
1. Introduction
Chronic myeloid leukemia (CML) is a clonal hematological neoplasm caused by a translocation between BCR and ABL1 genes, resulting the chimeric BCR-ABL1 gene encoding a constitutively active tyrosine kinase [Citation1]. Blocking of the biochemical activity of this protein produces clinical, cytogenetic, and molecular remissionsFootnote1 in persons with CML.
Substantial data indicate BCRABL1 is necessary and sufficient to cause CML (reviewed in [Citation2]). The ability of ionizing radiations to cause contiguous double-stand DNA breaks proximity of BCR and ABL1 in the interphase nucleus and detection of BCRABL1 transcripts in leukemia cells from atomic bomb survivors with CML. CML developing in these persons began when they were exposed to radiations from the A-bombs [Citation3,Citation4]. If we accept this hypothesis we can estimate (minimally) the interval from developing BCR-ABL1 to the diagnosis of chronic phase CML. These data suggest a latency of 2 to 30–40 years [Citation5]. Whether other mutations precede BCRABL1 in general or did so in the atomic bomb survivors developing CML is controversial but seems unlikely to be of fundamental import in the pathogenesis of the disease.
Biochemical targets of P210BCRABL1 are reviewed elsewhere [Citation6]. Tyrosine kinase-inhibitors (TKIs) such as imatinib, dasatinib, nilotinib, bosutinib, and ponatinib antagonize this aberrant biochemical activity resulting in a reduction in the size of the CML clone with attendant hematological, cytogenetic and molecular remissions. Some of these molecular remissions result in a ≤ 4.5 log reduction in numbers of CML cells. Whether persons with a negative polymerase chain reaction test for BCRABL1 transcripts have fewer or no residual CML cells is presently unknowable. However, for several reasons including the mechanism-of-action of TKIs, sampling error [Citation7], and limitations of the standard mRNA-based PCR assay (reviewed in reference [Citation8]), it is unlikely even prolonged TKI therapy results in eradication of all CML cells particularly the so-called CML stem cell in which the disease begins. For example, it is unlikely the CML stem cell neither transcribes BCRABL1 nor translates it. If so, how is it possible to stop TKI therapy after 2–3 years of deep molecular remission and not have recurrent leukemia for several years?
2. Hypotheses
A model of CML initiation, latency, evolution, diagnosis, TKI therapy, deep molecular response, TKI therapy discontinuation, and therapy-free remission (TFR) is shown in . One possibility (; upper path, right), exceedingly unlikely for reasons discussed previously, is TKI therapy directly eliminates the CML stem cell(s). Most kinetic models suggest it would take >20–25 years of TKI therapy to eradicate the CML clone [Citation9]. Another possibility is all CML cells are eliminated, not as a direct effect of TKI therapy but by clonal exhaustion (reviewed in reference [Citation10]). This might be a direct consequence of depleting the clone by continued pressure to proliferate, by loss of a critical, necessary microenvironmental or autocrine stimulus or a combination. However, the more likely possibility is CML stem cells remain after TKI therapy and that other mechanisms explain why CML does not recur in up to 50 percent of subjects with a deep molecular response for >2–3 years after discontinuing TKI therapy (TFR). These four mechanisms include: (1) latency; (2) immune-mediated control of residual CML cells; (3) exhaustion of the CML clone (perhaps best regarded as a form of elimination of the CML clone); and (4) stochastic events whereby cells biologically able to cause CML recurrence simply do not divide. This can also be regarded as one mechanism underlying latency but need not be identical. We discuss the likelihood of one or more of these mechanisms operate to produce TFR ().
Figure 1. Evolution of CML and effects of therapy.
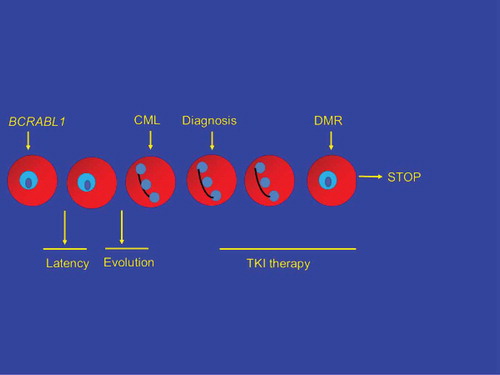
Figure 2. Direct eradication of CML.
