
1. Introduction
Proteolysis-targeting chimeras (PROTACs) have emerged as a novel therapeutic modality for the degradation of target proteins via the ubiquitin-proteasome system. Currently, more than a dozen PROTACs are under clinical trials. For example, ARV-110 (bavdegalutamide) and ARV-471 (ClinicalTrials.gov Identifiers: NCT03888612, NCT04072952) against androgen receptor and estrogen receptor in prostate and breast cancers, respectively, are in Phase 2 clinical trials. Structurally, PROTACs are chimeric molecules composed of two ligands – one for the protein of interest (POI) and one for a ubiquitin ligase (E3) – connected via a linker. Several E3 ligands for PROTACs have been developed so far, which include the ligands for the Von Hippel–Lindau (VHL), cereblon (CRBN), inhibitor of apoptosis proteins (IAPs), and mouse double minute 2 homolog (MDM2) E3 ubiquitin ligases. Structure–activity relationship studies of PROTACs often focus on the linker structures. For example, ARV-471 and ARV-110 contain short and rigid linkers with piperidine and piperazine moieties. Short lipophilic linkers with cyclic ionizable groups can increase cell-membrane permeability and solubility, and impart moderate metabolic stability [Citation1]. Structural rigidification of PROTACs may be necessary to optimize the geometry of the POI/PROTAC/E3 ligase ternary complex, thereby affecting PROTAC efficacy. Thus, chemical modifications are likely to be important in helping PROTACs meet absorption, distribution, metabolism, and excretion (ADME) requirements.
In contrast to small-molecule degraders such as thalidomide that generally show good cell permeability, most PROTACs show poor cell permeability due at least in part to a high molecular weight (usually over 800 Da); this is significantly higher than the ‘rule-of-5’ (Ro5) criteria of 500 Da, which is a guideline for orally active small-molecule drugs [Citation2]. Even though a slight reduction in molecular weight may be feasible, the smallest molecular weight of PROTACs is considered to be approximately 650 Da. Other molecular properties that are listed in Ro5 and Veber’s rules must also be considered in the development of PROTACs, such as logP, number of hydrogen bond donors (HBDs) and acceptors (HBAs), number of rotatable bonds, and polar surface area [Citation2,Citation3]. For example, the replacement of an amide bond with an ester reduces the HBDs and the polar surface area, which improves the membrane-permeability of the molecule [Citation4]. Solubility is also an important factor in oral bioavailability. It has been proposed that the aqueous solubility of compounds in the early phase of drug discovery should be >60 μg/mL [Citation5]; however, the solubility of PROTACs in water is generally poor and usually needs to be optimized. In the case of ARV-110 and ARV-471, the calculated topological polar surface areas (TPSAs) are 182.86 Å2 and 96.43 Å2, respectively, which are within the region of beyond-Ro5 drugs (50–200 Å2) [Citation6].
However, 2D descriptors such as logP and TPSA are insufficient to explain favorable permeability of compounds in beyond-Ro5 including high molecular weight oral drugs due to their folded conformations as chameleonicity. PROTAC classified as beyond-Ro5 may also have conformational flexibility and chameleonicity, which can propose both high cell permeability and aqueous solubility. To describe their characteristics, 3D descriptors such as radius of gyration and solvent-accessible 3D polar surface area (PSA) are important. The NMR analysis of PROTAC in chloroform mimicking the inside of cell membranes showed minimization of size and polarity of PROTAC with the formation of intramolecular and nonclassical hydrogen bonds, – interactions, and shielding of amide groups from solvent.
In the early phase of drug discovery, parallel artificial membrane permeability assay (PAMPA) and transcellular Caco-2 permeability assay are typically used to evaluate the passive transmembrane diffusion of PROTAC. PAMPA is used as a fast, low-cost, high-throughput screening tool, but there are cases where PROTACs for androgen receptor, which exhibits cell membrane permeability, could not be correctly assessed for permeability by the PAMPA method [Citation7]. Caco-2 assays can estimate permeability involved in active transport such as transporters-mediated mechanisms of efflux or cellular uptake and paracellular permeation, offering reliability for in vivo prediction of ADME mechanisms. However, low solubility in the assay buffers and nonspecific binding in Caco-2 assays may result in low recovery and/or inaccurate apparent permeability (Papp) values. Several approaches have been reported to increase the cell permeability of PROTACs as described below ( and ).
