
1. Introduction
One of the earliest and simplest forms of in silico predictive cheminformatics is the Quantitative Structure-Activity Relationship (QSAR). This algorithm aims to develop a mathematical model that establishes a relationship between the structural features and the biological activity [Citation1]. However, developing QSAR models can sometimes be challenging, especially when the number of experimentally known data points is limited [Citation2,Citation3]. In such cases, to avoid the statistical complexity, Read-Across has emerged as an alternative approach. However, one of the key drawbacks of the Read-Across approach is the lack of quantitative interpretability of the features. To combine the advantages of both QSAR and Read-Across, Banerjee and Roy [Citation4] developed statistical models using Read-Across-derived similarity and error-based measures serving as descriptors. This unconventional modeling approach was termed quantitative Read-Across Structure-Activity Relationship (q-RASAR) that encodes Read-Across-derived information into a statistical modeling framework. Over a relatively short period since its introduction, this technique has found use for modeling various pharmaceutical activity/toxicity/property endpoints [Citation4–12], environmental property endpoints [Citation13–29], and materials property-based endpoints [Citation30–35], and in most of the cases, the external predictivity was enhanced. Some case studies, additionally, reported enhancement in the internal validation metrics. lists various endpoints modeled using the q-RASAR approach. Within the q-RASAR paradigm, there is also a provision for modeling multiple endpoints simultaneously using the application of data fusion q-RASAR, which is somewhat similar to the multitask modeling approach using the perturbation-theory machine learning methodology [Citation36–38].
Table 1. Various endpoints modeled using the q-RASAR approach.
2. Selection of the important molecular features – a basic need for the computation of RASAR descriptors
A proper selection of the essential molecular features (descriptors) is an important aspect that determines the performance of the q-RASAR models. This is because the selected descriptor matrix is used to define the similarity among the compounds. This similarity is best defined when the modeler judiciously selects certain essential features from the complete feature pool using standard feature selection algorithms. If the similarity is defined based on the entire descriptor pool without feature selection, noise, inter-correlation, and redundancy will hinder the estimation of the actual similarity among compounds.
3. How to compute the RASAR descriptors
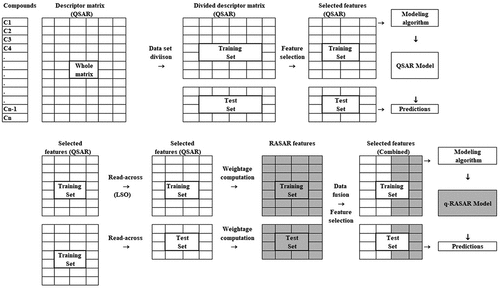
For the development of q-RASAR models, the basic pre-requisite is to compute the RASAR descriptors. This can be done using a simple Java-based RASAR descriptor calculator tool – RASAR-Desc-Calc-v3.0.3, which is freely available from https://sites.google.com/jadavpuruniversity.in/dtc-lab-software/home. This process is different from the standard procedure for the computation of conventional QSAR descriptors in:
Unlike the conventional approach, the RASAR descriptors are computed after dataset division and initial feature selection of the molecular descriptors. Therefore, the RASAR descriptors are computed separately for the training and test sets.
The standard molecular descriptors for a particular compound describe the chemical properties of the compound of interest. However, the RASAR descriptors describe the information of the close source neighbors for a particular query compound, and not the query compound itself.
The basic algorithm is that this tool computes the RASAR descriptors for the query set, using the chemical information from the source set. It is to be noted that the source set is always the training set of compounds that serve as the close source neighbors for a particular query compound. This information clarifies that when one intends to compute the RASAR descriptors for the test set, the training set file is used as the source set input file, while the test set file is used as the input for the query set. Similarly, when the training set RASAR descriptors are computed, the training set serves as both the source and query set inputs. This information has been reflected in for the better understanding of the readers. It is to be noted that in this particular case, a particular training set compound (serving as a query compound here) finds itself as the most similar compound in the list of close source compounds. In this case, the RASAR descriptor calculation is based on the subsequent nearest neighbors without taking the identical compound, thus removing the bias involved in the computation of the RASAR descriptors for the training set. This algorithm is termed Leave-Same-Out (LSO) [Citation14] where the same compound is not included in the RASAR descriptor computation. Please note that the LSO is automatically applied by the RASAR descriptor calculator tool, and the user does not require performing any additional operation. Additionally, this descriptor calculating tool requires a set of read-across-derived hyperparameters (σ, γ, and the number of close source compounds) associated with the best similarity algorithm. The user may either proceed with the default setting of the hyperparameters (σ = 1, γ = 1, number of close source compounds = 10) or use an optimized hyperparameter setting derived from Read-Across, which is mentioned in the next section.
