
ABSTRACT
Introduction
Anterior segment dysgeneses are congenital ocular anomalies that involve the cornea, iris, anterior chamber, iridicorneal angle structures, and ciliary body. Management highly varies and often depends on the extent of cornea and lens involvement and glaucoma diagnosis. A coordinated approach between pediatric ophthalmology, cornea, retina, and glaucoma specialists may be required to minimize complications and optimize results.
Areas covered
A review of the clinical findings of primary congenital glaucoma, congenital aniridia, Axenfeld-Rieger syndrome, Peters anomaly, sclerocornea, congenital ectropion uvea, and megalocornea/megalophthalmos will be followed by the current management of these diseases.
Expert opinion
For optimal outcomes, these diseases often require a multi-specialty approach incorporating glaucoma, cornea, and retina specialists with pediatric ophthalmologists. However, there is a critical shortage of pediatric ophthalmologists and few adult sub-specialists have an interest and desire to incorporate children into their practices. A greater emphasis on pediatric eye diseases during training and exposure to anterior segment dysgeneses is needed to provide the optimal care for these rare conditions.
1. Introduction
Anterior segment dysgeneses encompass a heterogenous group of congenital eye diseases in which there are varying degrees of abnormalities involving the cornea, iris, anterior chamber, iridicorneal angle structures, and ciliary body [Citation1,Citation2]. Management outcomes exhibit significant variability, largely contingent upon the extent of corneal and lens involvement as well as the presence of glaucoma [Citation1,Citation3]. In addition, it is imperative to treat other pediatric specific eye problems including amblyopia, strabismus, and refractive error, especially in unilateral cases, in order to optimize vision. Many cases show isolated ocular findings, but it is critical to recognize systemic associations which can be life threatening [Citation1,Citation2,Citation4].
Diseases under the anterior segment dysgenesis heading include primary congenital glaucoma (PCG), aniridia, Axenfeld-Rieger syndrome (ARS), iridogoniodysgenesis, Peters anomaly, sclerocornea, congenital ectropion uvea (CEU), anterior megalophthalmos, and megalocornea. The clinical findings of each of these diseases as well as management challenges will herein be discussed.
2. Primary congenital glaucoma (PCG)
PCG typically manifests within the initial 3 years of life and is marked by the classic triad of blepharospasm, photophobia, and epiphora [Citation5,Citation6]. These symptoms arise from elevated intraocular pressure (IOP), often linked to trabecular meshwork dysgenesis and/or Schlemm’s canal abnormalities. Increased IOP is commonly associated with corneal edema and Haabs striae (), but it is not necessarily a direct causative factor. However, it is important to note that many children may also present without buphthalmos or these classical signs [Citation7]. Gonioscopy, when not obscured by corneal edema, typically shows a scalloped iris that inserts into the trabecular meshwork [Citation5,Citation6]. Histology has demonstrated a variety of abnormalities including increased collagen in trabecular beam cores or within the trabecular spaces and displacement or absence of Schlemm’s canal [Citation8]. Further, in the 1950s, the concept of ‘Barkan’s membrane’ was introduced to describing the covering of the iridocorneal angle, and while optical coherence tomography (OCT) and ultrasound biomicroscopy have shown evidence of abnormal tissue or a hyperreflective membrane over the angle, this has never been confirmed histologically [Citation9,Citation10].
Unlike adults, increased IOP in infants and young children causes stretching of scleral and corneal tissues resulting in anterior and posterior segment lengthening (buphthalmos, ) with subsequent myopia [Citation11]. Compared to the sclera and corneal stroma and epithelium, Descemets membrane has limited distensibility and can tear (Haabs striae) with elevated IOP (, bottom row arrows) [Citation12]. In severe cases the elongation of the lens-iris diaphragm can result in lens dislocation. Optic cupping correlating with retinal nerve fiber layer loss is often present, but may not be immediately detectable due to corneal edema [Citation11]. Further, reversal of cupping, which does not represent restoration of retinal nerve fiber layer, but rather glial tissue hyperplasia or anterior movement of the lamina cribosa, is commonly seen in children () [Citation13]. Although this indicates IOP control, it can be deceptive of overall optic nerve health. Differentiating between the ‘color’ vs the ‘vessel’ cup shows the extent of the retina nerve fiber layer loss, which may not be able to be confirmed with formal visual field testing for at least a decade [Citation14].
Figure 1. Primary congenital glaucoma (PCG). (a) Two patients with PCG show buphthalmos and varying degrees of corneal edema (white arrows). Patient 2 also has a backwards ‘C’ shaped haabs striae (black arrows). (b) Optic nerve photographs of a patient with PCG at presentation (top row) and 4 years after obtaining IOP control from angle surgery (bottom row) show reversal of cupping (white arrowheads) especially in the left eye.
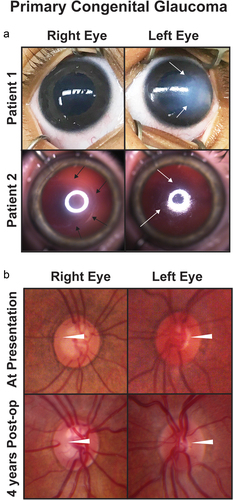
PCG is clinically divided into three subsets based on age of onset: neonatal, infantile, and late-onset. In neonatal onset, elevated IOP is diagnosed at birth or within 1 month of age while the infantile form presents between 1 and 24 months of age. Late onset, which may also be late recognized, is defined as presentation after 2 years of age [Citation15]. There is a fourth very rare entity termed spontaneously arrested or resolved PCG, in which there is clinical evidence of PCG (e.g. buphthalmos, Haabs striae, and optic nerve cupping). However, despite no history of surgical or medical treatment, the IOP is normal, and there is no disease progression [Citation16].
Angle surgery is the mainstay of PCG treatment. Surgical success tends to be less favorable in patients who present either at birth or after 1 year of age, as opposed to those who present between 2 and 12 months [Citation17–20]. However, surgical success may depend more on the severity of the disease rather than its age of onset or diagnosis. Traditional goniotomy is an ab interno approach that requires a gonioscopic view through the cornea [Citation18,Citation21]. Typically done either with a specialized knife or a 25-gauge needle, goniotomy allows for opening of 120–180º of the angle. Trabeculotomy ab externo is performed via a scleral flap in eyes with or without a gonioscopic view such that 120–180º of trabecular meshwork can be incised from Schlemm’s canal using trabeculotomes [Citation22]. Trabeculotomy can be combined with trabeculectomy, although whether this improves success is debated [Citation23]. More recently, angle surgery has been expanded to treat up to 360º of the trabecular meshwork [Citation24,Citation25]. This strategy was initially described by cannulating Schlemm’s canal with a polypropylene suture [Citation25]. Once passed 360º, each end of the suture is grasped to draw the suture into the anterior chamber, thereby incising the trabecular meshwork. A newer approach has used a lighted catheter to better visualize Schlemm’s canal and decrease risk of inadvertent passage posteriorly [Citation24]. The rate of 360º canalization of Schlemm’s canal in PCG widely ranges and is likely influenced by abnormal anatomy and surgeon experience [Citation26]. Studies have demonstrated a positive correlation between the degree of angle treatment and the success fate [Citation26–28]. While this may be due to the amount of trabecular meshwork that has been opened, it is also important to note that successful canalization suggests more anatomically intact angle structures [Citation26]. Thus, these eyes may have greater success with any angle surgery because of more normal anatomy.
If IOP remains elevated after angle surgery, trabecular meshwork bypass surgery is often required. Trabeculectomy utilizes the sclera to create a ‘trap door’ from which aqueous humor egresses through a sclerostomy at the base of the scleral flap. The fluid then forms a ‘bleb’ between the overlying conjunctiva and sclera from which the fluid is then absorbed through the extra ocular lymphatic and venous systems. Trabeculectomy surgery can be highly successful for obtaining IOP control, but has many pitfalls which are magnified in the pediatric population. The use of anti-fibrotics such as mitomycin C or 5-fluorouracil is often required to prevent scarring of the conjunctiva to the sclera that eliminates the bleb space. While this improves bleb survival, it sets up a life-long risk of potentially visually devastating infections [Citation29]. It is critical that the bleb be monitored as well as manipulated. Additional surgery may be required during the early post-operative period to adjust flow through the flap and optimize bleb formation [Citation30]. Long-term, the bleb may need adjustments to decrease the risk of infection and failure. As a result, in children trabeculectomy surgery has largely fallen out of favor for glaucoma drainage device (GDD) placement [Citation29–31].
