
ABSTRACT
Objectives
To investigate the mutational spectrum in the HBB gene in Arab patients with β-thal.
Methods
Authors searched five databases (PubMed, Science Direct, Scopus, Web of Science, and Google Scholar) from the time of inception until March 2020.
Results
The authors search strategy yielded 3,229 citations, of which 48 eligible studies captured. 105 mutations were captured, of these, 99 were shared between Arabs and other ethnic groups, six mutations were unique to Arabs (c.92 + 2 T > G, c.-240 G > A, c.150delC, c.420dupT, deletion of 192 bp spanning exon 1, intron 1, and the first two bases of exon 2 of HBB gene, and deletion of 9.6 kb, including exon 1 and intron 2 of HBB gene). The most common HBB gene mutations among Arabs were c.93–21 G > A, c.118 C > T, c.92 + 1 G > A, c.92 + 6 T > C, c.92 + 5 G > C, c.315 + 1 G > A, and c.27dupG. Consanguinity is high among Arab patients with β-thal. Migration into Arab countries led to allelic heterogeneity among Arab patients with β-thal.
Conclusion
Our findings present a platform for further genetic epidemiological studies for Arab patients with β-thal.
1. Introduction
Beta-thalassemia (β-thal, MIM # 613,985) is a chronic hemolytic anemia that is inherited in an autosomal recessive manner [Citation1]. It is characterized by reduced hemoglobin levels and red blood cell production [Citation2]. β-thal is caused by the reduction or absence of β-globin chains, which make a tetramer with the α-globin chains to produce hemoglobin. β0 mutations are those that completely inactivate the β gene, resulting in no β-globin production, other mutations allow the production of some β globin, and are classified according to the degree of quantitative reduction in the output of the β chains; as β+- or β++- (‘silent’) thalassemia [Citation3]. β-thal is characterized by variability in the clinical presentation that is determined by the prolonged reduction in β-globin chains, which results from mutations in the β-globin gene (HBB) [Citation1]. The HBB gene is positioned on the short arm of chromosome 11, with a size of 1.6 kb, and consists of three exons [Citation4]. To date, according to the Human Gene Mutation Database (HGMD®), more than 200 mutations were reported in the HBB gene that are associated with β-thal [Citation4]. The absence of β-globin chain production that leads to β0-thal is caused by initiation codon, splice-site junction, missense, nonsense, and frameshift mutations [Citation4]. On the other hand, reduction in β-globin chains which lead to β+-thal is caused by mutations in the promoter, and 5ʹ/3ʹ-UTRs [Citation4].
Worldwide, the number of babies born annually with β-thal has been estimated to be more than 40,000 [Citation5]. A total of 80 million are estimated to be carriers of β-thal [Citation5]. β-thal is a significant health problem in many countries and is most prevalent in low-income countries, including in the Mediterranean and southeast Asian regions. It is also broadly extended in the Indian subcontinent and Melanesia [Citation6,Citation7]. β-thal is also common in North America, Australia, and Northern Europe as a result of migration [Citation6]. In Arab countries, the carrier frequency ranges from 1% to 11% [Citation8]. Heterozygous carriers usually show no symptoms, where compound heterozygous and homozygous mostly lead to β-thal major that is common in Arab countries. On the other hand, β-thal intermedia results from the effect of genetic modulators with a modifying effect on the heterozygous or homozygous β-thal minor [Citation8].
In terms of genotype-phenotype correlations, β-thal is classified into four different groups: β-thal major (also known as Cooley’s anemia), β-thal intermedia, β-thal minor, and silent β-thal [Citation4]. Silent β-thal carriers are heterozygous for the β++ allele (β++/BN) and are characterized by normal-red blood cells and Hb A2 levels [Citation3]. The β-thal minor patients who inherit a single β0 or β+ allele are asymptomatic. However, their laboratory findings show increased Hb A2 levels and decreased microcytosis and hypochromia. The β-thal intermedia represents a clinical condition between the minor and major trait. Patients are either homozygous for the β+ allele (β+/β+) or compound heterozygous for β0 and β+ (β0/β+). The phenotypic spectrum of β-thal intermedia ranges from mild to severe anemia with occasional blood transfusions. The β-thal intermedia is observed in heterozygous patients with a single dominant β0 allele (β0/BN), homozygous for β0 (β0/β0) having alpha-thalassemia co-inheritance or in heterozygous with dominant β0 (β0/BN) or recessive β+ allele (β+/BN) with a-triplication co-inheritance. However, the genotype-phenotype correlation of β-thal intermedia is complex and remains to be fully elucidated [Citation9,Citation10]. The third and most severe form of thalassemia, referred to as β-thal major, is characterized by severe anemia and skeletal deformities. This form requires regular blood transfusions for survival. Most β-thal major cases are homozygous for the β0 allele (β0/β0), fewer cases are compound heterozygous (β+/β0) [Citation10].
