
ABSTRACT
Introduction
Variants in the two lens-expressed gap junction genes GJA3 and GJA8 are among the most common causes of inherited pediatric cataract. These two genes alone account for up to 18% of the cases, second only to the crystallin gene family.
Areas covered
All published cataract-associated variants in the GJA3 and GJA8 genes were reviewed for a role in pediatric cataract. Autosomal dominant inheritance was most frequently reported, alongside instances of reduced penetrance and autosomal recessive disease. Variant curation using the ACMG-AMP guidelines identified that many variants do not meet the modern standards for clinical interpretation of pathogenicity. There is broad phenotypic heterogeneity of cataract associated with gap junction gene variants. Pathogenic variants are located throughout both proteins with an enrichment in the N-terminal, first two transmembrane domains, and two extracellular loops.
Expert opinion
Nearly half the gap junction gene variants observed in cataract patients lack sufficient evidence of pathogenicity to form a useful clinical opinion. For many variants, this may be rectified over time as the variant is observed in additional patients but would be vastly accelerated by the generation of well-characterized and standardized functional data evaluating the specific effect of each variant on protein function.
1. Introduction
Pediatric cataract is characterized by the opacity of the crystalline lens of the eye with onset during childhood. It is also referred to as childhood or juvenile cataract, and as congenital cataract when the onset is at birth or in the first year of life. It is one of the most common causes of visual impairments in children, with a global prevalence of 4.24 per 10,000 births [Citation1]. The disorder is extremely heterogeneous, with up to half of cases demonstrating a genetic or inherited etiology [Citation2]. Dozens of genes have been shown to cause pediatric cataract [Citation3] with two of the most common being the lens-expressed gap junction genes GJA3 and GJA8 [Citation2] which encode the gap junction alpha-3 (connexin 46) and gap junction alpha-8 (connexin 50) proteins, respectively.
Gap junction proteins coordinate to form two types of channels. Hemichannels are created by the oligomerization of six connexin subunits and facilitate the molecular exchange between the extracellular fluid and the cytoplasm. The extracellular docking of two hemichannels results in the formation of gap junction channels that facilitate the movement of molecules between the cytoplasm of adjacent cells. Due to the avascular nature of the lens, gap junctions and hemichannels are critical structures in the maintenance of lens homeostasis, allowing the transport of ions and small molecules between lens fiber cells and assisting in the internal circulation of the lens [Citation4–6]. There are several mechanisms by which disruption of normal gap junctions and hemichannel function in the lens leads to cataract formation, but ultimately, most disease-causing variants result in impaired lens circulation. Specific mechanisms include impaired protein trafficking, reduced gap junction plaque formation or docking of hemichannels, impaired hemichannel gating, as well as lens Ca2+ homeostasis and biomineralization. These mechanisms and their relationship to specific genetic variants are reviewed elsewhere [Citation6–10].
The gap junction genes GJA3 and GJA8 were some of the first genes linked to pediatric and congenital cataracts. The GJA8 p.(Pro88Ser) variant was first reported by Shiels et al. in 1998 [Citation11], and a year later two families were reported by Mackay et al. [Citation12] to have the c.1137dup p.(Ser380Glnfs*88) and p.(Asn63Ser) changes in the GJA3 gene A recent review by Shiels and Hejtmancik [Citation13] analyzed data from the Cat-Map database [Citation3] and reported that 18% of the families with pediatric cataract have gap junction variants. This makes the gap junction gene family (with two genes relevant to cataract) second only to the crystallin gene family (consisting of 13 crystallin genes expressed in the lens), which accounts for 33% of the inherited pediatric cataracts. To assess this from a different perspective, we reviewed the proportion of GJA3 and GJA8 variants reported in studies assessing panels of known cataract causing genes in cohorts of patients with pediatric cataracts. We identified 19 such studies that assessed at least 14 cataract-associated genes and at least 18 families or cases, published between 2009 and 2022[Citation14–33]. On average 15% of the variants identified were in the GJA3 and GJA8 genes, comparable to the 18% reported in the Cat-Map database [Citation13]. However, there was a broad range with no observations of gap junction gene variants in three studies [Citation22,Citation27,Citation33] with up to 35–38% of the patients in other studies [Citation15,Citation18,Citation20]. This variability may be due to population differences or stochastic effects of small cohorts.
In this review, we collated all published cataract-associated variants within the GJA3 and GJA8 genes and reviewed the evidence for a role in pediatric cataract. We reviewed all papers referenced in PubMed, Web of Science and Embase (via Ovid) from the search string ‘GJA3’ or ‘GJA8’ and PubMed from the search string ‘genetics AND congenital cataract’ for information relating to genetic variants in either gene found in people with a diagnosis of pediatric cataract. We also included variants documented in the Cat-Map database for both genes [Citation3] and from any other publication identified through cross-citation in text. All articles were required to be accessible and in English, or could be translated to English, for review. One variant was excluded following discordance between the reported coding and amino acid changes (c.905T>C and p.Leu281Cys) [Citation34] as the intended disease-associated variant was unable to be resolved. Frameshift and insertion/deletion variants were assessed for correct nomenclature to ensure they conformed to the guidelines of the Human Genome Variation Society, with any changes outlined in . Protein topology and domains were obtained from UniProt for both GJA3 (reference number: Q9Y6H8) and GJA8 (reference number: P48165).
Table 1. GJA3 coding variants associated with childhood onset cataracts reported in the literature.
Table 2. GJA8 coding variants associated with childhood onset cataracts reported in the literature.
Since the first reports of cataract causing GJA3 and GJA8 variants in the late 1990s, we have identified 210 literature reported findings across 143 unique variants ( and ). Here, we review the evidence for a role for each variant in pediatric cataract and summarize the findings across both gap junction genes.
2. Inheritance patterns of GJA3 and GJA8 associated pediatric cataract
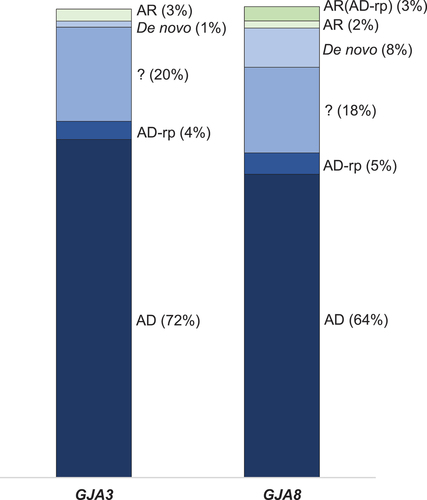
Autosomal dominant (AD) inheritance (a single copy of a variant is sufficient to cause disease) was the most commonly reported inheritance pattern for gap junction gene variants associated with pediatric cataract (). Other reported inheritance patterns that can be broadly included in the AD category were as follows: De novo inheritance where a variant arises in an affected individual and is not inherited from a parent, singletons without available family information carrying a single copy of the variant (AD inheritance is the most likely explanation), and reduced or incomplete penetrance (AD-rp) where the family displays AD inheritance but one or more variant carriers do not express the pediatric cataract phenotype. When considering this broader definition of AD inheritance, 97% of the GJA3 and 95% of the GJA8 variants are linked to cataract inherited in an AD manner. Autosomal recessive (AR) inheritance, when only homozygous variant carriers develop disease, is also reported in both genes (GJA3 3% and GJA8 2%) (). More complex inheritance patterns are also reported in several families with GJA8 variants, including three families containing both homozygous and heterozygous variant carriers with a cataract phenotype [Citation23,Citation107,Citation108] and a family with compound heterozygous variants that includes a heterozygous variant carrier with a cataract phenotype [Citation109] (indicated as AR(AD-rp) in ).
Figure 1. Reported inheritance patterns in families with variants in the GJA3 and GJA8 genes.
2.1. Autosomal dominant variants in the gap junction genes
In the GJA3 gene, 76 of the 78 published papers (97%) reported broad AD inheritance. This was further sub-categorized into classical AD (56 reports, 72%), heterozygous singleton (16 reports, 20%), de novo (1 report, 1%), or reduced penetrance (3 reports, 4%) (). A similar pattern was observed in the GJA8 gene. Of the 132 variant reports, 126 (95%) were broadly AD, with 85 (64%) variants displaying full autosomal dominant inheritance, 24 (18%) heterozygous singletons, 11 (8%) de novo, and six reports (5%) with reduced penetrance.
Across both genes, missense variants were most commonly responsible for AD pediatric cataracts. When assessed at the variant level, this was 45 (85%) of the 53 AD associated GJA3 variants while for GJA8 77 of the 81 (95%) broadly AD variants were missense (). In-frame insertion-deletion variants were only observed with AD inheritance in both genes. There were four reports of this variant type in GJA8: p.(Gly8_Leu11del) [Citation110], p.(His95_Ala96insYAVHY) [Citation111], p.(Lys131del) [Citation20], and p.(Glu144_Leu148del) [Citation112,Citation113]. All of these variants are located in the C-terminal cytoplasmic tail or central cytoplasmic loop of the GJA8 protein. In GJA3, a single in-frame insertion p.(Tyr272_Ala273insAsp) [Citation29] is located in the C-terminal cytoplasmic tail. The GJA3 gene contained seven frame-shift variants, all of which are in the N-terminal cytoplasmic tail. Six of these seven frame-shift variants were reported to have AD inheritance [Citation12,Citation114–119] with the most proximal of these being the p.(Ser256Glnfs*68) [Citation114] and the most distal the p.(Thr400Hisfs*68) [Citation119] change.