GDDs have been used in childhood glaucomas for over 25 years with varying levels of success [Citation32–34]. There are valved as well as non-valved implants and the size of the plates also differs which increases the complexity of determining their individual effectiveness. Both valved and non-valved implants demonstrate a range of success between 80 and 90% at 1-year, but long-term studies have shown that this decreases to 30–50% by 10-years. The valved Ahmed implants have the advantage of immediate IOP control with less risk of hypotony, but are often afflicted by a hypertensive phase which is associated with a lower rate of success compared to the non-valved Baerveldt device [Citation32–34].
Placement of the intraocular portion of the tube of the GDD in children can be challenging as these eyes are usually phakic, and there may be poor visualization due to corneal edema. Further, the tube in children may migrate anteriorly because of corneal and sclera elasticity, resulting in corneal decompensation [Citation32]. Tube trimming or sulcus or pars plana placement in pseudophakic and aphakic eyes may be warranted [Citation35]. In addition, the plates and overlying blebs, even in the superotemporal quadrant can cause or exacerbate strabismus and any part of the apparatus may become exposed increasing the risk of infection [Citation36]. While bleb encapsulation is frequently implicated as the leading cause of GDD failure, there remains a lack of consensus regarding subsequent management strategies [Citation37]. Bleb revision with removal of the capsule rarely yields long-term IOP control due to repeat encapsulation and in a non-valved implant may cause hypotony [Citation38]. A second GDD can also improve IOP control, but uses a different quadrant which may limit future options. A valved GDD may be exchanged for a non-valved GDD, thereby saving a second quadrant, but long-term success has not been published [Citation39]. Finally, cycloablation, either transcleral or endoscopic, can be effective especially in combination with GDDs to obtain IOP control [Citation40,Citation41].
While obtaining IOP control is the immediate priority in PCG, managing the consequences including corneal edema, refractive error, amblyopia, and strabismus is critical. The corneal edema in the vast majority of patients with PCG will resolve after the IOP is decreased, however, it may take months for complete resolution. In rare cases with corneal decompensation, Descemets stripping and endothelial keratoplasty is an option, but is challenging in young children due to the post-operative positioning requirement and higher rates of graft failure [Citation42]. Both astigmatism and myopia are common in PCG. Haabs striae are irreversible and can cause photophobia, irregular astigmatism, higher order aberrations, and disruption of the visual axis [Citation43]. In addition, buphthamos can cause axial lengths greater than 30 mm resulting in pathologic myopia, retinal detachment and lens dislocation. The severity of the astigmatism, myopia, and anisometropia should be considered when deciding the timing for prescribing glasses. Part time occlusion for amblyopia may be needed in asymmetric or unilateral disease, and strabismus surgery may also be recommended, but can be challenging if a GDD or trabeculectomy bleb is present.
The prompt diagnosis and treatment of PCG is essential for optimizing visual outcomes, which can range from 20/20 to no light perception. While the majority of cases respond to angle surgery, there are refractory cases that require multiple surgeries. Further, management of amblyopia, refractive error and strabismus is imperative.
3. Congenital aniridia
Congenital aniridia is typically considered an anterior segment dysgenesis due to its namesake of iris hypoplasia and its association with congenital cataracts and juvenile-onset glaucoma. However, in addition to the cornea and iris, congenital aniridia affects almost all eye structures including the optic nerve, fovea, and limbal stem cells [Citation44]. Early visual impairment is due to foveal hypoplasia and optic nerve abnormalities including dysplasia and hypoplasia. The foveal hypoplasia almost uniformly results in nystagmus and visual acuity between 20/50 and 20/200 [Citation45]. Aniriida îs often associated with congenital cataracts. However, due to iris hypoplasia, these cataracts are rarely visually significant during infancy and early childhood [Citation46,Citation47]. Indications for lensectomy include visual significance, impaired view to the posterior segment, or progressive lens dislocation. It is important to note that determining visual significance in aniridic patients may not be straightforward as there may not be an appreciable change in optotype visual acuity testing preoperatively or postoperatively, but rather the patient reports changes in visual function.
Cataract surgery in congenital aniridia can be challenging due to preexisting corneal opacification, capsular/zonular instability and altered anterior chamber dynamics due to iris hypoplasia. The use of capsular tension rings in congenital aniridia has been reported, nevertheless IOL dislocation requiring secondary removal as well as primary aphakia due to inability to place an IOL is not unusual [Citation48]. However, our recent study showed that aphakia in aniridic patients was associated with worse visual outcomes although there are other aspects such as corneal opacification, complicated intraoperative findings and contact lens intolerance that may attribute to this difference [Citation45]. Post-operatively, both limbal stem cell deficiency and endothelial cell decompensation can be exacerbated leading to worsened corneal opacification. Thus, the decision to proceed with cataract surgery involves multiple factors which are unique to congenital aniridia.
In congenital aniridia, glaucoma affects approximately half of affected individuals [Citation49,Citation50]. The most commonly stated mechanism of increased IOP is anterior rotation of the iris stump causing angle closure. As a result, there have been some advocates for prophylactic goniotomy to lower the iris root to prevent glaucoma [Citation49]. This has not been widely adopted as the procedure would need to be done prior to increased IOP, and there is the risk of damage to the lens and cornea. Further, gonioscopy does not always demonstrate a closed angle configuration, and thus there is also a component of angle dysgenesis [Citation51].
Treatment of increased IOP in congenital aniridia and many of the anterior segment dysgeneses other than PCG typically starts with ocular anti-hypertensive medications. While the majority of glaucoma medications have not been FDA approved for the use in children, beta-blockers, carbonic anhydrase inhibitors, prostaglandin analogues, and alpha adrenergic agonists are all commonly used in the pediatric population. As with adults, systemic absorption from eye drops is limited, but both systemic and local side effects can occur. The most commonly used beta-blocker, timolol, tends to be well-tolerated, although caution should be heeded in children with asthma or cardiac conditions due to risk of bronchospasm and bradycardia. Betaxolol, the selective beta-1 blocker, can be a substitute for asthmatics, but is often more difficult to obtain from commercial pharmacies. Dorzolamide is the only ocular hypertensive to be FDA approved for children, however, the stinging and irritation after instillation of the topical carbonic anhydrase inhibitors can be prohibitive, especially in young children. The prostaglandin analogues, latanoprost, travoprost, and bimatoprost have the easiest compliance with once daily dosing. Nevertheless, the side effects of eyelash growth, iris color change, eyelid skin changes, and periorbital fat atrophy can be seen in children and should be considered especially in unilateral cases and with prolonged use. Alpha adrenergic agonists are often the fourth line treatment as side effects of fatigue, irritability, and respiratory depression are common in children. Brimonidine should not be prescribed in children younger than 5 years of age while apraclonidine can be prudently used in infants, toddlers, and preschoolers. Regardless, parents/guardians should be educated as to the risk of these side effects and the need for careful monitoring especially with treatment initiation. Netarsudil, a Rho-kinase inhibitor, has been more recently approved for use in adults, and there is very limited data regarding efficacy and safety in children. Further, at this time, netarsudil typically incurs a large out-of-pocket expense for patients, that can limit its availability.
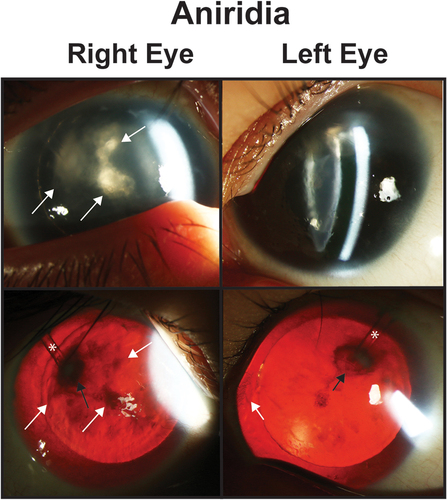
In congenital aniridia, it is important to note that there is a concern with topical medications, especially with carbonic anhydrase inhibitors and drops with the preservative benzalkonium chloride, in the context of limbal stem cell deficiency [Citation52]. Preservative-free eye drops should be used, especially in the presence of progressive keratopathy. Further, regular monitoring of the corneas is essential, and adjustments to treatments may be necessary in the presence of epitheliopathy. Ultimately, approximately half of patients with aniridic glaucoma require IOP-lowering surgery [Citation45,Citation52]. Angle surgery, either goniotomy or trabeculotomy, does not typically yield long-term IOP control which can be due to the closed angle formation or severe abnormalities of Schlemm’s canal and more posterior outflow channels [Citation45,Citation49,Citation53]. Trabeculectomy surgery shows varying degrees of success, but in aniridic eyes, there is a higher chance of a flat anterior chamber during the early post-operative phase because of iris hypoplasia [Citation45,Citation54]. This often requires refilling of the anterior chamber with viscoelastic either in the clinic or in the operating room. GDDs have largely supplanted trabeculectomies and success rates of GDDs in congenital aniridia vary between 60 and 90%. While this is greater than trabeculectomies (9–42%), many of these reports have fewer eyes with shorter follow-up [Citation45,Citation55,Citation56]. There are added challenges with GDDs in congenital aniridia. With the absence of iris, there is no protection of the lens either when making the sclerostomy or after implantation which can lead to a focal cataract (). In addition, with the corneal abnormalities including limbal stem cell deficiency and abnormal endothelium, it is critical to position the tube away from the cornea. The ideal placement is over the zonules in the periphery and when possible to place it in the sulcus between the iris root and ciliary body processes [Citation45]. In refractory cases, cycloablation can be useful, especially when combined with a GDD to provide outflow.
