
ABSTRACT
Introduction
Venous thromboembolism (VTE) is a complex disease that aggregates in families. Both acquired and genetic risk factors are important. Proper recognition and management of high-risk individuals are important.
Areas covered
The genetic risk factors for VTE, the clinical consequences, and future perspectives are summarized. Classical thrombophilia i.e., factor V Leiden (rs6025), the prothrombin G20210A mutation (rs1799963), deficiencies of antithrombin, protein C, and protein S and the recent findings from genome wide association studies (GWAS), transcriptome-wide association studies (TWAS), genetic risk score (GRS), VTE candidate genes, expression studies, animal studies, studies using next generation sequencing, pathway analysis, and clinical implications are discussed.
Expert opinion
Screening of inherited thrombophilia should be performed in special cases. Identification of strong risk variants might affect the management. The increasing number of genetic risk variants is likely to change management of VTE.
1. Introduction
Venous thromboembolism (VTE) is next to coronary heart disease (CHD) and stroke, the third most common cardiovascular disorder [Citation1–7]. The most frequent manifestations of VTE, includes superficial thrombophlebitis, deep vein thrombosis (DVT) of the lower extremities and pulmonary embolism (PE), though unusual manifestations also have been described: Budd–Chiari syndrome (hepatic vein occlusions) and thromboses of the vena cava and cerebral, portal, mesenteric, renal, and retinal veins [Citation1–7]. Lifetime risk of VTE is rather high and has been estimated to be 5–10%. VTE recurrence risk is also high [Citation1–8]. The risk of recurrent VTE is around 25% in 5 years.
VTE is a complex disorder with a multicausal origin [Citation6]. Acquired VTE risk factors include immobilization, surgery, trauma, pregnancy, puerperium, malignancy, and oral contraceptives. Twenty-five to 50% of patients with VTE have a spontaneous or unprovoked (idiopathic) VTE event with no identifiable transient risk factors depending on exact definition [Citation7,Citation8]. Patients with unprovoked VTE have especially high risk of recurrence after discontinuation of anticoagulant (AC) treatment [Citation7,Citation8].
VTE has also a strong genetic component with an estimated heritability of 40–60% in studies of families, twins, siblings, and half-siblings [Citation9–12]. That VTE clusters in families has been known for more than a century, reviewed by Zöller et al [Citation13]. Although rare deficiencies of natural anticoagulants antithrombin, protein C, and protein S was discovered among VTE patients early on it was not clear until Dahlbäck et al discovered resistance to activated protein C (APC-resistance) in 1993 that genetic factors were demonstrated to be common in VTE [Citation14,Citation15]. Soon APC-resistance was shown to be linked to a common F5 gene mutation named Factor V Leiden (rs6025) [Citation16–19]. Only two years later another common genetic variant associated with thrombosis was reported (prothrombin G20,210A or rs1799963) [Citation20]. Thrombophilic defects at these five gene loci are called classic thrombophilia [Citation21–23]. The present article will not only review classic thrombophilia but will also go beyond and focus on latest discoveries and future prospects of the genetic risk factors for VTE.
2. Classical thrombophilia
Thrombophilia is defined as an acquired or inherited condition with hypercoagulability that leads to an increased risk for VTE [Citation22]. Such hypercoagulable states involve either alterations in the vessel wall, the blood flow, and/or the composition of the blood (Virchow’s triad). Rudolf Virchow postulated this already in 1856. So far there are five classical inherited thrombophilias: antithrombin deficiency, protein C deficiency, protein S deficiency, resistance to activated protein C due to Factor V Leiden (rs6025), and the prothrombin mutation (prothrombin G20210A, rs1799963) [Citation21–25]. Thus, all inherited thrombophilias are either genetic defect in a natural anticoagulant (antithrombin, protein C, and protein S) or a coagulation factor (factor V and factor II) (). In , the more recent and weaker discovered VTE gene loci are marked with orange and the classical thrombophilia loci with red.
Figure 1. The Coagulations system and its regulating anticoagulant genes are depicted. The official HGNC (HUGO Gene Nomenclature Committee) abbreviations are used. Classical thrombophilia genes are marked with red and minor VTE risk genes are marked with orange
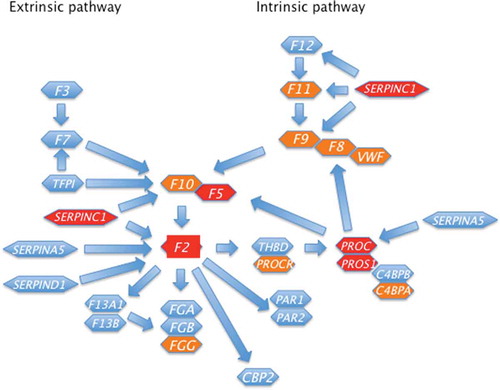
2.1. Deficiencies of the natural anticoagulants antithrombin, protein C, and protein S
Inherited deficiencies of the natural coagulation inhibitors antithrombin, protein C, and protein S are all strong risk factors for VTE [Citation23–25]. However, these deficiencies are rare (<1%) in the population. Typically, these disorders are due to different loss of function (LOF), i.e. gene inactivating, mutations in the SERPINC1, PROC, and PROS1 genes, respectively. Several hundreds of causative mutations have been described responsible for these defects. The most common mutations in the SERPINC1, PROC, and PROS1 genes are nonsynonymous point mutations (missense or nonsense) though deletions or insertions may occur [Citation25]. Moreover, dominant negative mutations (antimorphic mutations) with an altered gene product have been described in the SERPINC1 gene. The nature of the genetic defect and its functional consequences contribute to the heterogeneity of the clinical phenotype of classical thrombophilia. Due to the rareness of these variants, the risk of VTE has mainly been estimated in family studies. A tenfold or higher (especially antithrombin deficiency) increased risk for VTE in heterozygous carriers have been described. However, in studies among unselected consecutive patients with VTE, the risk associated with these deficiencies appears lower than in selected thrombophilic families [Citation26,Citation27]. Familial thrombophilia is believed to be the cause of additional defects that may as yet be unknown [Citation24,Citation25].