2.2. Gap junction variants with reduced penetrance
Variants in both the GJA3 and GJA8 genes have been reported with reduced, or incomplete, penetrance in families with pediatric cataract. The most striking of these is the c.227 G>A variant that results in a p.(Arg76His) change in the GJA3 protein. First reported in a large Australian family, the variant was identified following linkage analysis across known cataract genetic loci, at the time, and direct sequencing of the GJA3 gene [Citation120]. The variant segregated in all 21 affected individuals but was also identified in six unaffected relatives. Subsequently, a second report of this variant was observed in a multigenerational Danish family, where it segregated in all seven affected and one unaffected individual [Citation16]. The only other AD-rp variant reported in GJA3 was c.82G>A p.(Val28Met) observed in five affected and two unaffected individuals in an Indian family [Citation121], and a different variant at the same residue p.(Val28Leu) reported in a singleton [Citation114]. Interestingly, with the p.(Val28Met) variant, variable age of onset was observed in the family with three of the five affected individuals receiving a cataract diagnosis after 20 years of age and the two unaffected variant carriers in the youngest generation of the family.
In GJA8, c.73T>C p.(Trp25Arg) is reported with reduced penetrance in a small Australian family, where two affected and one unaffected individual carry the variant [Citation18]. In addition, there was a single unaffected variant carrier reported in a large Chinese family (31 affected individuals) with c.139G>A p.(Asp47Asn) [Citation122]. This p.(Asp47Asn) has been reported five additional times, all with standard AD inheritance, and has been functionally shown to impair the gap junction function by two independent groups [Citation123,Citation124]. The remaining four AD-rp variants () were identified in a single affected child and their unaffected parent. The clinical relevance of these variants is less clear.
2.3. Autosomal recessive variants in GJA3
In the GJA3 gene, there were two reports (3%) of pediatric cataract with AR inheritance (, ). A segregating c.950dup p.(His318Profs*8) variant was reported to segregate in a Pakistani family, identified via genome-wide homozygosity mapping [Citation125]. The three affected children had nuclear cataract phenotypes and secondary glaucoma following cataract surgery. More recently, in an African-American family trio with consanguineous history, a single affected female with nuclear and cortical cataracts was identified to be homozygous for a c.563A>G p.(Asn188Ser) missense change inherited from heterozygous parents [Citation126]. Located in the second extracellular loop of the GJA3 protein, other amino acid substitutions at this residue have been reported to cause autosomal dominant cataracts (, p.(Asn188Thr) and p.(Asn188Ile)). Functional assessment of this residue has identified impaired formation and dye transfer through mutant gap junctions with both the cataract-associated p.(Asn188Thr) and p.(Asn188Gln) (which has not yet been linked to disease) [Citation127], indicating the residue plays a functional role in connexon docking. The exact mechanism behind the p.(Asn188Ser) change causing autosomal recessive disease, rather than acting in a dominant negative manner causing autosomal dominant inheritance similar to other variants at this residue, requires further investigation. These variants provide some important insights into our ability to make assumptions about different amino acid changes at sites that have been previously reported to cause disease. We cannot assume that every residue change at a functionally important site will have the same functional or pathogenic outcome.
In the GJA3 gene, there was no clear link between variant type and inheritance pattern with one missense and one frameshift variant reported with AR inheritance to date. In total, seven frameshift variants are reported, all within the C-terminal tail region, which result in a nonsense sequence prior to a premature stop codon. The AR reported frameshift variant c.950dup p.(His318Profs*8) has the shortest nonsense sequence prior to the new stop codon [Citation125]. Some functional work has been performed to investigate the autosomal dominant c.1137dup p.(Ser380Argfs*88) change, which shows normal protein trafficking and plaque formation for a truncated p.Ser380Ter protein compared to protein with the additional nonsense sequence [Citation128]. Further work in that study identified some novel motifs in the nonsense sequence likely misdirected protein trafficking to other cellular regions [Citation128]; however, this may not be the case in all of these frameshift variants, and further study into all of them would be needed to fully elucidate the underlying disease-causing mechanisms.
2.4. Autosomal recessive variants in GJA8
In the GJA8 gene, there are five reports of families with affected homozygous variant carriers. However, the diagnosis of pediatric cataract in these families is not always restricted to those individuals. Only two of the five were counted here as fully AR (2%, , ). The first of these is an individual homozygous for a c.1273C>T stop-gain variant p.(Arg425*), but this report contains very little information regarding parental genotype and phenotypes and should be interpreted with caution [Citation15]. The second was an autosomal recessive c.607dup p.(Thr203Asnfs*47) change reported in two affected children in an Indian family, with heterozygous parents and relatives all presenting as unaffected [Citation129]. This variant resulted in the loss of the second extracellular domain through to the C-terminal protein tail, with both this variant and a truncated p.Thr203Ter protein shown to result in disrupted localization to the plasma membrane and retention in the endoplasmic reticulum [Citation130].
Three other variants have both homozygous and heterozygous variant carriers with cataract phenotypes in the same family, with varying levels of severity. In a small family reported by Ma et al. [Citation23], c.89dup p.(Ile31Hisfs*18) was observed in a daughter (homozygous) from consanguineous parents where only the affected mother (heterozygous) was available for assessment. In a Turkish family, two children with consanguineous parents were homozygous for a c.766dup p.(Ala256Glyfs*124) change, with two of the four heterozygous variant carriers also displaying a subtle nuclear cataract [Citation108]. Additional functional assessment of this frameshift variant demonstrated a reduced capacity to form gap junction plaques and transfer dyes to neighboring cells in comparison to both wildtype and a truncated p.(Ala256Ter) protein [Citation131]. The authors postulate that the new nonsense sequence created in the C-terminal tail region causes faster protein degradation, which was partially rescued following treatment with a proteasomal inhibitor [Citation131]. Finally, the only missense variant reported with homozygous individuals in GJA8, is p.(Val196Met) reported in a young female with a total cataract at 20 months from an Indian family with consanguineous parents [Citation107]. Five individuals in that family were heterozygous for the p.(Val196Met) change and all were reported to have healthy lenses, except for the mother who had non-vision impairing dot-like opacities diagnosed at 25 years of age. Dedicated in silico investigations indicate that this p.(Val196Met) change, in the second extracellular loop, does not alter the surface electrostatic potential or any of the other structural parameters that were assessed [Citation132], however detailed functional assessment of this variant has not yet been performed to validate those findings. A family, with compound heterozygous variants, has also been reported with a phenotype that mimics AR inheritance [Citation109]. Two affected siblings with congenital cataract both inherited GJA8 c.855del and c.1125del frameshift inducing variants in the C-terminal cytoplasmic tail. Mild nuclear opacities were identified in the father, heterozygous for c.1125del variant, whilst the mother was asymptomatic [Citation109] indicating a degree of reduced penetrance with these variants.
In comparison to the GJA3 gene, in GJA8 all the stop-gain and frameshift variants inducing an early stop codon are found in homozygous or compound heterozygous variant carriers. However, there is variability in the phenotypes between heterozygous and homozygous variant carriers and with only a small number of variants to assess, we cannot yet draw any firm associations between frameshift variants and AR inheritance in GJA8.
3. Phenotypes associated with GJA3 and GJA8 variants
No striking genotype–phenotype correlations were observed for either gene, with a variety of phenotypes spanning the full length of both genes, reinforcing the phenotypic heterogeneity that is well associated with pediatric cataracts [Citation3]. A description of the associated cataract phenotype was given in 76% of GJA3 and 69% of GJA8 variant reports. Studies not reporting a cataract phenotype typically either stated that a patient had ‘bilateral congenital cataracts’ or were drawing from historical patient records in which the data were unavailable. Nuclear cataract or nuclear component was the most commonly reported phenotype in both genes with 46% and 42% of GJA3 and GJA8 patients, respectively. The second most common description was lamellar or zonular with 17% for GJA3 and 9% for GJA8. A pulverulent, punctate, or dust-like appearance was reported for 31% of the GJA3 and 11% of GJA8 patients. The phenotypic heterogeneity extended to both missense and null (nonsense and frameshift) variants.
Other ocular and systemic phenotypes were reported for some variants. Only six GJA3 patients (8%) had other phenotypic information reported, including nystagmus, microphthalmia, microcornea, high myopia, secondary glaucoma, and one case of macrocephaly [Citation15]. A patient with a GJA3 p.(Leu97Arg) variant was reported to have congenital cataract and dominant optic atrophy, with the optic atrophy more likely caused by a OPA1 splicing variant [Citation133]. Comparatively, in GJA8 31% (41 reports) of cases had additional phenotypic information reported beyond the cataract phenotype. Most commonly this was microphthalmia and nystagmus, followed by microcornea, secondary glaucoma, coloboma, and strabismus. The GJA8 p.(Asp51Asn) residue is noteworthy, with all three reported cases having microphthalmia and either sclerocornea [Citation23] or a combination of anterior segment dysgenesis, persistent pupillary membrane, deep set eyes, corneal leukoma, buphthalmos, corectopia, and nystagmus [Citation134]. The p.(Gly94Arg) variant has been reported in two cases to cause no or rudimentary lenses alongside sclerocornea, coloboma, microphthalmia, glaucoma, and buphthalmos [Citation134–136]. Similarly, the p.(Gly94Glu) variant has been reported in two cases with cataract or disc-like lenses with additional sclerocornea, sunken eyeballs, and narrow palpebral fissures [Citation135–137]. Finally, a nearby p.(Val97Gly) variant is reported in a patient with microphthalmia, anterior segment dysgenesis, and secondary glaucoma [Citation134] while no phenotype information is reported for other individuals with this variant [Citation17].
