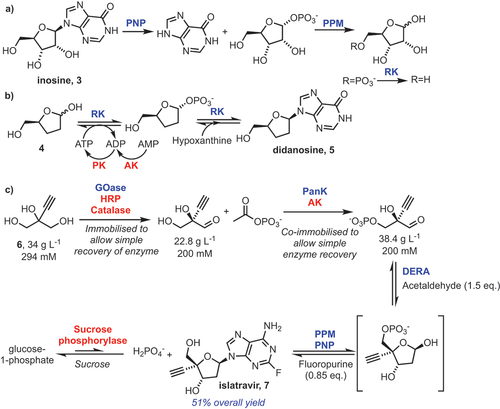
Initially, the three key enzymes in the cascade (ribokinase [RK], phosphopentomutase [PPM] and purine nucleoside phosphorylase [PNP], ) were identified and engineered to improve their activity toward the non-natural dideoxyribose 4 (). After optimizing the cascade to deliver 5, high concentrations of ATP were shown to be inhibitory to PPM, so an ATP recycling system was included (pyruvate kinase [PK], adenylate kinase [AK]). These enzymes convert the AMP and ADP that are produced during the RK phosphorylation of the sugar back into the ATP that is required for the RK step. This maintains sub-stoichiometric quantities of ATP, thereby avoiding inhibition of PMM.
Upon analysis of their cascade, the authors observed C1-phosphorylation activity for their engineered RK, effectively demonstrating a biosynthetic by-pass of the first step, and that removal of the engineered PPM from the cascade actually increased efficiency. The wild-type RK was from E. coli, and this was the first time that an RK had been shown to have this enzymatic activity. This work was limited by its practicable scale: the reactions were run on 1 mL scale, and the best yield for 5 was 44 μM (10 mg L−1), representing a low 4.4% HPLC yield. However, it provided inspiration for several further studies into non-natural nucleoside analogue synthesis [Citation17,Citation32].
4.2. Islatravir
Relatedly, researchers from Merck described in 2019 the evolution campaigns for five separate enzymes to realize a fully biocatalytic synthesis of experimental antiviral drug islatravir 7, starting from 2-ethynylglycerol 6 () [Citation17]. Enzyme engineering was performed on a galactose oxidase (GOase) to enable a stereoselective desymmetrization of 6, which proceeded in 80% ee, but overoxidation to the acid and subsequent removal of this undesired carboxylate enantiomer delivered an enantiopure aldehyde product in 99% ee. Additional engineering was conducted on a pantothenate kinase (PanK), an aldolase (DERA), and PPM/PNP. This remarkable feat of synthesis was performed in a single vessel, with sequential addition and removal of the enzymes and substrates and delivered 7 in 51% overall yield on half a gram scale. A key technology used here was enzyme immobilization. The GOase was immobilized, and the PanK and AK were co-immobilized. By converting these enzymes to heterogenous catalysts, filtration simplified the downstream processing by enabling removal of them prior to subsequent steps. The final product crystallized out of solution and could be recovered through filtration, obviating the need to immobilize the other enzymes. The use of sucrose phosphorylase was necessary in the final step to deliver 7, preventing the reverse reaction occurring by removing phosphate in situ. The authors did not disclose whether any traces of a 2’,3’-dideoxydidehydro by-product were formed.
4.3. Molnupiravir
Molnupiravir has recently been approved as a frontline treatment for SARS-CoV-2. As this is a non-native nucleoside analogue, a surge in demand for this raw material is anticipated. Consequently, the development of new synthetic routes toward its production are essential. A recent report from McIntosh et al. described the development of a three-step chemoenzymatic synthesis of molnupiravir from readily available starting materials in high yield () [Citation2]. As discussed in Section 1.3, developing biocatalytic routes to nucleoside analogues is often mired by the thermodynamic equilibrium between substrates and products. Alongside displacing the equilibrium through phosphate removal, the authors described several additional challenges: i) developing a biocatalytic nucleoside synthesis with a C5 esterified ribose ii) transfer of the heterobase to a non-natural sugar and iii) suitable conditions to install the oxime functionality at C4 of uridine.
Scheme 4. Chemoenzymatic synthesis of molnupiravir 12. MTRk = 5-S-methylthirbose kinase, UP = uridine phosphorylase, AcK = acetate kinase, PO = pyruvate oxidase, ATP = adenosine triphosphate.