Mutations causing inherited deficiencies of antithrombin, protein C or protein S may be associated with decreased plasma immunological and functional levels (Type I deficiency), whereas type II deficiency is associated with functionally abnormal protein molecules [Citation23–25]. Type II antithrombin deficiency is subdivided into three type: HBS (heparin binding site variants), RS (reactive site variants), and PE (variants giving pleiotropic effects). For protein C there exist just two types without subtypes: type I and type II deficiency. Protein S, the cofactor to protein C is a special case. Protein S exists in plasma as free or bound to C4b-binding protein (C4BP). Type I protein S deficiency is characterized by low levels of both free and total protein S. Type II is reserved for qualitative protein S deficiency, while type III deficiency is characterized by low free protein S and normal total protein S. It has been shown that the level of free protein S in plasma discriminates better between those with and without protein S deficiency than the level of total protein S.
2.2. F5 gene and resistance to activated protein C (APC-resistance)
Dahlbäck et al discovered resistance to activated protein C (APC-resistance) in 1993 as a common and strong risk factor for VTE [Citation14,Citation15]. Soon it become clear that in a majority of cases a single mutation (factor V Leiden, rs6025) in the factor V gene (F5) was the major cause of inherited APC-resistance [Citation16–19]. Factor V Leiden is a common gain‐of‐function (GOF) F5 gene mutation, i.e gene activating, with a prevalence of carriers among Caucasians of 5–10% [Citation16–19,Citation23–25]. Among patients with VTE it is found in 20–30%, and in around 50% of patients with familial thrombophilia [Citation23–25]. The variant leads to resistance to activated protein C (APC‐resistance) [Citation23–25]. The Arg506 to Gln mutation is located in the part of the gene encoding for one of the three cleavage sites in factor V (Arg306, Arg506, and Arg679), where APC inactivates factor Va [Citation23–25,Citation28]. The risk of thrombosis is 5‐fold increased in heterozygotes, and fiftyfold in homozygotes [Citation23–25].
More recent studies have identified a number of other GOF variants in the F5 gene among VTE patients, reviewed by Segers et al [Citation28]. However, except the R2 haplotype (rs6033, rs1800595, rs6030, rs6027) and A/G allele variants (rs6021, rs4524, rs4525, rs6032) these APC-resistance associated F5 gene variants are all rare [Citation28]. The rare variants Factor V Cambridge (rs118203906) and Factor V Hong Kong (rs118203905) both affect the APC cleavage site at Arg306 (or Arg534 when numbered from the starting Met including the signal peptide). More recently, some other rare F5 gene mutations such as FV Nara (Trp1920Arg) and FV Bonn (Ala512Val) that also cause APC-resistance and thrombosis have been identified [Citation29–32]. No F8 gene variants have been associated with APC-resistance.
2.3. Prothrombin G20210A (rs1799963)
A G-to-A transition at nucleotide 20210 (prothrombin G20210A, rs1799963) in the 3′‐untranslated part of the prothrombin gene (F2) causes increased prothrombin levels and an increased risk of VTE [Citation20]. This GOF mutation is also prevalent (around 2%) and almost only found in Caucasians. Carriers have a 2‐ to 3‐fold increased risk of venous thrombosis, and the variant is found in approximately 6% of patients with VTE [Citation23–25]. Homozygosity for the rs1799963 variant occurs though rarer than homozygosity for the rs6025 variant. However, the risk for VTE is high and has been reported to be 30 times increased for homozygotes for the rs1799963 variant [Citation23].
2.4. Antithrombin resistance due to prothrombin mutations
In analogy with the APC-resistance phenomena resistance to antithrombin has been reported in young patients with VTE [Citation33–36]. Mutations in the last exon of F2 with substitution of Arginine at residue 596 (Arg596Leu, Arg596Gln, Arg596Trp) have been identified as the cause of antithrombin resistance [Citation33–36]. However, these variants are rare and are not identified in gnomAD (https://gnomad.broadinstitute.org/, accessed 21 July 2020). A formal statistical association with VTE therefore remains to be proven.
2.5. Abnormal factor IX (Padua)
Simioni et al reported in 2009 a 25-year-old man with VTE and around eight times increased factor IX coagulation activity with normal factor IX level [Citation37]. This was due to a factor IX mutation with substitution of Arginine 338 for a Lysine (factor IX Padua). Recombinant factor IX–Arg338Lys had a specific activity that was 5- to 10- times increased compared with recombinant wild-type factor IX activity. However, factor IX Padua is just as the prothrombin variants causing antithrombin reistance rare and is not identified in gnomAD (https://gnomad.broadinstitute.org/, accessed 21 July 2020). A formal statistical association with VTE therefore remains to be proven also for FIX Padua.
2.6. Gene-gene interaction
As factor V Leiden and prothrombin 20210A both are common, compound heterozygotes are not extremely rare among individuals with deficiencies of antithrombin, protein C, and protein S. Individuals may have up to 30- to 100- times increased risk for VTE [Citation25]. However, these risks are estimated from family studies and might constitute an overestimation of the actual VTE risk. Moreover, the confidence intervals are wide. Whether newly created genetic risk scores (GRS) scores will interact in similar ways with classical thrombophilia remains to be determined.
3. New findings from genome wide association study
Genome wide association studies (GWAS) enable hypothesis-free methods for testing associations between loci (genetic regions) and diseases [Citation38]. Typically, hundreds of thousands to millions of genetic variants across the genomes of thousands of individuals are tested to identify genotype–phenotype associations. These associations have led to insights into the architecture of disease susceptibility for a number of diseases including VTE [Citation39–41]. Several novel VTE disease loci have been discovered and previously discovered VTE loci have been confirmed by GWAS studies, reviewed by Trégouët and Morange [Citation42].