Arabs are a major pan-ethnic group, and their union, the Arab League, comprises 22 countries [Citation11]. The Arab world has historically been the crossroad for different cultures that has significantly altered its ethnic composition, yielding a high degree of genetic heterogeneity [Citation12]. Given that β-thal is particularly associated with certain ethnic groups, and specific populations residing in particular geographic areas in the Arab world, genetic variations are believed to affect the phenotypic presentation of b-thal patients in a unique way that is different from other ethnic groups [Citation13]. Arabs are currently interested to understand the genetic architecture of what makes them susceptible to genetic diseases, and therefore started intensively to sequence the Arab genomes through national projects that aim to define the disease-causing genetic mutations for all genetic disorders. This started with Saudi Arabia [Citation14], followed by Qatar [Citation15], and currently, UAE sequencing their 1000 genomes [Citation16]. These projects are very important to determine the carrier frequency of β-thal among Arabs, and therefore will improve premarital genetic counseling and health care Arab patients with β-thal. Although β-thal-associated mutations have been extensively studied, the outcomes have been conflicting and inconsistent among different ethnic populations [Citation8,Citation17]. In addition, there has been relatively little attention devoted to comprehensively investigate the mutational spectrum in the HBB gene among Arab patients with β-thal. Therefore, in this systematic review, we aimed to summarize current evidence on the current spectrum of β-thal-related HBB mutations across the 22 Arab countries.
2. Materials and methods
2.1. Search strategy
A systematic review was performed of all peer-reviewed published research articles on Arab β-thal patients in the 22 Arab countries to identify the mutations in the HBB gene that is causative to β-thal in the Arab patients. Five literature databases (PubMed, Science Direct, Scopus, Web of Science, and Google Scholar) were searched to include all the relevant articles published in English from the time of inception until March 2020. We used the ‘Beta Thalassemia’ search term in combination with one of these terms: ‘genetic’ OR ‘mutation’ OR ‘variant,’ with the name of each of the 22 Arab countries. For examples ‘Beta Thalassemia’ AND ‘variant’ AND ‘Qatar.’ The eligible research articles were screened for the titles and abstracts.
2.2. Study selection
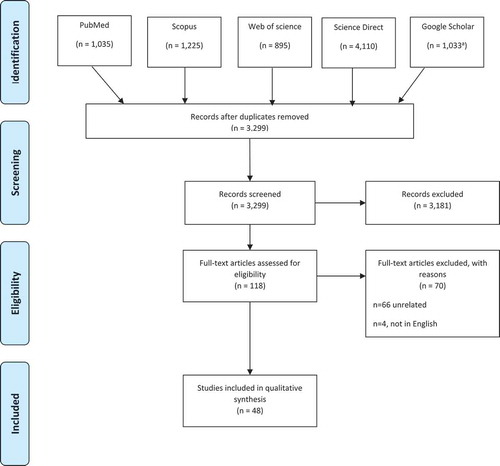
The selection of research articles was performed based on the following selection criteria: (1) original research articles published in peer-reviewed journals; (2) studies comprising Arab subjects residing in Arab countries who were diagnosed with β-thal; and (3) studies reporting HBB gene mutation data. The articles which failed to meet any of the inclusion criteria were excluded. The citations of the selected articles were exported to Endnote web X8, after which the duplicate articles were removed, and the remaining articles were assessed against the inclusion criteria ().