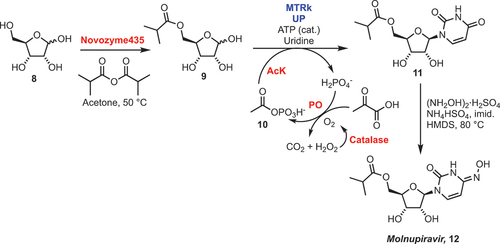
The first biocatalytic step used Novozyme435 (an immobilized lipase) to selectively esterify the primary C5-OH of d-ribose 8. This substrate was taken forward to a C1 phosphorylation. A 5-S-methylthioribose kinase (MTRk) was previously described as selectively phosphorylating the C1 position of a protected C5-thioether ribose derivative [Citation33]. Here, the wild-type MTRk from Klebsiella spp. was found to directly phosphorylate C1 of 9 and was subsequently engineered, with a 6-point mutant demonstrating further improved activity. This was quantified as a >100-fold improvement over the wild type, with a 10-fold reduction in enzyme loading (final loading 1 wt%), and a significant improvement in yield (from <10% to >90%). A uridine phosphorylase (UP) from E. coli provided a wild-type enzyme to then engineer a more effective UP for the glycosylation step. The 10-point variant afforded after several rounds of engineering demonstrated an 80-fold improvement over the wild-type, specifically with a 10-fold reduction in enzyme loading and a 6-fold increase in substrate concentration (11 mM to 67 mM). To address equilibrium issues associated with phosphate production, as well as the provision of ATP within the two-enzyme process, a pyruvate oxidase (PO) was implemented. This removed the phosphate generated by the action of UP and drove this equilibrium in the direction of 11. The inorganic phosphate by-product was then sequestered by the PO to produce the acetyl phosphate donor 10 that was necessary for the ATP regeneration. The donor 10 could be used by AcK to reform ATP, which meant it could be used in catalytic quantities. The synthesis was scaled to produce over 11 g of 12 in >99.5% purity (determined by quantitative NMR and UPLC) and 69% overall yield from 8. The reaction was run at 80 g L−1 substrate concentration, which is equivalent to 0.53 M.
4.4. Ribavirin, tecadenoson, and cladribine
An interesting strategy was recently developed by Ubiali, Speranza, and coworkers, who exploited the promiscuous nature of the purine nucleoside phosphorylase from Aeromonas hydrophilia (AhPNP) to synthesize several important nucleoside drugs [Citation34]. The same group had previously characterized this enzyme and optimized its expression in E. coli [Citation35], as well as demonstrating that it could be successfully immobilized and used in a 2 L process [Citation36]. A recent approach discussed the use of activated precursors for the glycosylation reaction. Specifically, methylation of the 7-position of guanosine, 2’-deoxyguanosine and guanosine arabinoside with methyl iodide delivered the 7-methylguanosine iodide salts. The transglycosylation with several bases afforded the nucleoside analogues ribavirin, tecadenoson, and cladribine in 67%, 49%, and 56% yields, respectively. The reactions were run with a 1:1 stoichiometry of donor to acceptor, showing that the obtained conversions were very impressive; the authors noted the 7-methylguanine by-product was not tolerated as a substrate for AhPNP, so the reverse reaction was completely unfavorable and helped the equilibrium. The guanosine arabinose derivative proved not to be a substrate for AhPNP, so this approach was not successful in their attempted synthesis of vidarabine. The reactions here were limited by scale (up to 20 mL) and substrate concentration (1 mM, so only 20 μmol per reaction). A similar approach was detailed by Mikailov and coworkers, with the synthesis of cladribine carried out on a 0.124 mmol scale (25 mg of product isolated) [Citation37].
5. Technology advances for enzymatic nucleoside analogue synthesis
5.1. Enzyme immobilization and flow biocatalysis
The syntheses described in Section 4 demonstrated whole-process optimization, with the use of immobilization to improve downstream processing and the implementation of accessory enzymes to remove phosphate and drive equilibria in the direction of the required products. An emerging area within bioprocess optimization is flow biocatalysis [Citation38,Citation39], whereby continuous reactors are used to improve the efficiency of a given process. Often, immobilized enzymes are applied in packed bed reactors to allow for improved enzyme productivity and simplified downstream processing [Citation40].