In 2019, Lindstrom et al and Klarin et al. published two large GWAS studies, respectively [Citation43,Citation44] (). Lindstrom et al meta-analyzed GWAS data for 30,234 VTE cases and 172,122 controls and tested the association between 12,923,718 SNPs and VTE. They identified 34 independent genetic signals for VTE risk from GWAS meta-analysis, of which 14 were novel associations. In the replicated loci together with loci identified by the other large GWAS by Klarin et al [Citation44] are presented. Klarin et al performed a discovery GWAS with testing of around 13 million DNA sequence variants for association with VTE (26,066 VTE cases and 624,053 controls) and replicated findings among 17,672 VTE cases and 167,295 controls. They identified 22 novel replicated loci shown in . The genome wide significant and replicated genes from the Lindstrom et al and Klarin et al GWAS are presented in according to potential linkage with VTE. For instance, the choline transporter SLC44A2 has been identified in red blood cell membrane [Citation45] and A4GALT is related to P1 blood group status [Citation46]. A gene variant (rs395967) of OSMR-AS1 has been associated with monocyte count [Citation47] and PEDP has been suggested to be involved in rheumatoid arthritis and immune response in tuberculosis [Citation48,Citation49]. Some loci involve different systems such as ABO gene. ABO gene is linked both to VWF (platelets) and factor VIII (coagulation) levels but also determine ABO blood group (erythrocytes). Whether all the ABO-associated risk is related to VWF and factor VIII levels remains to be determined. In are minor coagulation and anticoagulation GWAS loci marked with orange and classical thrombophilia loci marked with red. Several loci that reached genome wide significance but could not be replicated are shown in [Citation43,Citation44]. Whether these are spurious association or not remains to be determined.
Figure 2. Confirmed genes linked to venous thromboembolism (VTE) in family studies [Citation23–25] or genome wide association studies [Citation43,Citation44] grouped according to potential relation to VTE, i.e. anticoagulation, coagulation, platelets, erythrocytes, immune system, and unknown
![Figure 2. Confirmed genes linked to venous thromboembolism (VTE) in family studies [Citation23–25] or genome wide association studies [Citation43,Citation44] grouped according to potential relation to VTE, i.e. anticoagulation, coagulation, platelets, erythrocytes, immune system, and unknown](/cms/asset/166e6c05-11cb-4777-ba24-28b776c74f19/ierr_a_1804354_f0002_oc.jpg)
Table 1. Significant and replicated loci and variants in two large Genome wide association studies by Lindstrom et al [Citation43] and by Klarin et al [Citation44]
Table 2. Significant but not replicated loci and variants in two large Genome wide association studies by Lindstrom et al [Citation43] and by Klarin et al [Citation44]
Worth to mention is another recent GWAS of peripheral artery disease (PAD) including 31,307 PAD cases and 211,753 controls, by Klarin et al [Citation50]. For the first time they formally linked Factor V Leiden to an arterial disorder. Factor V Leiden was the second strongest variant associated with PAD with an odds ratio of 1.2 (95%CI 1.14–1.26, p value = 1.63E-12). Most interestingly, the Factor V Leiden variant’s effect estimate increased as PAD severity increased, with factor V Leiden carriers having a 62% increased risk of major amputation (OR = 1.62; 95%CI: 1.16–2.26). Moreover, no associations between Factor V Leiden and ischemic stroke or coronary heart disease were observed. This is in line with recent reports showing a protective effect of rivaroxaban against limb event in PAD [Citation51,Citation52].
4. Genetic risk score (GRS)
A drawback of GWAS studies is that most identified genetic variants are weak risk factors, i.e. with relative risks between 1.0 and 1.5. However, the identification of a large number of weak variants allows for creation of GRS [Citation43,Citation44]. Lindstrom et al created a GRS including 37 variants. Individuals belonging to the bottom fifth percentile had a 50% lower risk for VTE (OR = 0.51) and those among the top fifth percentile had a 3.2-fold greater odds ratio for VTE compared to the population in the middle range [Citation43]. Klarin et al created a 297 variants GRS for VTE that identified 5% of the population at a risk estimate similar to carriers of the factor V Leiden (rs6025) and prothrombin G20210A mutations [rs1799963] [Citation44].
5. Transcriptome-wide association studies (TWAS)
Transcriptome-wide association studies (TWAS) integrate genome-wide association studies (GWAS) and gene expression datasets to identify gene–trait associations [Citation53]. TWAS is thus an agnostic approach to prioritize causal genes at GWAS loci. Some loci will be non-causal, owing to sharing of expression quantitative trait loci (eQTL). TWAS is especially prone to spurious prioritization if expression data from non-trait-related tissues or cell types are used. Lindstrom et al [Citation43] used TWAS to identify 5 genetic loci with imputed gene expression levels differing between cases (n = 29 435) and controls (n = 157 769) of european ancestry in whole blood (SH2B3, SPSB1, RP11-747H7.3, RP4-737E23.2) and in liver (ERAP1). However, these loci remain to be confirmed.
6. High-throughput nucleotide sequencing
Another drawback of GWAS studies are that rare variants will not be detected. For instance, the variants causing deficiencies of antithrombin, protein C, and protein S are not detected by even very large GWAS studies [Citation43,Citation44]. The development of several so-called next generation sequencing (NGS) or High-Throughput Nucleotide Sequencing platforms is offering the possibility of generating fast, inexpensive and accurate genomic information, reviewed by Cunha et al [Citation54]. A few such studies have been published [Citation55–64]. These studies could be either agnostic [Citation61–64], i.e. whole exome sequencing (WES) or whole genome sequencing (WGS), or using NGS for re-sequencing of targeted candidate genes [Citation55–60]. A major problem is that the studied mutations are rare and even if studying mutation enrichment in genes a large study size will be necessary.
Worth to mention is the finding from a pilot study by Manderstedt et al using targeted sequencing of 17 candidate genes associated with VTE [Citation60]. Ninety-six VTE patients from the population-based Malmö Thrombophilia Study (MATS) were analyzed using an AmpliSeq strategy and Ion Torrent sequencing compared with data from public databases (gnomAD and SweGen). Determination of the nsyn/syn ratio (number of non-synonymous variants per non-synonymous site, nsyn, to the number of synonymous variants per synonymous site, syn) showed, as expected in VTE patients, an increase of non-synonymous relative to synonymous anticoagulant variants (SERPINC1, PROC, PROS1, PROCR, THBD) compared with controls (nsyn/syn, 0.95 vs. 0.68). In contrast, non-synonymous procoagulant (FGG, F2, F5, F9, F11, KNG1, GP6, VWF) variants (nsyn/syn, 0.31 vs. 0.63) showed a decrease in VTE patients compared to controls. This suggests that a deficit of non-synonymous variants in procoagulant genes is a potential novel mechanism contributing to VTE.