4. GJA3 and GJA8 variants in the ClinVar public repository
ClinVar is a public archive of human genetic variants with an accompanying interpretation of their association to a disease or phenotype [Citation138]. Submissions can be made from both clinical and research facilities following registration with the site. ClinVar was officially launched in 2013, and there has been increased reporting to this resource from both the research and clinical sectors following the release and uptake of the ACMG-AMP standards and guidelines for the interpretation of sequence variants [Citation139] by the American College of Medical Genetics and Association for Molecular Pathology (ACMG-AMP). The ACMG-AMP guidelines provide a series of criteria against which to assess a variant detected in a known disease-causing gene in a patient with the appropriate phenotype. Each criterion is assigned a strength level dependent on its capacity to distinguish benign from pathogenic evidence. The combination of criteria applied, along with the strength level, results in a classification of benign (B), likely benign (LB), variant of uncertain significance (VUS), likely pathogenic (LP), or pathogenic (P). A diagnostic benefit from genetic testing is achieved for patients when collective evidence enables a classification of LP or P. When variants are classified as VUS, they leave doubts over causality and can impede patients receiving a timely molecular diagnosis, if at all. There are clearly recognized benefits of a definitive molecular diagnosis including the ability to access accurate genetic counseling and recurrence risk prediction as well as determining if the cataract is isolated or part of a more complex syndrome [Citation140]. Similarly, obtaining sufficient evidence to classify variants as LB or B means variants can be discharged as candidates and focus can be directed to alternate genetic causes.
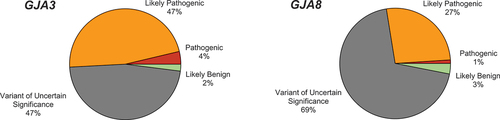
As of December 2022, there were 85 coding variants in GJA3 and 109 coding variants in GJA8 in ClinVar. For both genes, nearly half of the variants in ClinVar were classed as Variants of Uncertain Significance (VUS) with 47% and 50% of the variants in GJA3 and GJA8, respectively (). In both genes, 28% of coding variants were classified as either P or LP and 20–21% of the variants were classified as B or LB. Both genes also had a small set of variants with conflicting interpretations of pathogenicity, with different submitters coming to different conclusions. The classification reported in ClinVar is determined by the submitting department. The evidence used by the submitter to determine the classification is requested by ClinVar on submission but is not always included in detail. The quality and quantity of included evidence has improved in recent years, with justifications, ACMG-AMP criteria applied, and references to publications used as evidence more frequently included in the records. In ClinVar, a star system is used to guide the confidence behind the submissions, with variants that have undergone curation by an expert panel given the highest star rating. Neither the GJA3 nor the GJA8 gene has been officially curated by an expert panel.
Figure 2. Pathogenicity of ClinVar reported GJA3 and GJA8 coding variants.

We compared the literature reported variants collated through this review and assessed if they contained an entry in the ClinVar repository. Of the 55 GJA3 variants in , only 25 were present in ClinVar (45%). Similarly, for GJA8, only 35 of the 88 GJA8 variants in are recorded in ClinVar. Conversely, ClinVar contains an additional 60 GJA3 and 74 GJA8 unpublished coding variants, primarily reported from clinical settings (such as genetic testing laboratories) which have not been considered further in this review.
5. Reclassification of literature reported GJA3 and GJA8 variants
Over the last two and a half decades, the way variants are classified as disease causing or ‘pathogenic’ has adapted alongside the current availability of information and resources. Most notably, the publication of the ACMG-AMP standards and guidelines for the interpretation of sequence variants [Citation139] has had the biggest impact on the classification of disease-associated genetic variants. Whilst researchers reported variants as disease-causing with the best available information at the time and best intent for identifying the cause of disease for their patients and families, the evidence they used may no longer be sufficient to meet the standards required to claim a variant as definitively disease-causing.
Of the 210 variant reports identified by this review, 88 (42%) were published prior to the end of 2015 and would not have had the opportunity to utilize these guidelines. For the remaining 122 variant reports, there has been variable uptake of these guidelines for their classification by researchers, as well as numerous updated recommendations for their implementation over recent years. We examined the 143 unique variants, identified across the two genes, and conservatively reclassified them using the ACMG-AMP guidelines [Citation139] and the current recommendations for the use of each criterion (outlined in ) including the points-based system. In this modification to the original rules, points are assigned in the positive direction for pathogenic criteria and the negative direction for benign criteria and the sum of all points determines the classification [Citation141]. During this process, decisions were made on how best to apply each rule to these genes. Of particular note, we found that applying PVS1 for nonsense or frameshift variants following the detailed guidance provided [Citation142] was challenging given that it is not known if nonsense mediated decay is activated in these single exon genes, which is a key piece of knowledge required to follow these guidelines. Similarly, the application of criterion PM1 (variants in a mutational hotspot or critical well-established functional domain) required defining such domains. This was hampered by a lack of strong functional evidence evaluating the role of various domains in gap junction or hemichannel function. Ultimately, it was applied using data regarding the location of known disease-causing variants collated and assessed by Bai et al. [Citation143], which may result in circular arguments regarding the classification of variants in these regions that were used to define these regions. This is a limitation of the current approach and should be addressed for more accurate classifications in the future.
Table 3. ACMG-AMP classification criteria specifications.
5.1. Most literature reported variants classify as likely pathogenic or variants of uncertain significance under the ACMG-AMG guidelines
In GJA3, just over half of variants (51%) were classified as either P (4% n = 2) or LP (47% n = 26) (). Most of the remaining variants were VUS (47% n = 26), and one variant was classified as LB (2%). In contrast to this, only 27% of GJA8 variants were classified as either P (1% n = 1) or LP (27% n = 23) (). The majority of GJA8 variants classified as VUS (69% n = 61) and the remaining three (3%) were LB. None of the variants were classified as benign in either of the two genes.
Figure 3. ACMG-AMP classifications of literature reported GJA3 and GJA8 variants.
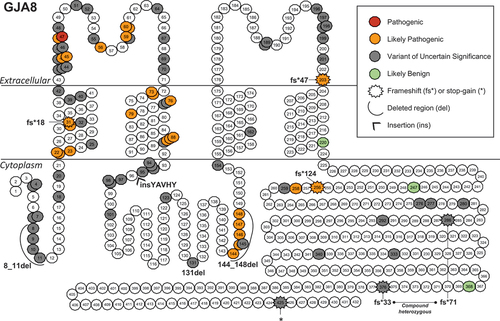
The literature reported variants were mapped onto schematics of the GJA3 (Gap junction alpha-3) and GJA8 (Gap junction alpha-8) proteins to visualize any trends in the location of pathogenic or benign variants (). Both proteins have a similar structure, consisting of a cytoplasmic N-terminal tail, four transmembrane domains connected by extracellular and cytoplasmic loops, and a cytoplasmic C-terminal tail. When the six subunits assemble into the connexon hemichannel, the transmembrane domains form a pore through which small molecules are transported across the membrane. In GJA3, the missense cataract-associated variants tend to impact the N-terminal cytoplasmic region, the first two transmembrane domains, and the two extracellular loops (). All seven frameshift variants were in the C-terminal tail region of GJA3, with only one other variant p.(Tyr272_273insAsp) in that region. In GJA8, a similar trend in the location of missense variants was observed, although with a higher proportion of them classifying as VUS. The three frameshift variants were distributed across the protein, a pair of frameshift variants with compound heterozygous inheritance and a single stop-gain variant were located in the C-terminal cytoplasmic tail. There was a higher number of insertion-deletion variants in GJA8 and all were classed as VUS with the exception of p.(Glu144_Leu148del), which was LP. The LB variants in GJA8 were all located in the C-terminal cytoplasmic tail or the final transmembrane domain and the sole GJA3 LB variant was a synonymous change p.(Val28=) at a residue located deep in the first transmembrane domain (not displayed in ).
Figure 4. Position of literature reported GJA3 variants.
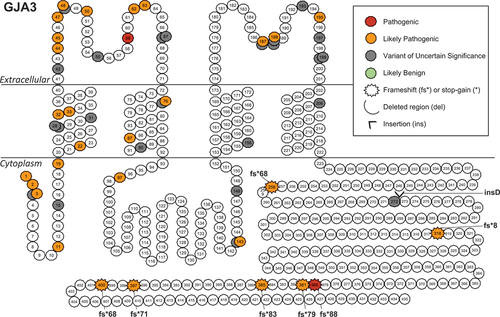
Figure 5. Position of literature reported GJA8 variants.
5.2. The importance of multiple observations and co-segregation evidence in variant classification
The use of co-segregation evidence (PP1) and proband counting (PS4) are valuable criteria when using the ACMG-AMP guidelines. Multiple observations of variants in independent cases can be a major contributor to moving variants from VUS to LP. When assessing the variants presented in and , 24% of GJA3 and 30% of GJA8 variants had more than one independent report. All variants with at least two reports resulted in the use of proband counting criterion PS4, albeit only at a supporting level for most variants. Across both genes, 74 variants qualified for PP1 (30 in GJA3 and 44 in GJA8). This criterion was applied at a moderate level 21 times and at a strong level 30 times. Multiple reports of a variant allowed application of PP1 at moderate or strong level on six occasions in GJA3 and 11 in GJA8. In GJA3, six variants moved from a VUS to LP classification and one from LP to P through PP1 and/or PS4 application due to multiple reports of the variant. Similarly, in GJA8, seven variants moved from the classification of VUS to LP, the p.(Asp47Asn) variant transitioned from LP to P with the use of PS4 at a moderate level.
Pediatric cataract is a rare disease with high genetic heterogeneity. As such, reports of the same variant in second or third unrelated individuals may never occur. However, with the increased uptake of clinical genetic testing for ocular diseases, there is an expected increase in the likelihood of observing variants in additional probands or families. Improved access to variant classification results from genetic testing using large-scale data sharing in appropriate repositories will provide enhanced capacity to build evidence toward LP classifications in both clinical and research-based variant assessments using PS4 and PP1.