Aniridic-associated keratopathy, which is found in up to 80% of individuals, is predominantly due to limbal stem cell deficiency, but can be worsened by endothelial decompensation () [Citation57,Citation58]. Pannus is often the earliest sign, which initially is not visually significant, but in late stages may involve the entire cornea. In addition, injury or surgery can lead to non-healing epithelial defects as well as trigger endothelial cell loss. Keratopathy is associated with poor visual prognosis as treatment options are limited [Citation45]. Limbal stem cell transplantation is possible in moderate keratopathy, but should ideally be a related donor to optimize HLA match and ABO compatibility [Citation59]. In addition, most cornea specialists advocate for post-operative life-long systemic immunosuppression. Prior studies of limbal stem cell transplantation have shown success rates between 0 and 70% as assessed through improvements in visual acuity and need for additional corneal surgeries [Citation45,Citation60]. This wide range may be due to disease severity at time of surgery as well as whether the stem cell transplantation was combined with other procedures, namely keratoplasty. Full thickness penetrating keratoplasty as well as partial thickness grafts (lamellar keratoplasty and Descemet stripping endothelial keratoplasty) all show high failure rates in congenital aniridia [Citation45,Citation57,Citation60,Citation61]. Keratoprostheses have become more main stream in congenital aniridia, but complications including glaucoma, retinal detachment, retroprosthetic membranes, prosthesis extrusion, endophthalmitis, and phthisis are prevalent [Citation62,Citation63]. It is important to note, that undergoing either penetrating keratoplasty or keratoprosthesis may be associated with worse visual outcomes, however, to get to the point where these corneal surgeries are recommended, these patients likely already had limited vision due to severe corneal involvement [Citation45]. Regardless, realistic exceptions as to visual outcomes, potential complications and the need for additional surgery need to be set with aniridic patients prior to undergoing corneal surgeries.
Figure 2. Aniridia. Slit lamp photos (top row) and backlit photos (bottom row) of a patient with aniridia shows keratopathy (white arrows) which is worse in the right eye the the left eye. In the right eye, the white arrows point to the same areas of keratopathy in both the slit lamp and backlit photographs. In the left eye, the white arrows points to a peripheral pannus that is more obvious in the backlit photograph. Both eyes have a superotemporal glaucoma drainage device (asterisk) which due to the proxmity to the lens has caused a focal cataract (black arrow).
While iris hypoplasia itself may have little direct effect on visual acuity, photophobia can be severe and secondarily decrease vision, especially when coupled with corneal abnormalities or aniridia-associated keratopathy [Citation64]. Artificial iris implants which are placed within the capsule, anterior to the intraocular lens during lens extraction, have varying degree of acceptance. Some studies have shown decreased light sensitivity with improvement in visual function [Citation64,Citation65]. Nevertheless, as described above, lensectomy in congenital aniridia can be challenging not only due to capsular instability, but also can trigger corneal decompensation.
Congenital aniridia is a challenging disease due to is widespread effects throughout the eye. Management can also be complicated by systemic issues such as Wilms Tumor, for which approximately 30% of cases of aniridia may have increased risk. Visual impairment starts early and often worsens because of cataract progression, glaucoma and keratopathy. The decision to intervene surgically is complex as cataract and glaucoma surgery may exacerbate corneal opacification, which by and large is the worse prognosticator for poor visual outcomes.
4. Axenfeld-Rieger syndrome and iridogoniodysgenesis
Axenfeld-Rieger syndrome (ARS) is often considered the classic anterior segment dysgenesis and is characterized by both iris and corneal abnormalities. By definition, ARS is the combination of Axenfeld anomaly, which is iris stands adherent to posterior embryotoxon (anterior displacement of Schwalbe’s line), and Rieger anomaly, which is iris hypoplasia associated with corectopia and/or pseudopolycoria () [Citation66,Citation67]. While the ocular structures underlie the majority of the morbidity of ARS, the syndrome also is associated with maxillary and mandibular hypoplasia, teeth abnormalities (e.g., oligodontia, microdontia), hearing loss, cardiac outflow anomalies, redundant umbilical tissue, and gastrointestinal defects [Citation68].
Figure 3. Axenfeld-Rieger Syndrome (ARS) and Iridogoniodysgenesis. Patients 1 and 2 have classic with posterior embryotoxon (white arrows) and iris hypoplasia leading to corectopia and pseudopolycoria. Both patients have superotemporal and inferornasal glaucoma drainage devices (asterisks). Patient 3 has prominent posterior embryotoxon (white arrows) and ectropion uvea with iris stromal atrophy (white arrowhead). Patient 4 has iridogoniodysgenesis associated with a FOXC1 mutation. The iris is hypoplasitic (black arrow) and a trabeculectomy with mitomycin C (black arrowhead) had previously been performed for glaucoma.
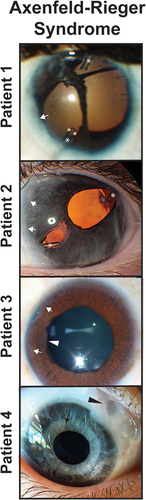
A challenge in ARS is the wide range of phenotypes, even within one family. Some affected individuals can have subtle findings such as 1–2 clock hours of Axenfled anomaly that may go undetected even by eye specialists. In contrast, other individuals within the same family can present with neonatal-onset glaucoma requiring urgent IOP-lowering surgery [Citation69]. Further, there is a subset of patients with iridogoniodysgenesis, who do not present with either Axenfeld or Rieger Anomaly, but show iris hypoplasia and immature angle structures ().
Glaucoma affects between 50 and 75% of patients with ARS [Citation66,Citation69,Citation70]. While there are iris strands that traverse the trabecular meshwork and connect to the posterior embryotoxon, these typically do not inhibit aqueous outflow or increase IOP. The source of increased IOP often originates from absence or dysgenesis of Schlemm’s canal and more posterior collecting channels [Citation68,Citation69,Citation71]. Topical ocular anti-hypertensives are the first-line treatment, however, more than two-thirds of individuals will require at least one glaucoma surgery [Citation69]. Angle surgery usually does not yield long-term IOP control in either iridogoniodsygenesis or ARS, in contrast to PCG [Citation69].
Most patients with ARS and glaucoma require trabecular meshwork bypass surgery, either trabeculectomy or GDD placement [Citation69]. Trabeculectomy surgery has been shown to have at least 50% success rate with some eyes having over 20 years of follow-up [Citation69,Citation72]. However like congenital aniridia, GDDs have largely supplanted trabeculectomies, despite few studies that have specifically focused on GDD success in ARS. Non-valved Baerveldt devices have shown higher success rates compared to valved Ahmed GDDs [Citation69]. Glaucoma in ARS often requires multiple surgeries to obtain IOP-control, and in refractory cases cycloablation can be useful [Citation69]. Nevertheless, especially in eyes with limited to no aqueous outflow, transcleral or endoscopic cycloablation often needs to be paired with an outflow surgery. Otherwise there is a fine line between glaucoma and hypotony such that multiple sessions of cycloablation may yield minimal effect, but subsequent GDD placement causes hypotony due to relative over-filtration.
The eye findings in ARS are critical to recognize as the diagnosis is often made by the ophthalmologist. Obtaining a comprehensive family history and examination of other family members is also helpful. From an eye standpoint, understanding the high risk for glaucoma and careful monitoring of IOP is imperative. Further, proper referral to genetics as well as other sub-specialists including pediatric dentistry, plastic surgery, cardiology, otolaryngology may be warranted.