7. Candidate VTE genes, expression studies, and animal studies
Anticoagulation and coagulation genes are natural candidate genes for VTE. For instance, several genetic variants have been found among VTE patients in the THBD gene [Citation65]. Some of these THBD genetic variants also exhibit reduced functional activity [Citation65]. Still, the THBD gene remains to be formally linked to VTE [Citation43,Citation44]. A Ser219Gly missense variant in the endothelial protein C receptor (EPCR), i.e. PROCR gene, has been linked to VTE [Citation66]. This has been confirmed in the GWAS study by Lindstrom et al [Citation43]. Many other genes involved in the regulation of the coagulation system has not been genetically linked to VTE such as tissue factor pathway inhibitor (TFPI), Heparin cofactor II (SERPIND1), protein Z (PROZ), and genes involved in the fibrinolytic system.
Mouse models have turned out to be useful for understanding the biology of VTE and testing new therapeutics [Citation67,Citation68]. Mouse models have been used to study the hypercoagulability caused by Factor V Leiden [Citation69]. Two recent mouse studies indicated GAS6/TAM pathway signaling might be involved in VTE and also that PCSK9 inhibitors may protect against VTE [Citation70,Citation71]. Thus, these genes and pathways might be worthwhile for further studies. Still, it is important to remember that there are differences between animal models and human studies. For instance, Erickson showed in a transgenic mice study that elevated levels of plasminogen activator inhibitor-1 (PAI-1) contribute to the development of venous but not arterial occlusions [Citation72]. However, so far PAI-1 (SERPINE1 gene) and other fibrinolytic genes like PLG, PLAT, PLAU, SERPINF2, SERPINB2, and CPB2 remains to be genetically linked to VTE risk in humans [Citation43,Citation44].
8. Pathways involved in the genetic pathogenesis of venous thromboembolism
All classical thrombophilias either involve the coagulation cascade (F2 and F5) or its regulation by anticoagulants, i.e. antithrombin and the protein C system [SERPINC1, PROC, PROS1, F5) [Citation73]. However, recent GWAS have expanded and revised the genetic architecture of VTE [Citation39–44,Citation74]. Though weaker associations GWAS shows that besides coagulation and the natural anticoagulants also platelets, erythrocytes, and inflammation are involved in the genetic pathogenesis of VTE ( and ). For some VTE loci identified in GWAS studies no evident link to VTE have been identified.
Pathway analysis is a way to better understand the findings from genetic studies. Pathway analysis may also identify novel VTE candidate genes for further study. ,b) present the result of GeneMANIA analysis of all confirmed genes linked to VTE (https://genemania.org/) [Citation75]. The GeneMANIA analysis is based on available genomics and proteomics data. Totally 587 network connections were identified. Twenty genes (filled circles) were identified and they were involved in coagulation (F3, F7, F2R), anticoagulation (TFPI, THBD, SERPINA5), platelets (GP5, GP9, GP1 BA, GP1BB, ADAMTS13), and immune system (ITGAD, KIR2DL1, KIR2DS2, KIR2DS1, PYCARD). Some genes had unknown potential relations to VTE (HDAC5, WHRN, USP7, FAM175A).
Figure 3A. GeneMANIA analysis [Citation75] showing 39 genes (dashed circles) linked to venous thromboembolism (dashed circles) [Citation23–25,Citation43,Citation44] and 20 related genes (filled circles) identified by GeneMANIA. Color of the 587 lines denotes different network connections: pathways (turquoise), co-expression (purple), physical interactions (red), co-localization (blue), shared protein domains (yellow), and genetic interaction (green). OSMR-AS1 and RP11-122C5.1 could not be found by GeneMANIA
![Figure 3A. GeneMANIA analysis [Citation75] showing 39 genes (dashed circles) linked to venous thromboembolism (dashed circles) [Citation23–25,Citation43,Citation44] and 20 related genes (filled circles) identified by GeneMANIA. Color of the 587 lines denotes different network connections: pathways (turquoise), co-expression (purple), physical interactions (red), co-localization (blue), shared protein domains (yellow), and genetic interaction (green). OSMR-AS1 and RP11-122C5.1 could not be found by GeneMANIA](/cms/asset/7c97b8c6-e6d1-4aa4-b64f-0cff143f8bca/ierr_a_1804354_f0003a_oc.jpg)
Figure 3B. GeneMANIA analysis [Citation75] showing the 587 connections between 39 genes (dashed circles) linked to venous thromboembolism (dashed circles) [Citation23–25,Citation43,Citation44] and 20 related genes (filled circles) identified by GeneMANIA. Only TSPAN15 has no connections to any other gene. The twenty identified genes by GeneMANIA involve coagulation (F3, F7, F2 R), anticoagulation (TFPI, THBD, SERPINA5), platelets (GP5, GP9, GP1 BA, GP1BB, ADAMTS13), immune system (ITGAD, KIR2DL1, KIR2DS2, KIR2DS1, PYCARD), and genes with unknown potential relation to VTE (HDAC5, WHRN, USP7, FAM175A)
![Figure 3B. GeneMANIA analysis [Citation75] showing the 587 connections between 39 genes (dashed circles) linked to venous thromboembolism (dashed circles) [Citation23–25,Citation43,Citation44] and 20 related genes (filled circles) identified by GeneMANIA. Only TSPAN15 has no connections to any other gene. The twenty identified genes by GeneMANIA involve coagulation (F3, F7, F2 R), anticoagulation (TFPI, THBD, SERPINA5), platelets (GP5, GP9, GP1 BA, GP1BB, ADAMTS13), immune system (ITGAD, KIR2DL1, KIR2DS2, KIR2DS1, PYCARD), and genes with unknown potential relation to VTE (HDAC5, WHRN, USP7, FAM175A)](/cms/asset/048d0de8-5f91-480c-9b75-d8b05bd13248/ierr_a_1804354_f0003b_oc.jpg)
9. Clinical implications
Thrombophilia screening is performed in many institutions. According to European Society of Cardiology (ESC) 2019 guidelines testing for thrombophilia (including the acquired risk factors antiphospholipid antibodies and lupus anticoagulant) could be considered in patients with VTE at a young age (e.g. aged <50 years) and in VTE patients without any identifiable risk factor, i.e. unprovoked or spontaneous VTE [Citation76]. With unprovoked means VTE occuring in the absence of any major reversible risk factor. A strong family history of VTE further increase the indication for thrombophilia testing. VTE patients who are carriers of some forms of major hereditary thrombophilia, notably those with confirmed deficiency of antithrombin, protein C, or protein S, and patients with homozygous factor V Leiden or homozygous prothrombin G20210A mutation, are potential candidates for indefinite anticoagulant treatment after a first episode of unprovoked VTE. Thrombophilia testing in these cases may help to tailor the regimen of the anticoagulant treatment over the long-term [Citation76]. ESC 2019 guidelines on the other hand point out that, no evidence of a clinical benefit of extended anticoagulant treatment is currently available for carriers of heterozygous factor V Leiden or prothrombin 20210A mutation. There exist clinical risk prediction models but the clinical value and the possible therapeutic implications of these models in the NOAC era are unclear [Citation76]. In addition to estimate the risk of recurrence, physicians also have to take into consideration the risk of bleeding. Incidence estimates from cohort studies conducted more than 15 years ago reported a 3% annual incidence of major bleeding in patients treated with VKAs (Vitamin K antagonists) [Citation76]. However, meta-analysis indicates a 40% lower major bleeding risk for novel oral anticoagulants (NOAC) compared with VKAs [Citation77]. In agreement with these meta-analysis Agnelli et al have shown that extended anticoagulation with apixaban at either a treatment dose (5 mg) or a thromboprophylactic dose (2.5 mg) twice daily reduced the risk of recurrent VTE without increasing the risk of major bleeding [Citation78]. More detailed reviews of the management of classical thrombophilia have been published by Campello and coworkers and is recommended for further reading [Citation79,Citation80].
