
1. Introduction
The similarity principle in medicinal chemistry postulates that structurally similar compounds exhibit similar bioactivities. For several decades, molecular fingerprints have served as an in-silico molecular representation of choice for early small molecule drug discovery-related tasks grounded in compound similarity such as virtual screening, assessing activity probabilities, analyzing chemical space, designing focused compound libraries, and providing features for predictive models describing quantitative structure activity or property relationships (QSAR or QSPR) [Citation1]. 2D fingerprints have been particularly popular because they are easy to generate and fast to compare using binary representations. Often the focus of 2D fingerprint similarity searches is the identification of molecules with activity against a specific target; however, conversely, poly-pharmacology can be explored as well searching for targets a compound may show activity against [Citation2]. Although new 2D fingerprints have been emerging that refine existing concepts such as the topological description of small molecules through characterizing its atomic environment [Citation3], no fundamentally new concepts have found broad acceptance in the past few years. Remaining most popular for similarity searches, 2D fingerprints still fall mostly into the classes of topological fingerprints capturing atomic features and connectivity of varying depths, circular fingerprints, structural keys capturing substructures, and pharmacophore fingerprints hashed into bit strings. A number of similarity measures can be employed to calculate similarities between compounds represented by bitstring fingerprints. While the Tanimoto coefficient calculating the ratio of the intersection and union of bits set to ‘1’ is arguably the most frequently used pairwise similarity measure, there are many others that have been established [Citation4]. What has changed in recent years are the ease of access to fingerprint-based similarity search methods, the increased use of 2D fingerprints in powerful machine learning and artificial intelligence methods, and the speed of performing similarity searches in large datasets ().
Figure 1. Enhancements in 2D fingerprint similarity searching.
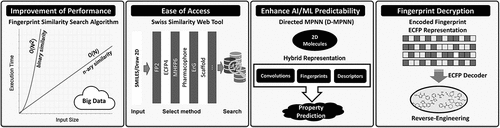
Although more complex and information-rich molecular representations such as 3D fingerprints exist, they rarely outperform 2D fingerprints [Citation5]. 3D fingerprints allow for encoding molecular features that are mostly inaccessible to 2D methods, such as describing non-bonding atoms close in space but topologically distant, stereochemistry, and conformational ensemble characteristics. These 3D features can add important additional molecular information and therefore have the potential to provide better similarity prediction performance. However, the often difficult identification and weighting of relevant conformations adds noise and complexity, often without performance gain. Comparing directly related extended 3D circular fingerprints (E3FP) and 2D circular fingerprints (E2FP) demonstrated better performance of the 3D method in bioactivity predictions; however, this example is a rare exception in 3D fingerprint superiority [Citation6]. 2D fingerprints remain largely popular due to their low sensitivity to feature noise and their proven success in finding diverse compounds with desired properties and activities. Of note are alternative molecular fingerprint representations such as biological or affinity fingerprints consisting of compound potencies against a panel of protein targets in combination with QSAR training that have generated promising results in scaffold hopping and virtual screening [Citation7].
2. Fingerprint enhancements
Performing fingerprint-based similarity searches against popular databases of commercially available compounds or compounds with bioactivity annotations has become more accessible for researchers without cheminformatics background. An update to the SwissSimilarity website now allows for fast searches against multiple popular databases for which several fingerprints have been precalculated [Citation8]. Equally beneficial is the availability of fingerprint calculators in open source software such as Open Babel [Citation9], PaDEL [Citation10], and RDKit (https://www.rdkit.org) that can be easily integrated in custom workflows or computer programming code. In addition, binary fingerprint-based open source QSPR models are easily accessible now [Citation11].
2D fingerprints have been used as descriptors in virtual screens for some time guiding early drug discovery efforts [Citation1]. It has been shown time and time again that ligand-based virtual screening methods such as those using 2D fingerprints-based similarity measures combined with data fusion techniques, consistently show equal or better performance than structure-based methods do for a number of drug targets [Citation12,Citation13]. More recently, a number of 2D fingerprints have been explored systematically in deep learning convolutional neural network models deployed for virtual screening [Citation14]. Molecular structures lend themselves to graph representations with atoms and bonds representing nodes and edges of graphs. They can be transferred into 2D matrices used as inputs of graph convolutional neural networks (GCN). GCN contain multiple layers where overlapping partial representations of an input graph are modulated using trainable filters (set of weights) that allow a molecular feature to be recognized independent of its location – a key concept of GCN. With the rise of molecular graph representations in deep neural networks, neural embedding concepts using low dimensional data vectors to represent data more efficiently in neural networks, have been extended to include multiple molecular fingerprints as recently demonstrated on a virtual screening exercise in search of selective CDK1 inhibitors [Citation15]. To overcome the short-range character of fingerprint representations, combining multiple fingerprints with other molecular descriptors has been a favored approach as recently demonstrated by Zhao et al. when searching for SARS-CoV‑2 3CLpro inhibitors identifying four natural products with antiviral activities [Citation16]. Although similarity alone can be used to perform database searches, virtual screening approaches typically deploy fingerprints as independent descriptors in machine learning algorithms. While there is a large spectrum of different machine learning methods ranging from random forest, Bayesian networks, to support vector machines and neural networks, the choice of the appropriate descriptors is often more important than the choice of the predictive algorithm. For instance, combining either circular fingerprints, path-based fingerprints, or substructure key fingerprints with an artificial neural network QSAR algorithm to predict biological activities of small molecule enzyme inhibitors resulted in vastly different performances for several targets [Citation17]. In one case, correlations between calculated and observed inhibition of angiotensin-converting enzyme activity varied between R2 = 0.08 and R2 = 0.41 depending on the fingerprint used. In another case of acetylcholine esterase inhibition, R2 values ranged from 0.04 to 0.43. It is therefore not surprising that combinations of substructure and topological representations into a conjoined 2D fingerprint have outperformed individual fingerprint representations across multiple algorithms including random forest and deep neural networks [Citation18]. When joining multiple fingerprints, the information density of the resulting merged molecular representation decreases. Different fingerprints encode different albeit usually overlapping aspects of molecules. Even for single fingerprint representations, some distinguishable features are more important than others. Feature selection methods are often employed to reduce the dimensionality of the fingerprint representation and thereby lower the propensity to overtrain models based on a limited number of training compounds. Following this observation, stacked deep belief networks with multiple 2D descriptors have been explored that determine the important features and also introduce new combinatorial descriptors thereby improving virtual screening performances [Citation19]. The process of selecting important features across multiple fingerprints is illustrated in .