5. Peters anomaly
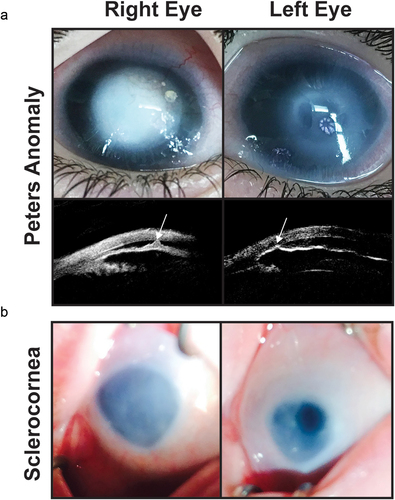
Peters anomaly is defined as a defect in the corneal stroma and endothelium resulting in a discrete corneal opacity (, Patient 1) [Citation73,Citation74]. Histologically, there is an absence of corneal endothelium underlying the opacity and adhesions between the cornea and iris (Type I) or between the cornea and lens (Type II) [Citation75]. Type I is more common and is not necessarily associated with cataract. Type II is more severe and the cataractous lens may be anteriorly displaced within a corneal staphyloma [Citation76].
Figure 4. Congenital corneal opacities. (a) Bilateral peters anomaly with a denser opacity in the right eye than the left eye. Ultrasound biomicroscopy demonstrates iridocorenal adhesions in the area of the corneal defect (white arrows). (b) Sclerocornea with microphthalmia showing a small hazy cornea with unclear demarcation of the limbus. The left eye has a superior iris opening which underlies an area of clearer cornea.
Management of the corneal opacity is central to the case of Peters anomaly [Citation75,Citation76]. Penetrating keratoplasty has been the traditional approach and has been the most common indication for full-thickness grafts in infants and young children. Nevertheless, the success rates of grafts are poor (50–70% at 1 year and 30% at 5–10 years) in Peters anomaly [Citation76–78]. For any corneal indication, the younger the age at time of penetrating keratoplasty, the more technically challenging the surgery is due to thinner corneas, scleral collapse, lens anteriorization and positive vitreous pressure. Further, there is greater inflammatory response post-operatively that increases the risk of early suture loosening that can result in infection and rejection. Additionally, in Peters anomaly, other abnormalities such as limbal stem cell deficiency, corneal staphyloma, microphthalmia, and aniridia can complicate both the surgery and increase the risk of graft rejection and failure [Citation74,Citation79,Citation80]. Keratoprostheses have been employed in Peters anomaly, especially after multiple failed penetrating keratoplasties [Citation76]. More so than traditional full-thickness grafts, keratoprostheses are commonly complicated with glaucoma, retinal detachment, retroprosthetic membranes, endophthalmitis, and even implant extrusion [Citation81]. Further, the life-long need for topical antibiotics and the inability to attempt other strategies post-keratoprosthesis, makes it difficult to advocate for its use as a primary procedure for Peters anomaly.
More recently, Descemets stripping with or without endothelial keratoplasty has been described [Citation82,Citation83]. The placement of an endothelial graft in an infant or young child is challenging given the strict post-operative positioning for graft adherence and is less likely successful in aphakic eyes. While there is less rejection or failure than a full-thickness graft, the need for long-term steroids can exacerbate IOP-control and cause a cataract. Removal of Descemets without endothelial graft placement has also been an approach which relies on endothelial cell hypertrophy and migration into the defect. This eliminates the need for a graft, but takes longer for improvement of corneal clarity and long-term results in these eyes with decreased endothelial cell counts from infancy are not widely reported [Citation82]. Lastly, an optical iridectomy to either widen the visual axis around the corneal abnormality or to create a second iris opening is feasible if there is an area of the cornea which is clear. While theoretically, an optical iridectomy may not yield as great of visual potential as a successful graft procedure, it does not carry the risk of rejection or failure nor increase the risk of glaucoma [Citation84,Citation85].
Glaucoma complicates more than 50% of eyes with Peters anomaly, with the majority of cases requiring multiple IOP-lowering surgeries [Citation76,Citation86]. It is important to note that obtaining accurate IOP in Peters anomaly can be challenging. The corneal abnormality has altered biomechanics which can make IOP measurements inaccurate. Other findings including corneal edema, scleral thinning or changes in axial length and optic nerve cupping may need to be followed more closely in these eyes. Like ARS, there may be absence or dysgenesis of Schlemm’s canal and posterior collecting channels. Further, the angle may show a closed configuration, although the prevalence of glaucoma did not seem to be exacerbated by penetrating keratoplasty surgery [Citation76].
As a result, angle surgery typically does not give long-term IOP control [Citation76,Citation86]. GDDs especially in combination with cycloablation can be effective, however, the anterior segment abnormalities may necessitate combined lensectomy and vitrectomy for posterior placement of the Intraocular portion of the tube [Citation35,Citation76]. Caution must be heeded, specifically in type II Peters anomaly, as the pars plana is often absent and the retina may have an anomalous insertion such that these eyes are at high risk for iatrogenic retinal breaks. Further, the corneal opacity also may require endoscopic visualization for vitrectomy surgery, which is not universally available [Citation87].
The management of Peters anomaly often requires input from cornea, glaucoma, retina, and pediatric ophthalmology specialists. A multi-disciplinary, coordinated approach may be key for minimizing complications and optimizing visual outcomes.
6. Sclerocornea
In contrast to Peters anomaly, sclerocornea is diffuse or peripheral opacification of the cornea due to absence of corneo-scleral demarcation and limbal stem cells (, Patient 2) [Citation3,Citation88]. Further, both Bowmans and Descemets layers may be abnormal or even absent, and the stromal collagen fibers can be irregular in diameter and disorganized in arrangement, decreasing the clarity of the tissue [Citation89]. The majority of these fibers are type I collagen, suggesting origination from the cornea and not the sclera where type III collagen is more common [Citation3,Citation90]. Further, blood vessels coming from the episclera and conjunctiva may be present within the anterior and mid-stroma [Citation89]. These corneal findings may also coexist with anterior segment abnormalities including iridocornal adhesions, iris hypoplasia with corectopia, posterior embryotoxon, trabeculodysgenesis, cataract, and congenital aphakia [Citation3]. In addition, sclerocornea is often seen in conjunction with the microphthalmia, coloboma, and anophthalmia (MAC) spectrum [Citation91].
The management of the corneal opacification in sclerocornea is dependent on the extent, laterality, and other associated ocular anomalies [Citation88]. Diffuse corneal involvement has similar challenges as Peters Anomaly. Penetrating keratoplasty is high risk for rejection and failure in infants and young children, especially if there is limbal stem cell deficiency [Citation74,Citation79,Citation80]. Success at 1 year ranges from 30–70% and continues to decrease over time [Citation78]. If there is evidence of an intact endothelial cell and Descemets layer by anterior segment OCT or ultrasound biomicroscopy, deep anterior lamellar keratoplasty (DALK) may be an option. While DALK decreases the risk of failure and rejection, the endothelial cell layer is often involved in many of these cases. Further, performing DALK in these eyes can be complicated by the tissue abnormalities leading to full thickness perforation and the need to convert to a full-thickness graft [Citation92]. If there is an area of the cornea that is more clear, an optical iridectomy can also be done to improve visual function [Citation84,Citation85]. In unilateral cases, amblyopia will be difficult to overcome and needs to be considered in the decision to attempt any surgery.
Due to both the shallow to flat anterior chamber and trabeculodysgenesis, individuals with sclerocornea are at high risk for glaucoma [Citation93]. Like Peters anomaly, obtaining accurate IOPs can be difficult. The appearance of the cornea and sclera, especially thinning due to buphthalmos, may be the main indication that IOP is elevated. While medical management should be attempted first, many of these eyes require IOP-lowering surgery. Angle surgery is often not feasible in these eyes due to a narrow or closed angle. GDD placement may be required, but like Peters anomaly, the intraocular placement of the tube may be difficult because of the shallow anterior chamber and poor view [Citation35,Citation87]. Thus, GDD placement may need to be combined with endoscopic lensectomy and vitrectomy for adequate visualization and placement of the tube more posteriorly [Citation87,Citation94].
Sclerocornea is a distinct disease from Peters anomaly, but has similar complexities in regards to management. Conservative management may be warranted especially in unilateral or mild cases. For bilateral cases, a multi-disciplinary approach involving cornea, glaucoma, retina and pediatric ophthalmology specialists is key for obtaining the best outcomes.