Table 2. Input file specifications for the computation of the RASAR descriptors.
4. Optimization of the read-across-derived hyperparameters
This can be done using the tool Auto_RA_Optimizer-v1.0 (available from https://sites.google.com/jadavpuruniversity.in/dtc-lab-software/home). This tool performs a grid search involving different combinations of the hyperparameters associated with the different similarity-based measures stated previously and reports the prediction performance. The user needs to divide the training set into a sub-training and a sub-test set (a.k.a. a calibration and a validation set), and these files should be the input for the tool. The set of hyperparameters generating the best (or among the best) prediction for the sub-test set should be considered as the optimized hyperparameter setting. This setting should be used for the computation of the RASAR descriptors. It should be noted that the user should not use the original training and test sets as input for the Auto_RA_Optimizer tool, since this concept does not adhere to the general concepts of hyperparameter optimization in Machine Learning. Additionally, the user may want to manually perform the grid search using the Read-Across-v4.2.2 (available from https://sites.google.com/jadavpuruniversity.in/dtc-lab-software/home) by taking the sub-training and sub-test sets as inputs and executing the tool for every possible combination of hyperparameters. This manual operation should also be done for classification-based RASAR modeling.
5. Data fusion and model development
Once the RASAR descriptors are computed for both the training and test sets, the user may want to fuse the RASAR descriptors with the original set of selected molecular features to generate a complete descriptor pool (data fusion). However, from the previous studies, it is observed that this step is essential in the case of q-RASAR [Citation7], for which the reason has already been explained in our previous publication [Citation13], but this step can be skipped in the case of classification-based RASAR (c-RASAR) [Citation6]. The complete descriptor pool should be further subjected to feature selection for the development of an array of statistical and machine-learning models. This feature selection algorithm should ideally be the same that was used to select the molecular features in the QSAR model for a comparable analysis.
A schematic representation of the modeling workflow for q-RASAR modeling and its difference with QSAR has been represented in .
Figure 1. A schematic representation of the modeling workflow for q-RASAR (LSO = leave-same-out).
6. Dos and don’ts during the q-RASAR model development
This section will point out different scenarios with possible errors during the q-RASAR model development with examples found from a few recent publications of different research groups.
q-RASAR is a hybrid modeling approach employing molecular descriptors (as used in QSAR) and similarity descriptors (as used in Read-Across). However, it is not an ensemble prediction approach. Srisongkram [Citation39] worked on predicting the skin cytotoxicity exerted by various compounds using chemometric modeling approaches. They have initially developed two QSAR models and two sets of Read-Across predictions using different descriptors and fingerprints. A final stacking regressor, in the form of a Random Forest, was used to generate predictions. This is an example of ensemble modeling where predictions from different models (here these are QSAR and Read-Across) are used as descriptors for a final stacking regression or a voting classification. It should be noted that this methodology is not a quantitative Read-Across Structure-Activity Relationship (q-RASAR), since this method is nothing but ensembling the predictions of Read-Across and QSAR. On the other hand, for the development of q-RASAR models, the basic pre-requisite is to compute the Read-Across-derived similarity and error-based descriptors that can be used in a modeling framework.