Figure 1. PROTAC delivery into cells and chemical structures of typical E3 ligands.
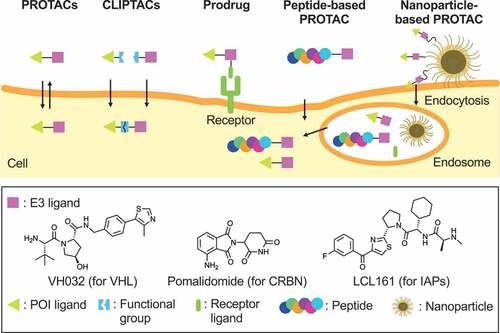
Table 1. Chemical structures of PROTACs described in this editorial.
2. Optimization of the E3 ligand – from peptide to small molecule
The molecular weight of a PROTAC may be reduced by using a small molecule, rather than peptide-based, E3 ligand. For example, a small-molecule ligand for VHL – a commonly used E3 ligase – is derived from a peptide-based VHL ligand. Peptide-based ligands are useful for interacting with target proteins by mimicking protein–protein interactions, which often occur on shallow surfaces. The first-generation VHL ligand was designed from the co-crystal structure of VHL and its natural ligand, hydroxylated hypoxia-inducible factor-1 (HIF-1). A 10-amino acid sequence from hydroxylated HIF-, which includes a crucial hydroxyproline (Hyp564) residue, was selected as a peptide ligand. However, the binding affinity of this ligand was in the micromolar range. To improve its affinity, the Ciulli’s group focused on the essential elements and acetylated N-terminus of the VHL ligand, thereby arriving at VH032, which shows higher binding affinity (Kd = 185 nM) and moderate lipophilicity [Citation8]. The low molecular weight of VH032 (473 Da) improved the usefulness of PROTACs that operate through VHL, and the chemical modification also improved cell membrane permeability [Citation8]. Subsequently, DT2216, PROTAC targeting B-cell lymphoma-extra large (Bcl-xL) containing the methylated VH032, was developed and is currently in Phase I clinical trial [Citation9]. In addition to the VHL ligand, non-peptidic E3 ligands, thalidomide-type compounds such as pomalidomide are used as cereblon ligands, and LCL161 and GDC-0152 are as inhibitor of apoptosis protein ligands.
3. Linker optimization
The drug-like properties of PROTACs can sometimes be dramatically improved by linker modifications that can change HBDs, lipophilicities, molecular weight, rotatable bonds, and polar surface area. The effect of HBDs of linkers on cell membrane permeability has been evaluated in bromodomains and extra-terminal domain (BET) degraders. The amide bond of BET degraders, MZ1 and ARV-771 was replaced with the ester group reducing one HBD and the lipophilicities, which improve PAMPA permeability, 0.01–0.1 and 0.2–0.3 (× 10−6 cm/s), and ALogP, 3.6–4.3 and 4.2–4.8, respectively, and showed improved degradation activity (pDC50) such as 7.2 ± 0.2 to 6.9 ± 0.2 and 7.4 ± 0.2 to 7.2 ± 0.2 [Citation4]. Analysis by classification of reported degraders showed that their efficacy of CRBN-based and VHL-based degraders correlates with an increasing number of rotatable bonds and molecular weight. The number of rotatable bonds is expected to increase with the widespread use of flexible PEG and alkyl linkers for PROTAC [Citation10]. On the other hand, it is possible to develop potent PROTACs, which have a relatively small molecular weight and a low number of rotatable bonds. Our group recently developed a linker-less PROTAC targeting hematopoietic prostaglandin D synthase (H-PGDS), PROTAC(H-PGDS)-7, using in silico design, which showed potent activity (DC50 = 17.3 pM) [Citation11]. PROTAC(H-PGDS)-7 shows small molecular weight, 743, and low rotatable bonds, 10, resulting in more drug-like properties. In addition, the linker moiety allows PROTACs to form folded conformations and decrease 3D polar surface area that correlates to high cell membrane permeability. For example, PEG linkers of PROTACs are more likely to fold structure than alkyl linkers [Citation12].