7. Congenital ectropion uvea (CEU)
CEU is a rare under-recognized form of anterior segment dysgenesis which can either be an isolated ocular finding or associated with systemic diseases including neurofibromatosis type I, Marfans syndrome, and facial hemihypertrophy [Citation95]. The main clinical characteristic is primary iris pigment epithelial hyperplasia with anterior displacement through the pupil and onto the iris stromal surface (, Left Eye) [Citation96]. Especially in infants and young children, this may initially appear to be anisocoria or irregular pupil shape. This is combined with a featureless iris which lacks crypts, furrows and rings and a dysgenic angle with a high scalloped iris insertion that does not extend above Schwalbe’s line [Citation97]. Histology has shown an endothelial-like cell layer that overlies the iris, and it is hypothesized that this is comprised of remnant primordial neural crest-derived cells that did not regress during development [Citation98].
Figure 5. Congenital ectropion uvea (CEU). A patient with unilateral congenital ectropion uvea (left eye) shows the iris epithelium hypertrophy (white arrow) around the pupil (white circle). The iris also shows stromal atrophy (black arrowhead) compared to the unaffected right eye. The left optic nerve is also more cupped than the right eye demonstrating the effect of increased IOP.
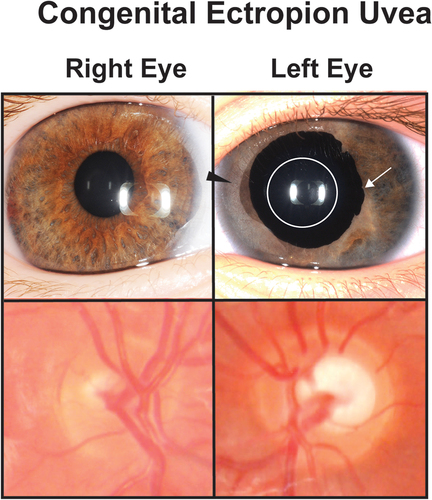
Glaucoma is almost universal in both diagnosis and need for IOP-lowering surgery, however, there is a difference in age of onset between bilateral and unilateral cases [Citation95,Citation99]. Individuals with bilateral CEU present with neonatal-onset glaucoma, which unlike PCG, does not respond to angle surgery [Citation100]. Further, infants with bilateral CEU present with worse corneal edema that less often resolves compared to PCG. In unilateral cases, the age of glaucoma onset is between early childhood to teenage years and may have an insidious onset resulting in late diagnosis [Citation95,Citation97]. The increase in IOP may be due to angle dysgenesis, which can be temporarily responsive to angle surgery. Nevertheless, gonioscopy and anterior segment optical coherence tomography show progressive angle closure that correlates with angle surgery failure, necessitating either trabeculectomy or GDD placement for IOP control [Citation97].
Although rare, CEU is an important entity to recognize in both the bilateral and unilateral forms. In cases of neonatal-onset bilateral CEU, angle surgery can be bypassed for GDD placement to obtain IOP control. In unilateral cases, infants and children may need further systemic work-up, but also need to be monitored carefully for signs of increased IOP.
8. Anterior megalophthalmos and megalocornea
Anterior megalophthalmos is the congenital, non-progressive enlargement of the anterior segment including the cornea, iris and ciliary body in the context of normal IOP while megalocornea is considered to be increased corneal diameter [Citation101]. However, the literature often uses the terms interchangeably as enlargement of the anterior segment including the ciliary body ring is reported in cases of megalocornea [Citation102]. In addition, posterior embryotoxon, cataract, iris hypoplasia, and lens dislocation as well as posterior findings including abnormal oral serrata, lattice degeneration, vitreous abnormalities, and retinal detachments have all been reported in anterior megalophthalmos and megalocornea [Citation103].
By definition the corneal diameter in these diseases is larger than 12.5 mm, and there is a decrease in the vitreous index (length of vitreous cavity/axial length) due to increased anterior chamber depth (). The average normal vitreous index is ~ 69% while in megalophthalmos and megalocornea this can be decreased to the mid-50s [Citation101]. Megalophthalmos and megalocornea may be mistaken for keratoglobus as all have enlarged corneal diameters. However, keratoglobus is considered a progressive disease with vision loss due to increasing pelllucid-like thinning and corneal protrusion, which can lead to corneal scarring and hydrops [Citation104].
Figure 6. Anterior megalophathlmos. Patient with anterior meglophathlmos whose corneas measured over 14 mm in horizontal diameter with no evidence of increased IOP or optic nerve changes. Ultrasound biomicroscopy showed a hyper deep anterior chamber and a thin central cornea.
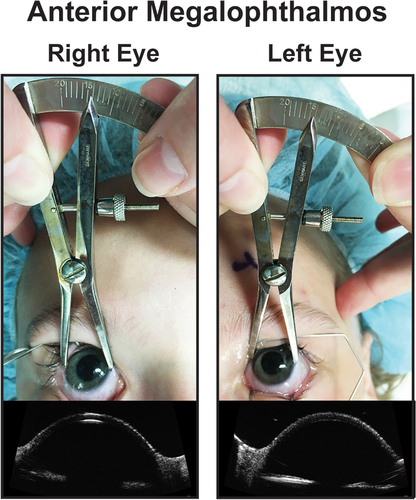
One challenge with anterior megalophthalmos and megalocornea is ruling out childhood glaucoma. An examination under anesthesia may be required. In addition, early onset cataract and lens dislocation is common because of the imbalance between the anterior and posterior segments [Citation101]. Lensectomy can be challenging due to altered fluid dynamics from the extremely deep anterior chamber, large capsular bag, and weakened zonules. Many eyes, especially with lens dislocation may require a capsular tension ring or sutured IOL or left aphakic [Citation102]. Further, careful examination of the pars plana is required as there may be abnormal anterior retinal insertion and retinal dialyses. Prophylactic cryotherapy or laser treatment should be considered if anterior retinal abnormalities are detected [Citation103].
9. Expert opinion
Overall anterior segment dysgeneses are rare with reported prevalence ranging from 1:20,000 (PCG in the United States) to 1:200,000 (ARS). While some of these diseases such as Peters anomaly, sclerocornea, and congenital aniridia are often diagnosed during infancy, others including PCG, ARS, CEU and megalophthalmos/megalocornea may be more subtle. However, these diseases can present unique management challenges that span multiple ophthalmologic subspecialties. A pediatric ophthalmologist should be involved in the care in order to address the visual rehabilitative needs including amblyopia, refractive error, and strabismus. While some pediatric ophthalmologists may be comfortable with angle surgery, most will need assistance with further glaucoma and cornea management. In addition, while these diseases are grouped together as affecting the anterior segment, many also have posterior involvement, specifically at the anterior retinal insertion. A retinal specialist may also be needed for safe placement of a posterior GDD or keratoprosthesis or to assist with an endoscopic approach. Thus, a coordinated multi-specialty approach is often required, yet there is a paucity of not only pediatric ophthalmologists but also specialists in glaucoma, cornea, and retina who are well-versed and truly interested in these diseases.
The specialization of ophthalmology often leans more toward adult care, and most ophthalmologists in the United States (U.S.) are less inclined to incorporate pediatric patients into their practice. This mind-set as well as limited medical student exposure to pediatric ophthalmology, may prevent trainees who have a strong interest in working with children from entering the specialty. This is further fueled by poor reimbursements especially with Medicaid, the greater clinic and OR time commitment that children require, and the comparatively lower salaries. The trickle down effect is that 5% or less of graduating ophthalmology residents in the U.S. choose to do a pediatric ophthalmology fellowship. This is a public health issue as there currently are not enough pediatric ophthalmologists for the U. S. population, and this is being further exacerbated as more practitioners are retiring than are being trained. More efforts need to be made at the medical student and resident levels to encourage interest in managing pediatric eye diseases. In addition, lobbying the state and federal governments to equalize payments for pediatric care compared to adults is imperative.
While increasing interest in working with children is needed, more training in pediatric diseases should be incorporated into glaucoma, cornea, and retina fellowships. These fellowships typically focus 99% of time and effort on adult diseases, which can create apathy toward pediatric conditions. The mantra ‘children are not little adults’ certainly applies, but is even more true in congenital diseases where the anatomy is inherently abnormal. As a result, good exposure and training, which is often lacking, is imperative for surgeons operating on eyes with anterior segment dysgenesis.
