
ABSTRACT
Introduction
Phage display technology is a well-established versatile in vitro display technology that has been used for over 35 years to identify peptides and antibodies for use as reagents and therapeutics, as well as exploring the diversity of alternative scaffolds as another option to conventional therapeutic antibody discovery. Such successes have been responsible for spawning a range of biotechnology companies, as well as many complementary technologies devised to expedite the drug discovery process and resolve bottlenecks in the discovery workflow.
Areas covered
In this perspective, the authors summarize the application of phage display for drug discovery and provide examples of protein-based drugs that have either been approved or are being developed in the clinic. The amenability of phage display to generate functional protein molecules to challenging targets and recent developments of strategies and techniques designed to harness the power of sampling diverse repertoires are highlighted.
Expert opinion
Phage display is now routinely combined with cutting-edge technologies to deep-mine antibody-based repertoires, peptide, or alternative scaffold libraries generating a wealth of data that can be leveraged, e.g. via artificial intelligence, to enable the potential for clinical success in the discovery and development of protein-based therapeutics.
1. Introduction
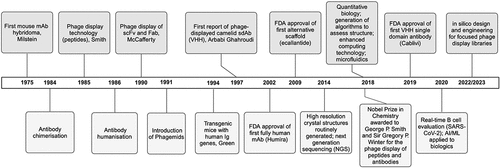
The implementation of in vitro display methodologies is well established for the discovery of therapeutic bioactive molecules, such as monoclonal antibodies (mAbs), nanobodies®, peptides, cyclic peptides, Anticalins®, and other biological scaffolds. Phage display was the first such in vitro technology initially devised for the display of peptides in the 1980s [Citation1], subsequently followed by the display of scFv (single chain variable fragments) and Fab (fragment antigen-binding regions) antibody fragments in the 1990s [Citation2–4] and then camelid VHH (variable domain of the heavy chain of a heavy chain only antibody) single domain antibodies [Citation5] and variable new antigen receptors (VNARs), which are the binding sites of the shark IgNAR variable single-domain antibodies [Citation6,Citation7]. VHH and VNAR antibody fragments have emerged as a biological format in more recent times; although considered by some as alternative scaffolds, they are nonhuman antibody fragments [Citation8]. depicts a historical timeline of peptide, antibody, and alternative scaffold discovery specifically highlighting key events in antibody platform developments, phage-derived therapeutics, and the implementation of cutting-edge technologies that have informed and accelerated this drug discovery process.
Figure 1. Protein-based biologics phage display timeline. This schematic shows a historical timeline of peptide, antibody, and alternative scaffold discovery with an emphasis on phage display where key events in antibody platform developments are highlighted, including the first approvals of phage-derived therapeutic protein-based biologics. Examples of the emergence and implementation of disruptive technologies that have radically accelerated drug discovery process are also indicated. Created with BioRender.com.
1.1. Phage display of antibodies
Phage display provides the means to generate protein molecules in a directed evolution manner artificially ‘in the test tube’ enabling the rapid identification of an enriched population of biomolecules that specifically target the antigen of interest. Crucially, phage display technology directly links phenotype to genotype via fusion of the peptide or antibody fragment to a coat protein on the bacteriophage virus particle. The most common format for the display of antibody fragments or scaffolds on phage is the genetic fusion of immunoglobulin variable chains or alternative scaffolds to the terminal phage pIII coat protein. This may be achieved either by direct engineering into the phage genome (the phage format) or by using a pIII-expressing plasmid in combination with a helper phage system (the phagemid format). For the phage format, all pIII protein molecules are expressed as fusions where 3–5 copies are displayed on the tip of bacteriophage (multivalent display). However, in most phagemid systems [Citation9], most of the pIII is derived from the helper phage and just a small proportion originates from the phagemid (less than one copy per phage on average), which leads to monovalent display. The advantage of the phage format is that it provides the potential to isolate binders with low to medium affinities, but higher affinities are required for the isolation of antigen binders when using the phagemid format.
M13 filamentous phage, including fd and f1, infects E. coli strains presenting the F pilus, which enables a fast and facile discovery process within a laboratory environment in just a few weeks. M13 belongs to the Ff group of filamentous phages that only infect E. coli expressing the F pilus where infection of the bacterium requires binding to the tip of the F pilus via the phage minor coat protein pIII (encoded by the g3p gene). The g3p display system is the most widely used and a phage vector carries all the genes needed for infection, replication, assembly, and budding while also incorporating the coat protein pIII-antibody fusion, resulting in multivalent display (3–5 copies). The phagemid vector system, by contrast, uncouples antibody display from phage propagation and is plasmid-based containing a bacterial origin of replication for propagation in E. coli and an F1 phage origin for replication and packaging of single-stranded DNA, as well as an antibiotic marker for the selection and propagation of the plasmid and molecular tags, e.g. c-Myc, that will assist with the detection and protein purification [Citation3,Citation10,Citation11]. While a key advantage of monoclonal display is to facilitate the routine selection of high affinity clones by avoiding the avidity effect during panning [Citation4,Citation12], there can also be a high wild-type phage background.
Coinfection of E. coli with a helper phage, such as M13KO7, is necessary to provide all the proteins for phage proliferation. Helper phage is packaged into virions at a lower efficiency than the phagemid, and this enables the majority of mature virions to contain phagemid, thus antibiotic selection of cells containing the phagemid with the biologic fusion is required. Only 1 in 10 phage particles will display the fusion protein and those that do will only contain a single copy [Citation13]. The M13KO7 helper phage system has been engineered further to improve antibody presentation where there is a deletion of the pIII gene, known as the hyperphage system [Citation14]. Hyperphages carry a deletion in the pIII gene and are generated in an E. coli packaging cell strain producing functional pIII, which is used to package a phage genome with a pIII deletion. The resulting hyperphage carry functional pIII on their surface but lack the pIII gene in their genome. These hyperphage can then be used to infect bacteria with a phagemid library. Each of the resulting phage particles presents several copies of the antibody, alternative scaffold, or peptide on its surface, thereby significantly increasing panning efficiency over 100-fold. Additional benefits include less antigen required for the panning process, use in cell panning, and an increase in phage ELISA sensitivity; this has been adopted as an industry standard.
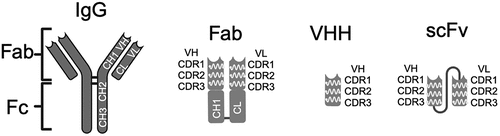
Phagemids are smaller than phage, so this allows for higher transformation efficiencies in the construction of large repertoire libraries and is best suited for the display of antibody fragments, such as scFv, Fab, and VHH, as well as alternative scaffolds, but can also be used to display peptides at a low valency; as phage vectors offer multivalent display, they are best suited for peptides and can be with either the g3p (3–5 copies) or g8p gene (~2700 copies) [Citation15]. The technique is widely used now for a variety of applications, one of the most significant being the generation of therapeutic biologics where large antibody fragments (scFv, Fab, or VHH, refer to ) or alternative scaffold repertoires derived from human sequences have been established, that yield functional binders with subnanomolar affinities [Citation16–20]. The empowerment that this combinatorial biochemical approach has given to the discovery of biologics has been internationally recognized with the joint award of the 2018 Nobel Prize in Chemistry to George P. Smith and Sir Gregory P. Winter for the phage display of peptides and antibodies, respectively.
Figure 2. Phage display antibody fragments. This schematic shows the structural differences between full length IgG molecules and Fab, scFv, and VHH antibody fragments used in the phage display of antibodies. Each format contains an antibody-binding region in the variable domain that is conferred by distinct sites known as complementarity-determining regions (CDRs), there are three CDRs within a variable region, and these are located toward the amino terminal. The Fab fragment (~50kDa) consists of two polypeptide chains corresponding to the variable heavy chain region (VH) linked to the first immunoglobulin constant-1 heavy chain domain (CH1) and the variable light chain domain (VL) linked to the light chain constant domain (CL); their association occurs via intramolecular interactions between residues on each polypeptide chain. The VH and VL can also be linked in a single polypeptide via a short Gly-Ser linker to form a scFv, which is smaller than a Fab fragment (~30kDa). The VHH fragment only consists of the heavy chain variable region and is the smallest antibody fragment shown here (~15kDa). Created with BioRender.com.
In addition, phage display has been shown to synergize with other display technologies, such as ribosome display and yeast display, which can be implemented for optimization of the protein’s drug-like properties and affinity maturation [Citation21,Citation22], as well other technologies such as next-generation sequencing (NGS), flow cytometry, and microfluidics to enhance throughput. The implementation of phage display has revolutionized therapeutic antibody discovery and development, initiating the establishment of multiple biotechnology companies where this was the core technology for generating human antibodies, for example, Cambridge Antibody Technology [Citation16], Dyax Corporation [Citation23], and MorphoSys [Citation24].
There are at least 19 approved mAbs and nanobody-based entities that have been derived from the application of phage display as summarized in . Other examples of current phage-derived antibodies that have attained late stage clinical development are shown in and include bimagrumab, which reached Phase 3 for the treatment of sporadic inclusion body myositis, but was subsequently discontinued for this indication for unspecified reasons, and is still active in Phase 2 for the treatment of obesity (targeting Activin Receptor Type 2B), utomilumab in Phase 2 for the treatment of breast cancer (targeting CD137, also known as 4-1BB), mavrilimumab in Phase 3 for the treatment of inflammation (targeting GM-CSFRα), and otilimab in Phase 3 studies evaluating clinical efficacy in the treatment of COVID-19 and COVID-19-related pneumonia (targeting GM-CSF). Other examples are shown in , as well as the most advanced alternative scaffolds, and a recent review provides a comprehensive overview of phage-derived mAbs in clinical development [Citation25]. Phage display is comparable to immunization methodologies with regards to the generation of high affinity binders, but can be used to isolate antibodies or other binder types to targets not easily found via hybridoma or single B-cell screening technologies, such as toxins and self-antigens.
Table 1. Examples of approved phage display-derived mAbs and nanobodies.
Table 2. Examples of phage display-derived mAbs, alternative scaffolds, and peptides that have progressed to clinical development.
As with small molecules, the discovery and development of novel biologics often begin with the screening of large libraries in which millions to billions of antibody (or antibody fragment) sequences are represented. When these libraries are diverse, with uniform representation among variants, and are free of liability motifs that can lead to poor pharmacokinetics or developability issues, hundreds of promising candidates can be found and carried forward for further characterization and optimization.
For therapeutic antibody discovery, antibody fragments containing the VH and VL regions (as scFv or Fab fragments) from naïve or synthetic repertoires or VHH fragments representing synthetic [Citation26], naïve [Citation27] or immune repertoires [Citation5,Citation28], respectively, have been the choice for display given the large size of an intact immunoglobulin molecule, and various examples are provided below. Further examples of each have been extensively reviewed in several recent publications [Citation25,Citation29–31]. The derivation of antibody libraries aims at the generation of different types of repertoires ranging from naïve repertoires from healthy volunteers, target-specific repertoires obtained from immunizing animals or vaccinated humans, as well as immune repertoires from disease conditions, to semisynthetic and designed fully synthetic libraries that can also encompass repertoires constructed to direct antibodies to specific target types, such as G protein-coupled receptors (GPCRs) [Citation32], and examples of these various repertoire libraries are described, as follows.
Repertoires of antibodies from nonimmunized donors have been considered a source from which it is possible to isolate highly specific antibodies to a wide range of antigens whereby heavy and light chains are randomly combined, and, due to the heavy and light chain rearrangement, the library diversity is dramatically increased beyond the natural heavy and light chain pairing. This approach generates specificities that do not exist in the natural repertoire enabling the identification of antibodies targeting any given antigen, including self-antigens. The derivation of these repertoires from naturally occurring human germline sequences ensures fully human sequences are used. A disadvantage of naïve repertoires is that discovered antibody clones often have lower affinities than those isolated from immune sources, therefore requiring optimization to enhance the affinity, which can be achieved using a variety of different display platforms that are compatible with phage display.
An early example of a naïve scFv library isolated mRNA from the B cells of lymphoid tissues from 43 nonimmunized donors, namely peripheral blood lymphocytes, tonsil tissue, and bone marrow. The VH and VL repertoires were amplified separately, and following assembly and preparation of the scFv fragments (using a (Gly4Ser)3 linker), the combinatorial repertoire was cloned into the phagemid pCANTAB6 and transfected into E. coli yielding 1.4 × 1010 transformants. The resulting library was then used for test selections against a variety of foreign and self-antigens, including haptens and the toxin doxorubicin, successfully generating specific binders to each of the antigens with affinities <10 nM [Citation16]. This scFv library has subsequently been expanded further using B cells from 160 donors derived from spleen and fetal liver tissue yielding a scFv phage library consisting of 1.29 × 1011 transformants [Citation18]. Similarly, one of the first commercial naïve Fab phage display libraries was generated using B cells derived from the peripheral blood lymphocytes isolated from four healthy donors, as well as those isolated from part of a tumor-free spleen removed from a gastric carcinoma patient. Heavy and light chain variable regions were amplified separately to maintain maximal diversity and then combined in a two-step cloning procedure into the bicistronic phagemid vector pCESI, which contained the constant domain regions, yielding 3.7 × 1010 independent Fab transformants [Citation23]. The resulting Fab library was evaluated using a variety of antigens, including tetanus toxoid, the breast cancer-associated MUC1 antigen, a hapten and three closely related glycoprotein hormones, yielding diverse panels of binders with low nanomolar double-digit affinities, as well as clones that were either specific or cross-reactive for the closely related glycoprotein hormone antigens.
While naïve antibody libraries are created for a large diversity of antibody sequences that can theoretically be used to isolate antibodies to any target, more focused libraries, such as those obtained from immunization, are enriched for a specific target, or utilizing antibody repertoires from autoimmune patients or volunteers previously exposed to infection or immunizations have also been generated [Citation33,Citation34]; refer to . Members of the camelid species are often immunized to prepare a focused or enriched immune library. Typically, IgG2 and IgG3 are chosen as the template from which to derive the VHH or nanobody fragments as they constitute the most abundant heavy chains in the blood system of camelids [Citation35]. In one of the first demonstrations of generating a VHH immune library, a dromedary camel was immunized simultaneously with tetanus toxoid and lysozyme, and following standard immunization procedures, peripheral blood lymphocytes were isolated from a blood sample. mRNA was isolated for cDNA synthesis, after which the VHH genes were amplified and prepared for cloning into the modified phagemid vector pHEN4 and ~ 500,000 transformants obtained. A duplicate library was constructed to increase the size of the repertoire for sampling (yielding ~107 transformants). Specific binders to tetanus toxoid could be isolated from both libraries, but only lysozyme specific binders were identified from the larger library, highlighting the important of the size of repertoire to provide the required diversity for identifying binders of interest. All binders were in the 10 nm-100 nM range [Citation5]. Further demonstration that the VHH format had utility in biologics discovery was provided with other immune libraries generated from immunization with α-amylase and erythrocyte carbonic anhydrase that yielded enzyme-specific active site binders generally with low double-digit affinities [Citation36]. Immune libraries are not restricted to the use of camelids as the source of antibody genes, and rabbits [Citation37], wild-type mice [Citation38,Citation39], and transgenic mice [Citation40] have also been used as immune repertoire sources for constructing immune antibody phage display libraries. However, this approach can have drawbacks related to variability and a general inability to scale. Direct PCR amplified repertoires are also uneven and present a source of sequence bias in immune-based libraries.
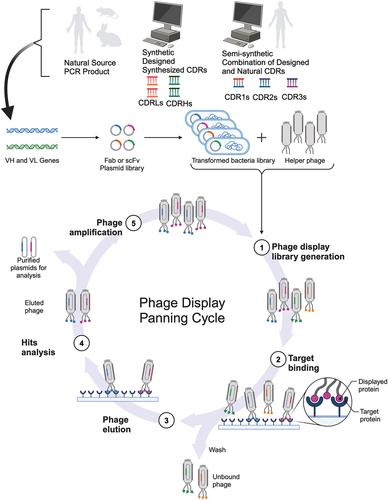
Figure 3. Types of phage libraries, construction, and panning process. This schematic depicts the different types of phage antibody libraries that can be constructed (top panel) and the iterative process of phage panning (bottom panel), which is equally applicable to the phage display of alternative scaffolds. The immune repertoire can be sourced from natural, semisynthetic, or fully synthetic variable genes and cloned into a phagemid vector. The resulting cloned repertoire is linked to gene3 for phage display, where E. coli is transformed with the phagemid library and then transfected with helper phage to produce the phage display library. This library is then incubated with the target antigen format to enrich for specific binders, where several washing steps are used to eliminate nonspecific binders and background. The remaining pool of phage binders are then eluted using a variety of methods (such as acidic pH or enzymatic cleavage of the linker between pIII and the antibody fragment) and used to transform E. coli for overnight amplification. This cycle is known as a round of selection. After three to five rounds of selection, single phage clones are evaluated for the specificity of their binding for the initial hit analysis and undergo DNA sequencing in parallel to determine the diversity of the positive phage antibody clone hits. Created with BioRender.com.
Advances in both DNA synthesis technology and synthetic biology have given researchers the power to design, produce, and screen custom semisynthetic and fully synthetic antibody libraries. There are many approaches to designing such libraries, and some examples are provided here. Whether starting with a known antibody sequence or taking a broader approach, bioinformatics can be applied to identify and eliminate potential sequence-related liabilities, and, more recently, computational tools have been harnessed to identify key loci in an antibody sequence where mutations are likely to impact antigen recognition [Citation41]. The resulting combinatorial variant library subsequently consists of antibody variants containing combinations of mutations, i.e. different amino acids can be encoded at a specific position and may alter the function or potency of the antibody clones [Citation42]. Since oligo pools can be used for generating these libraries, natural human CDRs can be directly folded into the library design. As a result, the synthetic antibody library is restricted only to sequences seen in nature and does not deviate from the natural limits of the antibody repertoire. Once designed, antibody libraries may then be constructed using DNA synthesis technology. Modern synthesis platforms leverage a myriad of approaches to create variant libraries that can generally be divided into direct and degenerate approaches.
Degenerate approaches rely on randomly incorporating nucleotides at specified locations within the growing oligonucleotide. Most common among these methods is the NNK approach, wherein the first and second positions of a codon may be occupied by any of the four nucleotide possibilities (A, T, G, C). The final position denoted by a K, the so-called wobble position, is limited to just two potential nucleotides (G, T). NNK synthesis can be extremely effective for directed evolution; however, its inefficiencies and bias-prone nature greatly limit its application in antibody development where redundancy of the genetic code may not provide additional value to the antibody library. This also means it is difficult to create uniform libraries where every variant antibody is equally represented. Another drawback with NNK synthesis is that it can produce stop codons, resulting in many truncated and nonfunctional antibodies [Citation43].