5.3. The GJA3 and GJA8 genes contain likely benign genetic variation observed in cataract patients
Both genes had at least one literature reported cataract-associated variant that we classified as LB after considering the ACMG-AMP guidelines in detail. The LB classification was predominantly due to the population minor allele frequency being greater than that expected for the disorder (rule BS1, ). In GJA3, this was the only synonymous variant included in this review, c.84G>A p.(Val28=) [Citation107] while in GJA8, there were three LB variants p.(Asn220Asp) [Citation134,Citation151], p.(Ile247Met)[Citation152–154], and p.(Glu368Gln) [Citation155,Citation156]. In the GJA8 gene, there were four variants classified as VUS due to the application of both pathogenic and benign rules, leading to an aggregate score of 0. These are a p.(Leu7Met) [Citation134], p.(Gly123Ser) [Citation26], p.(Leu292Gln) [Citation134], and p.(Gly333Arg) [Citation134]. These variants appear unlikely to contribute to disease and may be reclassified as benign in the coming years with additional evidence.
Two variants were observed in individuals harboring another reported genetic variant, which was more likely to be responsible for the phenotypes in those individuals. The first of these was in GJA8, p.(Ile247Met) which was classified as LB based on the population frequency. This variant was observed in a proband from a large family with a segregating likely disease-causing variant in the CRYBB1 gene [Citation152]. Secondly, the GJA8 p.(Leu292Gln) variant was observed alongside a heterozygous PAX2 p.(Ala177Glyfs*8) variant, which better matched the reported bilateral mild cataracts, optic nerve coloboma, nystagmus, photophobia, and small kidney phenotypes of the affected individual [Citation134]. Although the p.(Ile247Met) variant classifies as LB, it is unknown if the VUS p.(Leu292Gln) variant contributes to the phenotype observed in the second family. The reporting of benign variations observed in individuals with ocular phenotypes, in the literature and in appropriate databases, further assists with their interpretation over time and our understanding of functional regions in the proteins and their tolerance to change.
5.4. The role of functional assays in providing evidence of pathogenicity
A wide array of functional assays have been used to study gap junction proteins and the impact of specific variants on protein function. The ACMG-AMP guidelines allow for the inclusion of functional data in variant interpretation through criterion PS3 in the pathogenic direction when a clear functional effect is detected and BS3 in the benign direction when it is not. The original guidelines do not provide detailed guidance on applying these criteria; however, subsequent recommendations provide strict guidelines for assessing the quality of functional evidence and applying it at an appropriate level of strength when classifying variants [Citation157]. To include PS3/BS3 evidence, a functional assay should contain technical/biological replicates and both positive and negative controls, such as wildtype and empty vectors, respectively. The level of strength that can be applied is determined by the calculated odds of pathogenicity (OddsPath) with supporting, moderate, and strong evidence applied for OddsPath of >2.1, >4.3, or >18.7, respectively [Citation157]. OddsPath calculations are determined by the number of clinical validation controls (known pathogenic and benign variants) included in the assay or with known results from that assay, with those variants required to have achieved a classification of LP/P (or LB/B) without the use of PS3/BS3 criteria. Despite the availability of functional data for many variants, none of the reports satisfy these recommendations due to the absence of clinical validation controls. These controls perform a critical role in establishing the range of the assay outputs between functionally normal and functionally abnormal, which is needed for the confident interpretation of any functionally unknown ‘candidate’ variants and enables reproducibility of the data. The diversity of assays used to assess GJA3 and GJA8 variants to date means there are insufficient variants that have been studied using the same experimental protocols to establish baseline clinical control variants for any of the assays.
In this curation of variants, PS3 was used when animal models displayed a comparable phenotype from the same protein change as observed in the reporting family. Greater confidence accompanies the observation of cataract in an animal model as we can infer that the protein change sufficiently alters cellular biology to cause a comparable state of disease in the relevant tissue. In the GJA3 gene, the mouse model of the p.(Ser380Glnfs*88) variant [Citation158–160] provided evidence to move the variant from LP to P. In GJA8, the mouse model of the p.(Gly22Arg) variant [Citation161,Citation162] promoted that variant [Citation112] from VUS to LP and subsequently moved the p.(Gly22Ser) variant [Citation20,Citation29,Citation163] at the same location from VUS to LP through the use of the PM5 criterion, which is applied when another variant at that location has been classified as LP or P. Both variants demonstrate the impact of additional functional evidence on variant classification.
Several in vitro assays for the assessment of gap junction variants have been developed. The Xenopus model has been used to assess both hemichannel and gap junction functions; however, this assay requires dedicated setup and expertise and is not used by many laboratories. In this model, a wildtype or variant GJA3 or GJA8 complementary RNA (cRNA) is introduced into xenopus oocytes where it is translated into a protein which localizes to the plasma membrane. To assess gap junction functionality electrophysiological measurements are taken between paired cRNA injected oocytes to determine the degree of gap junction conductance. Hemichannel function is assessed in unpaired oocytes cultured under extracellular calcium concentrations that activate the hemichannels. These assays have been used to assess variants in both genes with examples such as GJA3 variants p.(Asp3Tyr), p.(Leu11Ser), p.(Thr19Met), p.(Asn63Ser), and p.(Ser380Glnfs*88) [Citation164–166] and GJA8 variants p.(Trp45Ser), p.(Gly46Val), and p.(Glu48Lys) [Citation167,Citation168]. All these GJA3 variants have been classified as LP or P without the use of PS3 and thus would count as clinical controls for this assay; however, additional assessment of known benign variants is required before this assay could be used to apply PS3 to novel variants.
In vitro assays using mammalian cell culture models are also used to assess hemichannel and gap junction functions. These assays primarily work by expressing wildtype or variant gap junction proteins in the cell type of choice and assessing the capacity of fluorescent dyes to move through these structures. Gap junction assessment typically requires a cell line stably expressing a transgene at high confluence. A gap junction permeable dye (such as neurobiotin) is injected directly into a cell using a microinjection apparatus, and the dye transfer to neighboring cells is measured [Citation131,Citation169,Citation170]. Like Xenopus oocyte assays, this technique requires a degree of skill and specialized equipment to facilitate. Alternatively, a scrape-load dye transfer assay uses a cut (scrape) across the confluent cell layer initiating dye uptake of both the gap junction permeable and impermeable dyes by damaged cells. The transfer of the gap junction permeable dye to neighboring (undamaged) cells is measured [Citation171–173]. Hemichannel assays assess the hemichannel permeable dye uptake (such as propidium iodide) from the surrounding environment, upon activation of the hemichannel with H2O2 [Citation173] or removal of extracellular calcium [Citation174–176]. Electrophysiological measurements of gap junction conductance can also be applied in the mammalian cell culture models [Citation124]. Protein localization or the appearance of gap junction plaques, via fluorescently tagged protein or antibody labeling, is commonly performed alongside most cell culture studies; however, interpretation of these findings is subjective. Finally, cell viability assays using standardized kits (such as MTT tetrazolium reduction assay) have been used to take a measure of cell survival in wildtype and variant transfected cells [Citation177,Citation178].
The classification information in show that the use of properly validated functional evidence (if positive) would influence the classification of many variants. There are a total of 41 variants across the two genes (14 in GJA3 and 29 in GJA8) that have scored 4 or 5 points, classifying them as VUS under the scheme used. Additional 1 or 2 points gained from the application of PS3 at the supporting (1 point) or moderate (2 point) level would re-classify these variants as LP, providing clear diagnostic benefit to variant carriers. To achieve this, standardized assays validated using appropriate clinical control variants must be developed and applied in a systematic way to all VUS and novel variants. The xenopus oocyte assay is the closest -with multiple LP or P variants already assessed but lacks benign controls. However, mammalian cell culture-based assays are more readily adaptable for moderate throughput use in most laboratories and may be better placed to rapidly advance the field.
6. Conclusion
Although inherited pediatric cataract is both genetically and phenotypically heterogeneous [Citation3] the lens expressed gap junction genes GJA3 and GJA8 are important causes of this disease. Both genes contain missense, nonsense, and frameshift variants that are clearly pathogenic in the context of cataract. There is a clustering of missense variants in the N-terminal tail and first two transmembrane domains of both proteins, but pathogenic variants are found throughout. Both autosomal dominant and recessive transmissions are reported, and reduced penetrance of dominant variants is seen in some families. Many of the disease-causing variants reported in the literature are not contained in the commonly used public repository ClinVar. The commonly used ACMG-AMP guidelines for the interpretation of clinically observed variants can be readily applied to gap junction variants in the context of pediatric cataract, and these strict criteria indicate that the evidence for pathogenicity for many literature reported variants do not meet modern standards for clinical interpretation of pathogenicity. It is very likely that the majority of reported variants do contribute to disease, but additional evidence is required to meet formal clinical interpretation guidelines used in a diagnostic setting.
7. Expert opinion
Variants in the two lens-expressed gap junction genes GJA3 and GJA8 are among the most common causes of inherited pediatric cataract. The crystallin gene family, which encodes structural proteins largely responsible for the high refractive index of the lens, is implicated more often [Citation2,Citation13], but consists of some 13 genes. The two gap junction genes are clearly important in lens biology. We are beginning to accurately determine the pathogenic nature of certain variants observed in cataract patients, but many variants within these genes need further investigation. Pediatric cataract is a rare disease, which means that variants that are commonly observed in the population can be ruled out as causative. However, with the advent of large-scale sequencing studies, it is evident that most genetic variation in humans is very rare [Citation179]. Therefore, merely the observation of a rare variant in one of these genes in a child with cataract is insufficient evidence to claim it as the cause of their disease. This is particularly true in a disease as heterogeneous as pediatric cataract when any one of the 50 other genes could be the cause. Historically, researchers have relied on evidence of the variant segregating with the cataract phenotype in a family to assign disease-causing status; however, in many cases, the variant was observed in small families with only one or two meioses, and the segregation was very likely to occur by chance. Only with large families or multiple families harboring the same variant can we accumulate sufficient robust evidence to assign pathogenicity.