4. CLIPTACs
In-cell click-formed proteolysis targeting chimaeras (CLIPTACs) are an attractive low-molecular weight PROTAC design [Citation13]. CLIPTACs are based on the intracellular construction of full PROTACs by a bio-orthogonal reaction, such as a click reaction, between two separate moieties. The combination of the bromodomain 4 (BRD4) ligand JQ1, functionalized with trans-cyclooctene, and E3 ligase ligand pomalidomide, functionalized with tetrazine, was used to form an active PROTAC targeting BRD4, JQ1-CLIPTAC, in cells.
5. Prodrugs: receptor-mediated delivery
Prodrugs are common in the field of small molecule-based drug design to improve cell permeability. Prodrugs have also been applied to PROTACs to overcome unfavorable cell permeability and pharmacokinetic properties. To increase the cell permeability of PROTACs, the polar surface was masked with lipophilic groups via cleavable bonds, such as esters, resulting in the development of an in vivo orally administrable degrader [Citation14]. The PROTAC was targeted by conjugating cell membrane receptor ligands for folate receptor α (FOLR1) [Citation15], human epidermal growth factor receptor 2 (HER2) and nucleolin. Liu and coworkers developed folate-ARV-771, containing folate, which degraded BRD4 in a FOLR1-dependent manner in cancer cells [Citation15]. In the case of HER2 and nucleolin, large molecules, such as antibodies [Citation16] and aptamers [Citation17], were conjugated with PROTACs, and these conjugated molecules were able to enter cells via endocytosis.
6. CPP conjugates
Cell-penetrating peptides (CPPs), such as oligoarginine and TAT peptide derived from HIV-1, have been used to transport molecules into the cell. The first PROTAC containing octa-arginine, (D-Arg)8, was reported by Sakamoto and coworkers, and targeted the androgen receptor (AR) [Citation18]. Our group developed a peptide-based PROTAC, LCL-stPERML-R7, targeting estrogen receptor α (ERα), which contained a cell-penetrating peptide [hepta-arginine, (L-Arg)7] to increase the cell permeability [Citation19]. In addition, side-chain stapling was introduced into the ERα-binding peptide to stabilize its helical structure, thereby enhancing the cell permeability and increasing the chemical stability. The stapled peptide-based PROTAC, LCL-stPERML-R7, showed moderate ERα-degradation activity.
7. Nanoparticles
In recent years, technologies of nanomedicine have been combined with advanced biomaterials and biomedical engineering to overcome the limitations of small molecule-based drugs. Nanoparticle-based delivery systems can transport cargos across biological barriers, while increasing circulation time and improving efficacy and safety. Nanoparticle-based PROTAC delivery systems can be separated into two categories: physical encapsulation and chemical conjugation [Citation20]. These nanoparticle-based delivery systems can enhance the stability and solubility of their cargo and have been applied to facilitate the cellular uptake of PROTACs. Nanoparticle-based systems can also increase the aqueous solubility of PROTACs and improve the physicochemical properties that are compromised by high hydrophobicity. A variety of nanoparticles, such as lipid, polymeric and inorganic, have been used as PROTAC delivery systems. SPNpro, developed by Zhang and coworkers, degrades immunosuppressive indoleamine 2,3-dioxygenase (IDO), thereby inhibiting tumor growth and metastasis via synergistic activation of effector T cells and photo-immunometabolic cancer therapy [Citation21].