The use of libraries synthesized with trinucleotide phosphoramidites (trimers) can be a good alternative to NNK libraries because this controlled approach, referred to as TRIM, incorporates blocks of three nucleotides at a time [Citation44]. The trinucleotide blocks can be synthesized through various means and are designed such that every desired codon is represented, whereas undesired codons, such as stop codons or those coding for cysteine, can be avoided, thereby enabling the generation of a larger number of mutated amino acid positions which increases the diversity of the resulting repertoire by reducing nonfunctional and poorly developable antibodies due to randomization. However, there is still the potential for truncation and liability motifs. The TRIM approach was further refined using a direct synthesis approach called Slonomics [Citation45] to engineer diversified CDR-L3 and CDR-H3 cassettes on selected fixed VH/VL framework pairs to create the fully synthetic Fab Ylanthia® library [Citation46].
Another direct approach used to create the next generation of phage libraries encompasses a semisynthetic strategy and one such example is the n-CoDeRTM scFv library, which is 2 × 109 variants in size, generated using a modular construction. The immune repertoire was sourced from lymph nodes from seven different donors, peripheral blood lymphocytes from nine different donors, spleen from one donor, and tonsil RNA from five donors. All six CDRs from donor repertoires were amplified and reassembled into a single light (DP-L3) and heavy chain (VH3-23) framework that is well-presented in the human repertoire and well-expressed in host cells [Citation47]. This approach circumvents loss in diversity due to low expression or the display level of certain germline genes. The library was validated against a range of haptens, peptides, carbohydrates, and proteins and has successfully isolated antibody clones with subnanomolar affinities; in fact, at least five mAbs generated using the n-CoDerTM library have entered clinical trials. The semisynthetic strategy has been refined further by grafting naturally occurring CDRs from human antibodies into frameworks based on well-behaved clinical antibodies, referred to as a Generation Three design [Citation48], in order to address the identification of highly developable therapeutic antibodies directly from the library, CDR-H3 sequences were amplified directly from B cells, while the remaining CDRs were sourced from a large next-generation sequencing dataset and all sequence liabilities eliminated. The resulting scFv library is 9 × 109 in size with the CDR diversity incorporated into 4 VH frameworks, 3 Vκ frameworks, and 1 Vλ framework [Citation48]. In addition, two in vitro display platforms, phage and yeast display, were implemented to isolate diverse antibodies to clinically relevant protein targets, yielding affinities down to the subnanomolar and picomolar range demonstrating that this approach has the potential to considerably reduce discovery timelines.
In the early days of antibody phage display, the initial focus was on generating large diverse repertoires (>1010 transformants) to identify high affinity functional binders, which was subsequently followed by addressing aspects of sequence liabilities via synthetic approaches to resolve developability and expression challenges that occurred with some naturally occurring antibody sequences and errors introduced by initial synthetic approaches, as well as removal of T cell epitopes, for example, by the selection of frameworks that exhibited optimal stability and expression. These early synthetic libraries focused on using selected frameworks restricting diversity to the CDRs [Citation49,Citation50], but were unable to robustly generate high affinity antibodies against a broad range of protein targets. Thus, as well as efforts to expand the diversity further in all the CDR loops, the range of well-behaved frameworks was increased to generate content that better reflected the natural repertoire observed in humans [Citation24,Citation51]. Modular design features enabling facile engineering, such as for affinity maturation, were incorporated [Citation46] along with a focus on antibody sequences (and therefore structure) that impacted on performance and function.
One avenue of refining synthetic library design has been to take a minimalist strategy and decrease randomization by using a single framework with a binary code to diversify the CDR-H3. In one such study, Tyr and Ser were selected as these have been found to be highly abundant in antibody binding sites [Citation52]. The initial library randomized only the VH CDR positions by binary code [Citation53] and then diversity expanded incrementally to the VL CDR positions, as well as nonparatope residues that may impact on CDR conformation and structure. Improvements in affinity (to single digit nanomolar) were observed for the model soluble antigen used (VEGF) where the tyrosine side chains were sufficient to mediate the majority of antigen interactions. However, a glycine residue in the CDR-H3 was determined to be critical for the paratope conformation enabling high affinity binding. A high throughput workflow process was established and pressure-tested using a panel of proteins to demonstrate that reduction in the complexity in the design of the library circumvented some of the common challenges encountered with natural antibodies that can stall high throughput processes, such as expression, purification, and sequence analysis, and could have potential utility for proteomic studies and identify features in CDR sequences that regulate antibody:antigen interactions. In fact, this approach has also been applied to the 10FN3 alternative scaffold [Citation54].
While Tyr/Ser binary libraries will produce high affinity antibodies for some antigens, this is not true for all protein classes, implying that greater diversity would be required for a broader range of antigens and wider applications, and may even need to be tailored to specific protein target classes with more complex structures. Thus, the minimalist synthetic CDR-H3 library strategy was explored further to generate antibodies with therapeutic potential [Citation55]. Trinucleotide synthesis technology [Citation56] was employed to introduce approx. equal quantities of all amino acids at each engineered position (other than cysteine) in order to evaluate the contribution of amino acid classes to antibody function. Therefore, libraries were constructed on a single framework scaffold, the clinically validated DP47 heavy chain and DP-K22 light chain, with either randomization of the CDR-H3 alone or the CDR-H3 and CDR-L3 and included incorporation of a range of loop lengths (in equal proportions rather than the parabolic distribution seen with the natural human antibody repertoire). These libraries were evaluated on a number of different types of antigen to determine the influence of the amino acid composition and CDR length on the output from the selections. It was observed that loop length and CDR-H3 stem composition (potentially stabilizing the loop) played a key role in the functional output, whereas diversification of CDR-L3 was not essential for library performance. This was further borne out by demonstrating proof of principle using delta-like ligand 4 (DLL4) as antigen and isolating high affinity binders with neutralization of the DLL4-NOTCH1 interaction yielding IC50 values below 10 nM (and equivalent to a benchmark antibody directed to DLL4). Broadening the diversity by the inclusion of nonparatope residues not only enhances affinity but is necessary for target specificity, as well as CDR-H3 loop length [Citation52] and the CDR-L3 loop length influences VH antigen interactions [Citation55].
Thus, semisynthetic and synthetic libraries incorporate design features such as a germline framework known to have favorable developability features. The most important aspect of diversity in any phage antibody library is that of the complementarity determining regions or CDRs, which confer antigen recognition and specificity. Many more examples of strategies applied to generate a robust phage display library for antibody discovery have been comprehensively reviewed in several recent publications [Citation25,Citation31] as well as the key attributes of historically important antibody libraries constructed for the identification of therapeutic antibodies [Citation31].
Direct synthesis has enabled a much higher degree of precision and efficiency. Through solid-phase synthesis along with miniaturized chemistry, it is possible to synthesize oligonucleotides without randomization. Specifically, DNA synthesis can now be reliably performed on a large scale [Citation57,Citation58], such that millions of custom oligonucleotides can be synthesized as oligo pools on chip surfaces making the generation of large libraries of deliberately designed antibodies [Citation59] that can be rapidly produced and screened in parallel. Significantly, such libraries need not be full-length antibodies as synthesis only needs to be restricted to antibody regions that contain CDR loops, such as VHH, scFv, and Fab antibody fragments. This approach opens the door to high-throughput screening opportunities. As with other synthesis methods, direct synthesis is not without its hurdles. Highly parallel direct oligonucleotide synthesis requires substantial infrastructure and automation. However, direct synthesis has greatly decreased the costs associated with DNA synthesis and will likely continue to do so as the technology advances [Citation60]. The synthetic approach to library construction can also lend itself to the successful generation of bispecifics, for example, by either using a common light chain strategy to link the two different targeting specificities conferred by the heavy chain [Citation61] or using ‘knob-in-hole’ engineering on the Fc domain to link the two different specificities [Citation62]. Smaller antibody fragments, such as VHHs, may also be combined into bispecifics and multispecifics to enable higher avidity binding to their target, as well as linkage to anti-albumin VHHs or fusion to Fc regions for half-life extension.
More recent efforts have been focused on synthetic library builds implementing strategies for diversity generation using techniques that speed up the construction process, as well as incorporating features, such as bar-coding, e.g. FlyCode® [Citation63], and NGS for expedient identification of important antibody clones thereby addressing one of the bottlenecks in the screening cascade. Increasing use is being made of in silico and AI-guided designs to address developability concerns, such as the avoidance of potential post-translational modifications, T cell helper epitopes, thereby further reducing timelines and costs. Language models can be used to suggest CDR sequences that mimic human-based sequences and from these generate new synthetic library designs [Citation64].
While it is possible to create synthetic libraries that offer greater diversity than natural repertoires [Citation65], there are differences between the roles of the different CDRs and their interactions with antigen. Structural analysis of antibody:antigen interactions using the bioinformatic tool ‘CDR Analyzer’ has shown that natural antibodies tend to possess multiple CDR interactions with the antigen interface, whereas synthetic antibodies exhibit antigen interactions mainly via CDR-H3 with far less contribution from CDR-H1 and CDR-H2 [Citation66]. This reflects certain types of interaction. For example, in natural antibodies, the CDR-H1 is involved in cation-pi interactions, the CDR-H2 is a significant source of salt bridges, and CDR-H3 has the most hydrogen bond interactions with the antigen interface, whereas in synthetic antibodies, it is the CDR-H3 that accounts for the majority of contact. Thus, while the objective may be to enhance affinity and potency, by focusing solely on engineering of CDR-H3, one potential drawback is impacting on other desired features such as expression, stability, and selectivity of the antibody.
There are a host of publicly available bioinformatic tools available that did not exist when the first commercial workhorse antibody libraries were generated but can be implemented for optimization or design for structural analysis, loop prediction, and profiling, such as PylgClassify for CDR structural classification [Citation67]; ABLooper for CDR loop prediction [Citation68]; Therapeutic Antibody Profiler [Citation69]; PLAbDAb, the patent and literature antibody database [Citation70]; and ImmuneBuilder for AI-based antibody structural prediction [Citation71] to name but a few. Computational epitope profiling using structural models has been shown to identify a broader diversity of antibodies that bind to the same epitope [Citation72], as well as correlations between naïve and antigen-exposed antibody repertoires [Citation73]. Coupled with NGS, these tools are enhancing the mining of complex antibody libraries, thereby facilitating the antibody discovery process [Citation74]. Such in silico applications consequently generate a plethora of data that needs capturing and analyzing. This has necessitated the establishment of cloud-based informatics for data management, e.g. AbXtractTM [Citation75] machine learning and artificial intelligence platforms.
1.2. Nanobodies® or VHH fragments
Nanobody® or VHH antibody fragments or nanobodies (15kDa in size) are among the smallest known antigen-binding antibody-based biologics and are derived from the naturally occurring heavy chain only antibodies in the Camelidae species. In addition to their small size, high solubility, high stability, and excellent tissue penetration in vivo, the modular format of these single domain antibodies (sdAb) render them amenable to the generation of bispecific (including HSA for half-life extension), bivalent or biparatopic biologics [Citation76], as well as multispecifics, fusion to Fc-domains, peptides, or toxins and can be conjugated to drug payloads and radionuclides. As with conventional antibody libraries, nanobody phage display libraries can be immune, synthetic, or naïve repertoires [Citation77]; however, other in vitro platform methodologies (ribosome display, yeast display, and bacterial display) are commonly being utilized for the in vitro display of VHH fragments.
VHH-biologics have been explored for their suitability as anti-tumor therapeutics [Citation78] and anti-infectives [Citation79,Citation80] with several VHH-based biologics or nanobodies® either approved or in clinical development for the treatment of immune-mediated and autoimmune diseases, as outlined below. Nanobodies® were first commercialized by Ablynx where the first marketed nanobody®, caplacizumab directed to von Willebrand factor, for the treatment of acquired thrombotic thrombocytopenia purpura (TTP) received global approval in 2018. Caplacizumab is a bivalent humanized nanobody® that targets the A1 domain of VWF, isolated from a llama immunized with recombinant A1 domain [Citation81] and inhibits the interaction between ultralarge VWF and platelet GpIb-IX-V, where the platelet receptor binding site on the A1 domain is cryptic and exposed under high shear conditions. Preclinical evaluation to confirm efficacy and safety profile was attained in a baboon model of acquired TTP where acute episodes of TTP were induced by administration of an ADAMTS13-inhibiting mAb. Caplacizumab completely prevented the rapid onset of severe thrombocytopenia and schistocytic hemolytic anemia. Brain CT scans and postmortem analysis did not reveal any sign of bleeding, suggesting that complete neutralization of VWF by ALX-0681 under conditions of thrombocytopenia was not linked with an excessive bleeding risk [Citation82].
Other examples where an immune VHH library has yielded a therapeutic candidate include gefurulimab which targets C5 and human serum albumin (HSA) and in clinical development for the treatment of immune-mediated disease [Citation83], ozoralizumab is a trivalent bispecific, which targets TNFα and HSA and marketed in Japan for the treatment of rheumatoid arthritis [Citation84] and sonelokimab, which is also a trivalent bispecific that targets IL-17A, IL-17B, and HSA and in Phase 2 studies for the treatment of psoriasis and hidradenitis suppurativa [Citation85]. Gefurulimab comprises an N-terminal albumin-binding VHH connected to a C-terminal C5-binding VHH via a flexible linker [Citation83] and blocks the cleavage of complement component 5 (C5) by its activating convertase into the biologically active C5a and C5b fragments that trigger the complement cascade. It can also inhibit complement activity supported by the rare C5 allelic variant featuring an R885H substitution in the macroglobulin7 domain of the antigen. Current clinical studies are assessing efficacy for the treatment of chronic disorders involving activation of the complement pathway and gefurulimab is currently in Phase 3 and Phase 2 studies for myasthenia gravis and Phase 1 studies for proteinuria and dermatomyositis.
1.3. Phage display of alternative scaffolds
Other protein display scaffolds have been developed in the field of phage display using similar phage-derived systems described above that are capable of specifically binding virtually any target. As well as the generation of useful research tools, some of them have already entered clinical trials; examples of such alternative scaffolds include the human Kunitz domain of human tissue factor pathway inhibitor [Citation86–88], Anticalins® [Citation89], albumin-binding domain-derived affinity protein (ADAPT) based on one of the albumin-binding domains (ABD) of streptococcal protein G [Citation90], a calcium-regulated affinity (CaRA) library based on the engineered B domain of staphylococcal protein [Citation91], Affibodies® based on the IgG binding Z domain of Protein A fold [Citation92], AdnectinsTM based on the 10Fn3 domain of human fibronectin [Citation93], avimers based on the A domains of human extracellular receptors such as LDL [Citation94] and iBodies® based on the Ig domain of human neural cell adhesion molecule 1 (NCAM) that has two binding region loops incorporated within the structure [Citation95]. In fact, ecallantide, a bradykinin 2 receptor antagonist (by virtue of its inhibition of plasma kallikrein) developed by Dyax Corporation for the treatment of acute attacks of Hereditary Angioedema (HAE) and based on the Kunitz domain, was the first engineered alternative scaffold to reach the market, obtaining FDA approval in 2009 [Citation96,Citation97]. These and other protein scaffolds that have been explored as novel alternatives to antibodies for their utility as tools, and therapeutics are extensively reviewed elsewhere [Citation98,Citation99]. Several examples of alternative scaffolds that have progressed to late-stage clinical development and to the market are described below.
1.3.1. 10FN3-Based Proteins: Monobodies and AdnectinsTM
Monobodies or AdnectinsTM are based on the tenth fibronectin type 3 (FN3) domain known as10FN3, where FN3 is a member of the immunoglobulin superfamily. 10FN3 is composed of seven β-strands, which forms a β-sandwich framework connected by six loops, three of which are variable (known as loops BC, DE, and FG) so can tolerate diversity and length in composition similar to antibody CDRs. As these structural features resemble those of the variable heavy chains of antibodies, 10FN3 has been engineered as a novel alternative scaffold that is 10 kDa in size.
The first 10FN3-based phage display library construction in the phagemid vector pAS38 was based on structural and sequence alignments between 10FN3, antibody variable domains, and other FN3 domains with five residues randomized in the BC loop along with five residues in the FG loop that was also shortened by three residues to reduce loop flexibility and improve its binding affinity and resulted in a library containing ~108 independent clones [Citation100]. Other components of the M13 phage, including the wild-type pIII, were produced using a helper phage in order to display less than one copy of the 10FN3-pIII fusion product on the surface. Proof of concept was attained by isolating binders specific to ubiquitin with low micromolar affinity. Library construction has since increased in complexity and design, such as the ratio of amino acid residues permitted in each selected position. A variety of these and display methodologies is described in further detail elsewhere [Citation93].
Phage display of monobodies has been explored for the structural determination of therapeutically relevant protein targets, such as exploring the switch II pocket conformation of KRAS(G12D). Here, the authors conducted four rounds of phage display before using yeast display to sort the enriched clones and perform affinity maturation [Citation101]. A crystal structure of the G12D selective monobody-KRAS costructure was resolved to 2.52 Å shedding light on the selectivity (or lack thereof) of peptidic and small molecule drug candidates (PBD: 1FNA) [Citation101]. While there is a challenge in delivery of biologics to intracellular targets, there is the potential to overcome this by using viruses, expression vectors, and mRNA constructs as genetically encoded agents.
The 10FN3-based products from the Koide phagemid libraries are referred to as monobodies to distinguish them from those generated by the Adnexus PROFusionTM methodology (AdnectinTM). While monobodies generated by phage display have largely been used for protein structural studies, the generation of AdnectinTM products for clinical applications has been accomplished by mRNA display (PROfusionTM), which is a covalent method of coupling nucleic acid to protein (via puromycin) and therefore will only be briefly outlined, as follows.
The original method used enzymatic linkage of the puromycin to the mRNA template [Citation102], but the fusion synthesis was optimized to utilize psoralen-crosslinked mRNA-puromycin templates [Citation103]. Templates were generated by randomizing 21 of 94 amino acids in FN3, by initiating in vitro transcription from a PCR-DNA library (1012 different sequences) [Citation104], thereby enabling a much larger repertoire to be sampled. An example of an AdnectinTM identified in using this system is lerodalcibep (LIB003), a recombinant fusion protein of a PCSK9-binding AdnectinTM and human serum albumin (for half-life extension), which acts as a lipid-lowering agent for the treatment of hypercholesterolemia and cardiovascular diseases and is currently being progressed in Phase 3 development for homozygous familial hypercholesterolemia.
1.3.2. Anticalins®
Anticalins® are an alternative protein scaffold, 20 kDa in size, based on the lipocalin structure, which encompasses a highly conserved β-Barrel, comprised of eight antiparallel β-strands with four hypervariable loops that are exposed at the surface that confer binding abilities, with a C terminal α-helix. The four loops form a cup-shaped ligand binding pocket in the structure of the Anticalin®, and diversity in the exposed regions of these loops is generated by combinatorial design (in a manner similar to antibody CDR engineering). The first published proof-of-concept study reported an Anticalin® from the Pieris brassicae butterfly species, the bilin-binding protein, directed to bind fluorescein [Citation105].