The introduction of the ACMG-AMP guidelines has revolutionized the interpretation of variants in a clinical setting. The intention of these guidelines was that they would be able to be applied consistently by different laboratories such that patients with the same variant would receive the same genetic test result. However, it soon became evident that the guidelines were open to interpretation themselves and have been applied inconsistently. This has been recognized by ClinGen, the body that oversees the ClinVar database, which has been working toward providing clear and detailed evidence-based guidance for each criterion, as well as establishing disease- and gene-specific expert panels to apply the criteria consistently to all variants within a given disease causing gene. No such panel has yet been established for cataract or gap junction genes, although this would be of benefit to the field. The utility of these guidelines for assessing the gap junction genes would be improved with more specific knowledge regarding the activation of nonsense mediated decay for the application of criterion PVS1 (for nonsense and frameshift variants) and specific functional evidence to define functional domains and mutational hotspots in both proteins for the application of PM1. Although many of the literature-based variant findings were published prior to ClinVar’s inception, many more recently reported variants are still not included in the database. By overlooking this repository as a means to share information, the genetics community is missing an opportunity to have a single point of contact for all disease-associated variants. A comprehensive resource would enable improved and consistent interpretation of gap junction variants moving forward, as well as a way to record and update new sources of evidence for each variant that alters its classification.
It is evident that variants of uncertain significance are a major challenge in the clinical interpretation of variants in these two genes. Such a classification is not clinically actionable and can leave a patient in limbo, unable to benefit from a molecular diagnosis nor rule out a genetic cause for their disease. The ACMG-AMP guidelines take a necessarily conservative approach to variant interpretation, meaning only variants with clear evidence are classified as likely pathogenic or pathogenic in a clinical setting where decisions will be made based on the genetic test result. In the case of the gap junction genes, nearly half the variants reported in the literature or in ClinVar are classified as VUS. Resolving as many of these variants as possible into likely benign or likely pathogenic categories, with high accuracy, should be a priority. The most rapid way to do this would be to develop well-validated in vitro functional assays that can be applied with moderate throughput to existing VUS and new variants as they are identified. Such assays exist and are reported in the literature, but lack the clinical validation required to apply them in this context. Generating this kind of clinical control data, along with publishing clear protocols that can be replicated in any standard molecular biology laboratory, should be a focus. If combined with accurate data sharing regarding the observation of variants in patients and families through repositories such as ClinVar for clinically observed variants, a large proportion of the VUS would be quickly reclassified and benefits to patients would accrue.
With gap junction gene-relevant specifications to the ACMG-AMP criteria, better communication and sharing of variant observations and interpretations, and readily available functional assays for a broad range of variants, we envisage that the majority of gap junction variants can be resolved to LP and LB in the next few years. This will provide clinically useful variant interpretation for patients and increase the molecular diagnostic rate for pediatric cataract. These genes could provide a framework to similarly improve genetic testing interpretation for other cataract genes.
Article highlights
Gap junction genes GJA3 and GJA8 are a common cause of pediatric cataract accounting for up to 18% of the inherited forms of the disease.
Variants in gap junction genes can cause cataract with autosomal dominant or autosomal recessive inheritance as well as with reduced penetrance.
There is broad phenotypic heterogeneity of cataract associated with gap junction gene variants.
Both missense and nonsense gap junction variants can lead to cataract with an enrichment of disease-causing variants in the first two transmembrane domains and extracellular loops of the proteins.
Most gap junction variants in cataract patients reported in the literature receive a formal classification of likely pathogenic or variant of uncertain significance according to the ACMG-AMP guidelines for variant classification.
There is a lack of functional data with appropriate pathogenic and benign controls for variants in the gap junction genes. Generating such data would allow the reclassification of many variants of uncertain significance to the clinically actionable likely pathogenic classification.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Wu X, Long E, Lin H, et al. Prevalence and epidemiological characteristics of congenital cataract: a systematic review and meta-analysis. Sci Rep. 2016 Jun 23;6(1):28564.
- Berry V, Georgiou M, Fujinami K, et al. Inherited cataracts: molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br J Ophthalmol. 2020 Oct;104(10):1331–1337.
- Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010 Oct 8;16:2007–2015. .
- Goodenough DA. The crystalline lens. A system networked by gap junctional intercellular communication. Semin Cell Biol. 1992 Feb;3(1):49–58.
- Mathias RT, Kistler J, Donaldson P. The lens circulation. J Membr Biol. 2007 Mar;216(1):1–16.
- Mathias RT, White TW, Gong X. Lens gap junctions in growth, differentiation, and homeostasis. Physiol Rev. 2010 Jan;90(1):179–206.
- Berthoud VM, Gao J, Minogue PJ, et al. Connexin mutants compromise the lens circulation and cause cataracts through biomineralization. Int J Mol Sci. 2020 Aug 13;21(16):5822.
- Berthoud VM, Ngezahayo A. Focus on lens connexins. BMC Cell Biol. 2017 Jan 17;18(Suppl 1):6.
- Beyer EC, Mathias RT, Berthoud VM. Loss of fiber cell communication may contribute to the development of cataracts of many different etiologies. Front Physiol. 2022;13:989524.
- Beyer EC, Ebihara L, Berthoud VM. Connexin mutants and cataracts. Front Pharmacol. 2013;4:43.
- Shiels A, Mackay D, Ionides A, et al. A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q [Article; Proceedings Paper]. Am J Hum Genet. 1998 Mar;62(3):526–532.
- Mackay D, Ionides A, Kibar Z, et al. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet. 1999 May;64(5):1357–1364.
- Shiels A, Hejtmancik JF. Inherited cataracts: genetic mechanisms and pathways new and old. Exp Eye Res. 2021 Aug;209:108662.
- Fan F, Luo Y, Wu J, et al. The mutation spectrum in familial versus sporadic congenital cataract based on next-generation sequencing. BMC Ophthalmol. 2020;20(1). DOI:10.1186/s12886-020-01567-x.
- Gillespie RL, O’Sullivan J, Ashworth J, et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology. 2014 Nov;121(11):2124–37.e1–2.
- Hansen L, Mikkelsen A, Nürnberg P, et al. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci. 2009 Jul;50(7):3291–3303.
- Jackson D, Malka S, Harding P, et al. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. American Journal of Medical Genetics, Part C: Seminars in Medical Genetics. 2020 01 Sep;184(3):578–589.
- Javadiyan S, Craig JE, Souzeau E, et al. High-throughput genetic screening of 51 pediatric cataract genes identifies causative mutations in inherited pediatric cataract in South Eastern Australia. G3 (Bethesda). 2017 Oct 5;7(10):3257–3268.
- Javadiyan S, Lucas SEM, Wangmo D, et al. Identification of novel mutations causing pediatric cataract in Bhutan, Cambodia, and Sri Lanka. Mol Genet Genomic Med. 2018 May 16;6(4):555–564.
- Jones JL, McComish BJ, Staffieri SE, et al. Pathogenic genetic variants identified in Australian families with paediatric cataract. BMJ Open Ophthalmol. 2022;7(1):e001064.
- Kumar M, Agarwal T, Kaur P, et al. Molecular and structural analysis of genetic variations in congenital cataract. Mol Vis. 2013;19:2436–2450.
- Li J, Leng Y, Han S, et al. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet J Rare Dis. 2018 Jun 18;13(1):94.
- Ma AS, Grigg JR, Ho G, et al. Sporadic and Familial congenital cataracts: mutational spectrum and new diagnoses using next-generation sequencing. Hum Mutat. 2016 Apr;37(4):371–384.
- Patel N, Anand D, Monies D, et al. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum Genet. 2017 Feb;136(2):205–225.
- Rechsteiner D, Issler L, Koller S, et al. Genetic analysis in a Swiss cohort of bilateral congenital cataract. JAMA Ophthalmol. 2021 Jul 1;139(7):691–700.
- Sun W, Xiao X, Li S, et al. Exome sequencing of 18 Chinese families with congenital cataracts: a new sight of the NHS gene. PLoS One. 2014;9(6):e100455.
- Zhai Y, Li J, Yu W, et al. Targeted exome sequencing of congenital cataracts related genes: broadening the mutation spectrum and genotype-phenotype correlations in 27 Chinese Han Families. Sci Rep. 2017 Apr 27;7(1):1219.
- Zhang XH, Wang JD, Jia HY, et al. Mutation profiles of congenital cataract genes in 21 northern Chinese families [Article]. Mol Vis. 2018;24:471–477.
- Fernandez-Alcalde C, Nieves-Moreno M, Noval S, et al. Molecular and genetic mechanism of non-syndromic congenital cataracts. Mutation Screening in Spanish Families [Article]. Genes. 2021Apr; 124: 13
- Bell S, Malka S, Lloyd IC, et al. Clinical Spectrum and genetic diagnosis of 54 consecutive patients aged 0-25 with bilateral cataracts. Genes (Basel). 2021 Jan 21;12(2):131.
- Kessel L, Bach-Holm D, Al-Bakri M, et al. Genetic disease is a common cause of bilateral childhood cataract in Denmark. Ophthalmic Genet. 2021 Dec;42(6):650–658.
- Ma A, Grigg JR, Flaherty M, et al. Genome sequencing in congenital cataracts improves diagnostic yield. Hum Mutat. 2021 Sep;42(9):1173–1183.
- Li D, Wang S, Ye H, et al. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population. Mol Vis. 2016;22:589–598.
- Kumar M, Agarwal T, Khokhar S, et al. Mutation screening and genotype phenotype correlation of α-crystallin, γ-crystallin and GJA8 gene in congenital cataract. Mol Vis. 2011 Mar 11;17:693–707.
- Yao K, Wang W, Zhu YN, et al. A novel GJA3 mutation associated with congenital nuclear pulverulent and posterior polar cataract in a Chinese family [Article]. Hum Mutat. 2011 Dec;32(12):1367–1370.