It is most likely that in the future weighted GRS using GWAS detected VTE risk variants may turn out to be useful for risk assessment instead of using individuals risk variants such as factor V Leiden or prothrombin 20210A mutation [Citation43,Citation44]. Moreover, high-throughput nucleotide sequencing is most useful for identifying individuals with mutations in the natural anticoagulant genes and should be considered to be included in thrombophilia assessment (i.e., SERPINC1, PROC, PROS1) [Citation60].
9.1. Combined oral contraceptives and classical thrombophilia
The Agency for Healthcare Policy and Research and the World Health Organization (WHO) advise patients with thrombogenic mutations to avoid combined oral contraceptives (COCs) [Citation83–86]. They recommend, for example, that ‘a personal history of VTE or a known thrombogenic mutation are conditions that represent an unacceptable health risk if combined hormonal contraception is used’ [Citation81–83]. However, present guidelines including WHO recommend against universal screening due to the low prevalence of thrombophilia and the high cost of screening [Citation83–86]. A systematic review by van Vlijmen et al indicates that the absolut VTE risk of heterozygosity for either the factor V Leiden or the prothrombin-G20210A mutation is modest, when no other risk factors are present (e.g., family history) [Citation87]. By contrast, the study discourages COC-use in women with antithrombin deficiency, protein C deficiency, protein S deficiency, double heterozygosity or homozygosity of factor V Leiden and prothrombin-G20210A mutation [Citation87]. It remains to be resolved whether a universal determination of GRS before COC-use could be useful for better risk assessment.
10. Conclusions
GWAS studies have added a number of weak loci to the strong classical five thrombophilia. Not only coagulation and anticoagulation but also platelets, erythrocytes, and the immune system and a number of genes with unknown linkage to VTE are involved in the genetic pathogenesis of VTE. GRS will be helpful instruments for risk assessment. Hopefully, even larger GWAS and especially WES and WGS in combination with bioinformatic tools will resolve the remaining heritability of VTE.
11. Expert opinion
The most important genetic risk factors for VTE (ie, classical thrombophilia) are all related to coagulation (F2 and F5 genes) or the anticoagulation system (SERPINC1, PROC, and PROS1 genes) ( and ). However, the recent two large GWAS studies added several novel gene loci all involved directly or indirectly in platelet biology, erythrocyte biology, or inflammation ( and ) [Citation43,Citation44,Citation74]. The GWAS study by Lindstrom et al also used Mendelian randomization and showed that some blood traits are causally associated with VTE risk [Citation43]. Thus, platelets that before having been implicated mostly in arterial thrombosis are also of importance for VTE [Citation43,Citation44,Citation74]. As erythrocytes also are constituents of thrombi it is possible that certain erythrocyte traits could increase the VTE risk [Citation88,Citation89]. For instance, in a large, population-based cohort study a graded independent association between red cell distribution width (RDW) and risk of first event of VTE was observed [Citation89]. This relationship was independent of potential confounders including hematological, multiple biological, socioeconomic, and lifestyle factors. Thus, evidence is accumulating that the formed elements in the blood need more attention from researchers in the field of VTE [Citation43,Citation44,Citation74,Citation88,Citation89]. Inflammatory disorders, such as infection and immune-mediated diseases, have been linked to VTE [Citation90]. However, the two recent GWAS studies are the first to link several genes involved in inflammation to VTE underlining the importance of inflammation in VTE pathogenesis [Citation43,Citation44,Citation74]. Whether inflammation per se is a driver of thrombosis or whether inflammation induced VTE is due to its effects on coagulation, anticoagulation, and platelets remains to be determined.
With increasing number of cases and controls more risk SNP for VTE will probably be identified in future GWAS studies. The effect size of these risk variants will probably all be minor. A natural development of these GWAS detected variants will therefore be the development and validation of separate GRS both for primary VTE and recurrent VTE. The combination of GRS with acquired risk factors will be useful for better risk assessment and tailored prophylaxis of both primary VTE and recurrent VTE. Another avenue will be the detection of novel risk loci due to enrichment of rare risk variants using WES and WGS technology. Though each variant is rare it is possible that together these variants will make a considerable contribution to the missing heritability of VTE.