The q-RASAR approach uses the same training and test sets as used in QSAR analysis.Gallagher and Kar [Citation23] developed a q-RASAR model to assess the toxicity of organic chemicals on Labeo rohita. They state that after the data fusion step, the authors have again divided the dataset into training and test sets using a random division approach. Although the authors mentioned that the composition of the training and test set was the same during the RASAR descriptor calculation, this step was unnecessary and error-prone. The composition of the training and test set should remain entirely the same throughout the modeling approach since the RASAR descriptors are computed based on the source compounds. (training set compounds)
The main input files for the RASAR descriptor computation are per where the RASAR descriptors for the test set should be generated taking the training set as the source set, and the test set as the query set, while the RASAR descriptors for the training set should be generated taking the training set as inputs for both the source and query sets. However, in the works of Pandey et al. [Citation25], this fact has been incorrectly represented, as the whole dataset is stated to be used as the query set.
The use of correct training and test set files and the application of the correct feature selection (as used in the case of QSAR) have correctly been shown in the works of Sun et al. [Citation11] and Chen et al. [Citation19].
7. Expert opinion
The concept of q-RASAR is a simple, yet highly conceptual algorithm that computes descriptors based on the properties of the close congeners of a particular query compound and not using the properties of that query compound itself. Therefore, it is highly important that the users need to understand that the training set is always the source set and the query molecules may belong to either the training or the test set, depending upon the input. It is to be remembered that the RASAR descriptor tool always calculates the RASAR descriptors for the query set, taking the training set compounds as the source set from where close source congeners are identified.
In terms of interpretability, it should be noted that the interpretation of the RASAR descriptors is relative. This means, unlike the conventional molecular descriptors where the descriptors describe the properties of a particular compound, the RASAR descriptors describe the properties of the close congeners of a particular query compound.
The RASAR descriptor calculator tool asks for a set of hyperparameters and the choice of similarity algorithm that is used to define the similarity between the source and query compounds. Typically, we recommend proceeding with the default setting of the hyperparameters (σ = 1, γ = 1, number of close source compounds = 10) when the dataset size is comparatively large and one may expect that the training set is sufficiently large enough to find at least 10 similar compounds for a particular query compound. We also recommend that the Gaussian Kernel similarity-based algorithm should be adopted when computing the RASAR descriptors using the default setting. The hyperparameter σ is associated with the Gaussian Kernel similarity algorithm while the hyperparameter γ is associated with the Laplacian Kernel similarity algorithm. The number of close congeners is common to all three similarity algorithms (i.e. Euclidean distance-based similarity, Gaussian Kernel similarity, and Laplacian Kernel similarity). If the modeler decides to optimize the hyperparameters, the training set may be divided into sub-training and sub-test sets, which may be fed into a grid search using the tool Auto_RA_Optimizer-v1.0. Additionally, the modeler may just select the best-performing similarity algorithm from the grid search results and then employ this similarity measure along with the corresponding default setting of the hyperparameters to compute the RASAR descriptors.
Another possibility where things can go wrong is when the user wants to analyze the applicability domain (AD) of the RASAR descriptor space or the fused data space and remove outliers just before the development of q-RASAR/c-RASAR models. While removal of the outliers/outside AD compounds from the test set is perfectly okay, the same is not straightforward for the training set. This is because the training set compounds are used as a source set for the computation of the RASAR descriptors, and the compound that is intended to be removed may be a member of the close source congeners for several query compounds. Therefore, when the user intends to remove such outliers from the training set, it is important to re-calculate the relevant RASAR descriptors using the same setting of the hyperparameters. This revised descriptor pool can now safely be used to develop q-RASAR/c-RASAR models.
Another issue is that the RASAR descriptor calculator tool may not run or may take more time to compute the RASAR descriptors when the size of the training and test sets are very large. Although the user can split the query set into different sections and input one section at a time to compute the RASAR descriptors for the query set, this is not applicable to the training set/source set since this will result in the improper identification of the close congeners as the tool will identify the required number of close source compounds from the limited sample size of the training set only. One should keep in mind that the training set/source set compounds should remain unaltered during the computation of the RASAR descriptors. Therefore, to tackle this issue, the user should allot a higher usable memory to the RASAR descriptor calculator tool. This can easily be done with the help of command prompt. In cases where the system specifications do not allow the allotment of a higher memory, alternative system may be tried.