There have been recent reports of nucleoside analogues being synthesized using continuous flow biocatalysis. Rinaldi et al. showed in 2020 two continuous systems for the transfer of bases, using either a one enzyme system for direct switching, or a two enzyme system for phosphorylation then addition of a purine [Citation41]. A more recent example from Benítez-Mateos and Paradisi reported that the PNP from Halomonas elongata could be immobilized on thiol-functionalized agarose microbeads (termed SH-AG) [Citation23]. This gave a recovered activity of 37% upon immobilization with a final activity of 6.9 U g−1 of support and enabled the continuous synthesis of five nucleoside analogues with yields generally around 20% with a 30 minute residence time.
Tamborini, Ubiali, and colleagues also presented a recent flow synthesis of the nucleoside analogue vidarabine [Citation42]. Using the AhPNP described above (Section 2.4) in concert with the uridine phosphorylase from Clostridium perfringens (CpUP), they tested immobilization on two different supports: a glyoxyl-functionalized agarose via condensation with surface amino groups, and EziG (a porous glass-resin which contains iron centers for coordination) via affinity binding of histidine-tagged proteins. In this instance, the EziG was found to be a much more effective support. Both enzymes had a 97% immobilization yield (the amount of enzyme immobilized from solution), whereas with the glyoxyl support the yield was only 40% for CpUP (100% for AhPNP). The recovered activity was better for both enzymes on EziG (14% vs 22% for CpUP, and 35% vs 52% for AhPNP). In practice, the enzymes were packed into a column (3 mL column volume) and run continuously (120 minute residence time) for 8 days. After this, they precipitated 1.1 g of vidarabine from the effluent in a 55% isolated yield.
To avoid the optimization of enzyme immobilization and selection of an appropriate support, Wagner et al. demonstrated that catalytically active inclusion bodies (CatIB) could be used as naturally occurring immobilized enzymes [Citation43]. Inclusion bodies are aggregated insoluble proteins that form as a consequence of incorrect folding and protein denaturing. Often these aggregates are disregarded; however, the authors here found that an adenosine 5′-monophosphate phosphorylase from Thermococcus kodakarensis (TkAMPase), which could enable transglycosylation of cytidine 5’-monophosphate (CMP), was primarily eluted in the insoluble fraction after protein purification (>80% of the protein). This precipitate was found to be active and could be used at higher temperatures (up to 90 °C) with retention of activity. The conversions were generally poor (no yield >26% for CMP at 60 °C), and the reactions were only run in the 96-well assay format, so were limited by scale. This does, however, offer a practically free immobilization method, and the resulting heterogenous catalysts are entirely biodegradable.
6. Conclusions
As interest continues to grow in using enzymatic processes for nucleoside and nucleotide synthesis, demand around competent analytical tools has also increased. As a result, improvements have been made for monitoring reactions involving nucleobase cleavage using spectral unmixing [Citation44]. Using a linear combination of absorption spectra (for a given set of compounds), deconvolution can track back to and monitor individual components, offering an HPLC-level of accuracy in analytical capability.
In addition, route efficiencies are beginning to be described for principal reactions within nucleoside synthesis, such as N-glycosylation (synthesis of β-nucleosides) [Citation45]. Using an E-factor analysis, the pitfalls of common synthetic methods (e.g., chromatography) and those of biocatalytic processes (e.g., unfavorable reaction equilibria) have been compared.
In summary, it is evident that the field of biocatalytic nucleoside analogue synthesis has made huge accomplishments in the last few years, typified by methodology developments using pyrimidine nucleoside phosphorylases and the landmark total syntheses of islatravir, didanosine, and molnupiravir. Despite these advances, however, there is still significant ground to be made compared to historical chemical synthesis approaches, which are ever capable of accessing diverse molecular space [Citation15], and effecting precise structural modifications to canonical structures [Citation46]. Future capability in rapidly accessing known drug motifs and evolving new pharmacophore space will surely be best accomplished through synergy, using enzymes and chemical synthesis in tandem to support the weaknesses inherent to each separate approach.