8. Expert opinion
PROTACs have several advantages over conventional drugs and have great potential as a therapeutic modality. The development of several orally available PROTACs is feasible. For the clinical development, the drug-like properties of PROTACs attract more attention. Even though oral bioavailability is not simply dependent on membrane permeability, there are PROTACs with moderate oral bioavailability even without meeting three parameters on Ro5 [Citation22]. In the point of lipophilicity, orally bioavailable PROTAC typically has high lipophilicity compared with conventional small-molecule drugs. The solubility and permeability of PROTACs could be significantly affected even by small structural modifications that can alter shape, flexibility/rigidity, pKa, etc., as other beyond Ro5 drugs. Recently developed PROTACs can be classified into several groups such as small molecule-, peptide- and nucleic acid-based molecules, with most compounds falling into the category of small molecules. However, most PROTACs are large in size because they have two ligands and a linker, and therefore, categorized in the beyond-Ro5 range. The reduction of molecular weight is often pursued to increase cell permeability and drug-likeness. Another approach to increase drug-likeness is to use an appropriate linker modification, which can impact cell permeability. For example, the use of a cationic and rigid linker, including piperidine and piperazine moieties, can improve rigidity, water solubility, and cell permeability. To estimate the cell permeability of PROTACs, several methods, such as PAMPA and Caco2 assays, and in silico calculations can be used. In addition to the chemical modification of the PROTACs to improve drug-likeness, the intracellular uptake of PROTACs can be increased by the use of membrane receptors. For peptide-based PROTACs, CPP conjugation, side-chain stapling, and cyclization, can increase cell permeability [Citation19]. Lipid nanoparticles are frequently used in the development of nucleic acid-based medicines (siRNAs, antisense, etc.), and such the lipid nanoparticles are expected to improve the cell permeability of PROTACs containing oligonucleotides. We think chemical modification of PROTACs to improve drug-likeness is the most promising approach to increase cell permeability for clinical application. Specifically, it is possible to improve cell membrane permeability of PROTACs by modifying its linker structures and binding styles (esterification, piperazine, cyclic amine linker, rigid linker) to conjugate the E3 ligand and the POI ligand. In addition, the assay system must be carefully selected to evaluate the permeability. As one efficient method, high-throughput screening of linkers was reported by synthesizing various PROTACs via amide formation between N-hydroxysuccinimide and the amine moiety, and evaluating permeability and degradation activity using relative binding affinities (RBAs) and the luminescent peptide tag, HiBiT, respectively [Citation23]. In the future, the combinations of analyzing big data on the permeability of PROTACs and predicting physicochemical properties is expected to be rational design with higher cell membrane permeability. Clinical application of larger PROTACs containing peptides and oligonucleotides is challenging at this moment; however, the combination with an appropriate drug delivery system will enable them to penetrate into cells efficiently, which will facilitate the clinical application of the PROTACs.
Abbreviation
proteolysis-targeting chimeras (PROTACs)
protein of interest (POI)
ubiquitin ligase (E3)
Von Hippel–Lindau (VHL)
cereblon (CRBN)
inhibitor of apoptosis proteins (IAPs)
mouse double minute 2 homolog (MDM2)
absorption, distribution, metabolism, and excretion (ADME)
rule-of-5 (Ro5)
hydrogen bond donors (HBDs)
hydrogen bond acceptors (HBAs)
topological polar surface areas (TPSAs)
polar surface area (PSA)
B-cell lymphoma-extra large (Bcl-xL)
bromodomains and extra-terminal domain (BET)
parallel artificial membrane permeability assay (PAMPA)
hypoxia-inducible factor-1 (HIF-1)
hematopoietic prostaglandin D synthase (H-PGDS)
Duchenne muscular dystrophy (DMD)
click-formed proteolysis targeting chimaeras (CLIPTACs)
bromodomain 4 (BRD4)
folate receptor α (FOLR1)
human epidermal growth factor receptor 2 (HER2)
cell-penetrating peptides (CPPs)
androgen receptor (AR)
estrogen receptor α (ERα)
indoleamine 2,3-dioxygenase (IDO)
relative binding affinities (RBAs)
Declaration of interest
M Naito belongs to the Social Cooperation Program of Targeted Protein Degradation supported by Eisai Co., Ltd. and serves as a scientific advisor to UBiENCE. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Cecchini C, Tardy S, Scapozza L. Linkers as game-changers in protac technology: emphasizing general trends in PROTAC pharmacokinetics for their rational design. Chimica. 2022;76(4):341–345.