Figure 2. Merging of multiple fingerprints.
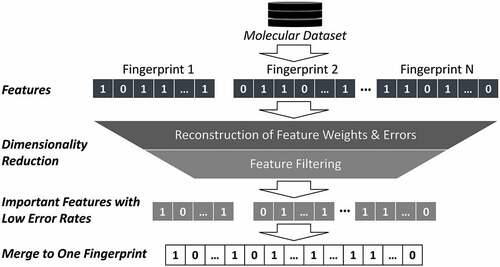
In recent years, fingerprints have been deployed as part of artificial intelligence algorithms. While compounds in generative models are often represented through graphs, sets of fragments, or text strings such as SMILES, the underlying fitness functions, often containing QSAR models, sometimes rely on 2D fingerprint representations [Citation20]. Molecular representations in predictive models depend on local versus global model applicability and the size of the data set. Pure fingerprint-based models perform well for smaller data sets of less than one thousand molecules. However, hybrid representations generalize better and exhibit higher performance in deep learning models exemplified by predicting molecular properties using directed message passing neural networks [Citation21]. Hence combining fingerprints with orthogonal molecular representation such as graph convolution is a promising way to enhance fingerprint utility.
As the databases used for virtual screening grow to tens and sometimes hundreds of millions of compounds, the speed of similarity searches becomes more important. To search in large databases, enhanced fingerprints such as MinHash encoding circular substructure have been introduced to speed up nearest neighbor searches [Citation22]. However, cross-comparing large datasets quickly becomes untenable as traditional binary compound comparisons using common similarity measures such as Tanimoto coefficients scale with the square of the number of compounds (O(N2)). A recently introduced n-ary method of comparing more than two compounds at a time, scales linearly with the number of compounds (O(N)) and can easily scale to comparing compound decks of millions of compounds, thereby greatly extending the reach of database similarity comparisons [Citation23]. To this end, extended (n-ary) similarity indices calculate similarities between n objects such as bitstrings or fingerprints. When comparing pairs of compounds, two corresponding bits either confer similarity (1–1), dissimilarity (0–1, 1–0), or coincidence (0–0). Instead, when comparing multiple compounds, Cn(k) counters are introduced, where k is the number of bits set to 1 at the respective bit position among n compounds. Depending on an a priori determined coincidence threshold (number of expected co-occurring bits set to 1), the Cn(k) counters are classified as similarity or dissimilarity counters and then used in analogy to pairwise comparators in all bitstring-based similarity indices such as Tanimoto.
Precompetitive data sharing among pharmaceutical companies without revealing the chemical structures of the compounds has been discussed for a long time but has also been hampered by fears of revealing compound identities through fingerprint representation. It was recently shown that up to 69% of a ChEMBL (https://www.ebi.ac.uk/chembl/) data set of more than 100,000 compounds could be reverse-engineered based on extended-connectivity fingerprints [Citation24]. It highlights the need to develop new 2D fingerprints with higher encryption to increase security aspects of cheminformatics solutions.