7. Expert opinion
Biocatalytic approaches to deliver simple nucleosides are not a recent achievement, however the advent of using this methodology to tackle more complex structural modifications certainly is. Advances in protein engineering, the discovery of new, promiscuous nucleoside processing enzymes, and our understanding of their reaction dynamics are beginning to strengthen the toolbox for exploring and achieving the synthesis of ever more complex targets.
It is sensible to consider this prospect of using enzymes to perform classical synthetic nucleoside-producing reactions in a broader context. For example, complex glycan synthesis has benefitted immeasurably from the development of glycosyltransferases to assemble (in part or in full) complex oligosaccharide targets for biological and biomedical applications [Citation47–51]. In addition, and comparatively to the example reviewed earlier for the provision of ribose 1-phosphates, the provision of the key sugar nucleotide building blocks to achieve this has also flourished [Citation52–56]. Similarly, looking to classical oligonucleotide synthesis [Citation57], the phosphoramidite method of assembling these essential sequences has been challenged recently not only by new chemical approaches [Citation58–60] but also by using enzyme platforms to achieve biopolymer synthesis [Citation61,Citation62]. Considering the enabling effect of introducing biocatalytic approaches into these fields, the future prospects for nucleoside analogue chemistry translate to one of significant potential.
However, many challenges remain. First and foremost, is the issue of scalability. Whilst specific examples of successfully developing scalable enzymatic cascade syntheses exist (vide supra), a wider development to transfer analytical to preparative capability is vital; technology developments, for example continuous flow processes, may help to surmount this challenge. When designing a biocatalytic process, there is a double-faceted optimization that is required: optimizing the enzyme, and the reaction it catalyzes. This often presents itself as an issue with regard to scaling a reaction; the time it takes to optimize enzyme production can leave little to do with the reaction itself. Approaches that improve expression levels and enzyme stability therefore play important roles in bioprocess optimization; here the use of immobilization may play a key role. Scaffold diversification is also a critical consideration. The evolution of broadly promiscuous nucleoside processing enzymes will enable rapid access to explore diverse pharmacophore space, from the perspectives of both the sugar ring and hetereobase components. Chemical synthesis will synergize here for some time as it remains the current tool to efficiently effect atom-specific structural modifications of key building blocks for further synthesis. Finally, contemplation of the requirements to utilize nucleosides and their analogues in nucleic acid chemistry requires that access to appropriate kinase (or similar) phosphorylation systems are robust (and potentially scalable), to provide the necessary nucleotide triphosphates.
From a technology and engineering perspective, recent reports of continuous biocatalytic nucleoside syntheses provide an intriguing snapshot of the future. The use of a process engineering approach is agnostic to the enzymatic system that is used, avoiding the requirement for protein engineering to improve enzyme performance. It also affords greater equilibrium control via substrate/product removal through continuous flow of the reaction mixture. This could remove the need for additional bespoke accessory enzymes to be researched (e.g. co-factor recycling systems) and provide a system to help with equilibrium displacement. While not all researchers have access to continuous reactor systems, such access does remove a prerequisite for advanced molecular biology skills to clone and design enzymes, an appropriate example of field of expertise synergy. Scientific disciplines will need to work in tandem (chemical vs biocatalytic approaches) and in some cases develop new relationships (e.g. to tackle scalability capabilities). When taken altogether, the discipline of enzymatic nucleoside analogue synthesis is primed for triumph. This historically privileged medicinal chemistry and drug discovery space ensures that the continued evolution of methods to improve their synthesis and structural diversity will endure.
Article highlights
There have been considerable advances in the biocatalytic synthesis of nucleoside analogues in recent times.
Use of nucleoside phosphorylases for transglycosylation reactions demonstrates a broadening of capability for the installation of nucleobases onto pentoses.
Enzymatic synthesis of ribose-modified scaffolds, with a particular focus on 2’- and 4’-modifications have developed, with a toolbox of biocatalysts now available to mediate such transformations.
The highly chemoselective nature of enzymes is also beginning to transform the way cascade chemical reactions can take place.
In particular, there have been significant achievements in effecting full biocatalytic syntheses of nucleoside analogue drugs. This includes didanosine, islatravir, and molnupiravir, which can now be synthesized with almost total biocatalytic routes.