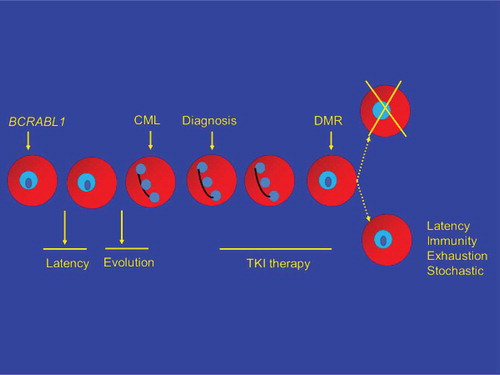
Figure 3. Indirect eradication or control of CML.
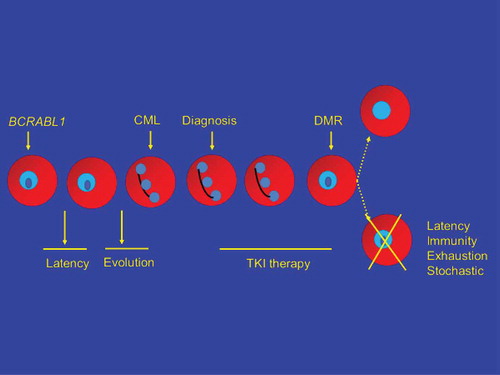
3. Latency
Data in atomic bomb survivors indicate latency, defined as the interval from the likely formation of BCRABL1 to diagnosis of CML is extraordinarily heterogeneous ranging from 2 to >3–40 years [Citation4,Citation5]. It could, of course, be longer in view of competing causes of death. Moreover, it is possible to roughly estimate how many persons with BCRABL1 in a cell with the biological ability to cause CML never developed it even after adjusting for competing cause of death and for surveillance biases. The implication is even though BCRABL1 in the right (or wrong) cell is necessary and sufficient to cause CML, CML does not develop in everyone with BCRABL1 in this cell, a finding consistent with reports of BCRABL1 transcripts in normal although the cell(s) transcribing the gene is unknown [Citation11–Citation13]. The conclusion of these data is it can take a very long time (or possibly forever given the limited human lifespan and competing causes of death) for clinical CML to evolve from a CML stem cell even if we assume a constant rate of cell division. The latter is most unlikely (see next). Other data from allotransplants in persons with CML indicate persons in remission for 5 years posttransplant have a slow, unrelenting risk of CML relapse after 5 and 10 more years with rates of 4 percent (95% confidence interval [CI], 3–5%) and 7 percent (5–9%) [Citation14]. Again, CML takes a long time to recur when numbers of CML cells are markedly reduced. Although part of latency can be contributed to by stochastic events such as the interval from acquiring BCRABL1 to the 1st asymmetrical division which gives rise to a cell able to cause CML, we distinguish latency from stochastic events (see next).
4. Immune-mediated mechanisms
There is considerable, perhaps unbridled enthusiasm for invoking the immune system in what could be termed unmeasurable residual disease after stopping TKI therapy. Several reports detail changes in immunity-related blood cells favoring this mechanism in persons with TFR [Citation15,Citation16].
Other reports correlate levels of immune cells with likelihood of achieving TFR (reviewed in reference [Citation17]). However, these associations should not be mistaken for cause-and-effect, which is an entirely different matter. CML begins in one cell and it is unclear why the immune system did not control CML before there were many CML cells requiring TKI therapy for disease control. One could speculate that TKI therapy stimulates anti-CML immunity but few data support this notion.
Is the immune system important in controlling CML? We think this unlikely. Children and adults with defective immunity have an increased risk of virus-related neoplasms and a few specific cancers such as melanoma, kidney cancer, and neuroblastoma but not CML. We reviewed data in >355,000 kidney transplant recipients with 1.4 × 10E+6 years at risk but found only 14 excess cases of CML [Citation18]. These data do not support the notion of a strong role for immune surveillance against CML. Whether it might operate in some persons alone or in cooperation with other mechanisms is uncertain.