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26.
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623.
- Klein VG, Bond AG, Craigon C, et al. Amide-to-ester substitution as a strategy for optimizing PROTAC permeability and cellular activity. J Med Chem. 2021;64(24):18082–18101.
- Kerns EH, Di L, Carter GT. In vitro solubility assays in drug discovery. Curr Drug Metab. 2008;9(9):879–885.
- Jimenez DG, Sebastiano MR, Caron G, et al. Are we ready to design oral PROTACs®? ADMET DMPK. 2021;9(4):243–254.
- Scott DE, Rooney TPC, Bayle ED, et al. Systematic investigation of the permeability of androgen receptor PROTACs. ACS Med Chem Lett. 2020;11(8):1539–1547.
- Galdeano C, Gadd MS, Soares P, et al. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel–Lindau (VHL) E3 ubiquitin ligase and the Hypoxia Inducible Factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem. 2014;57(20):8657–8663.
- Lv D, Pal P, Liu X, et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat Commun. 2021;12(1):6896.
- Maple HJ, Clayden N, Baron A, et al. Developing degraders: principles and perspectives on design and chemical space. MedChemComm. 2019;10(10):1755–1764.
- Yokoo H, Shibata N, Endo A, et al. Discovery of a highly potent and selective degrader targeting hematopoietic prostaglandin D synthase via in silico design. J Med Chem. 2021;64(21):15868–15882.
- Poongavanam V, Atilaw Y, Siegel S, et al. Linker-dependent folding rationalizes PROTAC cell permeability. J Med Chem. 2022;65(19):13029–13040.
- Lebraud H, Wright DJ, Johnson CN, et al. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci. 2016;2(12):927–934.
- Wei M, Zhao R, Cao Y, et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur J Med Chem. 2021;209:112903.
- Liu J, Chen H, Liu Y, et al. Cancer selective target degradation by folate-caged PROTACs. J Am Chem Soc. 2021;143(19):7380–7387.
- Dragovich PS, Pillow TH, Blake RA, et al. Antibody-mediated delivery of chimeric BRD4 degraders. part 1: exploration of antibody linker, payload loading, and payload molecular properties. J Med Chem. 2021;64(5):2534–2575.
- He S, Gao F, Ma J, et al. Aptamer-PROTAC conjugates (APCS) for tumor-specific targeting in breast cancer. Angew Chem Int Ed. 2021;60(43):23299–23305.
- Sakamoto KM, Kim KB, Kumagai A, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98(15):8554–8559.
- Yokoo H, Ohoka N, Takyo M, et al. Peptide stapling improves the sustainability of a peptide-based chimeric molecule that induces targeted protein degradation. Int J Mol Sci. 2021;22(16):8772.
- Chen Y, Tandon I, Heelan W, et al. Proteolysis-targeting chimera (PROTAC) delivery system: advancing protein degraders towards clinical translation. Chem Soc Rev. 2022;51(13):5330–5350.
- Zhang C, Zeng Z, Cui D, et al. Semiconducting polymer nano-PROTACs for activatable photo-immunometabolic cancer therapy. Nat Commun. 2021;12(1):2934.
- Pike A, Williamson B, Harlfinger S, et al. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discov Today. 2020;25(10):1793–1800.
- Hendrick CE, Jorgensen JR, Chaudhry C, et al. Direct-to-biology accelerates PROTAC synthesis and the evaluation of linker effects on permeability and degradation. ACS Med Chem Lett. 2022;13(7):1182–1190.