GWAS studies have identified many novel weak VTE variants [Citation43,Citation44]. With even larger study size even weaker VTE variants might be detected. The question is however, if there are more rare variants causing VTE in other genes than SERPINC1, PROC, and PROS1. An important tool for the success is the development of NGS enabling WES and WGS in combination with bioinformatic tools. Maybe in the near future knowledge of all genetic risk factors in an individual may lead to relevant genetic risk estimation. This will likely include GRS and also rare variants. However, it should be noted, that several acquired risk factors such as surgery and pregnancy confer even higher risks than most genetic variants associated with VTE. As acquired risk factors often are transient, they offer the best opportunity to reduce the burden of VTE by improved and individualized anticoagulant prophylaxis. In this context a better estimate of genetic risk may help for tailoring individualized anticoagulant prophylaxis. Risk scores including the total burden of acquired risk factors (even transient) and genetic risk burden must be the goal in order to tailor adequate treatment, prevention and prophylaxis.
Of special interest will be the research studying the importance of VTE risk factors such as FV Leiden and peripheral artery disease [Citation44,Citation50]. We early on reported about arterial thrombosis in APC-resistant patients [Citation91–93] and also among protein S deficient patients [Citation94], and now it seems that at least FV Leiden is an important graded risk factor for peripheral artery disease [Citation44,Citation50]. This will also be an important research field.
Article highlights
Venous thromboembolism is a complex disease that clusters in families with moderate to high heritability.
Five classical thrombilia exist: rs6025, rs1799963, and deficiencies of the natural anticoagulants antithrombin, protein C, and protein S
Recent GWAS have established a number of weak VTE variants and made GRS of large effect size possible.
Factor V Leiden is associated not only with VTE risk but also peripheral artery disease.
The agnostic methods WES and WGS will be important in future research to detect multiple rare variants in new gene loci.
Development of clinical risk scores including the total burden of acquired risk factors (even transient) and genetic risk burden might lead to tailored treatment and prophylaxis.
ESC 2019 guidelines recommend thrombophilia screeing for young VTE patients below 50 years of age and unprovoked VTE, especially in the presence of family history of VTE.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Heit JA. Epidemiology of venous thromboembolism. Nat Rev Cardiol. 2015;12:464–474.
- Donadini MP, Ageno W. Unusual site thrombosis. Semin Hematol. 2011;48:264–270.
- Hansson PO, Welin L, Tibblin G, et al. Deep vein thrombosis and pulmonary embolism in the general population. ‘The study of men born in 1913ʹ. Arch Int Med. 1997;157:1665–1670.
- Bell EJ, Lutsey PL, Basu S, et al. Lifetime risk of venous thromboembolism in two cohort studies. Am J Med. 2016;129:339.e19-26.
- Goldhaber SZ. Venous thromboembolism: epidemiology and magnitude of the problem. Best Pract Res Clin Haematol. 2012;25:235–242.
- Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353:1167–1173.
- White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107:I4–8.
- Ribeiro DD, Lijfering WM, Barreto SM, et al. Epidemiology of recurrent venous thrombosis. Braz J Med Biol Res. 2012;45:1–7.
- Heit JA, Phelps MA, Ward SA, et al. Familial segregation of venous thromboembolism. J Thromb Haemost. 2004;2:731–736.
- Tosetto A, Castaman G, Cappellari A, et al. The VITA project: heritability of resistance to activated protein C. Vincenza thrombophilia and arteriosclerosis. Thromb Haemost. 2000;84:811–814.
- Zöller B, Ohlsson H, Sundquist J, et al. A sibling based design to quantify genetic and shared environmental effects of venous thromboembolism in Sweden. Thromb Res. 2017;149:82–87.
- Larsen TB, Sørensen HT, Skytthe A, et al. Major genetic susceptibility for venous thromboembolism in men: a study of Danish twins. Epidemiology. 2003;14:328–332.
- Zöller B, Li X, Ohlsson H, et al. Family history of venous thromboembolism as a risk factor and genetic research tool. Thromb Haemost. 2015;114:890–900.
- Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci U S A. 1993;90:1004–1008.
- Svensson PJ, Dahlbäck B. Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med. 1994;330:517–522.
- Bertina RM, Koeleman BP, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67.
- Zöller B, Dahlbäck B. Linkage between inherited resistance to activated protein C and factor V gene mutation in venous thrombosis. Lancet. 1994;343:1536–1538.
- Voorberg J, Roelse J, Koopman R, et al. Association of idiopathic venous thromboembolism with single point-mutation at Arg506 of factor V. Lancet. 1994;343:1535–1536.
- Greengard JS, Sun X, Xu X, et al. Activated protein C resistance caused by Arg506Gln mutation in factor Va. Lancet. 1994;343:1361–1362.
- Poort SR, Rosendaal FR, Reitsma PH, et al. A common genetic variation in the 3ʹ-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–3703.
- Zöller B, Li X, Ohlsson H, et al. Epidemiology of familial aggregation of venous thromboembolism. Semin Thromb Hemost. 2016;42:821–832.
- Martinelli I, Bucciarelli P, Mannucci PM. Thrombotic risk factors: basic pathophysiology. Crit Care Med. 2010;38:S3–9.
- Mannucci PM, Franchini M. Classic thrombophilic gene variants. Thromb Haemost. 2015;114:885–889.
- Rosendaal FR, Reitsma PH. Genetics of venous thrombosis. J Thromb Haemost. 2009;7(Suppl 1):301–304.
- Zöller B, García de Frutos P, Hillarp A, et al. Thrombophilia as a multigenic disease. Haematologica. 1999;84:59–70.
- Heijboer H, Brandjes DPM, Büller HR, et al. Deficiencies of coagulation‐inhibiting and fibrinolytic proteins in outpatients with deep‐vein thrombosis. N Engl J Med. 1990;323:1512–1516.
- Koster T, Rosendaal FR, Briët E, et al. Protein C deficiency in a controlled series of unselected outpatients: an infrequent but clear risk factor for venous thrombosis (Leiden Thrombophilia Study). Blood. 1995;85:2756–2761.
- Segers K, Dahlbäck B, Nicolaes GA. Coagulation factor V and thrombophilia: background and mechanisms. Thromb Haemost. 2007;98:530–542.
- Cai H, Hua B, Fan L, et al. A novel mutation (g2172–>c) in the factor V gene in a Chinese family with hereditary activated protein C resistance. Thromb Res. 2010;125:545–548.