In cases of classification RASAR (c-RASAR) modeling, after computation of the RASAR descriptors, the modeler should remove SD_Activity, SE and CVact from the modeling analysis. These descriptors are the weighted standard deviation of the activity values of the close source congeners, the weighted standard error of the activity values of the close source congeners and the coefficient of variation of the activity values of the close source congeners. Since these descriptors deal with the observed response values of the close source congeners, they become irrelevant when the response values are graded and not quantitative values.
Additionally, indicator 1 and indicator 2 should not be considered for q-RASAR/c-RASAR modeling analysis since these are just notations for assessing the modelability of the dataset and are related to sm1 and sm2.
To summarize, it is highly essential for the users to completely understand the algorithm and concepts of q-RASAR/c-RASAR and use them carefully and judiciously to eliminate chances of errors. The readers may refer to the compilation on q-RASAR [Citation40] for an elborate understanding of the concepts and may also visit the RASAR webpage (https://sites.google.com/site/kunalroyindia/home/rasar) for the relevant concepts, publications, and videos.
Declaration of interest
The authors are the developers of the q-RASAR approach and the free tools (RASAR Descriptor Calculator and Auto RA Optimizer within a ‘q-RASAR’ project funded by Life Science Research Board, DRDO, India (LSRB/01/15001/M/LSRB-394/SH&DD/2022)). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
One reviewer declares employment with Charles River Laboratories. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Additional information
Funding
References
- Roy K, Kar S, Das RN. Understanding the basics of QSAR for applications in pharmaceutical sciences and risk assessment. Academic Press; 2015 Mar. doi: 10.1016/C2014-0-00286-9
- Novic M. Quantitative structure activity/toxicity relationship through neural networks for drug discovery or regulatory use. Curr Top Med Chem. 2023 Nov;23(29):2792–2804. doi: 10.2174/0115680266251327231017053718
- Gramatica P. Principles of QSAR modeling: comments and suggestions from personal experience. Int J Quant Str-Prop Rel. 2020 Jul;5(3):61–97. doi: 10.4018/IJQSPR.20200701.oa1
- Banerjee A, Roy K. First report of q-RASAR modeling toward an approach of easy interpretability and efficient transferability. Mol Divers. 2022 Jun;26(5):2847–2862. doi: 10.1007/s11030-022-10478-6
- Banerjee A, Roy K. Machine-learning-based similarity meets traditional QSAR: “q-RASAR” for the enhancement of the external predictivity and detection of prediction confidence outliers in an hERG toxicity dataset. Chemom Intell Lab Syst. 2023;237:104829. doi: 10.1016/j.chemolab.2023.104829
- Banerjee A, Roy K. Prediction-inspired intelligent training for the development of classification read-across structure–activity relationship (c-RASAR) models for organic skin sensitizers: assessment of classification error rate from novel similarity coefficients. Chem Res Toxicol. 2023 Aug;36(9):1518–1531. doi: 10.1021/acs.chemrestox.3c00155
- Banerjee A, Roy K. Read-across-based intelligent learning: development of a global q-RASAR model for the efficient quantitative predictions of skin sensitization potential of diverse organic chemicals. Environ Sci Processes Impacts. 2023 Sep;25(10):1626–1644. doi: 10.1039/D3EM00322A
- Pandey SK, Roy K. Development of a read-across-derived classification model for the predictions of mutagenicity data and its comparison with traditional QSAR models and expert systems. Toxicology. 2023;500:153676. doi: 10.1016/j.tox.2023.153676