5. Stochastic events
Hematopoietic stem cells, likely the cell in which CML begins, do not like to divide or do so only reluctantly. This makes evolutionary sense because the estimated 10E+4 hematopoietic stem cells in a normal person must sustain hematopoiesis for >80 years. Protection from external stimuli is essential to prevent exhausting the reservoir of hematopoietic stem cells. The process by which a hematopoietic stem cell decides to divide is unknown but is generally regarded as stochastic (reviewed in reference [Citation19]). The point is a hematopoietic stem cell with BCRABL1 is of no clinical relevance until the cell divides asymmetrically giving rise to a progeny capable of causing CML. Most data suggest this process occurs by chance and is best described a probability distribution much like the emission of a subatomic particle from a radionuclide. The point is that after stopping TKI therapy, the CML stem cell(s) might not divide for a long time (or perhaps forever or at least as the person with the cell lives). To extend the probability distribution of a hematopoietic stem cell dividing might be nonlinear. This process can be compared with the notion of latency. However, the distribution of CML in persons exposed to atomic bombs suggests different processes.
6. Summary
Almost 50 percent of persons with CML with a deep molecular remission for >2–3 years can stop TKI therapy without CML recurrence for several years. How TFR is achieved is unknown and controversial. We present data suggesting many mechanisms may operate. We think cancer latency and stochastic events are the most likely alone or combined but elimination of the leukemia clone by clonal exhaustion or immune mechanisms may also occur. It is also possible that different mechanisms, alone or combined, may operate in different persons with TFR.
Declaration of interest
A Hochhaus receives support from Novartis, Pfizer, Bristol-Myers Squibb, Incyte, Pfizer. RP Gale acknowledges support from the National Institute of Health Research (NIHR) Biomedical Research Centre funding scheme and is a part-time employee of Celgene Corp. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
One peer reviewer on this paper declares speaker honoraria from Novartis, Bristol-Myers Squibb, Pfizer, and Incyte. Peer reviewers have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed
Additional information
Funding
Notes
1. The concept of remission is widely used but its biological validity depends on the sensitivity and specificity of the assay used to define remission. Remission should not be confused with leukemia eradication because of the limited sensitivity of current assays.
References
- Hoyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017;129:21595–21606.
- Goldman JM, Melo JV. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464.
- Lukásová E, Kozubek S, Kozubek M, et al. Localization and distance between ABL and BCR genes in interphase nuclei of bone marrow cells of control donors and patients with chronic myeloid leukemia. Hum Genet. 1997;100:525–535.
- Aurer I, Juhbashi T, Sekine I, et al. Analysis of BCR/ABL abnormalities in mRNA from 20-year-old paraffin-embedded tissue for BCR/ABL rearrangement by polymerase chain reaction. Acta Haematol. 1993;90:5–7.
- Hsu WL, Preston DL, Soda M, et al. The incidence of leukemia, lymphoma and multiple myeloma among A-bomb survivors: 1950-2001. Radiat Res. 2013;179:361–382.
- Dhut S, Chaplin T, Young BD. BCR-ABL and BCR proteins: biochemical characterization and localization. Leukemia. 1990;4:745–750.
- Butturini A, Klein J, Gale RP. Modeling minimal residual disease (MRD)-testing. Leuk Res. 2003;27:293–300.
- Alikian M, Gale RP, Apperley JF, et al. Molecular technique for the personalized management of patients with chronic myeloid leukemia. Biomol Detect Qualif. 2017;11:4–20.
- Roeder I, Horn M, Glauche I, et al. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat Med. 2006;12:1181–1184.
- Ross DM, Hughes TP, Melo JV. Do we have to kill the last CML cell? Leukemia. 2011;25:193–200.
- Bose S, Deininger M, Gora-Tybor J, et al. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood. 1998 Nov 1;92(9):3362–3367.
- Biernaux C, Loos M, Sels A, et al. Detection of major BCR-ABL gene expression at a very level in blood cells of some healthy individuals. Blood. 1995;86:3118.
- Song J, Mercer D, Hu X, et al. Common leukemia-and lymphoma-associated genetic aberrations in healthy individuals. J Mol Diagn. 2011;13:213–219.
- Goldman JM, Majhail NS, Klein JP. Relapse and late mortality in 5-year survivors of myeloablative allogeneic hematopoietic cell transplantation for chronic myeloid leukemia in first chronic phase. J Clin Oncol. 2010;28:1888–1895.
- Ilander M, Olsson-Strömberg U, Schlums H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. 2017;31:1108–1116.
- Hughes A, Clarson I, Tang C, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood. 2017;129:1166–1176.
- Hughes A, Yong ASM. Immune effector recovery in chronic myeloid leukemia and treatment-free remission. Front Immunol. 2017;24(8):469.
- Gale RP, Opelz G. Is there immune surveillance against chronic myeloid leukemia? Possibly, but not much. Leuk Res. 2017;57:109–111.
- Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2:640–653.