3. Expert opinion
Similarity searching with 2D fingerprint representations of small molecules remains widely popular for virtual screening, predictive modeling, and chemical space navigation. There have been only few novel fingerprints of note introduced in recent years. Topological, circular, and substructure-coding binary representations remain the prevailing choice for 2D fingerprints. However, the applicability of fingerprints has been enhanced in recent years through better access, increased speed, and wider use as descriptors in predictive models. With the advent of deep learning and artificial intelligence methods applied to drug discovery, 2D fingerprints have become part of the molecular representations deployed there. As graph convolutional representations of small molecules have gained popularity in artificial intelligence approaches, combining 2D fingerprint descriptors among themselves or with orthogonal representations has proven to be superior in making deep learning models become more predictive. In generative models, 2D fingerprints serve as descriptors for QSAR models used as part of fitness functions informing reinforcement learning methods to automatically design novel molecules with improved properties. Comparing large database and navigating large chemical spaces using 2D fingerprint similarities has been hampered by the fact that pairwise comparisons scale with the square of the number of compounds. A new algorithms was introduced recently that alleviates this problem by scaling linearly with the number of compounds thereby allowing for millions of compounds to be cross compared. An area that still needs attention is the enablement of 2D fingerprint representations suitable for similarity assessments yet featuring encryption that enables precompetitive sharing of compound data without fear of reverse-engineering chemical structures.
Declaration of financial/other relationships
IM and YH are employees and shareholders of Alkermes, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Muegge I, Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discov. 2016;11(2):137–148.
- Ciriaco F, Gambacorta N, Alberga D, et al. Quantitative polypharmacology profiling based on a multifingerprint similarity predictive approach. J Chem Inf Model. 2021;61(10):4868–4876.
- Janela T, Takeuchi K, Bajorath J. Introducing a chemically intuitive core-substituent fingerprint designed to explore structural requirements for effective similarity searching and machine learning. Molecules. 2022;27(7):2331.
- Todeschini R, Consonni V, Xiang H, et al. Similarity coefficients for binary chemoinformatics data: overview and extended comparison using simulated and real data sets. J Chem Inf Model. 2012;52(11): 2884–2901.
- Gao K, Nguyen DD, Sresht V, et al. Are 2D fingerprints still valuable for drug discovery? Phys Chem Chem Phys. 2020;22(16):8373–8390.
- Axen SD, Huang XP, Caceres EL, et al. A simple representation of three-dimensional molecular structure. J Med Chem. 2017;60(17): 7393–7409.
- Skuta C, Cortes-Ciriano I, Dehaen W, et al. QSAR-derived affinity fingerprints (part 1): fingerprint construction and modeling performance for similarity searching, bioactivity classification and scaffold hopping. J Cheminform. 2020;12(1):39.
- Bragina ME, Daina A, Perez MAS, et al. The SwissSimilarity 2021 web tool: novel chemical libraries and additional methods for an enhanced ligand-based virtual screening experience. Int J Mol Sci. 2022;23(2): 811.
- O’Boyle NM, Banck M, James CA, et al. Open babel: an open chemical toolbox. J Cheminform. 2011;3:33.
- Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem. 2011;32(7):1466–1474.
- Venkatraman V. FP‑ADMET: a compendium of fingerprint‑based ADMET prediction models. J Cheminform. 2021;13(1):75.
- Zhao Y, Wang XG, Ma ZY, et al. Systematic comparison of ligand-based and structure-based virtual screening methods on poly (ADP-ribose) polymerase-1 inhibitors. Brief Bioinform. 2021;22(6):1–13.
- Zhang Q, Muegge I. Scaffold hopping through virtual screening using 2D and 3D similarity descriptors: ranking, voting, and consensus scoring. J Med Chem. 2006;49(5):1536–1548.
- Mendolia I, Contino S, Perricone U, et al. Convolutional architectures for virtual screening. BMC Bioinformatics. 2020;21(Suppl 8):310.
- Mendolia I, Contino S, De Simone G, et al. EMBER-embedding multiple molecular fingerprints for virtual screening. Int J Mol Sci. 2022;23(4):2156.
- Zhao J, Ma Q, Zhang B, et al. Exploration of SARS-CoV-2 3CL(pro) inhibitors by virtual screening methods, FRET detection, and CPE assay. J Chem Inf Model. 2021;61(12):5763–5773.
- Myint KZ, Wang L, Tong Q, et al. Molecular fingerprint-based artificial neural networks QSAR for ligand biological activity predictions. Mol Pharm. 2012;9(10):2912–2923.
- Xie L, Xu L, Kong R, et al. Improvement of prediction performance with conjoint molecular fingerprint in deep learning. Front Pharmacol. 2020;11:606668.
- Nasser M, Salim N, Hamza H, et al. Improved deep learning based method for molecular similarity searching using stack of deep belief networks. Molecules. 2020;26(1): 128.
- Perron Q, Mirguet O, Tajmouati H, et al. Deep generative models for ligand-based de novo design applied to multi-parametric optimization. J Comput Chem. 2022;43(10):692–703.
- Yang K, Swanson K, Jin W, et al. Analyzing learned molecular representations for property prediction. J Chem Inf Model. 2019;59(8):3370–3388.
- Probst D, Reymond JL. A probabilistic molecular fingerprint for big data settings. J Cheminform. 2018;10(1):66.
- Miranda-Quintana RA, Racz A, Bajusz D, et al. Extended similarity indices: the benefits of comparing more than two objects simultaneously. Part 2: speed, consistency, diversity selection. J Cheminform. 2021;13(1): 33.
- Le T, Winter R, Noé F and Clevert D. (2020). Neuraldecipher – reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structures. Chem. Sci., 11(38), 10378–10389. 10.1039/D0SC03115A