- Nogami K, Shinozawa K, Ogiwara K, et al. Novel FV mutation (W1920R, FVNara) associated with serious deep vein thrombosis and more potent APC resistance relative to FVLeiden. Blood. 2014;123:2420–2428.
- Guzmán N, Larama G, Ávila A, et al. Three novel variants in the coagulation factor V gene associated with deep venous thrombosis in Chilean patients with Amerindian ethnic background. Clin Chim Acta. 2015;444:24–28.
- Pezeshkpoor B, Castoldi E, Mahler A, et al. Identification and functional characterization of a novel F5 mutation (Ala512Val, FVBonn) associated with activated protein C resistance. Thromb Haemost. 2016;14:1353–1363.
- Miyawaki Y, Suzuki A, Fujita J, et al. Thrombosis from a prothrombin mutation conveying antithrombin resistance. N Engl J Med. 2012;366:2390–2396.
- Djordjevic V, Kovac M, Miljic P, et al. A novel prothrombin mutation in two families with prominent thrombophilia–the first cases of antithrombin resistance in a Caucasian population. J Thromb Haemost. 2013;11:1936–1939.
- Sivasundar S, Oommen AT, Prakash O, et al. Molecular defect of ‘Prothrombin Amrita’: substitution of arginine by glutamine (Arg553 to Gln) near the Na(+) binding loop of prothrombin. Blood Cells Mol Dis. 2013;50:182–183.
- Bulato C, Radu CM, Campello E, et al. New prothrombin mutation (Arg596Trp, Prothrombin Padua 2) associated with venous thromboembolism. Arterioscler Thromb Vasc Biol. 2016;36:1022–1029.
- Simioni P, Tormene D, Tognin G, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009;361:1671–1675.
- Tam V, Patel N, Turcotte M, et al. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20:467–484.
- Trégouët DA, Heath S, Saut N, et al. Common susceptibility alleles are unlikely to contribute as strongly as the FV and ABO loci to VTE risk: results from a GWAS approach. Blood. 2009;113:5298–5303.
- Buil A, Trégouët DA, Souto JC, et al. C4BPB/C4BPA is a new susceptibility locus for venous thrombosis with unknown protein S‐independent mechanism: results from genome‐wide association and gene expression analyses followed by case‐control studies. Blood. 2010;115:4644–4650.
- Heit JA, Armasu SM, McCauley BM, et al. Identification of unique venous thromboembolism‐susceptibility variants in African‐Americans. Thromb Haemost. 2017;117:758–768.
- Trégouët DA, Morange PE. What is currently known about the genetics of venous thromboembolism at the dawn of next generation sequencing technologies. Br J Haematol. 2018;180:335–345.
- Lindström S, Wang L, Smith EN, et al. Genomic and transcriptomic association studies identify 16 novel susceptibility loci for venous thromboembolism. Blood. 2019;134:1645–1657.
- Klarin D, Busenkell E, Judy R, et al. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat Genet. 2019;51:1574–1579.
- Bryk AH, Wiśniewski JR. Quantitative analysis of human red blood cell proteome. J Proteome Res. 2017;16:2752–2761.
- Westman JS, Stenfelt L, Vidovic K, et al. Allele-selective RUNX1 binding regulates P1 blood group status by transcriptional control of A4GALT. Blood. 2018;131:1611–1616.
- Yasukochi Y, Sakuma J, Takeuchi I, et al. Identification of nine novel loci related to hematological traits in a Japanese population. Physiol Genomics. 2018;50:758–769.
- Comertpay B, Gov E. Identification of key biomolecules in rheumatoid arthritis through the reconstruction of comprehensive disease-specific biological networks. Autoimmunity. 2020;53:156–166.
- Wang C, Li YY, Li X, et al. Serum complement C4b, fibronectin, and prolidase are associated with the pathological changes of pulmonary tuberculosis. BMC Infect Dis. 2014;14:52.
- Klarin D, Lynch J, Aragam K, et al. Genome-wide association study of peripheral artery disease in the million veteran program. Nat Med. 2019;25:1274–1279.
- Connolly SJ, Eikelboom JW, Bosch J, et al. Rivaroxaban with or without aspirin in patients with stable coronary artery disease: an international, randomised, double-blind, placebo-controlled trial. Lancet. 2018;391:205–218.
- Bonaca MP, Bauersachs RM, Anand SS, et al. Rivaroxaban in peripheral artery disease after revascularization. N Engl J Med. 2020 Mar 28 Epub ahead of print. DOI:10.1056/NEJMoa2000052.
- Wainberg M, Sinnott-Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51:592–599.
- Cunha ML, Meijers JC, Middeldorp S. Introduction to the analysis of next generation sequencing data and its application to venous thromboembolism. Thromb Haemost. 2015;114:920–932.
- Lotta LA, Wang M, Yu J, et al. Identification of genetic risk variants for deep vein thrombosis by multiplexed next-generation sequencing of 186 hemostatic/pro-inflammatory genes. BMC Med Genomics. 2012;5:7.
- Lotta LA, Tuana G, Yu J, et al. Next-generation sequencing study finds an excess of rare, coding single-nucleotide variants of ADAMTS13 in patients with deep vein thrombosis. J Thromb Haemost. 2013;11:1228–1239.
- Pagliari MT, Lotta LA, de Haan HG, et al. Next-generation sequencing and in vitro expression study of ADAMTS13 single nucleotidevariants in deep vein thrombosis. PLoS One. 2016;11:e0165665.
- Rühle F, Witten A, Barysenka A, et al. Rare genetic variants in SMAP1, B3GAT2, and RIMS1 contribute to pediatric venous thromboembolism. Blood. 2017;129:783–790.
- de Haan HG, van Hylckama Vlieg A, Lotta LA, et al. Targeted sequencing to identify novel genetic risk factors for deep vein thrombosis: a study of 734 genes. J Thromb Haemost. 2018;16:2432–2441.
- Manderstedt E, Lind-Halldén C, Svensson P, et al. Next-generation sequencing of 17 genes associated with venous thromboembolism reveals a deficit of non-synonymous variants in procoagulant genes. Thromb Haemost. 2019;119:1441–1450.