For human therapeutic applications, human-derived lipocalins, tear lipocalin (Tcl/Lcn1), and neutrophil-gelatinase associated lipocalin (NGAL/Lcn2), were employed as the scaffolds for Anticalin® library construction to reduce immunogenicity [Citation106,Citation107]. The latest Anticalins® phagemid library, Lcn2, was designed based on the crystal structure of native Lcn2 [Citation108] and cloned into the vector pNGAL108 [Citation107]. Targeted random mutagenesis of the binding site in the four variable loops was undertaken where trinucleotide-based DNA synthesis (specifically the Slonomics technique) was applied to allow better control of amino acid composition (and avoid stop codons) at the randomized positions [Citation107,Citation109]. The Lcn2 library also uses fusion with the full length minor coat protein g3 gene to ensure monovalent phage display. Subsequent refinement in the choice of amino acid positions for randomization was based on observations that the backbone conformation of the loops can wildly diverge from the native structure due to side chain replacements resulting in different unintended orientations of targeted amino acids residues as well as neighboring amino acid side chains. In addition, the loop regions in the parent library could also experience structural rearrangements. In the light of these key observations, careful selection of amino acid residues for randomization was performed to ensure the structural space within the ligand binding pocket was accessible to any given antigen. This resulting Anticalin® phage display library contains a single disulfide bridge and comprises 1.7 × 1010 independent clones.
Due to their relatively small size, first generation Anticalins® required strategies for half-life extension, such as pegylation, fusion to IgG or Fc fragment or albumin-binding domain. The latter is particularly pertinent for antagonism of a target receptor in autoimmune or inflammatory disease in order to circumvent receptor clustering and activation [Citation110]. Moreover, the modular properties of Anticalins® render them amenable to the generation of bispecific or multispecific fusion products that can encompass the linking of two independent Anticalins® [Citation111] or even to antibodies (MabcalinsTM) [Citation112].
Early examples of Anticalins® advanced into clinical study are PRS-050, which was a pegylated Anticalin® targeting VEGF-A in clinical development (completing Phase 1 studies) for the treatment of solid tumors, however, was discontinued as although the candidate demonstrated efficacy, it was not sufficiently differentiated over existing marketed VEGF products [Citation113]; another example is PRS-080, which was also pegylated and targeted hepcidin, in development (Phase 2) for the treatment of anemia, but this was also discontinued for strategic business reasons.
The most recent libraries have been successfully commercialized by Pieris Pharmaceuticals and are generating a number of therapeutic candidates currently in Phase 1 clinical studies, such as PRS-220 directed to CTGF (connective tissue growth factor) for the treatment of IPF, as well as several other Anticalins® in discovery and preclinical development. In particular, the bispecific MabcalinTM format is being extensively explored for immunomodulation, such as PRS-344 (Phase 2; ), an anti-PD-L1 antibody genetically fused to 4-1BB-targeting Anticalins® at each arm of the antibody’s C-terminal heavy chain; and BOS-342 (Phase 2), an antibody targeting glypican-3 linked to the 4-1BB Anticalin® via the Fc domain.
In addition, Anticalins® have been developed for in vivo imaging and use as theranostics; specific PSMA binders that can be employed for biomarker detection were identified from the human lipocalin 2 (Lcn2) scaffold library [Citation114] by using standard panning technique strategies on a variety of antigen formats. Enriched phagemid clones were then subcloned into the expression vector pNGAL98 [Citation109] in order to screen soluble preparations of Anticalins® for PSMA binding.
1.3.3. Affibodies®
The Affibody® protein scaffold is based on the Fc-binding domain (B-domain) of staphylococcal protein A with α-helix structure, 6 kDa in size. The B-domain comprises 58 amino acid residues, constituting a three α-helix bundle motif without disulfide bonds. The B-domain has been engineered and referred to as the Z domain or Affibody®, which originally was used to construct a combinatorial library by randomizing 13 soluble surface residues located in the first two helices [Citation115].
In this instance, the pKNl phagemid vector was created to encode for the OmpA signal peptide, the third helix of the wild-type Z domain followed by a 46 residue albumin-binding protein (originally incorporated to combat renal clearance, but used also for affinity purification in this example) linked in-frame to a truncated version (residues 249–406) of M13 phage minor coat protein III with the insertion of restriction digested α-helices 1 and 2 library PCR products situated between the signal peptide and the third helix to generate a combinatorial library. Following the cloning of the PCR-amplified library into a phagemid vector adapted for phage display of the mutants, DNA sequencing analysis indicated a random distribution of codons in the mutagenized positions and characterization analysis demonstrated that the adapted Affibody® scaffold could be secreted as a soluble protein with no detectable binding to human IgG compared to wild-type Z domain protein [Citation115].
The phagemid vector pAffi1 and phagemid library (a portion of Zlib2002, 3 × 109 members) were generated using the same strategy as above with 13 positions randomized through NNG/T degeneracy with the PCR amplicons cloned into the pUC119 phagemid vector into which the translation cassette from pKNI had been incorporated to create the pAffi1 library vector. After four rounds of selection, specific binders to amyloid-β peptides from this naïve library of randomized Affibody® molecules were identified, purified (by virtue of a C terminal hexahistidine tag) and characterized [Citation116]. This same library has also been used to generate binders to HER3 after four rounds of selection [Citation117]. The first-generation clone sequences of interest were then used to design an affinity maturation library employing staphylococcal surface display using FACS [Citation118], which resulted in several binders with subnanomolar affinity, as well as the ability to inhibit ligand binding to the receptor, although phage display has also been utilized to generate picomolar binders by affinity maturation yielding several candidate Affibodies® suitable for radioiodination and in vivo tumor imaging in animal models [Citation119].
Given their small size and high affinity, Affibodies® have been exploited for diagnostics and use in clinical imaging, such as PET and SPECT imaging, of tumors as they can be modified with imaging contrast agents, fluorescent labels and radioisotopes, such as the HER2-specific Affibody®, ABY-025/GE-226, which is labeled with 68Ga and being theranostically applied to monitor breast cancer metastases in an ongoing Phase2/Phase 3 clinical trial (NCT03655353). Phase 2 results reported thus far indicate that ABY-025 shows encouraging potential as a noninvasive indicator of in vivo HER2 receptor status in breast cancer and might predict early response to HER2-targeted therapies, prompting a wider study to confirm these observations [Citation120].
Moreover, Affibodies® are amenable to the creation of bispecific, e.g. HER2 and HER3 [Citation121], and multispecific molecules facilitated by virtue of their modular structure including genetic fusions, such as Albumod®, an albumin-binding domain (ABD) used for half-life extension [Citation122,Citation123] and the AffiMab, where an Affibody® can be fused to an antibody either to the N- or C-terminus of both the heavy and light chains, e.g. an anti-TNF such as adalimumab and an anti-IL-6 Affibody® [Citation124] or the anti-EGFR antibody cetuximab and an anti-HER3-specific Affibody® [Citation125], and demonstrate greater efficacy in this format rather than a combination of the two molecules. Other recent examples include EGFR-targeting Affibodies® fused to trastuzumab’s heavy or light chains [Citation126] and a PDGFRβ-binding Affibody® fused to each heavy chain of an Fc-silenced CD40 agonistic monoclonal antibody [Citation127] with several other AffiMabs in preclinical development [Citation128]. For the PDGFRβ-CD40 AffiMab, a cell-based assay was established to model the tumor microenvironment by co-culturing B cells (CD40 positive) with PDGFRβ-expressing HEK293T cells and engineering the level of target expression to quantify receptor density. This strategy may have a wider utility for assessing and developing novel bispecific biologic modalities in immuno-oncology settings [Citation129].
The most advanced Affibody® in therapeutic development for is an IL-17A inhibitor, izokibep (ABY-035), which has progressed to Phase 3 clinical studies for the treatment of several autoimmune diseases including psoriasis, spondyloarthritis, hidradenitis suppurativa, and uveitis. Preclinical evaluation studies demonstrate that izokibep, a IL-17 ligand trap comprising two IL-17A-specific Affibody® domains and one albumin-binding domain (18.6 kDa in size), selectively inhibits human IL-17A in vitro and in vivo with superior potency and efficacy relative to current benchmark anti-IL-17A mAbs [Citation130]. Study results from a Phase 1 trial (NCT0269014) have recently been published demonstrating the safety, pharmacokinetics, and preliminary efficacy of izokibep, when administered as single doses to healthy subjects and as single intravenous and multiple subcutaneous doses to patients with psoriasis [Citation130], providing further validation for the therapeutic potential of this alternative scaffold to generate multispecific biologics.
1.4. Peptide phage display
Various bacteriophage systems such as λ, T4, T7, and filamentous M13 have been developed; however, filamentous bacteriophages, especially M13, remain the most popular choice for peptide display, where the peptides are generally fused to the N terminus of either pIII (3–5 copies) [Citation131] or pVIII (up to 2700 copies) [Citation132] coat proteins and can be 6 to 43 amino acids in length [Citation133] and the peptides can be either linear [Citation134,Citation135] or cyclic where the peptides are constrained by a disulfide bond [Citation135,Citation136]. The resulting peptide phage display libraries may contain up to 1010 individual peptide sequences [Citation137], which can yield highly specific peptide binders with an affinity in the order of KD 3 nM to 5 µM [Citation138]. Peptide libraries have also been constructed using the D-amino acid configuration, so-called mirror image phage display, to confer resistance to proteolytic degradation by naturally occurring enzymes [Citation139,Citation140].
The majority of marketed peptide-based drugs are receptor agonists, e.g. GLP-1, that have been derived from endogenous peptides [Citation137]; however, the search for new drug targets has evolved to encompass more challenging approaches and peptide phage display libraries provide an opportunity for creating peptide antagonists as well as agonists [Citation141], such as cyclic and bimacrocyclic peptides [Citation142,Citation143], cell-penetrating peptides such as stapled peptides [Citation144,Citation145], knottins [Citation146,Citation147], and bispecific peptide-antibody fusions [Citation148] (refer to ). Intriguingly, the ultralong CDR-H3 region of bovine VH-only antibodies possesses a knottin-like structure and has been described as the smallest functional antibody fragment [Citation149], indicating that this alternative structure can also be considered an antibody-based entity. Such structured peptides tend to bind their targets with higher affinity compared to linear peptides as the side chain residues are held in preferred target-binding conformations.
To date, two peptides derived from phage display have been approved, namely ecallantide (targeting plasma kallikrein [Citation150]) approved in 2009 for the treatment of HAE and romiplostim (targeting the thrombopoietin receptor [Citation151]) approved in 2008 for the treatment of ITP; the latter comprises a peptide-Fc fusion or pepti-body. In addition, an affinity peptide ligand isolated from the Dyax peptide phage display library is routinely applied to the purification of recombinant Factor VIII [Citation152]. Examples of other therapeutic peptide drugs derived from phage display that have progressed to clinical development are shown in and include pepti-bodies, bicyclic toxin-conjugates, and bispecific/multivalent bicyclic peptide entities.
Peptide phage display can be used to identify ligands that bind to a host of protein and nonprotein targets; in addition, cell-specific and organ-specific targeting is feasible, such as to tumor vasculature [Citation153], brain [Citation154,Citation155], kidney [Citation155], lung [Citation156–158], and neuroblastoma [Citation159].
Moreover, organ-specific targeting can enable the mediation of drug delivery systems by peptides, e.g. liposomes [Citation160] and nanoparticles [Citation161], thereby enhancing therapeutic efficacy. In addition to use as a direct therapeutic molecule or for targeted drug delivery, the phage display of peptide ligands has several other applications, such as identification of disease-specific epitopes [Citation162], epitope mapping to rapidly assess potential B-cell and T-cell epitopes of antigens, which is key to vaccine design [Citation163,Citation164], as well as the identification of internalizing peptides via receptor-independent mechanisms that can target organelles and identify the peptide-ligand’s receptor, e.g. penetratin [Citation165], but is also cost-effective and compliments other resource intensive experimental methods used for this purpose, such as cocrystallization and NMR. Thus, peptide phage display libraries have a broad range of uses in the drug discovery process [Citation166].
As with antibody-based biologics, families of related peptides can be identified and one strategy for optimization is to build common motifs into a secondary library to select higher affinity peptides [Citation167,Citation168] with an improved KD <10 nM after such an affinity maturation step [Citation169]. If two peptide motifs bind at different sites on a target, it is even possible to join the peptides via a linker, such as PEG, creating a ‘biparatopic’ entity which also provides an avidity effect; such as, strategy was found to generate heterodimeric peptides with greater efficiency in blocking the VEGF-KDR interaction than either monomeric component peptide [Citation170]. While peptides offer lower cost of goods compared to antibodies, they also offer a similar level of specificity as antibodies and larger biologics, which is an advantage over small molecules, but often require an additional engineering step in order to optimize their pharmacokinetic profile and subsequent half-life extension using strategies as described for some of the smaller alternative scaffolds, such as fusion to the Fc domain of IgG, e.g. etanercept [Citation171]; HSA, e.g. albuinterferon [Citation172] and PEGylation, e.g. peginesatide [Citation173]. This conjugation approach is necessary for the application of therapeutic peptides for the treatment of chronic disease. In fact, the antibody-peptide conjugation strategy has been used to finesse the therapeutic modality of antibodies and peptides further, where such a bispecific combination can consist of two agonist molecules directed to two different targets or an agonist-acting ligand with an antagonist antibody, e.g. AMG-133, which combines a GLP-1 agonist peptide analogue with and GIP receptor antagonist antibody [Citation174].
While outside the focus of this review, it is worth mentioning that zinc finger protein phage display libraries have been used to identify novel DNA-binding proteins [Citation175,Citation176] that can have utility in research, as well as therapeutic applications.
2. Phage selection strategies
A summary of the key steps involved in the phage display selection process is provided in . The process of enrichment for target-specific phage clones is accomplished by biopanning over the antigen of interest using an initial over-representation of the library repertoire (typically 1012 phage particles). Antigen purity is intrinsic to a successful campaign, and a variety of different antigen types may be utilized, such as peptides (which are commonly conjugated to a carrier protein such as BSA or KLH) or purified recombinant protein, although cells overexpressing the target have also been successfully used as an antigen source. In this instance, stringent washing and depletion on non-target expressing cells is required to reduce the number of background binders. The source of purified protein needs to maintain the native structure and so for complex antigens with intricate secondary structure, in particular multipass membrane proteins, it may be necessary for specialist technologies, such as stabilization for thermostability [Citation177] or increased expression [Citation178,Citation179], formulation in detergents [Citation180], incorporation into nanodiscs [Citation181], virus-like particles [Citation182], proteoliposomes [Citation183], or the use of styrene-co-maleic acid lipid particles (SMALPs) [Citation184]. One drawback is that DNA and mRNA that are commonly used as antigen sources for in vivo immunization are not an antigen option for phage display (or indeed other in vitro display platforms).
Selections can be performed using a solid phase approach where surfaces can be coated with antigen in a variety of ways, the most common being direct adsorption to a plastic surface or via tags that interact with immobilized affinity agents coated to an ELISA plate well or a bead, and even direct coupling via chemistries to beads. The other selection strategy commonly used is liquid or soluble phase where the antigen can be biotinylated for selections in solution and the phage binder/antigen complexes are subsequently captured using streptavidin-coated magnetic beads. The soluble strategy is particularly useful for increasing the stringency of selection with each round of selection with the aim of isolating high affinity binders and is often used for affinity maturation or lead optimization activities.
Following incubation with the antigen, nonspecific phage clones or low affinity binders are eliminated by several rounds of washing and elution of the remaining bound phages (the output) from the antigen can be accomplished by a range of methods depending on which phage or phagemid or helper/hyperphage combination is employed, such as enzymatic digestion or change in pH or competition with free antigen. Acidic buffers, e.g. glycine or citric acid, or alkaline pH, e.g. triethylamine (TEA), is most commonly used for pH elution, but it is important to neutralize the resulting elution to maintain the infectivity and integrity of the phage. The enzymatic strategy uses a cleavage site, e.g. for trypsin, that has been introduced in between the pIII protein and antibody [Citation185], thus elution of phage is achieved by cleaving the c-Myc tag between the antibody and pIII protein. While pIII protein originating from the phagemid is trypsin-resistant, pIII protein in the helper phage is trypsin-sensitive rendering it noninfective, so this elution method also has the advantage of removing background infectivity (of bald and helper phage). The retrieved phage are then used to reinfect E. coli for an overnight propagation step and amplification via packaging with helper phage in the case of phagemid repertoires) to generate an enriched population of phage particles that are used as the input in the next round of selection.
Typically, 3–5 rounds of selection are conducted using an iterative process of panning, washing, and amplification of the resulting phage output pool to enrich for target-specific phage binders and reduce nonspecific background (refer to ). Enrichment of binders toward the antigen of interest can be evaluated by polyclonal phage ELISAs and used to identify selection rounds for further analysis on an individual clone basis to assess specificity of binding. Concurrently, DNA sequence analysis of the individual clones can be undertaken to determine the diversity of the specific binders.
The selection process can encompass different strategies to target particular properties, such as a favorable slow off-rate or pH sensitivity [Citation186,Citation187]; specific epitopes, such as by masking [Citation188]; competition by deselection to remove unwanted binders [Citation189]; and competition with a known ligand or binding interaction partner [Citation190]. Specific phage panning protocols can also be used to select for antibodies that can rapidly internalize on cells [Citation191]. In addition to de novo discovery of functional binders, phage display can also be applied to the structure-guided affinity maturation and lead optimization of candidate clones, for example, neutralizing antibodies to viral variants [Citation192], as well as humanization of mouse or rat derived antibodies, for example, as accomplished for adalimumab using a guided selection strategy [Citation193]. In fact, structural data on both target and antibody are becoming increasingly applied for the optimization of the candidate therapeutic [Citation194–196] and designing epitope-specific functional antibodies [Citation197]. A couple of recent developments in the application of phage display selection in more physiological conditions using in vitro and in vivo cellular settings are described below.
2.1. On-cell selections
Cells overexpressing membrane protein have been an established strategy to overcome the hurdle of difficult-to-express complex or challenging membrane protein receptors, such as GPCRs [Citation198,Citation199]. While the target protein is presented in a native context within the lipid bilayer of the cell membrane, the level of expression may be hard to control, may have low immunogenicity, and there are high levels of nonspecific background to contend with necessitating stringent counter-selection or depletion steps. Success may be target dependent, but nevertheless, antigen-expressing cells provide one solution to the bottleneck of antigen sourcing and historically have isolated functional antibodies, particularly when used in combination with other antigen formats [Citation200,Citation201].