- Addison PKF, Berry V, Holden KR, et al. A novel mutation in the connexin 46 gene (GJA3) causes autosomal dominant zonular pulverulent cataract in a Hispanic family [Article]. Mol Vis. 2006 Jul;12(88–90):791–795.
- Berry V, Ionides ACW, Pontikos N, et al. Whole-genome sequencing reveals a recurrent missense mutation in the Connexin 46 (GJA3) gene causing autosomal-dominant lamellar cataract. Eye (Lond). 2018 May 1;32(10):1661–1668.
- Hansen L, Yao W, Eiberg H, et al. The congenital “ant-egg” cataract phenotype is caused by a missense mutation in connexin46. Mol Vis. 2006 01 Sep;12:1033–1039.
- Santhiya ST, Kumar GS, Sudhakar P, et al. Molecular analysis of cataract families in India: new mutations in the CRYBB2 and GJA3 genes and rare polymorphisms. Mol Vis. 2010;16:1837–1847.
- Vidya NG, Rajkumar S, Vasavada AR. Genetic investigation of ocular developmental genes in 52 patients with anophthalmia/microphthalmia. Ophthalmic Genet. 2018 Jun;39(3):344–352.
- Jiang H, Jin Y, Bu L, et al. A novel mutation in GJA3 (connexin46) for autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2003 Oct 24;9:579–583.
- Guleria K, Sperling K, Singh D, et al. A novel mutation in the connexin 46 (GJA3) gene associated with autosomal dominant congenital cataract in an Indian family. Mol Vis. 2007 Sep 11;13:1657–1665.
- Yang Z, Li Q, Ma X, et al. Mutation analysis in Chinese families with autosomal dominant hereditary cataracts. Curr Eye Res. 2015;40(12):1225–1231.
- Zhou Z, Hu S, Wang B, et al. Mutation analysis of congenital cataract in a Chinese family identified a novel missense mutation in the connexin 46 gene (GJA3). Mol Vis. 2010 Apr 21;16:713–719.
- Bennett TM, Shiels A. A recurrent missense mutation in GJA3 associated with autosomal dominant cataract linked to chromosome 13q. Mol Vis. 2011;17:2255–2262.
- Ma MF, Li LB, Pei YQ, et al. Use of high-throughput targeted exome sequencing in genetic diagnosis of Chinese family with congenital cataract. Int J Ophthalmol. 2016;9(5):650–654.
- Ma Z, Zheng J, Yang F, et al. Two novel mutations of connexin genes in Chinese families with autosomal dominant congenital nuclear cataract [Article]. Br J Ophthalmol. 2005 Nov;89(11):1535–1537.
- Yang G, Xing B, Liu G, et al. A novel mutation in the GJA3 (connexin46) gene is associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2011 Apr 27;17:1070–1073.
- Guo Y, Yuan L, Yi J, et al. Identification of a GJA3 mutation in a Chinese family with congenital nuclear cataract using exome sequencing. Indian J Biochem Biophys. 2013 Aug;50(4):253–258.
- Li B, Liu Y, Liu Y, et al. Identification of a GJA3 mutation in a large family with bilateral congenital cataract. DNA Cell Biol. 2016 Mar;35(3):135–139.
- Hu Y, Gao L, Feng Y, et al. Identification of a novel mutation of the gene for gap junction protein α3 (GJA3) in a Chinese family with congenital cataract. Mol Biol Rep. 2014 Jul;41(7):4753–4758.
- Bennett TM, Mackay DS, Knopf HL, et al. A novel missense mutation in the gene for gap-junction protein alpha 3 (GJA3) associated with autosomal dominant “nuclear punctate” cataracts linked to chromosome 13q. Mol Vis. 2004 Jun 11;10:376–382.
- Wang L, Chen YH, Chen XL, et al. Further evidence for P59L mutation in GJA3 associated with autosomal dominant congenital cataract [Article]. Indian J Ophthalmol. 2016 Jul;64(7):508–512.
- Guleria K, Vanita V, Singh D, et al. A novel “pearl box” cataract associated with a mutation in the connexin 46 (GJA3) gene. Mol Vis. 2007 Jun 4;13:797–803.
- Zhang L, Qu X, Su S, et al. A novel mutation in GJA3 associated with congenital Coppock-like cataract in a large Chinese family. Mol Vis. 2012 26 Jul;18:2114–2118.
- Yuan L, Guo Y, Yi J, et al. Identification of a novel GJA3 mutation in congenital nuclear cataract. Optom Vis Sci. 2015 Mar;92(3):337–342.
- Ding X, Wang B, Luo Y, et al. A novel mutation in the connexin 46 (GJA3) gene associated with congenital cataract in a Chinese pedigree. Mol Vis. 2011;17:1343–1349.
- Rees MI, Watts P, Fenton I, et al. Further evidence of autosomal dominant congenital zonular pulverulent cataracts linked to 13q11 (CZP3) and a novel mutation in connexin 46 (GJA3). Hum Genet. 2000 Feb;106(2):206–209.
- Li Y, Wang J, Dong B, et al. A novel connexin46 (GJA3) mutation in autosomal dominant congenital nuclear pulverulent cataract. Mol Vis. 2004 Sep 14;10:668–671.
- Zhang X, Wang L, Wang J, et al. Coralliform cataract caused by a novel connexin46 (GJA3) mutation in a Chinese family. Mol Vis. 2012;18:203–210.
- Zhang M, Lv H, Huang C, et al. Targeted exome sequencing identified a novel GJA3 gene missense mutation causes autosomal dominant congenital cataract in a large Chinese family. Int J Clin Exp Med. [2017 30 Mar];10(3):5143–5151.
- Wang KJ, Zhu SQ. A novel p.F206I mutation in Cx46 associated with autosomal dominant congenital cataract. Mol Vis. 2012;18:968–973.
- Lefter M, Vis JK, Vermaat M, et al. Next generation HGVS nomenclature checker. Bioinformatics. 2021 Feb 4;37(18):2811–2817.
- Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016 Oct 6;99(4):877–885.
- Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014 Mar;46(3):310–315.
- Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019 Jan 8;47(D1):D886–d894.
- Zhang L, Liang Y, Zhou Y, et al. A missense mutation in GJA8 encoding connexin 50 in a Chinese pedigree with autosomal dominant congenital cataract. Tohoku J Exp Med. 2018 Feb;244(2):105–111.
- Jabbarpour N, Saei H, Jabbarpoor Bonyadi MH, et al. Identification of novel cis-mutations in the GJA8 gene in a 3-generation Iranian family with autosomal dominant congenital nuclear cataract. Ophthalmic Genet. 2022;21:1–6.
- Mackay DS, Bennett TM, Culican SM, et al. Exome sequencing identifies novel and recurrent mutations in GJA8 and CRYGD associated with inherited cataract. Hum Genomics. 2014;8:19.
- Willoughby CE, Arab S, Gandhi R, et al. A novel GJA8 mutation in an Iranian family with progressive autosomal dominant congenital nuclear cataract [Article]. J Med Genet. 2003 Nov;40(11):4.
- Wang K, Wang B, Wang J, et al. A novel GJA8 mutation (p.I31T) causing autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2009;15:2813–2820.
- Dang FT, Yang FY, Yang YQ, et al. A novel mutation of p.F32I in GJA8 in human dominant congenital cataracts [Article]. Int J Ophthalmol. 2016 Nov;9(11):1561–1567.
- Sun WM, Xiao XS, Li SQ, et al. Mutational screening of six genes in Chinese patients with congenital cataract and microcornea [Article]. Mol Vis. 2011 Jun;17(168–69):1508–1513.
- Guo R, Huang D, Ji J, et al. A novel mutation GJA8 NM_005267.5: c.124G>A, p.(E42K) causing congenital nuclear cataract. BMC Ophthalmol. 2022 15 Apr;22(1):172.
- Mohebi M, Chenari S, Akbari A, et al. Mutation analysis of connexin 50 gene among Iranian families with autosomal dominant cataracts [Article]. Iran J Basic Med Sci. 2017 Mar;20(3):288–293.
- Devi RR, Vijayalakshmi P. Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea [Article]. Mol Vis. 2006 Mar;12(21–22):190–195.
- Zhang H, Chen Z, He K, et al. Unique presentation of congenital cataract concurrent with microcornea, microphthalmia plus posterior capsule defect in monozygotic twins caused by a novel GJA8 mutation. Eye (Lond). 2019 Apr;33(4):686–689.
- Vanita V, Singh JR, Singh D, et al. A novel mutation in GJA8 associated with jellyfish-like cataract in a family of Indian origin [Article]. Mol Vis. 2008 Feb;14(38–40):323–326.
- Minogue PJ, Tong JJ, Arora A, et al. A mutant connexin50 with enhanced hemichannel function leads to cell death [Article]. Invest Ophthalmol Vis Sci. 2009 Dec;50(12):5837–5845.
- Li JY, Wang QW, Fu QY, et al. A novel connexin 50 gene (gap junction protein, alpha 8) mutation associated with congenital nuclear and zonular pulverulent cataract [Article]. Mol Vis. 2013;19:767–774.
- Yan N, Xiao L, Hou C, et al. X-linked inheritances recessive of congenital nystagmus and autosomal dominant inheritances of congenital cataracts coexist in a Chinese family: a case report and literature review. BMC Med Genet. 2019 Mar 19;20(1):41.
- Wang L, Luo Y, Wen W, et al. Another evidence for a D47N mutation in GJA8 associated with autosomal dominant congenital cataract [Article]. Mol Vis. 2011 Sep;17(258–59):2380–2385.
- Liang C, Liang H, Yang Y, et al. Mutation analysis of two families with inherited congenital cataracts [Article]. Mol Med Rep. 2015 Sep;12(3):3469–3475.