- Kumar V, Banerjee A, Roy K. Machine learning-based q-RASAR approach for the in silico identification of novel multi-target inhibitors against Alzheimer’s disease. Chemom Intell Lab Syst. 2024 Feb;245:105049. doi: 10.1016/j.chemolab.2023.105049
- Kumar V, Banerjee A, Roy K. Breaking the barriers: machine-learning-based c-RASAR approach for accurate blood–brain barrier permeability prediction. J Chem Inf Model. 2024 May;64(10):4298–4309. doi: 10.1021/acs.jcim.4c00433
- Sun G, Bai P, Fan T, et al. QSAR and chemical read-across analysis of 370 potential MGMT inactivators to identify the structural features influencing inactivation potency. Pharmaceutics. 2023 Aug;15(8):2170. doi: 10.3390/pharmaceutics15082170
- Khatun S, Dasgupta I, Islam R, et al. Unveiling critical structural features for effective HDAC8 inhibition: a comprehensive study using quantitative read-across structure–activity relationship (q-RASAR) and pharmacophore modeling. Mol Divers. 2024. doi: 10.1007/s11030-024-10903-y
- Banerjee A, Roy K. On some novel similarity-based functions used in the ML-based q-RASAR approach for efficient quantitative predictions of selected toxicity end points. Chem Res Toxicol. 2023 Feb;36(3):446–464. doi: 10.1021/acs.chemrestox.2c00374
- Banerjee A, Kar S, Pore S, et al. Efficient predictions of cytotoxicity of TiO2-based multi-component nanoparticles using a machine learning-based q-RASAR approach. Nanotoxicology. 2023 Feb;17(1):78–93. doi: 10.1080/17435390.2023.2186280
- Chatterjee M, Roy K. “Data fusion” quantitative read-across structure-activity-activity relationships (q-RASAARs) for the prediction of toxicities of binary and ternary antibiotic mixtures toward three bacterial species. J Hazard Mater. 2023;459:132129. doi: 10.1016/j.jhazmat.2023.132129
- Chatterjee M, Banerjee A, Tosi S, et al. Machine learning - based q-RASAR modeling to predict acute contact toxicity of binary organic pesticide mixtures in honey bees. J Hazard Mater. 2023;460:132358. doi: 10.1016/j.jhazmat.2023.132358
- Ghosh S, Chatterjee M, Roy K. Quantitative read-across structure-activity relationship (q-RASAR): a new approach methodology to model aquatic toxicity of organic pesticides against different fish species. Aquat Toxicol. 2023;265:106776. doi: 10.1016/j.aquatox.2023.106776
- Ghosh S, Roy K. Quantitative read-across structure-activity relationship (q-RASAR): a novel approach to estimate the subchronic oral safety (NOAEL) of diverse organic chemicals in rats. Toxicol. 2024;505:153824. doi: 10.1016/j.tox.2024.153824
- Chen S, Sun G, Fan T, et al. Ecotoxicological QSAR study of fused/non-fused polycyclic aromatic hydrocarbons (FNFPAHs): assessment and priority ranking of the acute toxicity to Pimephales promelas by QSAR and consensus modeling methods. Sci Tot Environ. 2023 Jun;876:162736. doi: 10.1016/j.scitotenv.2023.162736
- Sobanska AW. In silico assessment of risks associated with pesticides exposure during pregnancy. Chemosphere. 2023;329:138649. doi: 10.1016/j.chemosphere.2023.138649
- Yang L, Tian R, Li Z, et al. Data driven toxicity assessment of organic chemicals against Gammarus species using QSAR approach. Chemosphere. 2023 Jul;328:138433. doi: 10.1016/j.chemosphere.2023.138433
- Yang S, Kar S. First report on chemometric modeling of tilapia fish aquatic toxicity to organic chemicals: toxicity data gap filling. Sci Tot Environ. 2024;907:167991. doi: 10.1016/j.scitotenv.2023.167991
- Gallagher A, Kar S. Unveiling first report on in silico modeling of aquatic toxicity of organic chemicals to Labeo rohita (Rohu) employing QSAR and q-RASAR. Chemosphere. 2024 Feb;349:140810. doi: 10.1016/j.chemosphere.2023.140810
- Ghosh V, Bhattacharjee A, Kumar A, et al. q-RASTR modelling for prediction of diverse toxic chemicals towards T. pyriformis. SAR QSAR Environ Res. 2023;35(1):11–30. doi: 10.1080/1062936X.2023.2298452