On-cell selections have been evaluated further to offer a means of replicating the in vivo environment by the use of multiple cell types. This approach was initially applied to several complex integral membrane proteins, namely tetraspanin CD151, carbonic anhydrase 9, and integrin-α11 [Citation202] and termed CellectSeq. Pools of phage antibodies enriched for target specific binders by in situ cell selections were subjected to NGS analysis to identify antigen-specific antibodies by in silico analysis. Resulting clones of interest were expressed as Fab fragments and reformatted to IgG for purification to validate their binding profiles on cell surface expressed antigen. More recently, the technique has been refined to incorporate microfluidics described as µCellect [Citation203]. The principle is to perform rapid on-cell phage display in a highly stringent heterogeneous mixture of cells to quickly remove nonspecific binders. Microfluidics are applied to separate antigen-presenting cells with high throughput and specificity. Machine learning is then implemented to analyze the resulting phage antibody sequences from isolated pools to determine structural trends that contribute to binding affinity and narrow down panels of potential candidates for further characterization based on enrichment and clustering analysis. Software tools like embeddings aid in clustering of millions of sequences and allow the researcher to maximize sequence diversity in the sequences pulled from phage pannings. Proof of concept has been attained with the GPCR FZD7 identifying antibodies with picomolar affinity [Citation203].
2.2. In vivo selections
An ambitious and emerging selection strategy to identify best-in-class therapeutic antibodies against challenging or novel disease targets has explored the utility of injecting phage libraries into animals to identify phage antibodies binding to specific tissues using the same principles as with cell and organ-targeting peptide phage display [Citation204]. This takes the selection conditions into the complexity of biological systems in a disease context while also aiming for favorable pharmacokinetic profiles of the antibodies that are retrieved after tissue harvest. Incubation time and route of administration are critical as phage degradation is thought to occur rapidly mediated by phagocytic cells as well as the B-cell response [Citation205]. Initially trialed using peptide phage display libraries, in vivo selections enabled the identification of receptor-ligand pairing in various disease contexts. In fact, in vivo selections have even been evaluated in humans with the role of IL-11 R in prostate cancer being validated in this manner [Citation206].
Subsequent work using in vivo selections in animal disease models has led to the identification of biomarkers relating to atherosclerosis [Citation207], including galectin-3, which is considered a biomarker of coronary and carotid atherosclerosis [Citation208], as well as developments in diabetes research where scFv antibodies specific to ɑ-islet cells and β-islet cells can be used in in vivo imaging and determining β-cell mass in patients [Citation209]. The identification of several tumor-specific antibodies has been achieved in this manner using both human cancer patients and an orthotopic mouse model [Citation210,Citation211]. Another exciting option for this approach is the application of in vivo phage selections to isolate antibodies that can traverse the blood brain barrier and thereby offer the opportunity to identify new targets that could be harnessed for drug delivery to the CNS [Citation212]. The same group have also been using in vivo selections to generate ADCs [Citation213]. While the full potential of this technique has yet to be realized, the examples described here suggest that the methodology may offer a route to personalized medicine, expand the target landscape further and deconvolute the role of receptors and their ligands in disease.
3. The role of next-generation sequencing in antibody discovery
NGS has revolutionized the antibody discovery workflow and is routinely implemented now in the screening cascade as it greatly enhances throughput to identify specific enriched antibody phage clones and thereby aid lead selection. As well as assisting the process of original VH/VL pairing from scFv [Citation214] and Fab formats [Citation215], by coupling the CDR DNA sequencing data of initial snapshot sampling of target-specific clones with confirmed functional activity, it is also possible for the researcher to deep-mine those selection outputs that have produced the most diverse functional antibody clones to identifying further related CDR lineages. Furthermore, NGS sampling on a large scale can be undertaken on pools of phage antibody clones to identify clusters of antibody sequences indicating enrichment [Citation32]. These resulting clusters of antibody clones can then be tested for their ability to modulate the target of interest and this potentially enables the retrieval of rare clones that may not have been otherwise detected via the standard screening cascade because entire phage pools can be subjected to sequencing.
While NGS generates a huge increase in the number of reads that can be conducted in parallel, these are smaller than full length construct reads produced by Sanger sequencing, but nevertheless, the advantages are speed, cost, and is less labor-intensive, where the full-length antibody sequence can be constructed from the short contiguous reads that overlap. NGS analysis is also routinely applied to the evaluation of the quality of a library, the analysis of changes in heavy chain germline usage, the evaluation of CDR diversity, in particular CDR-H3 composition and length distribution, and is offered as a service by a host of contract research organizations (CROs).
NGS platforms that have been commonly applied to short-read sequencing include the original Illumina (Solexa) system [Citation214], Roche’s 454 system [Citation46], which has since been discontinued, the Ion Torrent PMG platform [Citation216], and MiSeq (also an Illumina system) [Citation217]. More recently, the Illumina MiSeq platform was employed to assess the diversity of the sheep immunoglobulin repertoire [Citation218]. NGS methodology has evolved to provide longer read sequencing with high accuracy and is typified by the Pacific Biosciences and Oxford Nanopore technologies. A detailed analysis of the strategies and platforms used for NGS in antibody discovery is beyond the scope of this article, but there are several excellent reviews focusing on this topic [Citation219–222].
4. Other applications for phage display antibody discovery and generation of therapeutic molecules
4.1. Targeting venoms and toxins
Antibody generation against targets otherwise systemically toxic, e.g. venoms (snakes, spiders, cone snails, scorpions, jellyfish, bees) or other toxins (bacterial toxins, such as botulinum, cholera; plant toxins, such as atropine from Belladona or nightshade species, calcium oxalate from Ornithogalum species, cardiac glycosides from the oleander species) can be facilitated by in vitro methodologies such as phage display [Citation223]. In addition, various drugs are often involved with poisonings and toxicities, either after accidental overdosing or deliberate consumption, e.g. digoxin and opioids.
A precedent for the neutralization of toxins and venoms by antibodies is provided by an ovine anti-digoxin Fab polyvalent preparation used to treat oleander poisoning. The anti-toxin Fab fragment markedly reduced case fatality and its absence resulted in a three-fold rise in deaths [Citation224]. However, due to high production costs, this treatment for plant toxin poisoning was withdrawn in 2002 [Citation224]. However, this antidigoxin Fab fragment is still used for the treatment of digoxin toxicity [Citation225], marketed as DigiFab® and DigiBind®. Other examples of antibody fragment antidotes are the ovine polyvalent ViperaTAb® for combating cardiotoxins present in viper venom, Anavip® (N American pit viper), an equine (Fab’)2 fragment, which has a longer half-life and CroFab® (N American pit viper).
All these treatments have been dependent on animal-derived (horse and sheep) polyclonal antibodies. However, despite their life-saving importance and avoidance of life-long disability, there are global shortages, production is difficult and expensive with the cost having increased over the past few decades rendering treatment unaffordable to most people in the developing world where envenomation is more frequent. As such, snake bites are considered a neglected tropical disease. Prompt access to antivenoms in rural settings is challenging due to lack of transportation, cold chain, and health care logistics. Antivenoms have conventionally been derived from immunizing large mammals (usually horses or sheep) with venom, followed by the purification of polyclonal antibodies from the blood plasma. However, the polyclonal preparation itself will vary from batch to batch impacting the level of efficacy, and, due to the nonhuman content, patients can often suffer side effects due to serum sickness or anaphylaxis, and multiple doses are required for effective neutralization adding to the cost burden. In addition, this is a heterogeneous preparation that also contains a high level of non-neutralizing antibodies.
Recombinant antivenoms based on oligoclonal mixtures of human IgG antibodies produced by CHO cell cultivation may be a key to obtaining better envenoming therapies in a more cost-effective manner [Citation226] and enable the neutralization of hemorrhagic, myotoxic, neurotoxic, and cytotoxic activities of venom in a single mixture.
In vitro display systems, such as phage display, can enable this strategy to generate oligoclonal mixtures of antibodies or a single monovalent antibody, and indeed, several groups have generated neutralizing antibodies to venoms and toxins in this manner, such as black mamba venom oligoclonals [Citation227], Shiga toxin type 2 neutralizing Fab fragment [Citation228], and an IgG against α-cobratoxin that was derived from phage display and shown to prevent lethality [Citation229]. Moreover, the latter was the first report to demonstrate neutralization of whole snake venom by a single recombinant monoclonal antibody.
Antivenom nanobodies have also been generated by the immunization of llamas followed by phage display of the resulting immune repertoire [Citation230,Citation231]. Other small single-domain alternative scaffolds, such as AdnectinsTM, Affibodies®, and Anticalins®, have also been proposed as potential candidates for antivenom/antitoxin development [Citation232]. Due to their high stability and low production costs, these could present an attractive alternative for envenomation treatment in geographically remote areas and poorer countries where maintaining the cold chain is a challenge [Citation233]. Recombinant scorpion antivenoms have also been generated [Citation234], as well as C. difficile toxin A/B neutralizing antibodies [Citation235], anti-rabies scFvs [Citation236,Citation237], anti-Bacteroides fragilis endotoxin nanobodies [Citation238], and anti-diptheria toxin neutralizing antibodies [Citation239]. The facile methodologies that are used to engineer these smaller antibody formats also enable simple optimization of properties such as high affinity and neutralization properties. Moreover, production of these antibody formats is straightforward in E. coli generating correctly folded protein at a far lower cost than mammalian expression systems.
4.2. Targeting pHLA/MHC-presented peptides on T cell receptors
Tebentafusp is a bispecific peptide-HLA-directed CD3 T cell engager that targets gp100 [Citation240] recently approved for the treatment of uveal melanoma (and currently is in Phase 3 clinical studies for cutaneous melanoma), which was generated by the application of phage display. The stability of the recombinant T-cell receptor (TCR) was achieved by introducing an interchain disulfide bond into the interface between the TCR constant domains. This strategy is applicable to a wide range of different TCRs and enables the display on the surface of bacteriophage [Citation241]. Tebentafusp (IMCgp100) is a fusion protein containing a modified form of human TCR specific for the gp100 antigen and fused to an anti-CD3ε single chain antibody fragment, with anti-neoplastic activity. The drug candidate targets and binds to the tumor-associated antigen (TAA) gp100 presented on the melanoma tumor cell; the anti-CD3ε fragment moiety binds to CD3ε-expressing T lymphocytes, thereby selectively cross-linking tumor cells and T-lymphocytes. It leads to the recruitment of cytotoxic T lymphocytes (CTLs) to the T lymphocyte/tumor cell aggregates and results in CTL-mediated death of gp100-expressing melanoma cancer cells. Tebentafusp is indicated for the treatment of HLA-A *02:01-positive adult patients with unresectable or metastatic uveal melanoma and advanced cutaneous melanoma.
4.3. Epitope identification for vaccine development
Historically, vaccines usually comprise inactivated or attenuated forms of pathogens, but these can risk either not eliciting a sufficient robust immune response or can cause side effects. A recent review has highlighted the important role of phage display in identifying epitopes and mimotopes for vaccine development [Citation242,Citation243]. Specific epitopes have the potential to be engineered in the vaccine of interest to efficiently generate an immune response in the recipient. Random peptide sequences or even gene fragments have been displayed and screened using sera from patients or vaccinated individuals to identify pathogen epitopes for viruses, e.g. severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [Citation244], influenza [Citation245]; bacteria, e.g. Mycobacterium tuberculosis [Citation246]; and parasites, e.g. Plasmodium falciparum [Citation247]. The process is high throughput, cost effective, and can be customized to the pathogen in question enabling a viable alternative to the traditional route of vaccine development. Epitope identification by phage display has also enabled the generation of diagnostic assays for infectious diseases, as well as the development of vaccines. This strategy also offers the opportunity to directly use phage-based vaccines directly from the analysis following endotoxin removal [Citation248], although this vaccine format has yet to be approved. The identification of key epitopes also assists the wider field of therapeutic discovery encompassing monoclonal antibodies (for example, to block an interaction), small molecules (for example, to disrupt the pathogen life cycle), and gene therapy (for example, by integrating a key epitope into the host cells, a sustained neutralizing immune response can be attained).
4.4. ORFeome phage display
A relatively recent and elegant refinement of phage display for epitope mapping and identification utilizes the open reading frames (ORFs) representing a pathogen’s genome displayed on the surface of phage particles in order to identify immunogenic epitopes. ORFeome display has an advantage of being able to detect immunogenic proteins independent of the transcriptome or proteome, as it does not rely on cDNA or protein extracts, and is not limited to surface proteins. These ORFeome libraries display all possible proteins encoded by a pathogen, bacteria, or virus on the phage surface. Consequently, immunogenic proteins can be selected using disease-related immunoglobulins from patient serum to isolate the protein interaction partner. One of the first studies described used this methodology to identify immunogenic salivary gland proteins from ticks that are recognized by human immune sera [Citation249]. Ticks are vectors for a variety of different pathogens, including Borrelia burgdorferi, the causative agent of Lyme disease, which is a major public health concern in many countries as there is currently no available vaccine. The study was able to select the metalloprotease MP1 as a potential candidate for vaccine development. Subsequently, this methodology has been extended to many other pathogens, such as Listeria [Citation250,Citation251], Leptospira [Citation252], SARS-CoV-2 [Citation244], as well as biomarker discovery [Citation253]. The resulting peptides yielded from such investigations can be used to develop diagnostic assays or vaccines and evaluate protein:protein interactions [Citation254].
5. SARS-CoV-2 as a case study example of the response to the pandemic with the rapid generation of antiviral mAbs using phage display
The response of the antibody discovery and drug development community to the COVID-19 viral pandemic encompassed widespread efforts involving multiple strategies of antibody discovery, from immunization of transgenic mice with viral proteins [Citation255], to screening patient sera and B cells [Citation256,Citation257] and in vitro display platforms [Citation258,Citation259] to rapidly identify neutralizing antibodies that could either be used prophylactically or to treat infected patients while effective vaccines were being developed, as well as for the generation of rapid antigen test assays [Citation260]. This also included a large contribution from the phage display community and provides excellent exemplification for the rapid expedient discovery of diverse neutralizing mAbs from large-scale human antibody phage display libraries [Citation51,Citation261]. Combinations of these were also applied, e.g. constructing phage display libraries from convalescing patient immune repertoires [Citation262], but all methods were able to isolate potent antibodies against this newly emerging virus.
The Spike surface glycoprotein mediates cell entry where the S1 subunit comprises an N-terminal domain (NTD) and a receptor-binding domain (RBD), which interacts with the host cell receptor angiotensin-converting enzyme 2 (ACE2). Spike is the primary target of neutralizing antibodies against SARS-CoV-2 where three distinct epitope bins have been identified that retain activity against other virus variants. The majority of the mAbs yielded from all these campaigns target the RBD of the S1 subunit of SARS-CoV-2 spike protein, but other neutralizing mAbs have also been identified that bind the NTD as well as mAbs directed to S2 [Citation261].
On the whole, potent neutralizers were identified from convalescent patients and immunized animals; however, at least, a couple of studies have demonstrated that it has also been possible to isolate highly potent neutralizing antibodies with low nanomolar affinity from human naïve antibody libraries that were equivalent or superior in their efficacy compared to the immune routes [Citation263,Citation264]. This is most likely a reflection of the carefully designed semisynthetic nature of these libraries that have also been optimized for diversity and downstream developability [Citation48,Citation265]. For one of these examples, yeast display was implemented in a few more rounds of selection to identify ultra-potent (IC50 < 2 ng/ml) human neutralizing antibodies [Citation264]. In addition, these examples also underscore the importance of how very potent antibodies can be discovered against a newly emerging viral pathogen because only the antigen sequence was required rather than a reliance on donor or patient materials for the original immune source (e.g. B cells after immunization or recovery). In fact, one of the antibodies identified from a human naïve phage display library in one of the above examples, STE90-C11 or COR-101, was progressed into a Phase 1b/Phase 2 clinical trial (NCT04674566). This exemplifies the expediency of phage display to generate potent SARS-CoV-2 neutralizers versus isolating antibodies from convalescing patients or immunization of animals.
Another challenge was the identification of candidates that retained binding to SARS-CoV-2 variants Alpha, Beta, Gamma, Kappa, and Delta; however, when the virus mutated further to generate more variants of interest/concern, such as Omicron, the neutralization efficiency of these mAbs was reduced. This necessitated the engineering of existing neutralizing mAbs that can be used as therapeutic candidates where optimization has increased affinity, potency, and reduced viral escape pathways by broadening the neutralization capacity across the increasing range of viral variants [Citation266–268]. Other strategies to combat viral escape include targeting more conserved regions where there is less mutational load [Citation269], the use of mAb cocktails or bispecifics covering several epitopes [Citation270,Citation271]. In fact, phage display-based affinity maturation could be applied to developing mAbs for the treatment of many different viral infections to reduce viral escape pathways as well as appropriate affinity maturation in vaccine immunization to aid resistance to viral variation [Citation267]. Thus, phage display can also be harnessed for rapid optimization and affinity maturation engineering in addition to the discovery of de novo protein-based biologics.
6. Conclusion
Phage display is an established in vitro display system for the generation of functional biological entities with the protein-based therapeutic pipeline expanding further with antibodies, peptides, and alternative scaffolds derived from this platform in addition to the traditional immunization route for generating antibodies. The advances gained through AI-assisted in silico design, structural biology, assay miniaturization, and throughput of sample handling serve to enhance the breadth of epitope coverage, the identification of rare and potent functional biologics, as well as significantly reduce timelines compared to those from 20 years ago. Furthermore, the combination of phage display with other display platforms, in addition to harnessing the techniques and methods outlined, coupled with creative strategies to thoroughly sample a repertoire or library reinforces the optimism and potential for success in identifying first-in-class and best-in-class biologic modalities.
7. Expert opinion
While this review has focused on phage display, there are several other in vitro display platforms that are actively applied in therapeutic antibody and alternative scaffold discovery, including ribosome display which is a cell-free system producing in vitro protein – mRNA complexes [Citation272] and commonly used for scFv [Citation273], VHH [Citation274], and DARPin® libraries [Citation275] as well as affinity maturation [Citation276]; yeast display which is a cell surface display system [Citation277] commonly used for IgG and VHH [Citation278] as well as affinity maturation [Citation279]; mammalian cell surface display [Citation280], and covalent DNA display methodologies such as CIS display [Citation281]. Similarly, these platforms may also incorporate the various aspects of in silico design and developability used for phage display libraries [Citation278,Citation282], as well as AI and machine learning [Citation278] and due to synergy between the platforms they are often combined in a therapeutic campaign.
All these in vitro display methods require DNA sequence analysis to identify unique lead candidates in the screening cascade. Historically, output diversity involved random colony picking and Sanger sequencing, which required full length reads of the resulting antibody fragments. However, the advent of NGS has provided a more cost-effective means to evaluate the diversity of enriched binders, e.g. by enabling a much deeper analysis of unique CDR-H3 or other binding motif clusters and broadening the number of enriched clones sampled in a much faster timeline [Citation283].