Moving forward, technology improvements for nucleoside synthesis using enzyme immobilization and flow capability have begun to emerge as enabling technologies for nucleoside biosynthesis, as they can help to overcome key problems, including enzyme inhibition and thermodynamic equilibrium issues.
This box summarizes key points contained in the article.
Conflict of Interest
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Eastman RT, Roth JS, Brimacombe KR, et al. Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent Sci. 2020;6(5):672–683.
- McIntosh JA, Benkovics T, Silverman SM, et al. Engineered Ribosyl-1-Kinase enables concise synthesis of molnupiravir, an antiviral for COVID-19. ACS Cent Sci. 2021;7(12):1980–1985.
- Serra I, Bavaro T, Cecchini DA, et al. A comparison between immobilized pyrimidine nucleoside phosphorylase from Bacillus subtilis and thymidine phosphorylase from Escherichia coli in the synthesis of 5-substituted pyrimidine 2′-deoxyribonucleosides. J Mol Catal B Enzym. 2013;95:16–22.
- Visser DF, Rashamuse KJ, Hennessy F, et al. High-yielding cascade enzymatic synthesis of 5-methyluridine using a novel combination of nucleoside phosphorylases. Biocatal Biotransfor. 2010;28(4):245–253.
- Turner NJ. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol. 2009;5(8):567–573.
- Arnold FH. Directed evolution: bringing new chemistry to life. Angewandte Chemie Int Ed Engl. 2018;57(16):4143–4148.
- Savile CK, Janey JM, Mundorff EC, et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science. 2010;329(5989):305–309.
- Zhang P, Iding H, Cedilote M, et al. A practical synthesis of (2R)-3,5-di-O-benzoyl-2-fluoro-2-C-methyl-D-ribono-γ-lactone. Tet Asymm. 2009;20(3):305–312.
- Xiang DF, Bigley AN, Desormeaux E, et al. Enzyme-Catalyzed kinetic resolution of chiral precursors to antiviral prodrugs. Biochem. 2019;58(29):3204–3211.
- Slagman S, Fessner W-D. Biocatalytic routes to anti-viral agents and their synthetic intermediates. Chem Soc Rev. 2020;50:1968–2009.
- Fateev IV, Kostromina MA, Abramchik YA, et al. Multi-Enzymatic cascades in the synthesis of modified nucleosides: comparison of the thermophilic and mesophilic pathways. Biomol. 2021;11(4):586.
- Kamel S, Weiß M, Klare HFT, et al. Chemo-enzymatic synthesis of α-D-pentofuranose-1-phosphates using thermostable pyrimidine nucleoside phosphorylases. Mol Catal. 2018;458:52–59.
- Hellendahl KF, Kamel S, Wetterwald A, et al. Human deoxycytidine kinase is a valuable biocatalyst for the synthesis of nucleotide analogues. Catalysts. 2019;9(12):997.
- Kaspar F, Seeger M, Westarp S, et al., Diversification of 4′-Methylated nucleosides by nucleoside phosphorylases. ACS Catal. 11(17): 10830–10835. 2021.
- Meanwell M, Silverman SM, Lehmann J, et al. A short de novo synthesis of nucleoside analogs. Science. 2020;369(6504):725–730.
- Kaspar F, Giessmann RT, Hellendahl KF, et al. General principles for yield optimization of nucleoside phosphorylase‐catalyzed transglycosylations. ChemBioChem. 2020;21(10):1428–1432.
- Huffman MA, Fryszkowska A, Alvizo O, et al., Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science. 366(6470): 1255–1259. 2019.
- Kaspar F, Giessmann RT, Neubauer P, et al., Thermodynamic reaction control of nucleoside phosphorolysis. Adv Synth Catal. 362(4): 867–876. 2020.
- Kaspar F, Neubauer P, Kurreck A. The peculiar case of the hyper-thermostable pyrimidine nucleoside phosphorylase from thermus thermophilus. ChemBioChem. 2021;22(8):1385–1390.
- Hellendahl KF, Kaspar F, Zhou X, et al. Optimized biocatalytic synthesis of 2‐selenopyrimidine nucleosides by transglycosylation. ChemBioChem. 2021;22(11):2002–2009.
- Lin L, Sheng J, Huang Z. Nucleic acid X-ray crystallography via direct selenium derivatization. Chem Soc Rev. 2011;40(9):4591–4602.