- Pandey NK, Murmu A, Banjare P, et al. Integrated predictive QSAR, read across, and q-RASAR analysis for diverse agrochemical phytotoxicity in oat and corn: a consensus-based approach for risk assessment and prioritization. Environ Sci Pollut Res. 2024 Jan;31(8):12371–12386. doi: 10.1007/s11356-024-31872-7
- Khan K, Jillella GK, Gajewicz-Skretna A. Integrated modeling of organic chemicals in tadpole ecotoxicological assessment: exploring Qstr, Q-Rasar, and intelligent consensus prediction techniques. SSRN. 2024. doi: 10.2139/ssrn.4724872
- Banjare P, Singh R, Pandey NK. In silico soil degradation and ecotoxicity analysis of veterinary pharmaceuticals on terrestrial species: first report. Toxicol Res. 2024;13(1):tfae020. doi: 10.1093/toxres/tfae020
- Das S, Samal A, Ojha PK. Chemometrics-driven prediction and prioritization of diverse pesticides on chickens for addressing hazardous effects on public health. J Hazard Mater. 2024;471:134326. doi: 10.1016/j.jhazmat.2024.134326
- Jiang J-R, Cai W-X, Chen Z-F, et al. Prediction of acute toxicity for chlorella vulgaris caused by tire wear particle-derived compounds using quantitative structure-activity relationship models. Water Res. 2024;256:121643. doi: 10.1016/j.watres.2024.121643
- Banerjee A, Gajewicz-Skretna A, Roy K. A machine learning q-RASPR approach for efficient predictions of the specific surface area of perovskites. Mol Inform. 2023;42:2200261. doi: 10.1002/minf.202200261
- Ghosh S, Chatterjee M, Roy K. Predictive quantitative read-across structure–property relationship modeling of the retention time (log tR) of pesticide residues present in foods and vegetables. J Agric Food Chem. 2023;71(24):9538–9548. doi: 10.1021/acs.jafc.3c01438
- Pore S, Banerjee A, Roy K. Machine learning-based q-RASPR modeling of power conversion efficiency of organic dyes in dye-sensitized solar cells. Sust Energy Fuels. 2023 Jun;7(14):3412–3431. doi: 10.1039/D3SE00457K
- Pandey SK, Banerjee A, Roy K. Machine learning-based q-RASPR predictions of detonation heat for nitrogen-containing compounds. Mater Adv. 2023 Oct;4(22):5797–5807. doi: 10.1039/D3MA00535F
- Pore S, Banerjee A, Roy K. Application of machine learning-based read-across structure-property relationship (RASPR) as a new tool for predictive modelling: prediction of power conversion efficiency (PCE) for selected classes of organic dyes in dye-sensitized solar cells (DSSCs). Mol Inform. 2024;43:e202300210. doi: 10.1002/minf.202300210
- Pandey SK, Roy K. Predicting performance and stability parameters of energetic materials (EMs) using the machine learning-based q-RASPR approach. Energy Adv. 2024;3:1293–1306. doi: 10.1039/D4YA00215F
- Kleandrova VV, Cordeiro MNDS, Speck-Planche A. Optimizing drug discovery using multitasking models for quantitative structure–biological effect relationships: an update of the literature. Expert Opi Drug Discov. 2023 Aug;18(11):1231–1243. doi: 10.1080/17460441.2023.2251385
- Kleandrova VV, Cordeiro MNDS, Speck-Planche A. Current in silico methods for multi-target drug discovery in early anticancer research: the rise of the perturbation-theory machine learning approach. Future Med Chem. 2023 Sep;15:1647–1650. doi: 10.4155/fmc-2023-0241
- Kleandrova VV, Speck-Planche A. PTML modeling for pancreatic cancer research: in silico design of simultaneous multi-protein and multi-cell inhibitors. Biomedicines. 2022 Feb;10:491. doi: 10.3390/biomedicines10020491
- Srisongkram T. Ensemble quantitative read-across structure–activity relationship algorithm for predicting skin cytotoxicity. Chem Res Toxicol. 2023 Dec;36(12):1961–1972. doi: 10.1021/acs.chemrestox.3c00238
- Roy K, Banerjee A. q-RASAR: a path to predictive cheminformatics. NY: Springer; 2024. doi: 10.1007/978-3-031-52057-0