From a glaucoma perspective, approaches not typically employed in adults, may be warranted in children such as combining a Stage 1 Baerveldt with angle surgery, especially if goniotomy or trabeculotomy is not expected to yield long-term IOP control. In addition, combining strabismus surgery with GDD placement, in particular Baerveldts, should be considered if there is preexisting strabismus, which may be working against optimal visual outcomes from an amblyopia standpoint or causing cosmetic or social concerns for the patient and parents/guardians. Cornea surgery in children also has many pitfalls, especially regarding penetrating keratoplasty. Suture removal must be done much earlier in children than adults as suture loosening leads to inflammation, neovascularization, and subsequent graft failure over a quicker timeframe compared to adults. Retinal surgery may also not be straightforward due to abnormal anterior retinal insertion and the robust proliferative vitreoretinopathy response in children. While preventing iatrogenic retinal breaks is always important, in eyes with anterior segment dysgenesis and congenital diseases it is critical to recognize any inherently abnormal anatomy. The differences in surgeries and the recognition of bigger picture challenges in children are not widely taught making it such that most specialists are not comfortable with these complex pediatric diseases.
Many of the diseases categorized as anterior segment dysgenesis are complex and require multiple surgeries. For best outcomes, it is important that the specialists communicate and coordinate with each other and have a good understanding of the disease and anatomy. This is unfortunately not the case in many instances as there are few pediatric glaucoma, pediatric cornea, and pediatric retina specialists. More emphasis on pediatric diseases during training is needed to inspire the newer generations of ophthalmologists to gain an appreciation and passion for treating children and complex congenital anomalies.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22(5):314–324. doi: 10.1097/ICU.0b013e328349412b
- Ito YA, Walter MA. Genomics and anterior segment dysgenesis: a review. Clin Exp Ophthalmol. 2014;42(1):13–24. doi: 10.1111/ceo.12152
- Harissi-Dagher M, Colby K. Anterior segment dysgenesis: Peters anomaly and sclerocornea. Int Ophthalmol Clin. 2008;48(2):35–42. doi: 10.1097/IIO.0b013e318169526c
- Ma AS, Grigg JR, Jamieson RV. Phenotype-genotype correlations and emerging pathways in ocular anterior segment dysgenesis. Hum Genet. 2019;138(8–9):899–915. doi: 10.1007/s00439-018-1935-7
- Faiq M, Sharma R, Dada R, et al. Genetic, biochemical and clinical insights into primary congenital glaucoma. J Curr Glaucoma Pract. 2013;7(2):66–84. doi: 10.5005/jp-journals-10008-1140
- Ko F, Papadopoulos M, Khaw PT. Primary congenital glaucoma. Prog Brain Res. 2015;221:177–189.
- Kaushik S, Dubey S, Choudhary S, et al. Anterior segment dysgenesis: insights into the genetics and pathogenesis. Indian J Ophthalmol. 2022;70(7):2293–2303. doi: 10.4103/ijo.IJO_3223_21
- Tawara A, Inomata H. Developmental immaturity of the trabecular meshwork in congenital glaucoma. Am J Ophthalmol. 1981;92(4):508–525. doi:10.1016/0002-9394(81)90644-9
- Barkan O. Pathogenesis of congenital glaucoma: gonioscopic and anatomic observation of the angle of the anterior chamber in the normal eye and in congenital glaucoma. Am J Ophthalmol. 1955;40(1):1–11. doi:10.1016/0002-9394(55)92114-0
- Gupta V, Chaurasia AK, Gupta S, et al. In vivo analysis of angle dysgenesis in primary congenital, juvenile, and adult-onset open angle glaucoma. Invest Ophthalmol Vis Sci. 2017;58(13):6000–6005. doi: 10.1167/iovs.17-22695
- Karaconji T, Zagora S, Grigg JR. Approach to childhood glaucoma: a review. Clin Exp Ophthalmol. 2022;50(2):232–246. doi:10.1111/ceo.14039
- de Oliveira RC, Wilson SE. Descemet’s membrane development, structure, function and regeneration. Exp Eye Res. 2020;197:108090. doi: 10.1016/j.exer.2020.108090
- Glaser TS, Go MS, Kelly MP, et al. Intraoperative mounted optical coherence tomography findings following reversal of optic nerve head cupping in childhood glaucoma. Am J Ophthalmol. 2022;243:109–117. doi: 10.1016/j.ajo.2022.08.003
- Ely AL, El-Dairi MA, Freedman SF. Cupping reversal in pediatric glaucoma–evaluation of the retinal nerve fiber layer and visual field. Am J Ophthalmol. 2014;158(5):905–915. doi: 10.1016/j.ajo.2014.07.030
- Walton DS, Nagao K, Yeung HH, et al. Late-recognized primary congenital glaucoma. J Pediatr Ophthalmol Strabismus. 2013;50(4):234–238. doi: 10.3928/01913913-20130423-02
- Alayu KS, Shibeshi MA, Alemu AM. Spontaneously arrested bilateral primary congenital glaucoma: a case report from Ethiopia. Ethiop J Health Sci. 2022;32(2):463–466. doi: 10.4314/ejhs.v32i2.27
- McPherson SDJ, McFarland D. External trabeculotomy for developmental glaucoma. Ophthalmol. 1980;87(4):302–305. doi: 10.1016/S0161-6420(80)35233-0
- Gramer E, Tausch M, Kraemer C. Time of diagnosis, reoperations and long-term results of goniotomy in the treatment of primary congenital glaucoma: a clinical study. Int Ophthalmol. 1996;20(1–3):117–123. doi: 10.1007/BF00212957
- Walton DS, Katsavounidou G. Newborn primary congenital glaucoma. J Pediatr Ophthalmol Strabismus. 2005;42:333–341. doi: 10.3928/01913913-20051101-01
- Ghate D, Wang X. Surgical interventions for primary congenital glaucoma. Cochrane Database Syst Rev. 2015;1:Cd008213. doi: 10.1002/14651858.CD008213.pub2
- Barkan O. Surgery of congenital glaucoma: review of 196 eyes operated by goniotomy. Am J Ophthalmol. 1953;36(11S):1523–1534.
- Allen L, Burian HM. Trabeculotomy ab externo. A new glaucoma operation: technique and results of experimental surgery. Am J Ophthalmol. 1962;53:19–26.