The drug target landscape encompasses many types of challenging targets, such as GPCRs, ion channels, transporters, and other multispanning membrane proteins, where access to suitable yields of physiologically relevant antigen presents the first bottleneck encountered. Increasing efforts are focusing on the rational design of antibodies directed to particular epitopes on a target antigen by using combinations of structural data (e.g. from a crystal structure of the target and/or the antibody fragment) and predictions generated by bioinformatics (e.g. from amino acid sequences and modeling algorithms, such as Rosetta and AlphaFold). Others have generated structural dynamics-sensitive druggability probes based on the carefully controlled kinetics of selected proteases to inform the identification of potential antibody epitopes via microfluidics. This strategy has been applied to the TRPV1 ion channel to identify conformationally sensitive modulating antibodies and apoptosis-triggering mutant-selective KRAS antibodies [Citation284]. The latter example will necessitate the use of other technology for intracellular delivery, which in this case is achieved by oncolytic viral delivery. Other techniques are used to determine binder partner epitopes, including conformational changes, hydrogen-deuterium exchange [Citation285], and plasma-induced modification of biomolecules [Citation286]. In addition, the protease-probe methodology relies on the use of peptides that represent the binding interface as antigen and, as such, may risk limited structure similarity with the native target. However, the authors suggest that this approach could be further refined by presentation of the native target incorporated in lipids or a membrane scaffold protein (e.g. a nanodisc) and applying the epitope-related peptides in the screening cascade [Citation284]. Nevertheless, these approaches underscore the importance of understanding the target structure, dynamics, and biological role in disease context.
The emergence of high throughput FACS sorting, as well as the even greater automated processing of samples afforded by microfluidics and microcapillaries, has also been empowering for drug discovery of biologics, particularly for the screening of antigen-reactive B cells, but also for phage antibody screening. In fact, microfluidic technology for specific antibody screening has been successfully integrated with several platforms, i.e. hybridoma technology, phage display, single B cell antibody screening, yeast display, antibody expression, and cell-free protein synthesis [Citation287], demonstrating its broad application and the refinement of microfluidic devices over the past decade, some of which are even now commercially available. For phage display, antigen immobilized to beads or even cells over-expressing the target of interest may be used in such a strategy [Citation204]. This technique recently has been further refined to screen and sort in a single round high-affinity phage-displayed antibodies against virus glycoproteins by undertaking electrophoresis under two alternating electrical fields (lateral and vertical) through an agarose gel ‘functionalized’ with the respective antigen, where the antigen of interest is linked to agarose gel via a heterobifunctional photo-crosslinker. Antigen-specific antibodies are captured at several outlet channels with high affinity phage antibodies concentrated at the channels nearest to the inlet port and lower affinity phage antibodies separated at channels furthest from the application site [Citation288]. Such sample handling capacity will vastly increase the number of selection campaigns that can be undertaken in parallel in a rapid and cost-effective fashion, such as the ‘multiplex’ approach adopted to identify functional VHHs directed to the FSH receptor by encompassing a variety of antigen formats [Citation289]. Consequently, high throughput methods have been devised for conversion to full length IgG to solve the reformatting bottleneck [Citation290], as well as Fc-fusion generation and purification [Citation291].
Finally, several limitations and resolutions created to mitigate against drawbacks not previously discussed elsewhere in this review are outlined, as follows. While phage display has several advantages over other platforms, such as providing a fast and cheap route to the discovery of biologics and being able to generate large library repertoires, there have been several challenges that have been solved to optimize this display methodology, such as employing bioinformatic methods (and more recently, AI/ML methods) to avoid base bias, amino acids at ‘unnatural’ positions, sequence liability, and good expression profiles. Expression bias and folding errors can be problematic as the E. coli bacterial host shows a preference in expressing proteins rich in certain amino acids (methionine and lysine) and is not able to fold complex immunoglobulins nor provide critical post-translational modifications, such as disulfide bonds. On the other hand, phage display methodology is better at correctly displaying smaller antibody fragments, such as scFv, VHHs, followed by Fab fragments [Citation290]. However, scFvs are prone to aggregation leading to false positives and antibody fragments often require reformatting to full-length IgG, which can also lead to a high drop-out rate for scFv hits. Thus, as previously mentioned, some of these limitations have been countered by using alternative scaffolds with better folding properties or by combining phage display with a eukaryotic system, such as yeast display, with later rounds of selection to solve some of the expression problems and post-translational processing.
Phage display technology can be used to generate antibodies without host immunization (for example, in the situation where antigen is limiting), and bacteriophages are fairly resilient to external factors such as pH and high temperatures, which enables the selection conditions to be exquisitely manipulated to push the selection pressures in a certain direction that is not possible with in vivo antibody generation. However, it is useful to generate a focused library from host immunization where the latter can take advantage of the use of DNA or mRNA as antigen.
Developability nowadays is more commonly introduced at the stage of creating a phage library repertoire streamlining the drug discovery and development process. Many AI/ML methodologies are now being applied in combination with wet lab work to resolve this important aspect in candidate optimization at as early a stage as is possible, addressing biochemical and physical properties (e.g. introducing pH sensitivity or avoiding negative attributes such as hydrophobicity or aggregation) of the molecule to ensure good manufacturability and patient administration (e.g. ensuring low viscosity at high concentrations for subcutaneous delivery). As an extension of this, potential immunogenicity is still an ongoing problem to be solved across all biologic-generating platforms. While there has been an increase in the range of designs and novel formats of protein-based therapeutics, the ability to predict the immunogenicity risk and risk mitigation has not kept pace. Thus, there is a strong requirement here to be able to more reliably assess immunogenicity, and AI/ML provides the potential to develop better tools, for example, by mining clinical data.
Another challenge for the antibody field is addressing the target space to identify appropriate novel disease targets/novel epitopes and validating them. Cytoplasmic delivery [Citation292] and blood-brain-barrier shuttles [Citation293] are still challenges yet to be robustly solved and would open the target space further. Internalising phage vectors have been created to explore the possibility of accessing the intracellular environment as well as targeting specific organelles [Citation294,Citation295]. In addition, efforts continue to enhance phage display techniques to identify high affinity antibodies directed to challenging targets [Citation296].
Notably, language-model-guided affinity maturation is being applied with increasing frequency [Citation297] and, looking to the future, the combination of target structural knowledge with AI being harnessed for the optimization of lead molecules, as well as being leveraged for de novo discovery is a highly active area of focus in the industry. An example of this is provided by a C5aR1 case study where DNA synthesis and library assembly technology were combined with structural-based machine learning to design a targeted anti-C5aR1 discovery library to identify several highly potent and selective lead antibodies against C5aR1 [Citation298]. In this example, the design of a C5aR1 focused library utilized existing structural information to predict solvent exposed regions of the GPCR. The exposed surface was then divided into smaller epitopes using a neural network trained on a structural repository of antibody-antigen interaction to identify epitopes with high probability of antibody interaction. Using these discrete epitopes, an algorithm generated a large pool of millions of candidate antibody structures with unique binding loops. This collection of structures was finally triaged down to ~ 10,000 structures using neural network models, which selects candidates with favorable biophysical characteristics. The C5aR1 focused phagemid library was designed using the predicted CDRs from the ~ 10,000 candidates to generate a diversity of 1 × 109. Cell-based panning selections were conducted, and phage pools from later rounds of panning were characterized by long-read NGS to obtain paired VH and VL sequences. Clones were selected for IgG reformatting using bioinformatics to assess abundance of clones, enrichment across rounds and clustering. A total of 96 clones with 45 different HCDR3 sequences were selected and produced as IgG molecules. Several of the candidates identified from this study are also shown to have functionally antagonistic effects on C5aR1-mediated signaling in cell-based assays. One of these promising leads was further optimized resulting in multiple clones with functional potency comparable to the clinical lead [Citation298].
Phage display can also be used to generate molecular sensing probes that enable the diagnosis, prognosis, and monitoring of disease via their interaction with cell-secreted and cell-associated biomarkers, such as PSA and PSMA in prostate cancer [Citation299]. This provides the potential for the development of portable biosensors at the patient screening-clinic interface and point-of-care for an earlier diagnosis rather than the delay encountered when sending samples to a dedicated laboratory for analysis [Citation300]. There is the potential to develop more blood biomarkers for an earlier diagnosis of Alzheimer’s disease [Citation301], as well as developing biosensors for theranostic use [Citation302] as has been undertaken extensively for pathogen, infectious virus, and biotoxin detection [Citation300].
In summary, the combination of in vitro phage display of large immune repertoires with microfluidics handling, miniaturization of sampling, NGS, and bioinformatics to empower the handling and analysis of enormous data sets is revolutionizing the protein-based biologics discovery workflow. The vast increase in computing power and capacity is fundamental to enable machine learning and effective data management. With these advances increasing being employed, it will be illuminating to see what achievements are made in the next few years and how further technology refinements will impact the therapeutic biologics pipeline and the diversity of the different types of protein-based biologics therein. The widespread application of phage display continues to hold great potential across the spectrum of biologics discovery and development, not only for the generation of therapeutics, but other important areas that support the development of biological drugs including diagnostics, biomarkers, and target identification/validation, where it is apparent that current AI/ML applications are complimentary to the current methods used for protein-based drug discovery process rather than being used as a replacement.
Article highlights
Phage display is routinely implemented to rapidly generate a diversity of different biologic formats and alternative scaffolds for the therapeutic pipeline, where phagemid vectors are the most commonly used route for therapeutic discovery.
Phage display is compatible with other in vitro platforms, e.g. ribosome and yeast display, and can also be combined with immunization strategies to produce focused phage antibody-based libraries representing enriched immune repertoires.
Several peptides, pepti-bodies, and monoclonal antibodies derived from phage display are now marketed and many others are in late-stage clinical development, including alternative scaffolds.
Phage display methodology has been further developed to harness the potential of on-cell selections and in vivo selections, including in animal disease models, as well as MHC/peptide complexes presented on T cell receptors, the identification of biomarkers which are important for the drug development process, applied to epitope identification for vaccine development and further refined for ORFeome analysis, which enables the identification of immunogenic epitopes of a pathogen without the requirement for protein extracts or production for the analysis.
The establishment of a number of disruptive technologies, such as NGS, in silico technologies, microfluidics, bioinformatics and computing power, machine learning (ML), and artificial intelligence (AI), are complimentary to the deployment of phage display in the drug discovery process and are now frequently applied in a parallel workflow, thereby significantly augmenting and expediting the discovery, optimization, and design of biologics.
Abbreviations used
| AI | = | Artificial intelligence |
| ADCs | = | Antibody drug conjugates |
| BSA | = | Bovine serum albumin |
| CD137 | = | Cluster of differentiation-137 |
| CNS | = | Central nervous system |
| COVID-19 | = | Coronavirus disease 2019 |
| ELISA | = | Enzyme-linked immunosorbent assay |
| FACS | = | Fluorescence-activated cell sorting |
| FDA | = | The United States Food and Drug Administration |
| FZD7 | = | Frizzled-7 |
| GCSF | = | Granulocyte-colony stimulating factor |
| GLP-1 | = | Glucagon-like peptide-1 |
| GIP | = | Gastric inhibitory polypeptide |
| GM-CSF | = | Granulocyte macrophage-colony stimulating factor |
| GM-CSFRα | = | Granulocyte macrophage-colony stimulating factor receptor alpha |
| GpI-b-IX-V | = | Glycoprotein Ib-IX-V membrane receptor complex |
| HLA | = | Human leukocyte antigens |
| IgNAR | = | Shark new antigen receptor |
| IL-11 R | = | Interleukin-11 receptor |
| KLH | = | Keyhole limpet hemocyanin |
| KRAS | = | A GTPase gene product of Kirsten rat sarcoma virus oncogene |
| LDL | = | Low-density lipoprotein |
| MHC | = | Major histocompatibility complex |
| MUC1 | = | Mucin 1 |
| NMR | = | Nuclear magnetic resonance |
| PET | = | Positron emission tomography |
| SPECT | = | Single-photon emission computed tomography |
| SPR | = | Surface plasmon resonance |
| TRIM | = | Trinucleotide mutagenesis |
| TRPV1 | = | Transient receptor potential vanilloid 1 |
Declaration of interest
AK Sato is an employee of Twist Bioscience. Meanwhile, CJ Hutchings is an independent consultant who provides consultancy services to Abalone Bio, AbCellera Biologics, Abilita Bio, AdAlta, Aureka Biotechnologies, Confo Therapeutics, DJS Antibodies, Immuto Scientific, Kyowa Kirin, MAbsolve, Maxion Therapeutics, Nxera Pharma UK, OmniAb, and Twist Bioscience. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–1317. doi: 10.1126/science.4001944
- McCafferty J, Griffiths AD, Winter G, et al. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348(6301):552–554. doi: 10.1038/348552a0
- Breitling F, Dübel S, Seehaus T, et al. A surface expression vector for antibody screening. Gene. 1991;104(2):147–153. doi: 10.1016/0378-1119(91)90244-6
- Barbas CF 3rd, Kang AS, Lerner RA, et al. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc Natl Acad Sci USA. 1991;88:7978–7982. doi: 10.1073/pnas.88.18.7978
- Arbabi Ghahroudi M, Desmyter A, Wyns L, et al. Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 1997;414(3):521–526. doi: 10.1016/S0014-5793(97)01062-4
- Stanfield RL, Dooley H, Verdino P, et al. Maturation of shark single-domain (IgNAR) antibodies: evidence for induced-fit binding. J Mol Biol. 2007;367(2):358–372. doi: 10.1016/j.jmb.2006.12.045
- Buschhaus MJ, Becker S, Porter AJ, et al. Isolation of highly selective IgNAR variable single-domains against a human therapeutic Fc scaffold and their application as tailor-made bioprocessing reagents. Protein Eng Des Sel. 2019;32(9):385–399. doi: 10.1093/protein/gzaa002
- Könning D, Kolmar H. Beyond antibody engineering: directed evolution of alternative binding scaffolds and enzymes using yeast surface display. Microbe Cell Fact. 2018;17(1):17; 32. doi: 10.1186/s12934-018-0881-3
- Qi H, Lu H, Qiu HJ, et al. Phagemid vectors for phage display: properties, characteristics and construction. J Mol Biol. 2012;417(3):129–143. doi: 10.1016/j.jmb.2012.01.038
- Dübel S, Breitling F, Fuchs P, et al. A family of vectors for surface display and production of antibodies. Gene. 1993;128(1):97–101. doi: 10.1016/0378-1119(93)90159-Z
- Söderlind E, Simonsson AC, Borrebaeck CA. Phage display technology in antibody engineering: design of phagemid vectors and in vitro maturation systems. Immunol Rev. 1992;130(1):109–124. doi: 10.1111/j.1600-065X.1992.tb01523.x
- Lowman HB, Bass SH, Simpson N, et al. Selecting high-affinity binding proteins by monovalent phage display. Biochemistry. 1991;30(45):10832–10838. doi: 10.1021/bi00109a004
- Hoogenboom HR, de Bruïne AP, Hufton SE, et al. Antibody phage display technology and its applications. Immunotechnology. 1998;4(1):1–20. doi: 10.1016/S1380-2933(98)00007-4
- Rondot S, Koch J, Breitling F, et al. A helper phage to improve single-chain antibody presentation in phage display. Nat Biotechnol. 2001;19(1):75–78. doi: 10.1038/83567
- Barbas CF 3rd. Recent advances in phage display. Current Opinion In Biotechnology. 1993;4(5):526–530. doi: 10.1016/0958-1669(93)90072-5
- Vaughan TJ, Williams AJ, Pritchard K, et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nature. 1996;14(3):309–314. doi: 10.1038/nbt0396-309