- Gunda P, Manne M, Adeel SS, et al. Detection of c.139G > A (D47N) mutation in GJA8 gene in an extended family with inheritance of autosomal dominant zonular cataract without pulverulent opacities by exome sequencing [Article]. J Genet. 2018 Sep;97(4):879–885.
- Li J, Xia CH, Wang E, et al. Screening, genetics, risk factors, and treatment of neonatal cataracts. Birth Defects Res. 2017 Jun 1;109(10):734–743.
- Shen C, Wang J, Wu X, et al. Next-generation sequencing for D47N mutation in Cx50 analysis associated with autosomal dominant congenital cataract in a six-generation Chinese family. BMC Ophthalmol. 2017 May 19;17(1):73.
- Berry V, Mackay D, Khaliq S, et al. Connexin 50 mutation in a family with congenital “zonular nuclear” pulverulent cataract of Pakistani origin. Hum Genet. 1999 Jul-Aug;105(1–2):168–170.
- Hadrami M, Bonnet C, Veten F, et al. A novel missense mutation of GJA8 causes congenital cataract in a large Mauritanian family. Eur J Ophthalmol. 2019 Nov;29(6):621–628.
- Ding N, Chen ZY, Song XD, et al. Novel mutation of GJA8 in autosomal dominant congenital cataracts [Article]. Ann Transl Med. 2020 Sep;8(18):7.
- Li D, Xu C, Huang D, et al. Identification and functional analysis of a novel missense mutation in GJA8, p.Ala69Thr. BMC Ophthalmol. 2020 Nov 20;20(1):461.
- Wang KJ, Da Wang J, Chen DD, et al. Characterization of a p.R76H mutation in Cx50 identified in a Chinese family with congenital nuclear cataract. J Formosan Med Assoc. 2020 2020Jan 01;119(1, Part 1):144–149.
- Vanita V, Hennies HC, Singh D, et al. A novel mutation in GJA8 associated with autosomal dominant congenital cataract in a family of Indian origin [Article]. Mol Vis. 2006 Oct;12(136–38):1217–1222.
- Ge XL, Zhang YL, Wu YM, et al. Identification of a novel GJA8 (Cx50) point mutation causes human dominant congenital cataracts [Article]. Sci Rep. 2014;4:6.
- Arora A, Minogue PJ, Liu X, et al. A novel GJA8 mutation is associated with autosomal dominant lamellar pulverulent cataract: further evidence for gap junction dysfunction in human cataract [Article]. J Med Genet. 2006 Jan;43(1):7.
- Vanita V, Singh JR, Singh D, et al. A mutation in GJA8 (p.P88Q) is associated with “balloon-like” cataract with Y-sutural opacities in a family of Indian origin [Article]. Mol Vis. 2008 Jun;14(138):1171–1175.
- Jin A, Zhao Q, Liu S, et al. Identification of a new mutation p.P88L in Connexin 50 associated with dominant congenital cataract. Front Cell Dev Biol. 2022;10:794837.
- Ren M, Yang XG, Dang XJ, et al. Exome sequencing identifies a novel mutation in GJA8 associated with inherited cataract in a Chinese family [Article]. Graefes Arch Clin Exp Ophthalmol. 2017 Jan;255(1):141–151.
- Hansen L, Yao W, Eiberg H, et al. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci. 2007 Sep;48(9):3937–3944.
- Ponnam SPG, Ramesha K, Matalia J, et al. Mutational screening of Indian families with hereditary congenital cataract [Article]. Mol Vis. 2013;19:1141–1148.
- Li X, Si N, Song Z, et al. Clinical and genetic findings in patients with congenital cataract and heart diseases. Orphanet J Rare Dis. 2021 May 31;16(1):242.
- Hu S, Wang B, Zhou Z, et al. A novel mutation in GJA8 causing congenital cataract-microcornea syndrome in a Chinese pedigree. Mol Vis. 2010;16:1585–1592.
- Prokudin I, Simons C, Grigg JR, et al. Exome sequencing in developmental eye disease leads to identification of causal variants in GJA8, CRYGC, PAX6 and CYP1B1 [Article]. Eur J Hum Genet. 2014 Jul;22(7):907–915.
- Gao XB, Cheng J, Lu CL, et al. A novel mutation in the connexin 50 Gene (GJA8) associated with autosomal dominant congenital nuclear cataract in a Chinese family [Article]. Curr Eye Res. 2010 Jul;35(7):597–604.
- Chen JH, Qiu J, Chen H, et al. Rapid and cost-effective molecular diagnosis using exome sequencing of one proband with autosomal dominant congenital cataract. Eye (Lond). 2014 Dec;28(12):1511–1516.
- Yan M, Xiong C, Ye SQ, et al. A novel connexin 50 (GJA8) mutation in a Chinese family with a dominant congenital pulverulent nuclear cataract [Article]. Mol Vis. 2008 Mar;14(46–53):418–424.
- Chen C, Sun Q, Gu MM, et al. A novel Cx50 (GJA8) p.H277Y mutation associated with autosomal dominant congenital cataract identified with targeted next-generation sequencing [Article]. Graefes Arch Clin Exp Ophthalmol. 2015 Jun;253(6):915–924.
- Ponnam SP, Ramesha K, Matalia J, et al. Mutational screening of Indian families with hereditary congenital cataract. Mol Vis. 2013;19:1141–1148.
- Schmidt W, Klopp N, Illig T, et al. A novel GJA8 mutation causing a recessive triangular cataract [Article]. Mol Vis. 2008 May;14(101–02):851–856.
- Lin Y, Chen X, Liang C, et al. Novel compound heterozygous variant of GJA8 gene in two siblings with congenital cataract mimics an autosomal recessive trait. Eur J Ophthalmol. 2022;19:11206721221132874.
- Astiazaran MC, Garcia-Montano LA, Sanchez-Moreno F, et al. Next generation sequencing-based molecular diagnosis in familial congenital cataract expands the mutational spectrum in known congenital cataract genes [Article]. Am J Med Genet A. 2018 Dec;176(12):2637–2645.
- Cui XK, Zhou Z, Zhu KK, et al. A novel Cx50 Insert mutation from a Chinese congenital cataract family impairs its cellular membrane localization and function [Article]. DNA Cell Biol. 2018 May;37(5):449–456.
- Wang X, Wang D, Wang Q, et al. Broadening the mutation spectrum in GJA8 and CHMP4B: novel missense variants and the associated phenotypes in six Chinese han congenital cataracts families. Front Med (Lausanne). 2021;8:713284.
- Min HY, Qiao PP, Asan, et al. targeted genes sequencing identified a novel 15 bp deletion on GJA8 in a Chinese family with autosomal dominant congenital cataracts [Article]. Chin Med J. 2016 Apr;129(7):860–+.
- Berry V, Ionides A, Pontikos N, et al. Whole Exome sequencing reveals novel and recurrent disease-causing variants in lens specific gap junctional protein encoding genes causing congenital cataract. Genes (Basel). 2020 May 6;11(5):512.
- Sun W, Xiao X, Li S, et al. Mutation analysis of 12 genes in Chinese families with congenital cataracts. Mol Vis. 2011;17:2197–2206.
- Sun Y, Man J, Wan Y, et al. Targeted next-generation sequencing as a comprehensive test for Mendelian diseases: a cohort diagnostic study. Sci Rep. 2018 Aug 3;8(1):11646.
- Li S, Zhang J, Cao Y, et al. Novel mutations identified in Chinese families with autosomal dominant congenital cataracts by targeted next-generation sequencing. BMC Med Genet. 2019 Dec 16;20(1):196.
- Zhou D, Ji H, Wei Z, et al. A novel insertional mutation in the connexin 46 (gap junction alpha 3) gene associated with autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2013;19:789–795.
- Cui XK, Zhu KK, Zhou Z, et al. A novel frameshift mutation in CX46 associated with hereditary dominant cataracts in a Chinese family. Int J Ophthalmol. 2017;10(5):684–690.
- Burdon KP, Wirth MG, Mackey DA, et al. A novel mutation in the Connexin 46 gene causes autosomal dominant congenital cataract with incomplete penetrance. J Med Genet. 2004 Aug;41(8):e106.
- Devi RR, Reena C, Vijayalakshmi P. Novel mutations in GJA3 associated with autosomal dominant congenital cataract in the Indian population. Mol Vis. 2005 Oct 11;11:846–852.
- He W, Li X, Chen JJ, et al. Genetic linkage analyses and Cx50 mutation detection in a large multiplex Chinese family with hereditary nuclear cataract [Article]. Ophthalmic Genet. 2011 Mar;32(1):48–53.
- Arora A, Minogue PJ, Liu X, et al. A novel connexin50 mutation associated with congenital nuclear pulverulent cataracts. J Med Genet. 2008 Mar;45(3):155–160.
- Rubinos C, Villone K, Mhaske PV, et al. Functional effects of Cx50 mutations associated with congenital cataracts. Am J Physiol Cell Physiol. 2014 Feb 1;306(3):C212–20.
- Micheal S, Niewold ITG, Siddiqui SN, et al. Delineation of novel autosomal recessive mutation in GJA3 and autosomal dominant mutations in GJA8 in Pakistani congenital cataract families. Genes (Basel). 2018 Feb 20;9(2):112.
- Hassan AY, Yousaf S, Levin MR, et al. Novel homozygous missense variant in GJA3 connexin domain causing congenital nuclear and cortical cataracts. Int J Mol Sci. 2021 Dec 27;23(1).
- Schadzek P, Schlingmann B, Schaarschmidt F, et al. The cataract related mutation N188T in human connexin46 (hCx46) revealed a critical role for residue N188 in the docking process of gap junction channels. Biochim Biophys Acta. 2016 Jan;1858(1):57–66.