- Kaspar F, Wolff DS, Neubauer P, et al. pH-independent heat capacity changes during phosphorolysis catalyzed by the pyrimidine nucleoside phosphorylase from geobacillus thermoglucosidasius. Biochem. 2021;60(20):1573–1577.
- Benítez‐Mateos AI, Paradisi F. Sustainable flow‐synthesis of (Bulky) nucleoside drugs by a novel and highly stable nucleoside phosphorylase immobilized on reusable supports. ChemSusChem. 2021. https://doi.org/10.1002/cssc.202102030
- Visser DF, Hennessy F, Rashamuse J, et al. Stabilization of Escherichia coli uridine phosphorylase by evolution and immobilization. J Mol Catal B Enzym. 2011;68(3–4):279–285.
- Xie X, Huo W, Xia J, et al. Structure–activity relationship of a cold-adapted purine nucleoside phosphorylase by site-directed mutagenesis. Enzyme Microb Tech. 2012;51(1):59–65.
- Alexeev CS, Kulikova IV, Gavryushov S, et al. Quantitative prediction of yield in transglycosylation reaction catalyzed by nucleoside phosphorylases. Adv Synth Catal. 2018;360(16):3090–3096.
- Heba Y, Sarah W, Viola R, et al. Efficient biocatalytic synthesis of dihalogenated purine nucleoside analogues applying thermodynamic calculations. Molecules. 2020;25(4):934.
- Arco JD, Fernández-Lucas J. Purine and pyrimidine salvage pathway in thermophiles: a valuable source of biocatalysts for the industrial production of nucleic acid derivatives. Appl Microbiol Biot. 2018;102(18):7805–7820.
- Nyhan WL. Nucleotide synthesis via salvage pathway. In: eLS. John Wiley&Sons, Ltd (Ed.); 2014. doi:https://doi.org/10.1002/9780470015902.a0001399.pub3.
- Birmingham WR, Starbird CA, Panosian TD, et al., Bioretrosynthetic construction of a didanosine biosynthetic pathway. Nat Chem Biol. 10(5): 392–399. 2014.
- Englund JA, Baker CJ, Raskino C, et al. Zidovudine, didanosine, or both as the initial treatment for symptomatic HIV-infected children. New Engl J Medicine. 1997;336(24):1704–1712.
- Nawrat CC, Whittaker AM, Huffman MA, et al. Nine-step stereoselective synthesis of islatravir from deoxyribose. Org Lett. 2020;22(6):2167–2172.
- Albers E. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′‐methylthioadenosine. IUBMB Life. 2009;61(12):1132–1142.
- Rabuffetti M, Bavaro T, Semproli R, et al., Synthesis of ribavirin, tecadenoson, and cladribine by enzymatic transglycosylation. Catalysts. 9(4): 355. 2019.
- Ubiali D, Serra CD, Serra I, et al. Production, characterization and synthetic application of a purine nucleoside phosphorylase from Aeromonas hydrophila. Adv Synth Catal. 2012;354(1):96–104.
- Serra I, Daly S, Alcantara AR, et al. Redesigning the synthesis of vidarabine via a multienzymatic reaction catalyzed by immobilized nucleoside phosphorylases. RSC Adv. 2015;5(30):23569–23577.
- Drenichev MS, Alexeev CS, Kurochkin NN, et al., Use of nucleoside phosphorylases for the preparation of purine and pyrimidine 2′‐deoxynucleosides. Adv Synth Catal. 360(2): 305–312. 2018.
- Britton J, Majumdar S, Weiss GA. Continuous flow biocatalysis. Chem Soc Rev. 2018;47(15):5891–5918.
- Santis PD, Meyer L-E, Kara S. The rise of continuous flow biocatalysis – fundamentals, very recent developments and future perspectives. React Chem Eng. 2020;5:2155–2184.
- Thompson MP, Peñafiel I, Cosgrove SC, et al. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org Process Res Dev. 2019;23(1):9–18.
- Rinaldi F, Fernández-Lucas J, de la FD, et al. Immobilized enzyme reactors based on nucleoside phosphorylases and 2′-deoxyribosyltransferase for the in-flow synthesis of pharmaceutically relevant nucleoside analogues. Bioresour Technol. 2020;307:123258.