- Mandal AK, Gothwal VK, Khanna R. Combined trabeculotomy-trabeculectomy for primary congenital glaucoma: long-term experience from a tertiary referral centre in a developing nation. Acta Ophthalmol. 2022;100(2):e439–e447. doi: 10.1111/aos.14984
- Sarkisian SRJ. An illuminated microcatheter for 360-degree trabeculotomy [corrected] in congenital glaucoma: a retrospective case series. J AAPOS. 2010;14(5):412–416. doi: 10.1016/j.jaapos.2010.07.010
- Beck AD, Lynn MJ, Crandall J, et al. Surgical outcomes with 360-degree suture trabeculotomy in poor-prognosis primary congenital glaucoma and glaucoma associated with congenital anomalies or cataract surgery. J AAPOS. 2011;15(1):54–58. doi: 10.1016/j.jaapos.2010.12.002
- Rojas C, Bohnsack BL. Rate of complete catheterization of Schlemm’s canal and trabeculotomy success in primary and secondary childhood glaucomas. Am J Ophthalmol. 2020;212:69–78. doi: 10.1016/j.ajo.2019.11.029
- Al-Hazmi A, Awad A, Zwaan J, et al. Correlation between surgical success rate and severity of congenital glaucoma. Br J Ophthalmol. 2005;89(4):449–453. doi: 10.1136/bjo.2004.047761
- Yalvac IS, Satana B, Suveren A, et al. Success of trabeculotomy in patients with congenital glacuoma operated on within 3 months of birth. Eye (Lond). 2007;21(4):459–464. doi: 10.1038/sj.eye.6702223
- Waheed S, Ritterband DC, Greenfield DS, et al. Bleb-related ocular infection in children after trabeculectomy with mitomycin C. Ophthalmol. 1997;104(12):2117–2120. doi: 10.1016/S0161-6420(97)30051-7
- Jabeen S, Noorani S, Memon MN, et al. Success rate of augmented trabeculectomy in primary congenital glaucoma. J Pediatr Ophthalmol Strabismus. 2022;59(3):180–186. doi: 10.3928/01913913-20211027-01
- Beck AD, Freedman SF, Kammer J, et al. Aqueous shunt devices compared with trabeculectomy with mitomycin-C for children in the first two years of life. Am J Ophthalmol. 2003;136(6):994–1000. doi: 10.1016/S0002-9394(03)00714-1
- Netland PA, Walton DS. Glaucoma drainage implants in pediatric patients. Ophthalmic Surg Lasers Imaging. 1993;24(11):723–729. doi: 10.3928/1542-8877-19931101-04
- Jacobson A, Besirli CG, Bohnsack BL. Outcomes of Baerveldt glaucoma drainage devices in pediatric eyes. J Glaucoma. 2021 Dec 21;21(1). doi: 10.1186/s12886-021-01827-4
- Jacobson A, Bohnsack BL. Ologen augmentation of Ahmed valves in pediatric glaucomas. J AAPOS. 2022;26(3):e1–122.e126. doi: 10.1016/j.jaapos.2022.02.009
- Ozgonul C, Besirli CG, Bohnsack BL. Combined vitrectomy and glaucoma drainage device implantation surgical approach for complex pediatric glaucomas. J AAPOS. 2017;21(2):121–126. doi: 10.1016/j.jaapos.2017.02.001
- Talsania SD, Nallasamy N, Lee AR, et al. Risk factors for strabismus following glaucoma drainage device implantation for refractory childhood glaucoma. J AAPOS. 2019;23(3):.e145.141–.e145.146. doi: 10.1016/j.jaapos.2019.02.005
- Lavin MJ, Franks WA, Wormald RP, et al. Clinical risk factors for failure in glaucoma tube surgery. A comparison of three tube designs. Arch Ophthalmol. 1992;110(4):480–485. doi: 10.1001/archopht.1992.01080160058030
- Shah AA, WuDunn D, Cantor LB. Shunt revision versus additional tube shunt implantation after failed tube shunt surgery in refractory glaucoma. Am J Ophthalmol. 2000;129(4):455–460. doi: 10.1016/S0002-9394(99)00410-9
- Zuo W, Lesk MR. Surgical outcome of replacing a failed Ahmed glaucoma valve by a baerveldt glaucoma implant in the same quadrant in refractory glaucoma. J Glaucoma. 2018;27(5):421–428. doi: 10.1097/IJG.0000000000000912
- Sood S, Beck AD. Cyclophotocoagulation versus sequential tube shunt as a secondary intervention following primary tube shunt failure in pediatric glaucoma. J AAPOS. 2009;13(4):379–383. doi: 10.1016/j.jaapos.2009.05.006
- Glaser TS, Mulvihill MS, Freedman SF. Endoscopic cyclophotocoagulation (ECP) for childhood glaucoma: a large single-center cohort experience. J AAPOS. 2019;23(2):.84.e81–.84.e87. doi: 10.1016/j.jaapos.2018.10.014
- Huang SC, Soong HK, Brenz RM, et al. Problems associated with penetrating keratoplasty for corneal edema in congenital glaucoma. Ophthalmic Surg. 1989;20(6):399–402. doi: 10.3928/1542-8877-19890601-05
- Hu Y, Fang L, Guo X, et al. Corneal configurations and high-order aberrations in primary congenital glaucoma. J Glaucoma. 2018;27(12):1112–1118. doi: 10.1097/IJG.0000000000001049
- Samant M, Chauhan BK, Lathrop KL, et al. Congenital aniridia: etiology, manifestations and management. Expert Rev Ophthalmol. 2016;11(2):135–144. doi: 10.1586/17469899.2016.1152182
- Jacobson A, Mian SI, Bohnsack BL. Clinical outcomes and visual prognostic factors in congenital aniridia. BMC Ophthalmol. 2022;22(1):235.
- Netland PA, Scott ML, Boyle JW, et al. Ocular and systemic findings in a survey of aniridia subjects. J AAPOS. 2011;15(6):562–566. doi: 10.1016/j.jaapos.2011.07.009
- Chang JW, Kim JH, Kim S-J, et al. Congenital aniridia: long-term clinical course, visual outcome, and pronostic factors. Korean J Ophthalmol. 2014;28(6):479–485. doi: 10.3341/kjo.2014.28.6.479
- Wang JD, Zhang JS, Xiong Y, et al. Congenital aniridia with cataract: case series. BMC Ophthalmol. 2017;17(1):115. doi: 10.1186/s12886-017-0503-6
- Swanner JC, Walton DS, Chen TC. Prevention of aniridic glaucoma with goniosurgery. Int Ophthalmol Clin. 2004;44(1):67–71. doi:10.1097/00004397-200404410-00008
- Balikov DA, Jacobson A, Prasov L. Glaucoma syndromes: insights into glaucoma genetics and pathogenesis from monogenic syndromic disorders. Genes (Basel). 2021;12(9):1403. doi: 10.3390/genes12091403
- Bajwa A, Burstein E, Grainger RM, et al. Anterior chamber angle in aniridia with and without glaucoma. Clin Ophthalmol. 2019;13:1469–1473. doi: 10.2147/OPTH.S217930
- Muñoz-Negrete FJ, Teus MA, García-Feijoó J, et al. Aniridic glaucoma: an update. Arch Soc Esp Oftalmol (Engl Ed). 2021;96(Suppl 1):52–59. doi: 10.1016/j.oftal.2020.11.005
- Adachi M, Dickens CJ, Hetherington JJ, et al. Clinical experience of trabeculotomy for the surgical treatment of aniridic glaucoma. Ophthalmol. 1997;104(12):2121–2125. doi: 10.1016/S0161-6420(97)30041-4
- Durai I, Pallamparthy S, Puthuran GV, et al. Outcomes of glaucoma drainage device implantation and trabeculectomy with mitomycin C in glaucoma secondary to aniridia. Am J Ophthalmol. 2021;227:173–181. doi: 10.1016/j.ajo.2021.03.008
- Arroyave CP, Scott IU, Gedde SJ, et al. Use of glaucoma drainage devices in the management of glaucoma associated with aniridia. Am J Ophthalmol. 2002;135:155–159. doi: 10.1016/S0002-9394(02)01934-7
- Demirok GS, Eksioglu U, Yakin M, et al. Short- and long-term results of glaucoma valve implantation for aniridia-related glaucoma: a case series and literature review. Turk J Ophthalmol. 2019;49:183–187. doi: 10.4274/tjo.galenos.2019.07348
- Latta L, Figueiredo FC, Ashery-Padan R, et al. Pathophysiology of aniridia-associated keratopathy: developmental aspects and unanswered questions. Ocul Surf. 2021;22:245–266. doi: 10.1016/j.jtos.2021.09.001
- van Velthoven AJH, Utheim TP, Notara M, et al. Future directions in managing aniridia-associated keratopathy. Surv Ophthalmol. 2023;68(5):940–956. doi: 10.1016/j.survophthal.2023.04.003
- Cheung AY, Sarnicola E, Kurji KH, et al. Cincinnati protocol for preoperative screening and donor selection for ocular surface stem cell transplantation. Cornea. 2018;37(9):1192–1197. doi: 10.1097/ICO.0000000000001662
- Holland EJ, Djalilian AR, Schwartz GS. Management of aniridic keratopathy with keratolimbal allograft: a limbal stem cell transplantation technique. Ophthalmol. 2003;110(1):125–130. doi: 10.1016/S0161-6420(02)01451-3
- Kremer I, Rajpal RK, Rapuano CJ, et al. Results of penetrating keratoplasty in aniridia. Am J Ophthalmol. 1993;115(3):317–320. doi: 10.1016/S0002-9394(14)73581-0
- Nascimento ESR, Shen LQ, Chiou CA, et al. Glaucoma management in patients with aniridia and boston type 1 keratoprosthesis. Am J Ophthalmol. 2019;207:258–267. doi: 10.1016/j.ajo.2019.06.018
- Bonneau S, Tong CM, Yang Y, et al. The treatment of end-stage corneal disease: penetrating keratoplasty compared with Boston type 1 keratoprosthesis. Graefes Arch Clin Exp Ophthalmol. 2022;260(9):2781–2790. doi: 10.1007/s00417-022-05646-1
- Romano D, Bremond-Gignac D, Barbany M, et al. Artificial iris implantation in congenital aniridia: a systematic review. Surv Ophthalmol. 2022;68(4):794–808. doi: 10.1016/j.survophthal.2022.11.001
- Rickmann A, Szurman P, Januschowski K, et al. Long-term results after artificial iris implantation in patients with aniridia. Graefes Arch Clin Exp Ophthalmol. 2016;254(7):1419–1424. doi: 10.1007/s00417-016-3292-3
- Shields MB, Buckley E, Klintworth GK, et al. Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Surv Ophthalmol. 1985;29(6):387–409. doi: 10.1016/0039-6257(85)90205-X
- Ozeki H, Shirai S, Ikeda K, et al. Anomalies associated with Axenfeld–Rieger syndrome. Graefes Arch Clin Exp Ophthalmol. 1999;237(9):730–734. doi: 10.1007/s004170050304
- Michels K, Bohnsack BL. Ophthalmological manifestations of axenfeld-rieger syndrome: current perspectives. Clin Ophthalmol. 2023;17:819–828. doi:10.2147/OPTH.S379853
- Zepeda EM, Branham K, Moroi SE, et al. Surgical outcomes of glaucoma associated with Axenfeld–Rieger syndrome. BMC Ophthalmol. 2020;20:172. doi: 10.1186/s12886-020-01417-w
- Reis LM, Maheshwari M, Capasso J, et al. Axenfeld-Rieger syndrome: more than meets the eye. J Med Genet. 2022;60(4):368–379. doi: 10.1136/jmg-2022-108646
- Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc. 1983;81:736–784.