- Hutchings C, Carmen S, Lennard S. Generation of naive human antibody libraries. In: Kontermann R, Dübel S, editors. Antibody Engineering. Berlin (HB): Springer Lab Manuals. Springer; 2001. p. 93–108. doi: 10.1007/978-3-662-04605-0_6
- Lloyd C, Lowe D, Edwards B, et al. Modelling the human immune response: performance of a 1011 human antibody repertoire against a broad panel of therapeutically relevant antigens. Protein Eng Des Sel. 2009;22(3):159–168. doi: 10.1093/protein/gzn058
- Hoet RM, Cohen EH, Kent RB, et al. Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat Biotechnol. 2005;23(3):344–348. doi: 10.1038/nbt1067
- Sabir JS, Atef A, El-Domyati FM, et al. Construction of naïve camelids VHH repertoire in phage display-based library. C R Biol. 2014;337(4):244–249. doi: 10.1016/j.crvi.2014.02.004
- Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d
- Barbas CF 3rd. Phage display: a laboratory manual. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2001. doi: 10.1006/abio.2001.5085
- de Haard HJ, van Neer N, Reurs A, et al. A large non-immunized human fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J Biol Chem. 1999;274(26):18218–18230. doi: 10.1074/jbc.274.26.18218
- Knappik A, Ge L, Honegger A, et al. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Bio. 2000;296(1):57–86. doi: 10.1006/jmbi.1999.3444
- Alfaleh MA, Alsaab HO, Mahmoud AB, et al. Phage display derived monoclonal antibodies: from bench to bedside. Front Immunol. 2020;11:1986. doi: 10.3389/fimmu.2020.01986
- Moutel S, Bery N, Bernard V, et al. NaLi-H1: a universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. Elife. 2016;5:e16228. doi: 10.7554/eLife.16228
- Olichon A, de Marco A. Preparation of a naïve library of camelid single domain antibodies. Methods Mol Biol. 2012;911:65–78. doi: 10.1007/978-1-61779-968-6_5
- Yang H, Vasylieva N, Wang J, et al. Precise isolation and structural origin of an ultra-specific nanobody against chemical compound. J Hazard Mater. 2023;458:131958. doi: 10.1016/j.jhazmat.2023.131958
- Song BPC, Ch’ng ACW, Lim TS. A review of phage display: a jack-of-all-trades and master of most biomolecule display. Int J Biol Macromol. 2024;256(Pt 2):128455. doi: 10.1016/j.ijbiomac.2023.128455
- França RKA, Studart IC, Bezerra MRL, et al. Progress on phage display technology: tailoring antibodies for cancer immunotherapy. Viruses. 2023;15(9):1903. doi: 10.3390/v15091903
- Zhang Y. Evolution of phage display libraries for therapeutic antibody discovery. MAbs. 2023;15(1):2213793. doi: 10.1080/19420862.2023.2213793
- Liu Q, Garg P, Hasdemir B, et al. Functional GLP-1R antibodies identified from a synthetic GPCR-focused library demonstrate potent blood glucose control. MAbs. 2021;13(1):1893425. doi: 10.1080/19420862.2021.1893425
- Tikunova N, Dubrovskaya V, Morozova V, et al. The neutralizing human recombinant antibodies to pathogenic orthopoxviruses derived from a phage display immune library. Virus Res. 2012;163(1):141–150. doi: 10.1016/j.virusres.2011.09.008
- Throsby M, van den Brink E, Jongeneelen M, et al. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLOS ONE. 2008;3(12):e3942. doi: 10.1371/journal.pone.0003942
- Hamers-Casterman C, Atarhouch T, Muyldermans S, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446–448. doi: 10.1038/363446a0
- Lauwereys M, Arbabi Ghahroudi M, Desmyter A, et al. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. Embo J. 1998;17(13):3512–3520. doi: 10.1093/emboj/17.13.3512
- Nguyen TTH, Lee JS, Shim H. Construction of Rabbit Immune Antibody Libraries. Methods Mol Biol. 2023;2702:93–106. doi: 10.1007/978-1-4939-7447-4_7
- Rojas G, Almagro JC, Acevedo B, et al. Phage antibody fragments library combining a single human light chain variable region with immune mouse heavy chain variable regions. J Biotechnol. 2002;94(3):287–298. doi: 10.1016/S0168-1656(01)00432-1
- Jahromi ZM, Salmanian AH, Rastgoo N, et al. Isolation of BNYVV coat protein-specific single chain Fv from a mouse phage library antibody. Hybridoma (Larchmt). 2009;28(5):305–313. doi: 10.1089/hyb.2009.0004
- Teng Y, Young JL, Edwards B, et al. Diverse human VH antibody fragments with bio-therapeutic properties from the Crescendo Mouse. N Biotechnol. 2020;55:65–76. doi: 10.1016/j.nbt.2019.10.003
- Krawczyk K, Dunbar J, Deane CM. Computational tools for aiding rational antibody design. Methods Mol Biol. 2017;1529:399–416. doi: 10.1007/978-1-4939-6637-0_21
- Peterson SM, Juliana CA, Hu CF, et al. Optimization of a glucagon-like peptide 1 receptor antagonist antibody for treatment of hyperinsulinism. Diabetes. 2023;72(9):1320–1329. doi: 10.2337/db22-1039
- Pinheiro VB. [cited 2023 Nov 12]. Available from: https://pinheirolab.com/2020/04/28/dna-library-synthesis-for-directed-evolution/
- Prassler J, Thiel S, Pracht C, et al. HuCAL PLATINUM, a synthetic fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol. 2011;413(1):261–278. doi: 10.1016/j.jmb.2011.08.012
- Van den Brulle J, Fischer M, Langmann T, et al. A novel solid phase technology for high-throughput gene synthesis. Biotechniques. 2008;45(3):340–343. doi: 10.2144/000112953
- Tiller T, Schuster I, Deppe D, et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs. 2013;5(3):445–470. doi: 10.4161/mabs.24218
- Söderlind E, Strandberg L, Jirholt P, et al. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat Biotechnol. 2000;18(8):852–856. doi: 10.1038/78458
- Teixeira AAR, Erasmus MF, D’Angelo S, et al. Drug-like antibodies with high affinity, diversity and developability directly from next-generation antibody libraries. MAbs. 2021;13(1):1980942. doi: 10.1080/19420862.2021.1980942
- Sidhu SS, Li B, Chen Y, et al. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol. 2004;338(2):299–310. doi: 10.1016/j.jmb.2004.02.050
- Barbas CF 3rd, Bain JD, Hoekstra DM, et al. Semisynthetic combinatorial antibody libraries: a chemical solution to the diversity problem. Proc Natl Acad Sci USA. 1992;89(10):4457–4461. doi: 10.1073/pnas.89.10.4457
- Rothe C, Urlinger S, Löhning C, et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J Mol Biol. 2008;376(4):1182–1200. doi: 10.1016/j.jmb.2007.12.018
- Fellouse FA, Esaki K, Birtalan S, et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373(4):924–940. doi: 10.1016/j.jmb.2007.08.005
- Fellouse FA, Li B, Compaan DM, et al. Molecular recognition by a binary code. J Mol Biol. 2005;348(5):1153–1162. doi: 10.1016/j.jmb.2005.03.041
- Koide A, Gilbreth RN, Esaki K, et al. High-affinity single-domain binding proteins with a binary-code interface. Proc Natl Acad Sci, USA. 2007;104(16):6632–6637. doi: 10.1073/pnas.0700149104
- Mahon CM, Lambert MA, Glanville J, et al. Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J Mol Biol. 2013;425(10):1712–1730. doi: 10.1016/j.jmb.2013.02.015
- Virnekäs B, Ge L, Plückthun A, et al. Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994;22(25):5600–5607. doi: 10.1093/nar/22.25.5600
- Hughes RA, Ellington AD. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic Biology. Cold Spring Harb Perspect Biol. 2017;9(1):a023812. doi: 10.1101/cshperspect.a023812
- Kuiper BP, Prins RC, Billerbeck S. Oligo pools as an affordable source of synthetic DNA for cost-effective library construction in protein- and metabolic pathway engineering. Chembiochem. 2022;23(7):e202100507. doi: 10.1002/cbic.202100507
- Yuan TZ, Garg P, Wang L, et al. Rapid discovery of diverse neutralizing SARS-CoV-2 antibodies from large-scale synthetic phage libraries. MAbs. 2022;14(1):2002236. doi: 10.1080/19420862.2021.2002236
- Meiser LC, Antkowiak PL, Koch J, et al. Reading and writing digital data in DNA. Nat Protoc. 2020;15(1):86–101. doi: 10.1038/s41596-019-0244-5
- Van Blarcom T, Lindquist K, Melton Z, et al. Productive common light chain libraries yield diverse panels of high affinity bispecific antibodies. MAbs. 2018;10(2):256–268. doi: 10.1080/19420862.2017.1406570
- Atwell S, Ridgway JB, Wells JA, et al. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270(1):26–35. doi: 10.1006/jmbi.1997.1116
- Egloff P, Zimmermann I, Arnold FM, et al. Engineered peptide barcodes for in-depth analyses of binding protein libraries. Nat Methods. 2019;16(5):421–428. doi: 10.1038/s41592-019-0389-8
- Shuai RW, Ruffolo JA, Gray JJ. IgLM: infilling language modeling for antibody sequence design. Cell Syst. 2023;14(11):979–989.e4. doi: 10.1016/j.cels.2023.10.001
- Harel Inbar N, Benhar I. Selection of antibodies from synthetic antibody libraries. Arch Biochem Biophys. 2012;526(2):87–98. doi: 10.1016/j.abb.2011.12.028
- Burkovitz A, Ofran Y. Understanding differences between synthetic and natural antibodies can help improve antibody engineering. MAbs. 2016;8(2):278–287. doi: 10.1080/19420862.2015.1123365
- Adolf-Bryfogle J, Xu Q, North B, et al. PyIgClassify: a database of antibody CDR structural classifications. Nucleic Acids Res. 2015;43(Database issue):D432–8. doi: 10.1093/nar/gku1106
- Abanades B, Georges G, Bujotzek A, et al. ABlooper: fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics. 2022;38(7):1877–1880. doi: 10.1093/bioinformatics/btac016
- Raybould MIJ, Marks C, Krawczyk K, et al. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci, USA. 2019;116(10):4025–4030. doi: 10.1073/pnas.1810576116
- Abanades B, Olsen TH, Raybould MIJ, et al. The patent and literature antibody database (PLAbDab): an evolving reference set of functionally diverse, literature-annotated antibody sequences and structures. Nucleic Acids Res. 2024;52(D1):D545–D551. doi: 10.1093/nar/gkad1056
- Abanades B, Wong WK, Boyles F, et al. ImmuneBuilder: deep-learning models for predicting the structures of immune proteins. Commun Biol. 2023;6(1):575. doi: 10.1038/s42003-023-04927-7
- Spoendlin FC, Abanades B, Raybould MIJ, et al. Improved computational epitope profiling using structural models identifies a broader diversity of antibodies that bind to the same epitope. Front Mol Biosci. 2023;10:1237621. doi: 10.3389/fmolb.2023.1237621
- DeKosky BJ, Lungu OI, Park D, et al. Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc Natl Acad Sci, USA. 2016;113(19):E2636–45. doi: 10.1073/pnas.1525510113
- Vaisman-Mentesh A, Wine Y. Monitoring Phage Biopanning by Next-Generation Sequencing. Methods Mol Biol. 2018;1701:463–473. doi: 10.1007/978-1-4939-7447-4_26
- Available from: https://www.specifica.bio/abxtract/
- Conrath KE, Lauwereys M, Wyns L, et al. Camel single-domain antibodies as modular building units in bispecific and bivalent antibody constructs. J Biol Chem. 2001;276(10):7346–7350. doi: 10.1074/jbc.M007734200
- Muyldermans S. A guide to: generation and design of nanobodies. FEBS J. 2021;288(7):2084–2102. doi: 10.1111/febs.15515
- Bannas P, Hambach J, Koch-Nolte F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol. 2017;8:1603. doi: 10.3389/fimmu.2017.01603
- Vanlandschoot P, Stortelers C, Beirnaert E, et al. Nanobodies®: new ammunition to battle viruses. Antiviral Res. 2011;92(3):389–407. doi: 10.1016/j.antiviral.2011.09.002
- Hultberg A, Temperton NJ, Rosseels V, et al. Llama-derived single domain antibodies to build multivalent, superpotent and broadened neutralizing anti-viral molecules. PLOS ONE. 2011;6(4):e17665. doi: 10.1371/journal.pone.0017665
- Ulrichts H, Silence K, Schoolmeester A, et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood. 2011;118(3):757–765. doi: 10.1182/blood-2010-11-317859
- Callewaert F, Roodt J, Ulrichts H, et al. Evaluation of efficacy and safety of the anti-VWF nanobody ALX-0681 in a preclinical baboon model of acquired thrombotic thrombocytopenic purpura. Blood. 2012;120(17):3603–3610. doi: 10.1182/blood-2012-04-420943
- Jindal S, Pedersen DV, Gera N, et al. Characterization of the bispecific VHH antibody gefurulimab (ALXN1720) targeting complement component 5, and designed for low volume subcutaneous administration. Mol Immunol. 2024;65:29–41. doi: 10.1016/j.molimm.2023.12.004
- Tanaka Y. Ozoralizumab: first Nanobody® therapeutic for rheumatoid arthritis. Expert Opin Biol Ther. 2023;23(7):579–587. doi: 10.1080/14712598.2023.2231344
- Papp KA, Weinberg MA, Morris A, et al. IL17A/F nanobody sonelokimab in patients with plaque psoriasis: a multicentre, randomised, placebo-controlled, phase 2b study. Lancet. 2021;397(10284):1564–1575. doi: 10.1016/S0140-6736(21)00440-2
- Roberts BL, Markland W, Siranosian K, et al. Protease inhibitor display M13 phage: selection of high-affinity neutrophil elastase inhibitors. Gene. 1992;121(1):9–15. doi: 10.1016/0378-1119(92)90156-J
- Dennis MS, Lazarus RA. Kunitz domain inhibitors of tissue factor-factor VIIa. I. Potent inhibitors selected from libraries by phage display. J Biol Chem. 1994;269(35):22137–22144. doi: 10.1016/S0021-9258(17)31766-0
- Dennis MS, Herzka A, Lazarus RA. Potent and selective Kunitz domain inhibitors of plasma kallikrein designed by phage display. J Biol Chem. 1995;270(43):25411–25417. doi: 10.1074/jbc.270.43.25411
- Skerra A. ‘Anticalins’: a new class of engineered ligand-binding proteins with antibody-like properties. J Biotechnol. 2001;74(4):257–275. doi: 10.1016/S1389-0352(01)00020-4
- Möller M, Jönsson M, Lundqvist M, et al. An easy-to-use high-throughput selection system for the discovery of recombinant protein binders from alternative scaffold libraries. Protein Eng Des Sel. 2023;36:gzad011. doi: 10.1093/protein/gzad011
- Jönsson M, Scheffel J, Larsson E, et al. CaRA - a multi-purpose phage display library for selection of calcium-regulated affinity proteins. Nat Biotechnol. 2022;72:159–167. doi: 10.1016/j.nbt.2022.11.005
- Hjelm LC, Dahlsson Leitao C, Ståhl S, et al. Selection of affibody molecules using phage display. Cold Spring Harb Protoc. 2023 Jul 25. doi: 10.1101/pdb.prot108399
- Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24(1–2):3–9. doi: 10.1093/protein/gzq097
- Jeong KJ, Mabry R, Georgiou G. Avimers hold their own. Nat Biotechnol. 2005;23(12):1493–1494. doi: 10.1038/nbt1205-1493
- Griffiths K, Dolezal O, Cao B, et al. i-bodies, Human single domain antibodies that antagonize chemokine receptor CXCR4. J Biol Chem. 2016;291(24):12641–12657. doi: 10.1074/jbc.M116.721050
- Stolz LE, Horn PT. Ecallantide: a plasma kallikrein inhibitor for the treatment of acute attacks of hereditary angioedema. Drugs Today (Barc). 2010;46(8):547–555. doi: 10.1358/dot.2010.46.8.1507205
- Thompson CA. FDA approves kallikrein inhibitor to treat hereditary angioedem. Am J Health Syst Pharm. 2010;67(2):93. doi: 10.2146/news100005
- Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15(1):14–27. doi: 10.1110/ps.051817606
- Luo R, Liu H, Cheng Z. Protein scaffolds: antibody alternatives for cancer diagnosis and therapy. RSC Chem Biol. 2022;3(7):830–847. doi: 10.1039/D2CB00094F
- Koide A, Bailey CW, Huang X, et al. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284(4):1141–1151. doi: 10.1006/jmbi.1998.2238
- Akkapeddi P, Hattori T, Khan I, et al. Exploring switch II pocket conformation of KRAS(G12D) with mutant-selective monobody inhibitors. Proc Natl Acad Sci, USA. 2023;120(28):e2302485120. doi: 10.1073/pnas.2302485120
- Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci. 1997;94(23):12297–12302. doi: 10.1073/pnas.94.23.12297
- Kurz M, Gu K, Lohse PA. Psoralen photo-crosslinked mRNA-puromycin conjugates: a novel template for the rapid and facile preparation of mRNA-protein fusions. Nucleic Acids Research. 2000;28(18):83e–83. doi: 10.1093/nar/28.18.e83
- Liu R, Barrick JE, Szostak JW, et al. Optimized synthesis of RNA-protein fusions for in vitro protein selection. Methods Enzymol. 2000;318:268–293. doi: 10.1016/s0076-6879(00)18058-9
- Beste G, Schmidt FS, Stibora T, et al. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc Natl Acad Sci, USA. 1999;96:1898–1903. doi: 10.1073/pnas.96.5.1898
- Rothe C, Skerra A. Anticalin® proteins as therapeutic agents in human diseases. BioDrugs. 2018;32(3):233–243. doi: 10.1007/s40259-018-0278-1
- Gebauer M, Schiefner A, Matschiner G, et al. Combinatorial design of an anticalin directed against the extra-domain b for the specific targeting of oncofetal fibronectin. J Mol Biol. 2013;425(4):780–802. doi: 10.1016/j.jmb.2012.12.004
- Goetz DH, Holmes MA, Borregaard N, et al. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10(5):1033–1043. doi: 10.1016/S1097-2765(02)00708-6
- Gebauer M, Skerra A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012;503:157–188. doi: 10.1016/B978-0-12-396962-0.00007-0
- Siegemund M, Oak P, Hansbauer EM, et al. Pharmacokinetic engineering of OX40-blocking anticalin proteins using monomeric plasma half-life extension domains. Front Pharmacol. 2021;12:759337. doi: 10.3389/fphar.2021.759337
- Schlehuber S, Skerra A. Duocalins: engineered ligand-binding proteins with dual specificity derived from the lipocalin fold. Biol Chem. 2001;382(9):1335–1342. doi: 10.1515/BC.2001.166
- Wachter S, Angevin T, Bubna N, et al. Application of platform process development approaches to the manufacturing of Mabcalin™ bispecifics. J Biotech. 2023;377:13–22. doi: 10.1016/j.jbiotec.2023.10.003
- [cited 2024 Feb 6]. Available from: https://www.pieris.com/investors/sec-filings/all-sec-filings/content/0001193125-15-109883/0001193125-15-109883.pdf
- Barinka C, Ptacek J, Richter A, et al. Selection and characterization of anticalins targeting human prostate-specific membrane antigen (PSMA). Protein Eng Des Sel. 2016;29(3):105–115. doi: 10.1093/protein/gzv065
- Nord K, Nilsson J, Nilsson B, et al. A combinatorial library of an α-helical bacterial receptor domain. Protein Eng. 1995;8(6):601–608. doi: 10.1093/protein/8.6.601
- Grönwall C, Sjöberg A, Ramström M, et al. Affibody-mediated transferrin depletion for proteomics applications. Biotech J. 2007;2(11):1389–1398. doi: 10.1002/biot.200700053
- Kronqvist N, Malm M, Göstring L, et al. Combining phage and staphylococcal surface display for generation of ErbB3-specific affibody molecules. Protein Eng Des Sel. 2011;24(4):385–396. doi: 10.1093/protein/gzq118
- Kronqvist N, Löfblom J, Severa D, et al. Simplified characterization through site-specific protease-mediated release of affinity proteins selected by staphylococcal display. FEMS Microbiol Lett. 2008;278(1):128–136. doi: 10.1111/j.1574-6968.2007.00990.x
- Orlova A, Magnusson M, Eriksson TL, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521
- Alhuseinalkhudhur A, Lindman H, Liss P, et al. A phase II study of 68Ga-ABY-025 PET for non-invasive quantification of HER2 expression in breast cancer. In: Proceedings of the 2021 San Antonio Breast Cancer Symposium; 2021 Dec 7-10; San Antonio, TX. Philadelphia (PA): AACR; Cancer Res. 2022;82(4 Suppl): Abstract nr P3-02-06).