- Tamborini L, Previtali C, Annunziata F, et al., An enzymatic flow-based preparative route to vidarabine. Molecules. 25(5): 1223. 2020.
- Kamel S, Walczak MC, Kaspar F, et al. Thermostable adenosine 5′-monophosphate phosphorylase from Thermococcus kodakarensis forms catalytically active inclusion bodies. Sci Rep. 2021;11(1):16880.
- Kaspar F, Giessmann RT, Westarp S, et al. Spectral unmixing‐based reaction monitoring of transformations between nucleosides and nucleobases. ChemBioChem. 2020;21(18):2604–2610.
- Kaspar F, Stone MRL, Neubauer P, et al. Route efficiency assessment and review of the synthesis of β-nucleosid via N-glycosylation of nucleobases. Green Chem. 2020;23(1):37–50.
- Guinan M, Huang N, Hawes CS, et al. Chemical synthesis of 4′-thio and 4′-sulfinyl pyrimidine nucleoside analogues. Org Biomol Chem. 2021. https://doi.org/10.1039/d1ob02097h.
- Wang Z, Chinoy ZS, Ambre SG, et al. A general strategy for the chemoenzymatic synthesis of asymmetrically branched N-glycans. Science. 2013;341(6144):379–383.
- Šardzík R, Green AP, Laurent N, et al. Chemoenzymatic synthesis of o-mannosylpeptides in solution and on solid phase. J Am Chem Soc. 2012;134(10):4521–4524.
- Rexer T, Laaf D, Gottschalk J, et al. Enzymatic Synthesis of Glycans and Glycoconjugates. Adv Biochem Eng Biotech. 2020;175:231–280.
- Gottschalk J, Elling L. Current state on the enzymatic synthesis of glycosaminoglycans. Curr Opin Chem Biol. 2021;61:71–80.
- Xu Y, Masuko S, Takieddin M, et al. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science. 2011;334(6055):498–501.
- Ahmadipour S, Beswick L, Miller GJ. Recent advances in the enzymatic synthesis of sugar-nucleotides using nucleotidylyltransferases and glycosyltransferases. Carbohydr Res. 2018;469:38–47.
- Ahmadipour S, Pergolizzi G, Rejzek M, et al. Chemoenzymatic synthesis of c6-modified sugar nucleotides to probe the GDP- d -mannose dehydrogenase from Pseudomonas aeruginosa. Org Lett. 2019;21(12):4415–4419.
- Gantt RW, Peltier-Pain P, Cournoyer WJ, et al. Using simple donors to drive the equilibria of glycosyltransferase-catalyzed reactions. Nat Chem Biol. 2011;7(10):685–691.
- Chen Y, Thon V, Li Y, et al. One-pot three-enzyme synthesis of UDP-GlcNAc derivatives. Chem Commun. 2011;47(38):10815–10817.
- Beswick L, Ahmadipour S, Dolan JP, et al. Chemical and enzymatic synthesis of the alginate sugar nucleotide building block: GDP-D-mannuronic acid. Carbohydr Res. 2019;485:107819.
- Zhou X, Kiesman WF, Yan W, et al. Development of kilogram-scale convergent liquid-phase synthesis of oligonucleotides. J Org Chem. 2021. https://doi.org/10.1021/acs.joc.1c01756.
- Huang Y, Knouse KW, Qiu S, et al. A P(V) platform for oligonucleotide synthesis. Science. 2021;373(6560):1265–1270.
- Knouse KW, Flood DT, Vantourout JC, et al. Nature Chose phosphates and chemists should too: how emerging P(V) methods can augment existing strategies. ACS Cent Sci. 2021;7(9):1473–1485.
- Wunnava S, Dirscherl CF, Výravský J, et al. Acid‐catalyzed RNA‐oligomerization from 3’,5’‐cGMP. Chem Eur J. 2021;27(70):17581–17585.
- Lee HH, Kalhor R, Goela N, et al. Terminator-free template-independent enzymatic DNA synthesis for digital information storage. Nat Commun. 2019;10(1):2383.
- Palluk S, Arlow DH, de Rond, et al. De novo DNA synthesis using polymerase-nucleotide conjugates. Nat Biotechnol. 2018;36(7):645–650.