- Minogue PJ, Liu X, Ebihara L, et al. An aberrant sequence in a connexin46 mutant underlies congenital cataracts. J Biol Chem. 2005 Dec 9;280(49):40788–40795.
- Ponnam SP, Ramesha K, Tejwani S, et al. Mutation of the gap junction protein alpha 8 (GJA8) gene causes autosomal recessive cataract. J Med Genet. 2007 Jul;44(7):e85.
- Somaraju Chalasani ML, Muppirala M, Gp SP, et al. A cataract-causing connexin 50 mutant is mislocalized to the ER due to loss of the fourth transmembrane domain and cytoplasmic domain. FEBS Open Bio. 2013;3(1):22–29.
- Minogue PJ, Beyer EC, Berthoud VM. A connexin50 mutant, CX50fs, that causes cataracts is unstable, but is rescued by a proteasomal inhibitor. J Biol Chem. 2013 Jul 12;288(28):20427–20434.
- Sarkar D, Ray K, Sengupta M. Structure-function correlation analysis of connexin50 missense mutations causing congenital cataract: electrostatic potential alteration could determine intracellular trafficking fate of mutants. Biomed Res Int. 2014;2014:673895.
- Moon D, Park HW, Surl D, et al. Precision medicine through next-generation sequencing in inherited eye diseases in a Korean Cohort. Genes (Basel). 2021 Dec 23;13(1):27.
- Ceroni F, Aguilera-Garcia D, Chassaing N, et al. New GJA8 variants and phenotypes highlight its critical role in a broad spectrum of eye anomalies [Article]. Hum Genet. 2019 Sep;138(8–9):1027–1042.
- Ma AS, Grigg JR, Prokudin I, et al. New mutations in GJA8 expand the phenotype to include total sclerocornea. Clin Genet. 2018 Jan;93(1):155–159.
- Ma A, Yousoof S, Grigg JR, et al. Revealing hidden genetic diagnoses in the ocular anterior segment disorders [Article]. Genet Med. 2020 Oct;22(10):1623–1632.
- Zhou L, Sun X, Wang X, et al. Identification and functional analysis of two GJA8 variants in Chinese families with eye anomalies. Mol Genet Genomics. 2022 Nov;297(6):1553–1564.
- Landrum MJ, Chitipiralla S, Brown GR, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020 Jan 8;48(D1):D835–d844.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015 May;17(5):405–424.
- Burdon KP. The utility of genomic testing in the ophthalmology clinic: a review. Clin Exp Ophthalmol. 2021 Aug;49(6):615–625.
- Tavtigian SV, Harrison SM, Boucher KM, et al. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020 Oct;41(10):1734–1737.
- Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018 Nov;39(11):1517–1524.
- Bai D, Wang J, Li T, et al. Differential domain distribution of gnomAD- and disease-linked connexin missense variants. Int J Mol Sci. 2021 Jul 22;22(15):7832.
- Kelly MA, Caleshu C, Morales A, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018 Mar;20(3):351–359.
- Burdon KP, Graham P, Hadler J, et al. Specifications of the ACMG/AMP variant curation guidelines for myocilin: recommendations from the clingen glaucoma expert panel. Hum Mutat. 2022 Oct 11;43(12):2170–2186.
- Sequence Variant Interpretation Working Group. Sequence Variant Interpretation Recommendation for Absence/Rarity (PM2) - Version 1.0. 2020.
- Sequence Variant Interpretation Working Group. Sequence variant interpretation recommendation for de novo criteria (PS2 & PM6) - Version 1.1. 2021.
- Pejaver V, Byrne AB, Feng B-J, et al. Evidence-based calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for clinical use of PP3/BP4 criteria. bioRxiv. 2022. Doi:10.1101/2022.03.17.484479.
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019 Jan 24;176(3):535–548.e24.
- Biesecker LG, Harrison SM. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018 Dec;20(12):1687–1688.
- Kuo DS, Sokol JT, Minogue PJ, et al. Characterization of a variant of gap junction protein α8 identified in a family with hereditary cataract. PLoS One. 2017;12(8):e0183438.
- Reis LM, Tyler RC, Muheisen S, et al. Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes [Article]. Hum Genet. 2013 Jul;132(7):761–770.
- Polyakov AV, Shagina IA, Khlebnikova OV, et al. Mutation in the connexin 50 gene (GJA8) in a Russian family with zonular pulverulent cataract [Letter]. Clin Genet. 2001 Dec;60(6):476–478.
- Graw J, Schmidt W, Minogue PJ, et al. The GJA8 allele encoding CX50I247M is a rare polymorphism, not a cataract-causing mutation [Article]. Mol Vis. 2009 Sep;15(200):1881–1885.
- Senthil Kumar G, Dinesh Kumar K, Minogue PJ, et al. The E368Q Mutant Allele of GJA8 is associated with congenital cataracts with intrafamilial variation in a South Indian Family. Open Access Journal of Ophthalmology. 2016;1(1).
- Zahid A, Muazzam A, Mustafa S, et al. A Novel gap junction Alpha 8 (GJA8) mutation associated with a congenital cataract patient in Pakistan [Article]. Pak J Zool. 2017 Aug;49(4):1365–1372.
- Brnich SE, Abou Tayoun AN, Couch FJ, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019 Dec 31;12(1):3.
- Berthoud VM, Minogue PJ, Yu H, et al. Connexin46fs380 causes progressive cataracts. Invest Ophthalmol Vis Sci. 2014 Aug 7;55(10):6639–6648.
- Gao J, Minogue PJ, Beyer EC, et al. Disruption of the lens circulation causes calcium accumulation and precipitates in connexin mutant mice. Am J Physiol Cell Physiol. 2018 Apr 1;314(4):C492–c503.
- Minogue PJ, Gao J, Zoltoski RK, et al. Physiological and optical alterations precede the appearance of cataracts in Cx46fs380 Mice. Invest Ophthalmol Vis Sci. 2017 Aug 1;58(10):4366–4374.
- Chang B, Wang X, Hawes NL, et al. A Gja8 (Cx50) point mutation causes an alteration of alpha 3 connexin (Cx46) in semi-dominant cataracts of Lop10 mice. Hum Mol Genet. 2002 Mar 1;11(5):507–513.
- Runge PE, Hawes NL, Heckenlively JR, et al. Autosomal dominant mouse cataract (Lop-10). Consistent differences of expression in heterozygotes. Invest Ophthalmol Vis Sci. 1992 Oct;33(11):3202–3208.
- Ye Y, Wu M, Qiao Y, et al. Identification and preliminary functional analysis of two novel congenital cataract associated mutations of Cx46 and Cx50. Ophthalmic Genet. 2019 Oct;40(5):428–435.
- Pal JD, Liu X, Mackay D, et al. Connexin46 mutations linked to congenital cataract show loss of gap junction channel function. Am J Physiol Cell Physiol. 2000 Sep;279(3):C596–602.
- Tong JJ, Minogue PJ, Kobeszko M, et al. The connexin46 mutant, Cx46T19M, causes loss of gap junction function and alters hemi-channel gating. J Membr Biol. 2015 Feb;248(1):145–155.
- Tong JJ, Sohn BC, Lam A, et al. Properties of two cataract-associated mutations located in the NH2 terminus of connexin 46. Am J Physiol Cell Physiol. 2013 May 1;304(9):C823–32.
- Banks EA, Toloue MM, Shi Q, et al. Connexin mutation that causes dominant congenital cataracts inhibits gap junctions, but not hemichannels, in a dominant negative manner. J Cell Sci. 2009 Feb 1;122(Pt 3):378–388.
- Tong JJ, Minogue PJ, Guo W, et al. Different consequences of cataract-associated mutations at adjacent positions in the first extracellular boundary of connexin50. Am J Physiol Cell Physiol. 2011 May;300(5):C1055–64.
- Ren Q, Riquelme MA, Xu J, et al. Cataract-causing mutation of human connexin 46 impairs gap junction, but increases hemichannel function and cell death. PLoS One. 2013;8(9):e74732.
- Thomas BC, Minogue PJ, Valiunas V, et al. Cataracts are caused by alterations of a critical N-terminal positive charge in connexin50. Invest Ophthalmol Vis Sci. 2008 Jun;49(6):2549–2556.
- Li H, Jiang H, Rong R, et al. Identification of GJA3 p.S50P Mutation in a Chinese Family with Autosomal Dominant Congenital Cataract and Its Underlying Pathogenesis. DNA Cell Biol. 2020 Oct;39(10):1760–1766.
- Chen L, Su D, Li S, et al. The connexin 46 mutant (V44M) impairs gap junction function causing congenital cataract. J Genet. 2017 Dec;96(6):969–976.
- Shi W, Riquelme MA, Gu S, et al. Connexin hemichannels mediate glutathione transport and protect lens fiber cells from oxidative stress. J Cell Sci. 2018 Mar 21;131(6).
- Yao Y, Zheng X, Ge X, et al. Identification of a novel GJA3 mutation in a large Chinese family with congenital cataract using targeted exome sequencing. PLoS One. 2017;12(9):e0184440.
- Zhu Y, Yu H, Wang W, et al. A novel GJA8 mutation (p.V44A) causing autosomal dominant congenital cataract. PLoS One. 2014;9(12):e115406.
- Li L, Fan DB, Zhao YT, et al. GJA8 missense mutation disrupts hemichannels and induces cell apoptosis in human lens epithelial cells. Sci Rep. 2019 16 Dec;9(1):19157.
- Yu Y, Wu M, Chen X, et al. Identification and functional analysis of two novel connexin 50 mutations associated with autosome dominant congenital cataracts. Sci Rep. 2016 May 24;6(1):26551.
- Su D, Yang Z, Li Q, et al. Identification and functional analysis of GJA8 mutation in a Chinese family with autosomal dominant perinuclear cataracts. PLoS One. 2013;8(3):e59926.
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020 May;581(7809):434–443.