- Cunha MLR, Meijers JCM, Rosendaal FR, et al. Whole exome sequencing in thrombophilic pedigrees to identify genetic risk factors for venous thromboembolism. PLoS One. 2017;12:e0187699.
- Halvorsen M, Lin Y, Sampson BA, et al. Whole exome sequencing reveals severe thrombophilia in acute unprovoked idiopathic fatal pulmonary embolism. EBioMedicine. 2017;17:95–100.
- Lee EJ, Dykas DJ, Leavitt AD, et al. Whole-exome sequencing in evaluation of patients with venous thromboembolism. Blood Adv. 2017;1:1224–1237.
- Lindström S, Brody JA, Turman C, et al. A large-scale exome array analysis of venous thromboembolism. Genet Epidemiol. 2019;43:449–457.
- Kunz G, Ohlin AK, Adami A, et al. Naturally occurring mutations in the thrombomodulin gene leading to impaired expression and function. Blood. 2002;99:3646–3653.
- Dennis J, Johnson CY, Adediran AS, et al. The endothelial protein C receptor (PROCR) Ser219Gly variant and risk of common thrombotic disorders: a HuGE review and meta-analysis of evidence from observational studies. Blood. 2012;119:2392–2400.
- Diaz JA, Saha P, Cooley B, et al. Choosing a mouse model of venous thrombosis. Arterioscler Thromb Vasc Biol. 2019;39:311–318.
- Cleuren AC, van Vlijmen BJ, Reitsma PH. Transgenic mouse models of venous thrombosis: fulfilling the expectations? Semin Thromb Hemost. 2007;33:610–616.
- Cui J, Eitzman DT, Westrick RJ, et al. Spontaneous thrombosis in mice carrying the factor V Leiden mutation. Blood. 2000;96:4222–4226.
- Paciullo F, Momi S, Gresele P. PCSK9 in haemostasis and thrombosis: possible pleiotropic effects of PCSK9 inhibitors in cardiovascular prevention. Thromb Haemost. 2019;119:359–367.
- Law LA, Graham DK, Di Paola J, et al. GAS6/TAM pathway signaling hemostasis and thrombosis. Front Med (Lausanne). 2018;5:137.
- Erickson LA, Fici GJ, Lund JE, et al. Development of venous occlusions in mice transgenic for the plasminogen activator inhibitor-1 gene. Nature. 1990;346:74–76.
- Dahlbäck B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol. 2005;25:1311–1320.
- Zöller B. Genetics of venous thromboembolism revised. Blood. 2019;134:1568–1570.
- Franz M, Rodriguez H, Lopes C, et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46:W60–4.
- Konstantinides SV, Meyer G, Becattini C, et al. 2019 ESC guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS). Eur Heart J. 2020;41:543–603.
- Kakkos SK, Kirkilesis GI, Tsolakis IA. Editor’s Choice - efficacy and safety of the new oral anticoagulants dabigatran, rivaroxaban, apixaban, and edoxaban in the treatment and secondary prevention of venous thromboembolism: a systematic review and meta-analysis of phase III trials. Eur J Vasc Endovasc Surg. 2014;48:565–575.
- Agnelli G, Buller HR, Cohen A, et al. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368:699–708.
- Campello E, Spiezia L, Simioni P. Diagnosis and management of factor V Leiden. Expert Rev Hematol. 2016;9:1139–1149.
- Campello E, Spiezia L, Adamo A, et al. Thrombophilia, risk factors and prevention. Expert Rev Hematol. 2019;12:147–158.
- Vernon E, Hiedemann B, Bowie BH. Economic evaluations of thrombophilia screening prior to prescribing combined oral contraceptives: a systematic and critical review. Appl Health Econ Health Policy. 2017;15:583–595.
- Centers for Disease Control and Prevention (CDC). U.S. selected practice recommendations for contraceptive use, 2016. MMWR Recomm Rep Morb Mortal Wkly Rep. 2016;65(4):1–66.
- World Health Organization. Medical eligibility criteria for contraceptive use. 5th Geneva: WHO. 2015. [cited 2020 Jul 20]. Available from: http://apps.who.int/iris/bitstream/10665/181468/1/9789241549158_eng.pdf?ua=1
- Wu O, Robertson L, Twaddle S, et al. Screening for thrombophilia in high-risk situations: a meta analysis and cost-effectiveness analysis. Br J Haematol. 2005;131:80–90.
- Grody WW, Griffin JH, Taylor AK, et al. American College of Medical Genetics consensus statement on factor V Leiden mutation testing. Genet Med. 2001;3:139–148.
- Centers for Disease Control. US medical eligibility criteria (US MEC) for contraceptive use, 2016. [cited 2020 Jul 20] Available from:https://www.cdc.gov/reproductivehealth/contraception/mmwr/mec/summary.html
- van Vlijmen EF, Wiewel-Verschueren S, Monster TB, et al. Combined oral contraceptives, thrombophilia and the risk of venous thromboembolism: a systematic review and meta-analysis. J Thromb Haemost. 2016;14:1393–1403.
- Gersh KC, Nagaswami C, Weisel JW. Fibrin network structure and clot mechanical properties are altered by incorporation of erythrocytes. Thromb Haemost. 2009;102:1169–1175.
- Zöller B, Melander O, Svensson P, et al. Red cell distribution width and risk for venous thromboembolism: a population-based cohort study. Thromb Res. 2014;133:334–339.
- Zöller B, Li X, Sundquist J, et al. Autoimmune diseases and venous thromboembolism: a review of the literature. Am J Cardiovasc Dis. 2012;2:171–183.
- Lindblad B, Svensson PJ, Dahlbäck B. Arterial and venous thromboembolism with fatal outcome and resistance to activated protein C. Lancet. 1994;343:917.
- Holm J, Zöller B, Svensson PJ, et al. Myocardial infarction associated with homozygous resistance to activated protein C. Lancet. 1994;344:952–953.
- Zöller B, Svensson PJ, He X, et al. Identification of the same factor V gene mutation in 47 out of 50 thrombosis-prone families with inherited resistance to activated protein C. J Clin Invest. 1994;94:2521–2524.
- Zöller B, Berntsdotter A, García de Frutos P, et al. Resistance to activated protein C as an additional genetic risk factor in hereditary deficiency of protein S. Blood. 1995;85:3518–3523.