- Malm M, Bass T, Gudmundsdotter L, et al. Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension. Biotechnol J. 2014;9(9):1215–1222. doi: 10.1002/biot.201400009
- Jonsson A, Dogan J, Herne N, et al. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng Des Sel. 2008;21(8):515–527. doi: 10.1093/protein/gzn028
- Jussing E, Lu L, Grafström J, et al. [68Ga]ABY-028: an albumin-binding domain (ABD) protein-based imaging tracer for positron emission tomography (PET) studies of altered vascular permeability and predictions of albumin-drug conjugate transport. EJNMMI Res. 2020;10(1):106. doi: 10.1186/s13550-020-00694-2
- Yu F, Gudmundsdotter L, Akal A, et al. An affibody-adalimumab hybrid blocks combined IL-6 and TNF-triggered serum amyloid A secretion in vivo. MAbs. 2014;6(6):1598–1607. doi: 10.4161/mabs.36089
- LaFleur DW, Abramyan D, Kanakaraj P, et al. Monoclonal antibody therapeutics with up to five specificities: functional enhancement through fusion of target-specific peptides. MAbs. 2013;5(2):208–218. doi: 10.4161/mabs.23043
- Volk AL, Mebrahtu A, Ko BK, et al. Bispecific antibody molecule inhibits tumor cell proliferation more efficiently than the two-molecule combination. Drugs RD. 2021;21(2):157–168. doi: 10.1007/s40268-021-00339-2
- Mega A, Mebrahtu A, Aniander G, et al. A PDGFRB- and CD40-targeting bispecific AffiMab induces stroma-targeted immune cell activation. MAbs. 2023;15(1):2223750. doi: 10.1080/19420862.2023.2223750
- Available from: https://www.abclon.com/en/
- Mebrahtu A, Aniander G, Mega A, et al. Co-culture platform for tuning of cancer receptor density allows for evaluation of bispecific immune cell engagers. N Biotechnol. 2024;79:120–126. doi: 10.1016/j.nbt.2023.12.012
- Klint S, Feldwisch J, Gudmundsdotter L, et al. Izokibep: preclinical development and first-in-human study of a novel IL-17A neutralizing Affibody molecule in patients with plaque psoriasis. MAbs. 2023;15(1):2209920. doi: 10.1080/19420862.2023.2209920
- Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249(4967):386–390. doi: 10.1126/science.1696028
- Greenwood J, Willis AE, Perham RN. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J Mol Biol. 1991;220(4):821–827. doi: 10.1016/0022-2836(91)90354-9
- McConnell SJ, Uveges AJ, Fowlkes DM, et al. Construction and screening of M13 phage libraries displaying long random peptides. Mol Divers. 1996;1(3):165–176. doi: 10.1007/BF01544954
- Cortese R, Monaci P, Nicosia A, et al. Identification of biologically active peptides using random libraries displayed on phage. Curr Opin Biotech. 1995;6(1):73–80. doi: 10.1016/0958-1669(95)80012-3
- Clackson T, Wells JA. In vitro selection from protein and peptide libraries. Trends Biotechnol. 1994;12(5):173–184. doi: 10.1016/0167-7799(94)90079-5
- Sato AK, Sexton DJ, Morganelli LA, et al. Development of mammalian serum albumin affinity purification media by peptide phage display. Biotech Prog. 2002;18(2):182–192. doi: 10.1021/bp010181o
- Ladner RC, Sato AK, Gorzelany J, et al. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9(12):525–529. doi: 10.1016/S1359-6446(04)03104-6
- Huang L, Sexton DJ, Skogerson K, et al. Novel peptide inhibitors of angiotensin-converting enzyme 2. J Biol Chem. 2003;278(18):15532–15540. doi: 10.1074/jbc.M212934200
- Schumacher TN, Mayr LM, Minor DL Jr, et al. Identification of D-peptide ligands through mirror-image phage display. Science. 1996;271(5257):1854–1857. doi: 10.1126/science.271.5257.1854
- Malhis M, Funke SA. Mirror-image phage display for the selection of D-amino acid peptide ligands as potential therapeutics. Curr Protoc. 2024;4(2):e957. doi: 10.1002/cpz1.957
- Sato AK, Viswanathan M, Kent RB, et al. Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol. 2006;17(6):638–642. doi: 10.1016/j.copbio.2006.10.002
- Heinis C, Rutherford T, Freund S, et al. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5(7):502–507. doi: 10.1038/nchembio.184
- Simonetti L, Ivarsson Y. Genetically encoded cyclic peptide phage display libraries. ACS Cent Sci. 2020;6(3):336–338. doi: 10.1021/acscentsci.0c00087
- Miki T, Namii K, Seko K, et al. Pattern enrichment analysis for phage selection of stapled peptide ligands. Chem Sci. 2022;13(43):12634–12642. doi: 10.1039/D2SC04058A
- Guerlavais V, Sawyer TK, Carvajal L, et al. Discovery of sulanemadlin (ALRN-6924), the first cell-permeating, stabilized α-helical peptide in clinical development. J Med Chem. 2023;66(14):9401–9417. doi: 10.1021/acs.jmedchem.3c00623
- Smith GP, Patel SU, Windass JD, et al. Small binding proteins selected from a combinatorial repertoire of knottins displayed on phage. J Mol Biol. 1998;277(2):317–332. doi: 10.1006/jmbi.1997.1621
- Lui BG, Salomon N, Wüstehube-Lausch J, et al. Targeting the tumor vasculature with engineered cystine-knot miniproteins. Nat Commun. 2020;11(1):295. doi: 10.1038/s41467-019-13948-y
- Wang L, Xu J, Kong Y, et al. Engineering a novel antibody-peptide bispecific fusion protein against MERS-CoV. Antibodies (Basel). 2019;8(4):53. doi: 10.3390/antib8040053
- Huang R, Warner Jenkins G, Kim Y, et al. The smallest functional antibody fragment: ultralong CDR H3 antibody knob regions potently neutralize SARS-CoV-2. Proc Natl Acad Sci USA. 2023;120(39):e2303455120. doi: 10.1073/pnas.2303455120
- Lehmann A. Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Exp Opin Biol Ther. 2008;8(8):1187–1199. doi: 10.1517/14712598.8.8.1187
- Molineux G, Newland A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: from bench to bedside. Br J Hematol. 2010;150(1):9–20. doi: 10.1111/j.1365-2141.2010.08140.x
- Nixon AE, Sexton DJ, Ladner RC. Drugs derived from phage display: from candidate identification to clinical practice. MAbs. 2014;6(1):73–78. doi: 10.4161/mabs.27240
- Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279(5349):377–380. doi: 10.1126/science.279.5349.377
- Drin G, Rousselle C, Scherrmann JM, et al. Peptide delivery to the brain via adsorptive-mediated endocytosis: advances with SynB vectors. AAPS Pharm Sci. 2002;4(4):E26. doi: 10.1208/ps040426
- Pasqualini R, Ruoslahti E. Organ targeting in vivo using phage display peptide libraries. Nature. 1996;380(6572):364–366. doi: 10.1038/380364a0
- Giordano RJ, Edwards JK, Tuder RM, et al. Combinatorial ligand-directed lung targeting. Proc Am Thorac Soc. 2009;6(5):411–415. doi: 10.1513/pats.200903-014AW
- Driessen WH, Bronk LF, Edwards JK, et al. On the synergistic effects of ligand-mediated and phage-intrinsic properties during in vivo selection. Adv Genet. 2010;69:115–133. doi: 10.1016/S0065-2660(10)69005-0
- Staquicini DI, Barbu EM, Zemans RL, et al. Targeted phage display-based pulmonary vaccination in mice and non-human primates. Med. 2021;2(3):321–342. doi: 10.1016/j.medj.2020.10.005
- Loi M, Di Paolo D, Soster M, et al. Novel phage display-derived neuroblastoma-targeting peptides potentiate the effect of drug nanocarriers in preclinical settings. J Control Release. 2013;170(2):233–241. doi: 10.1016/j.jconrel.2013.04.029
- Chang DK, Lin CT, Wu CH, et al. A novel peptide enhances therapeutic efficacy of liposomal anti-cancer drugs in mice models of human lung cancer. PLOS ONE. 2009;4(1):e4171. doi: 10.1371/journal.pone.0004171
- Gray BP, Li S, Brown KC. From phage display to nanoparticle delivery: functionalizing liposomes with multivalent peptides improves targeting to a cancer biomarker. Bioconjug Chem. 2013;24(1):85–96. doi: 10.1021/bc300498d
- Saw PE, Song EW. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell. 2019;10(11):787–807. doi: 10.1007/s13238-019-0639-7
- Parmley SF, Smith GP. Filamentous fusion phage cloning vectors for the study of epitopes and design of vaccines. Adv Exp Med Biol. 1989;251:215–218. doi: 10.1007/978-1-4757-2046-4_21
- Perham RN, Terry TD, Willis AE, et al. Engineering a peptide epitope display system on filamentous bacteriophage. FEMS Microbiol Rev. 1995;17(1–2):25–31. doi: 10.1111/j.1574-6976.1995.tb00184.x
- Rangel R, Guzman-Rojas L, le Roux LG, et al. Combinatorial targeting and discovery of ligand-receptors in organelles of mammalian cells. Nat Commun. 2012;3:788. doi: 10.1038/ncomms1773
- Wu CH, Liu IJ, Lu RM, et al. Advancement and applications of peptide phage display technology in biomedical science. J Biomed Sci. 2016;23:8. doi: 10.1186/s12929-016-0223-x
- Fairbrother WJ, Christinger HW, Cochran AG, et al. Novel peptides selected to bind vascular endothelial growth factor target the receptor-binding site. Biochemistry. 1998;37(51):17754–17764. doi: 10.1021/bi981931e
- Fleming TJ, Sachdeva M, Delic M, et al. Discovery of high-affinity peptide binders to BLyS by phage display. J Mol Recognit. 2005;18(1):94–102. doi: 10.1002/jmr.722
- Landon LA, Zou J, Deutscher SL. Is phage display technology on target for developing peptide-based cancer drugs? Curr Drug Discov Technol. 2004;1(2):113–132. doi: 10.2174/1570163043335108
- Sato AK, Sexton DJ, Dransfield DT, et al. KDR and VEGF/KDR binding peptides and their use in diagnosis and therapy. WO03074005. 2003.
- Rich SJ, Bello-Quintero CE. Advancements in the treatment of psoriasis: role of biologic agents. J Manag Care Pharm. 2004;10(4):318–325. doi: 10.18553/jmcp.2004.10.4.318
- Balan V, Nelson DR, Sulkowski MS, et al. A Phase I/II study evaluating escalating doses of recombinant human albumin-interferon-alpha fusion protein in chronic hepatitis C patients who have failed previous interferon-alpha-based therapy. Antivir Ther. 2006;11(1):35–45. doi: 10.1177/135965350601100111
- Fishbane S, Roger SD, Martin E, et al. Peginesatide for maintenance treatment of anemia in hemodialysis and nondialysis patients previously treated with darbepoetin alfa. Clin J Am Soc Nephrol. 2013;8(4):538–545. doi: 10.2215/CJN.03440412
- Véniant MM, Lu SC, Atangan L, et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat Metab. 2024;6(2):290–303. doi: 10.1038/s42255-023-00966-w
- Rebar EJ, Greisman HA, Pabo CO. Phage display methods for selecting zinc finger proteins with novel DNA-binding specificities. Methods Enzymol. 1996;267:129–149. doi: 10.1016/s0076-6879(96)67010-4
- Wu H, Yang WP, Barbas CF 3rd. Building zinc fingers by selection: toward a therapeutic application. Proc Natl Acad Sci, USA. 1995;92(2):344–348. doi: 10.1073/pnas.92.2.344
- Hutchings CJ, Cseke G, Osborne G, et al. Monoclonal anti-β1-adrenergic receptor antibodies activate G protein signaling in the absence of β-arrestin recruitment. MAbs. 2014;6(1):246–261. doi: 10.4161/mabs.27226
- Mileni M. Modified membrane spanning proteins and methods for the preparation and use thereof. US 0123956. 2016.
- Schütz M, Schöppe J, Sedlák E, et al. Directed evolution of G protein-coupled receptors in yeast for higher functional production in eukaryotic expression hosts. Sci Rep. 2016;6:21508. doi: 10.1038/srep21508
- Scott DJ, Kummer L, Egloff P, et al. Improving the apo-state detergent stability of NTS1 with CHESS for pharmacological and structural studies. Biochim Biophys Acta. 2014;1838(11):2817–2824. doi: 10.1016/j.bbamem.2014.07.015
- Gardill B, Huang J, Tu L, et al. Nanodisc technology facilitates identification of monoclonal antibodies targeting multi-pass membrane proteins. Sci Rep. 2020;10(1):1130. doi: 10.1038/s41598-020-58002-w
- Doranz BJ, Willis S. Lipoparticles comprising proteins, methods of making, and using the same. US 8574590. 2004.
- Suharni, Nomura Y, Arakawa T, et al. Proteoliposome-based selection of a recombinant antibody fragment against the human M2 muscarinic acetylcholine receptor. Monoclon Antib Immunodiagn Immunother. 2014;33(6):378–385. doi: 10.1089/mab.2014.0041
- Sharma P, Plant M, Lam SK, et al. Kinetic analysis of antibody binding to integral membrane proteins stabilized in SMALPs. BBA Adv. 2021;1:100022. doi: 10.1016/j.bbadva.2021.100022
- Kristensen P, Winter G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des. 1998;3(5):321–328. doi: 10.1016/S1359-0278(98)00044-3
- Dobson CL, Devine PW, Phillips JJ, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 2016;6(1):38644. doi: 10.1038/srep38644
- Tulika T, Pederson RW, Rimbault C, et al. Phage display assisted discovery of a pH-dependent anti-α-cobratoxin antibody from a natural variable domain library. Protein Sci. 2023 Oct;32(12):e4821. doi: 10.1002/pro.4821
- Bracken CJ, Lim SA, Solomon P, et al. Bi-paratopic and multivalent human VH domains neutralize SARS-CoV-2 by targeting distinct epitopes within the ACE2 binding interface of Spike. bio Rxiv. 2020. doi: 10.1101/2020.08.08.242511
- Parsons HL, Earnshaw JC, Wilton J, et al. Directing phage selections towards specific epitopes. Protein Eng. 1996;9(11):1043–1049. doi: 10.1093/protein/9.11.1043
- Zeng X, Li L, Lin J, et al. Isolation of a human monoclonal antibody specific for the receptor binding domain of SARS-CoV-2 using a competitive phage biopanning strategy. Antib Ther. 2020;3(2):95–100. doi: 10.1093/abt/tbaa008
- Fitting J, Blume T, Ten Haaf A, et al. Phage display-based generation of novel internalizing antibody fragments for immunotoxin-based treatment of acute myeloid leukemia. MAbs. 2015;7(2):390–402. doi: 10.1080/19420862.2015.1007818
- Peissert F, Pedotti M, Corbellari R, et al. Adapting neutralizing antibodies to viral variants by structure-guided affinity maturation using phage display technology. Glob Chall. 2023;7(10):2300088. doi: 10.1002/gch2.202300088
- Jespers LS, Roberts A, Mahler SM, et al. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology. 2004;12(9):899–903. doi: 10.1038/nbt0994-899
- Lowe DC, Gerhardt S, Ward A, et al. Engineering a high-affinity anti-IL-15 antibody: crystal structure reveals an α-helix in VH CDR3 as key component of paratope. J Mol Biol. 2011;406(1):160–175. doi: 10.1016/j.jmb.2010.12.017
- Cannon DA, Shan L, Du Q, et al. Experimentally guided computational antibody affinity maturation with de novo docking, modelling and rational design. PLoS Comput Bio. 2019;15(5):e1006980. doi: 10.1371/journal.pcbi.1006980
- Nelson MH, Fritzell S, Miller R, et al. The bispecific tumor antigen-conditional 4-1BB x 5T4 agonist, ALG.APV-527, mediates strong T-Cell activation and potent antitumor activity in preclinical studies. Mol Cancer Ther. 2023;22(1):89–101. doi: 10.1158/1535-7163.MCT-22-0395
- Nimrod G, Fischman S, Austin M, et al. Computational design of epitope-specific functional antibodies. Cell Rep. 2018;25(8):2121–2131. doi: 10.1016/j.celrep.2018.10.081
- Grihalde ND, Collins CA, Pellacani AU, et al. GLP-1 receptor agonist and allosteric modulator monoclonal antibodies and uses thereof. US0275288. 2006.
- Hermann T, Bueltmann A. Antibodies targeting specifically human CXCR2. US0060347. 2016.
- Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs. 2010;2(6):594–606. doi: 10.4161/mabs.2.6.13420
- Hutchings CJ, Koglin M, Olson WC, et al. Opportunities for therapeutic antibodies directed at G protein-coupled receptors. Nat Rev Drug Discov. 2017;16(9):661. doi: 10.1038/nrd.2017.173
- Kelil A, Gallo E, Banerjee S, et al. CellectSeq: In silico discovery of antibodies targeting integral membrane proteins combining in situ selections and next-generation sequencing. Commun Biol. 2021;4(1):561. doi: 10.1038/s42003-021-02066-5
- Philpott DN, Gomis S, Wang H, et al. Rapid on-cell selection of high-performance human antibodies. ACS Cent Sci. 2022;8(1):102–109. doi: 10.1021/acscentsci.1c01205
- André AS, Moutinho I, Jnr D, et al. In vivo Phage Display: A promising selection strategy for the improvement of antibody targeting and drug delivery properties. Front Microbiol. 2022;13:962124. doi: 10.3389/fmicb.2022.962124
- Zou J, Dickerson MT, Owen NK, et al. Biodistribution of filamentous phage peptide libraries in mice. Mol Biol Rep. 2004;31(2):121–129. doi: 10.1023/B:MOLE.0000031459.14448.af
- Zurita AJ, Troncoso P, Cardó-Vila M, et al. Combinatorial screenings in patients: the interleukin-11 receptor alpha as a candidate target in the progression of human prostate cancer. Cancer Res. 2004;64(2):435–439. doi: 10.1158/0008-5472.CAN-03-2675
- Deramchia K, Jacobin-Valat MJ, Vallet A, et al. In vivo phage display to identify new human antibody fragments homing to atherosclerotic endothelial and subendothelial tissues. Am J Pathol. 2012;180(6):2576–2589. doi: 10.1016/j.ajpath.2012.02.013
- Hemadou A, Fontayne A, Laroche-Traineau J, et al. In vivo human single-chain fragment variable phage display-assisted identification of Galectin-3 as a new biomarker of atherosclerosis. J Am Heart Assoc. 2021;10(19):e016287. doi: 10.1161/JAHA.120.016287
- Ueberberg S, Schneider S. Phage library-screening: a powerful approach for generation of targeting-agents specific for normal pancreatic islet-cells and islet-cell carcinoma in vivo. Regul Pept. 2010;160(1–3):1–8. doi: 10.1016/j.regpep.2009.11.017
- Krag DN, Shukla GS, Shen GP, et al. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006;66(15):7724–7733. doi: 10.1158/0008-5472.CAN-05-4441
- van Lith SA, Roodink I, Verhoeff JJ, et al. In vivo phage display screening for tumor vascular targets in glioblastoma identifies a llama nanobody against dynactin-1-p150Glued. Oncotarget. 2016;7(44):71594–71607. doi: 10.18632/oncotarget.12261
- Aguiar SI, Dias JNR, André AS, et al. Highly specific blood-brain barrier transmigrating single-domain antibodies selected by an In vivo phage display screening. Pharmaceutics. 2021;13(10):1598. doi: 10.3390/pharmaceutics13101598
- André AS, Dias JNR, Aguiar SI, et al. Rabbit derived VL single-domains as promising scaffolds to generate antibody–drug conjugates. Sci Rep. 2023;13(1):4837. doi: 10.1038/s41598-023-31568-x
- Ravn U, Gueneau F, Baerlocher L, et al. By-passing in vitro screening–next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010;38(21):e193. doi: 10.1093/nar/gkq789
- Barreto K, Maruthachalam BV, Hill W, et al. Next-generation sequencing-guided identification and reconstruction of antibody CDR combinations from phage selection outputs. Nucleic Acids Res. 2019;47(9):e50. doi: 10.1093/nar/gkz131
- D’Angelo S, Kumar S, Naranjo L, et al. From deep sequencing to actual clones. Protein Eng Des Sel. 2014;27(10):301–307. doi: 10.1093/protein/gzu032
- Yang W, Yoon A, Lee S, et al. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp Mol Med. 2017;49(3):e308. doi: 10.1038/emm.2017.22
- Park M, de Villavicencio Diaz TN, Lange V, et al. Exploring the sheep (Ovis aries) immunoglobulin repertoire by next generation sequencing. Mol Immunol. 2023;156:20–30. doi: 10.1016/j.molimm.2023.02.008
- Fischer N. Sequencing antibody repertoires: the next generation. MAbs. 2011;3(1):17–20. doi: 10.4161/mabs.3.1.14169
- Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–351. doi: 10.1038/nrg.2016.49
- Rouet R, Jackson KJL, Langley DB, et al. Next-generation sequencing of antibody display repertoires. Front Immunol. 2018;9:118. doi: 10.3389/fimmu.2018.00118
- Hu T, Chitnis N, Monos D, et al. Next-generation sequencing technologies: An overview. Hum Immunol. 2021;82(11):801–811. doi: 10.1016/j.humimm.2021.02.012
- Unkauf T, Miethe S, Fühner V, et al. Generation of recombinant antibodies against toxins and viruses by phage display for diagnostics and therapy. Adv Exp Med Biol. 2016;917:55–76.