- Alsheikheh A, Klink J, Klink T, et al. Long-term results of surgery in childhood glaucoma. Graefes Arch Clin Exp Ophthalmol. 2007;245(2):195–203. doi: 10.1007/s00417-006-0415-2
- Shigeyasu C, Yamada M, Mizuno Y, et al. Clinical features of anterior segment dysgenesis associated with congenital corneal opacities. Cornea. 2012;31(3):293–298. doi: 10.1097/ICO.0b013e31820cd2ab
- Kurilec JM, Zaidman GW. Incidence of Peters anomaly and congenital corneal opacities interfering with vision in the United States. Cornea. 2014;33(8):848–850. doi:10.1097/ICO.0000000000000182
- Bhandari R, Ferri S, Whittaker B, et al. Peters anomaly: review of the literature. Cornea. 2011;30(8):939–944. doi: 10.1097/ICO.0b013e31820156a9
- Dolezal KA, Besirli CG, Mian SI, et al. Glaucoma and cornea surgery outcomes in Peters anomaly. Am J Ophthalmol. 2019;208:367–375. doi: 10.1016/j.ajo.2019.08.012
- Rao KV, Fernandes M, Gangopadhyay N, et al. Outcome of penetrating keratoplasty for Peters anomaly. Cornea. 2008;27(7):749–753. doi: 10.1097/ICO.0b013e31816fe9a7
- Lowe MT, Keane MC, Coster DJ, et al. The outcome of corneal transplantation in infants, children, and adolescents. Ophthalmol. 2011;118(3):492–497. doi: 10.1016/j.ophtha.2010.07.006
- Yang LL, Lambert SR. Peters’ anomaly. A synopsis of surgical management and visual outcome. Ophthalmol Clin North Am. 2001;14(3):467–477. doi:10.1016/S0896-1549(05)70245-5
- Chang JW, Kim JH, Kim SJ, et al. Long-term clinical course and visual outcome associated with Peters’ anomaly. Eye (Lond). 2012;26(9):1237–1242. doi: 10.1038/eye.2012.128.
- Aldave AJ, Kamal KM, Vo RC, et al. The Boston type I keratoprosthesis: improving outcomes and expanding indications. Ophthalmol. 2009;116(4):640–651. doi: 10.1016/j.ophtha.2008.12.058
- Liu YC, Soh YQ, Kocaba V, et al. Selective endothelial removal: a case series of a phase I/II surgical trial with long-term follow up. Front Med. 2022;9:901187. doi: 10.3389/fmed.2022.901187
- Ramappa M, Chaurasia S, Mohamed A, et al. Selective endothelialectomy in Peters anomaly: a novel surgical technique and its clinical outcomes in children. Cornea. 2022;41(12):1477–1486. doi: 10.1097/ICO.0000000000003134
- Spierer O, Cavuoto KM, Suwannaraj S, et al. Outcome of optical iridectomy in Peters anomaly. Graefes Arch Clin Exp Ophthalmol. 2018;256(9):1679–1683. doi: 10.1007/s00417-018-4000-2
- Rajagopal RN, Fernandes M. Peters anomaly: novel non-invasive alternatives to penetrating keratoplasty. Semin Ophthalmol. 2023;38(3):275–282. doi:10.1080/08820538.2023.2176238
- Yang LL, Lambert SR, Lynn MJ, et al. Surgical management of glaucoma in infants and children with Peters’ anomaly: long-term structural and functional outcome. Ophthalmol. 2004;111(1):112–117. doi: 10.1016/j.ophtha.2003.02.002
- Jacobson A, Besirli CG, Bohnsack BL. Outcomes of combined endoscopic vitrectomy and posteriorly placed glaucoma drainage devices in pediatric patients. BMC Ophthalmol. 2022;22(1):149. doi: 10.1186/s12886-022-02373-3
- Nischal KK. Genetics of congenital corneal opacification–impact on diagnosis and treatment. Cornea. 2015;34 Suppl(10):S24–34. doi: 10.1097/ICO.0000000000000552
- Ma DH, Yeh LK, Chen HC, et al. Epithelial phenotype in total sclerocornea. Mol Vis. 2014;20:468–479.
- Kanai A, Wood TC, Polack FM, et al. The fine structure of sclerocornea. Investig Ophthalmol Vis Sci. 1971;10(9):687–694.
- Quiroz-Casian N, Chacon-Camacho OF, Barragan-Arevalo T, et al. Sclerocornea-microphthalmia-aphakia complex: description of two additional cases associated with novel FOXE3 mutations and review of the literature. Cornea. 2018;37(9):1178–1181. doi: 10.1097/ICO.0000000000001655
- Sharma N, Agarwal R, Jhanji V, et al. Lamellar keratoplasty in children. Surv Ophthalmol. 2020;65(6):675–690. doi: 10.1016/j.survophthal.2020.04.002
- Plaisancié J, Ragge NK, Dollfus H, et al. FOXE3 mutations: genotype-phenotype correlations. Clin Genet. 2018;93(4):837–845. doi: 10.1111/cge.13177
- Thompson AC, Thompson MO, Lim ME, et al. Microphthalmia, dermal aplasia, and sclerocornea syndrome: endoscopic cyclophotocoagulation in the management of congenital glaucoma. J Glaucoma. 2018;27(1):e7–e10. doi: 10.1097/IJG.0000000000000812
- Jacobson A, Moroi SE, Bohnsack BL. Characteristics and outcomes of glaucoma associated with congenital ectropion uvea. Am J Ophthalmol. 2022;241:1–8. doi:10.1016/j.ajo.2021.08.023
- Dowling JL, Albert DM, Nelson LB, et al. Primary glaucoma associated with iridotrabecular dysgenesis and ectropion uveae. Ophthalmol. 1985;92(7):912–921. doi: 10.1016/S0161-6420(85)33935-0
- Wang GM, Thuente D, Bohnsack BL. Angle closure glaucoma in congenital ectropion uvea. Am J Ophthalmol Case Rep. 2018;10:215–220. doi: 10.1016/j.ajoc.2018.03.009
- Edward DP, Morales J, Bouhenni RA, et al. Congenital ectropion uvea and mechanisms of glaucoma in neurofibromatosis type 1. Ophthalmol. 2012;119:1485–1494. doi: 10.1016/j.ophtha.2012.01.027
- Ritch R, Forges M, Hetherington J, et al. Congenital ectropion uveae with glaucoma. Ophthalmol. 1984;91:326–331. doi: 10.1016/S0161-6420(84)34288-9
- Kaushik S, Choudhary S, Kaur A, et al. Neonatal-onset congenital ectropion uveae may be caused by a distinct CYP1B1 pathologic variant. Am J Ophthalmol. 2022;239:54–65. doi: 10.1016/j.ajo.2022.01.014
- Ong APC, Zhang J, Vincent AL, et al. Megalocornea, anterior megalophthalmos, keratoglobus and associated anterior segment disorders: a review. Clin Exp Ophthalmol. 2021;49(5):477–497. doi: 10.1111/ceo.13958
- Messina M, Ross AR, Pocobelli G, et al. Cataract surgery with intraocular lens implantation in 3 brothers with megalocornea: long-term follow-up. J Cataract Refract Surg. 2018;44(3):399–402. doi: 10.1016/j.jcrs.2018.01.020
- Kumawat D, Alam T, Sahay P, et al. Ocular abnormalities and complications in anterior megalophthalmos: a case series. Eye (Lond). 2019;33(5):826–832. doi: 10.1038/s41433-018-0329-3
- Rathi VM, Murthy SI, Bagga B, et al. Keratoglobus: an experience at a tertiary eye care center in India. Indian J Ophthalmol. 2015;63(3):233–238. doi: 10.4103/0301-4738.156927