- Eddleston M, Senarathna L, Mohammed F, et al. Deaths due to the lack of an affordable antitoxin for plant poisoning. Lancet. 2003;362(9389):1041–1044. doi: 10.1016/S0140-6736(03)14415-7
- Sinclair AJ, Hewick DS, Johnston PC, et al. Kinetics of digoxin and anti-digoxin antibody fragments during treatment of digoxin toxicity. Br J Clin Pharmacol. 1989;28(3):352–356. doi: 10.1111/j.1365-2125.1989.tb05437.x
- Kini RM, Sidhu S, Laustsen AH. Biosynthetic oligoclonal antivenom (BOA) for snakebite and next-generation treatments for snakebite victims. Toxins (Basel). 2018;10(12):534. doi: 10.3390/toxins10120534
- Laustsen AH, Karatt-Vellatt A, Masters EW, et al. In vivo neutralization of dendrotoxin-mediated neurotoxicity of black mamba venom by oligoclonal human IgG antibodies. Nat Commun. 2018;9(1):4957. doi: 10.1038/s41467-018-07480-8
- Luz D, Gómez FD, Ferreira RL, et al. The deleterious effects of shiga toxin type 2 are neutralized in vitro by FabF8: Stx2 recombinant monoclonal antibody. Toxins (Basel). 2021;13(11):825. doi: 10.3390/toxins13110825
- Ledsgaard L, Laustsen AH, Pus U, et al. In vitro discovery of a human monoclonal antibody that neutralizes lethality of cobra snake venom. MAbs. 2022;14(1):2085536. doi: 10.1080/19420862.2022.2085536
- Richard G, Meyers AJ, McLean MD, et al. In vivo neutralization of α-cobratoxin with high-affinity llama single-domain antibodies (VHHs) and a VHH-Fc antibody. PLoS One. 2013;8(7):e69495. doi: 10.1371/journal.pone.0069495
- Bailon Calderon H, Yaniro Coronel VO, Cáceres Rey OA, et al. Development of nanobodies against hemorrhagic and myotoxic components of bothrops atrox snake venom. Front Immunol. 2020;11:655. doi: 10.3389/fimmu.2020.00655
- Jenkins TP, Fryer T, Dehli RI, et al. Toxin neutralization using alternative binding proteins. Toxins (Basel). 2019;11(1):53. doi: 10.3390/toxins11010053
- Jenkins TP, Laustsen AH. Cost of Manufacturing for recombinant snakebite antivenoms. Front Bioeng & Biotech. 2020;8:703. doi: 10.3389/fbioe.2020.00703
- Alonso Villela SM, Kraïem-Ghezal H, Bouhaouala-Zahar B, et al. Production of recombinant scorpion antivenoms in E. coli: current state and perspective. Appl Microbiol Biotechnol. 2023;107(13):4133–4152. doi: 10.1007/s00253-023-12578-1
- Raeisi H, Azimirad M, Nabavi-Rad A, et al. Application of recombinant antibodies for treatment of Clostridioides difficile infection: current status and future perspective. Front Immunol. 2022;13:972930. doi: 10.3389/fimmu.2022.972930
- Boruah BM, Liu D, Ye D, et al. Single domain antibody multimers confer protection against rabies infection. PLOS ONE. 2013;8(8):e71383. doi: 10.1371/journal.pone.0071383
- Turki I, Hammami A, Kharmachi H, et al. Engineering of a recombinant trivalent single-chain variable fragment antibody directed against rabies virus glycoprotein G with improved neutralizing potency. Mol Immunol. 2014;57(2):66–73. doi: 10.1016/j.molimm.2013.08.009
- Guo Y, Ouyang Z, He W, et al. Screening and epitope characterization of diagnostic nanobody against total and activated Bacteroides fragilis toxin. Front Immunol. 2023;14:1065274. doi: 10.3389/fimmu.2023.1065274
- Wenzel EV, Bosnak M, Tierney R, et al. Human antibodies neutralizing diphtheria toxin in vitro and in vivo. Sci Rep. 2020;10(1):571. doi: 10.1038/s41598-019-57103-5
- Li Y, Moysey R, Peter E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23(3):349–354. doi: 10.1038/nbt1070
- Boulter JM, Glick M, Todorov P, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;6(9):707–711. doi: 10.1093/protein/gzg087
- Palma M. Epitopes and mimotopes identification using phage display for vaccine development against infectious pathogens. Vaccines (Basel). 2023;11(7):1176. doi: 10.3390/vaccines11071176
- Sato AK. Phage libraries. In: Morrow W, Sheikh N, Schmidt C, Davies D, editors. Vaccinology: principles and practice. Wiley; 2012. doi: 10.1002/9781118345313.ch11
- Ballmann R, Hotop SK, Bertoglio F, et al. ORFeome phage display reveals a major immunogenic epitope on the S2 Subdomain of SARS-CoV-2 spike protein. Viruses. 2022;14(6):1326. doi: 10.3390/v14061326
- Khurana S, Wu J, Verma N, et al. H5N1 virus-like particle vaccine elicits cross-reactive neutralizing antibodies that preferentially bind to the oligomeric form of influenza virus hemagglutinin in humans. J Virol. 2011;85(21):10945–10954. doi: 10.1128/JVI.05406-11
- Wang L, Deng X, Liu H, et al. The mimic epitopes of Mycobacterium tuberculosis screened by phage display peptide library have serodiagnostic potential for tuberculosis. Pathog Dis. 2016;74(8):ftw091. doi: 10.1093/femspd/ftw091
- Eda S, Sherman IW. Selection of peptides recognized by human antibodies against the surface of Plasmodium falciparum-infected erythrocytes. Parasitology. 2005;130(Pt 1):1–11. doi: 10.1017/S0031182004006328
- Palma M. Aspects of phage-based vaccines for protein and epitope immunization. Vaccines (Basel). 2023;11(2):436. doi: 10.3390/vaccines11020436
- Becker M, Felsberger A, Frenzel A, et al. Application of M13 phage display for identifying immunogenic proteins from tick (Ixodes scapularis) saliva. BMC Biotechnol. 2015;15:43. doi: 10.1186/s12896-015-0167-3
- Mendonça M, Moreira GM, Conceição FR, et al. Fructose 1,6-bisphosphate aldolase, a novel immunogenic surface protein on listeria species. PLOS ONE. 2016;11(8):e0160544. doi: 10.1371/journal.pone.0160544
- Moreira GMSG, Köllner SMS, Helmsing S, et al. Pyruvate dehydrogenase complex—enzyme 2, a new target for Listeria spp. detection identified using combined phage display technologies. Sci Rep. 2020;10(1):15267. doi: 10.1038/s41598-020-72159-4
- Ramli SR, Moreira GMSG, Zantow J, et al. Discovery of Leptospira spp. seroreactive peptides using ORFeome phage display. PLOS Negl Trop Dis. 2019;13(1):e0007131. doi: 10.1371/journal.pntd.0007131
- Heine PA, Ballmann R, Thevarajah P, et al. Biomarker Discovery by ORFeome Phage Display. Methods Mol Biol. 2023;2702:543–561. doi: 10.1007/978-1-0716-3381-6_27
- Li W. ORF phage display to identify cellular proteins with different functions. Methods. 2012;58(1):2–9. doi: 10.1016/j.ymeth.2012.07.013
- Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;69(6506):1014–1018. doi: 10.1126/science.abd0831
- Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369(6506):1010–1014. doi: 10.1126/science.abd0827
- Jones BE, Brown-Augsburger PL, Corbett KS, et al. The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates. Sci Transl Med. 2021;3(593):eabf1906. doi: 10.1126/scitranslmed.abf1906
- Bell BN, Powell AE, Rodriguez C, et al. Neutralizing antibodies targeting the SARS-CoV-2 receptor binding domain isolated from a naïve human antibody library. Protein Sci. 2021;30(4):716–727. doi: 10.1002/pro.4044
- Chen X, Gentili M, Hacohen N, et al. A cell-free nanobody engineering platform rapidly generates SARS-CoV-2 neutralizing nanobodies. Nat Commun. 2021;12(1):5506. doi: 10.1038/s41467-021-25777-z
- Cantera JL, Cate DM, Golden A, et al. Screening antibodies raised against the spike glycoprotein of SARS-CoV-2 to support the development of rapid antigen assays. ACS Omega. 2021;6(31):20139–20148. doi: 10.1021/acsomega.1c01321
- Hastie KM, Li H, Bedinger D, et al. Defining variant-resistant epitopes targeted by SARS-CoV-2 antibodies: a global consortium study. Science. 2021;374(6566):472–478. doi: 10.1126/science.abh2315
- Bertoglio F, Fühner V, Ruschig M, et al. A SARS-CoV-2 neutralizing antibody selected from COVID-19 patients binds to the ACE2-RBD interface and is tolerant to most known RBD mutations. Cell Rep. 2021;6(4):109433. doi: 10.1016/j.celrep.2021.109433
- Bertoglio F, Meier D, Langreder N, et al. SARS-CoV-2 neutralizing human recombinant antibodies selected from pre-pandemic healthy donors binding at RBD-ACE2 interface. Nat Commun. 2021;12(1):1577. doi: 10.1038/s41467-021-21609-2
- Ferrara F, Erasmus MF, D’Angelo S, et al. A pandemic-enabled comparison of discovery platforms demonstrates a naïve antibody library can match the best immune-sourced antibodies. Nat Commun. 2022;13(1):462. doi: 10.1038/s41467-021-27799-z
- Kügler J, Wilke S, Meier D, et al. Generation and analysis of the improved human HAL9/10 antibody phage display libraries. BMC Biotechnol. 2015;15:10. doi: 10.1186/s12896-015-0125-0
- Rouet R, Mazigi O, Walker GJ, et al. Potent SARS-CoV-2 binding and neutralization through maturation of iconic SARS-CoV-1 antibodies. MAbs. 2021;13(1):1922134. doi: 10.1080/19420862.2021.1922134
- Zhao F, Keating C, Ozorowski G, et al. Engineering SARS-CoV-2 neutralizing antibodies for increased potency and reduced viral escape pathways. iScience. 2022;25(9):104914. doi: 10.1016/j.isci.2022.104914
- Zhao F, Yuan M, Keating C, et al. Broadening a SARS-CoV-1-neutralizing antibody for potent SARS-CoV-2 neutralization through directed evolution. Sci Signal. 2023;16(798):eabk3516. doi: 10.1126/scisignal.abk3516
- Fedry J, Hurdiss DL, Wang C, et al. Structural insights into the cross-neutralization of SARS-CoV and SARS-CoV-2 by the human monoclonal antibody 47D11. Sci Adv. 2021;7(23):eabf5632. doi: 10.1126/sciadv.abf5632
- Starr TN, Greaney AJ, Dingens AS, et al. Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016. Cell Rep Med. 2021;2(4):100255. doi: 10.1016/j.xcrm.2021.100255
- Yuan TZ, Lucas C, Monteiro VS, et al. A synthetic bispecific antibody capable of neutralizing SARS-CoV-2 delta and omicron. bio Rxiv. 2022. doi: 10.1101/2022.01.04.474803
- Hanes J, Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci, USA. 1997;94(10):4937–4942. doi: 10.1073/pnas.94.10.4937
- Hanes J, Jermutus L, Weber-Bornhauser S. Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc Natl Acad Sci, USA. 1998;5(24):14130–14135. doi: 10.1073/pnas.95.24.14130
- Bencurova E, Pulzova L, Flachbartova Z, et al. A rapid and simple pipeline for synthesis of mRNA-ribosome-V(H)H complexes used in single-domain antibody ribosome display. Mol Biosyst. 2015;11(6):1515–1524. doi: 10.1039/C5MB00026B
- Schilling J, Schöppe J, Plückthun A. From DARPins to LoopDARPins: novel LoopDARPin design allows the selection of low picomolar binders in a single round of ribosome display. J Mol Biol. 2014;426(3):691–721. doi: 10.1016/j.jmb.2013.10.026
- Thom G, Minter R. Optimization of CAT-354, a therapeutic antibody directed against interleukin-13, using ribosome display. Methods Mol Biol. 2012;805:393–401. doi: 10.1007/978-1-61779-379-0_22
- Adimab LLC. Rationally designed synthetic antibody libraries and uses therefor. WO2009036379. 2009.
- Arras P, Yoo HB, Pekar L, et al. AI/ML combined with next-generation sequencing of VHH immune repertoires enables the rapid identification of de novo humanized and sequence-optimized single domain antibodies: a prospective case study. Front Mol Biosci. 2023;10:1249247. doi: 10.3389/fmolb.2023.1249247
- Koide A, Koide S. Affinity maturation of single-domain antibodies by yeast surface display. Methods Mol Biol. 2012;911:431–443. doi: 10.1007/978-1-61779-968-6_26
- Parthiban K, Perera RL, Sattar M, et al. A comprehensive search of functional sequence space using large mammalian display libraries created by gene editing. MAbs. 2019;11(5):884–898. doi: 10.1080/19420862.2019.1618673
- Odegrip R, Coomber D, Eldridge B, et al. CIS display: In vitro selection of peptides from libraries of protein-DNA complexes. Proc Natl Acad Sci, USA. 2004;101(9):2806–2810. doi: 10.1073/pnas.0400219101
- Dyson MR, Masters E, Pazeraitis D, et al. Beyond affinity: selection of antibody variants with optimal biophysical properties and reduced immunogenicity from mammalian display libraries. MAbs. 2020;12(1):1829335. doi: 10.1080/19420862.2020.1829335
- Erasmus MF, Ferrara F, D’Angelo S, et al. Insights into next generation sequencing guided antibody selection strategies. Sci Rep. 2023;13(1):18370. doi: 10.1038/s41598-023-45538-w
- Trkulja CL, Jungholm O, Davidson M, et al. Rational antibody design for undruggable targets using kinetically controlled biomolecular probes. Sci Adv. 2021;7(16):eabe6397. doi: 10.1126/sciadv.abe6397
- Ow SY, Kapp EA, Tomasetig V, et al. HDX-MS study on garadacimab binding to activated FXII reveals potential binding interfaces through differential solvent exposure. MAbs. 2023;15(1):2163459. doi: 10.1080/19420862.2022.2163459
- Minkoff BB, Blatz JM, Choudhury FA, et al. Plasma-generated OH radical production for analyzing three-dimensional structure in protein therapeutics. Sci Rep. 2017;7(1):12946. doi: 10.1038/s41598-017-13371-7
- Sun H, Hu N, Wang J. Application of microfluidic technology in antibody screening. Biotech J. 2022;17(8):e2100623. doi: 10.1002/biot.202100623
- Sanluis-Verdes A, Peñaherrera A, Torán JL, et al. Selection of phage-displayed antibodies with high affinity and specificity by electrophoresis in microfluidic devices. Electrophoresis. 2023;44(9–10):864–872. doi: 10.1002/elps.202200187
- Zehnaker A, Vallet A, Gourdon J, et al. Combined multiplexed phage display, high-throughput sequencing, and functional assays as a platform for identifying modulatory VHHs targeting the FSHR. Int J Mol Sci. 2023;4(21):15961. doi: 10.3390/ijms242115961
- Xiao X, Douthwaite JA, Chen Y, et al. A high-throughput platform for population reformatting and mammalian expression of phage display libraries to enable functional screening as full-length IgG. MAbs. 2017;9(6):996–1006. doi: 10.1080/19420862.2017.1337617
- Fisher SM, Trail D, Hecht RI. The high throughput purification of Fc-fusion proteins. Process Biochemistry. 2006;41:2473–2476. doi: 10.1016/j.procbio.2006.06.010
- Gaston J, Maestrali N, Lalle G, et al. Intracellular delivery of therapeutic antibodies into specific cells using antibody-peptide fusions. Sci Rep. 2019;9(1):18688. doi: 10.1038/s41598-019-55091-0
- Peng X, Liu X, Kim JY, et al. Brain-penetrating peptide shuttles across the blood-brain-barrier and extracellular-like space. Bioconjug Chem. 2023;34(12):2319–2336. doi: 10.1021/acs.bioconjchem.3c00446
- Hong FU, Castro M, Linse K. Tumor specifically internalizing peptide ‘HN-1’: targeting the putative receptor retinoblastoma-regulated discoidin domain receptor 1 involved in metastasis. World J Clin Oncol. 2022;13(5):323–338. doi: 10.5306/wjco.v13.i5.323
- Rangel R, Dobroff AS, Guzman-Rojas L, et al. Targeting mammalian organelles with internalizing phage (iPhage) libraries. Nat Protoc. 2013;8(10):1916–1939. doi: 10.1038/nprot.2013.119
- Chung DH, Kong S, Young NJ, et al. Rare antibody phage isolation and discrimination (RAPID) biopanning enables identification of high-affinity antibodies against challenging targets. Commun Biol. 2023;6(1):1036. doi: 10.1038/s42003-023-05390-0
- Hie BL, Shanker VR, Xu D, et al. Efficient evolution of human antibodies from general protein language models. Nat Biotechnol. 2024;42(2):275–283. doi: 10.1038/s41587-023-01763-2
- Maxwell MA, Wang L, Safavi C, et al. Discovery and optimization of novel complement component 5a receptor 1 antagonist antibodies. In: Conference poster presentation, Antibody Engineering & Therapeutics. San Diego; 2023 Dec.
- Petrenko VA. Phage Display’s prospects for early diagnosis of prostate cancer. Viruses. 2024;16(2):277. doi: 10.3390/v16020277
- Wang M, Pang S, Zhang H, et al. Phage display derived probes in biosensing. Trends Analyt Chem. 2024;173:117629. doi: 10.1016/j.trac.2024.117629
- San Segundo-Acosta P, Montero-Calle A, Fuentes M, et al. Identification of Alzheimer’s disease autoantibodies and their target biomarkers by phage microarrays. J Proteome Res. 2019;18(7):2940–2953. doi: 10.1021/acs.jproteome.9b00258
- Han L, Liu P, Petrenko VA, et al. A label-free electrochemical impedance cytosensor based on specific peptide-fused phage selected from landscape phage library. Sci Rep. 2016;6:22199. doi: 10.1038/srep22199