Figure 1. Flow diagram of the process for study selection. aFor the Google Scholar search, only the most relevant records were retained, as n = 44,217 for the total-identified records
2.3. Data extraction
The data selection was done independently by three scientists (AA, AK, and SY) and finally edited by the senior author (HZ), the technical revision was done by an experienced hematologist (MY). The eligible articles were fully screened, and the genetic mutations relevant data, the number of patients/chromosomes/alleles examined, the reported clinical phenotype was captured ( and Supplementary ). Additionally, two human hemoglobin variant databases were utilized to check for the frequency of the detected mutations in different ethnic groups and the related clinical phenotypes. These two databases were: IthaGenes (https://www.ithanet.eu/db/ithagenes) and HbVar: (http://globin.bx.psu.edu/hbvar/menu.html). Moreover, HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), EVS (https://evs.gs.washington.edu/EVS/) and LOVD (https://www.lovd.nl/) databases were used to check for the uniqueness of the mutations in the Arab populations.
Table 1. Beta-thalassemia HBB gene mutations unique to Arabs
Table 2. Beta-thalassemia common mutations in the HBB gene among Arab countries shared with other ethnic groups
3. Results
3.1. Search findings
Our search strategy resulted in 8,298 citations; of which 3,229 remained after removing the duplicates. According to the inclusion criteria mentioned earlier in the Method section, 3,181 citations were excluded (). The remaining 118 articles were considered eligible, of which 70 articles were excluded, 66 were irrelevant and four were not in the English language. The remaining 48 articles were included in the systematic analysis.
3.2. Molecular genetics findings
The data of the captured HBB gene mutations that are unique for Arab populations are summarized in , the shared mutations that are circulated among Arabs and other ethnic groups are listed in . The most common mutations that are circulated among Arabs are shown in . Furthermore, any reported data about the patient hemoglobin level, transfusion frequency, and presence of complications were collected. A total of 105 mutations in the HBB were reported in patients from 20 Arab countries (, and Supplementary ).
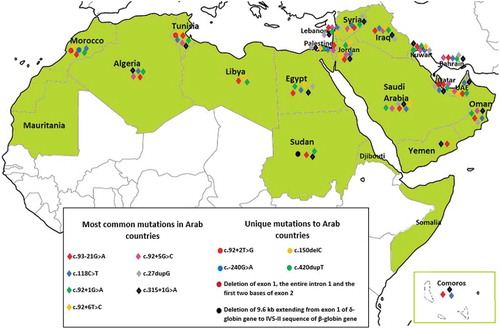
Figure 2. Distribution of HBB mutations in Arab countries. The green color indicates Arab countries. The mapping was limited to the most common and unique mutations only, the entire list of mutations are list in Supplementary : Beta-thalassemia HBB gene shared mutations between Arabs and other ethnic groups Note: The map is extracted from the free map product (http://english.freemap.jp/item/africa/africa_1.html)/Africa’s regional Thumbnail, under the Creative Commons Attribution 3.0 unported (CCBY3.0) license
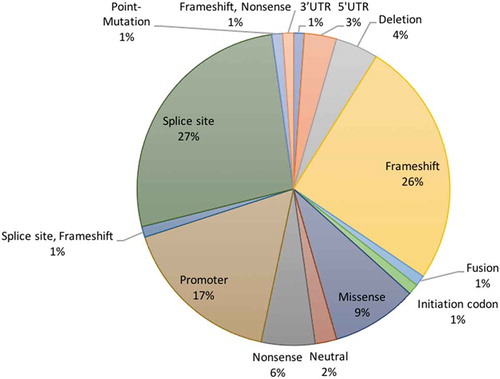
Most of the HBB gene mutations were splice site (27%) and frameshift (26%) mutations (). The other mutations were promoter (17%), missense (9%), nonsense (6%), deletion (4%) mutations, while the remaining (11%) mutations were 5ʹ-UTR, 3ʹ-UTR, fusion, or combined frameshift and nonsense mutations ().
Figure 3. Types of HBB gene mutations associated with β-thal in the Arab countries
Six of the 105 captured mutations were unique to Arabs c.92 + 2 T > G, c.-240 G > A, c.150delC, c.420dupT, deletion of 192 bp within the β-globin gene spanning exon 1, intron 1, and the first two bases of exon 2 and deletion of 9.6 kb extending from exon 1 of the δ-globin gene to IVS-II sequence of a β-globin gene ( and ), 99 mutations was shared with other ethnic groups (), of which seven were identified to be the most frequent in Arab countries ( and ).
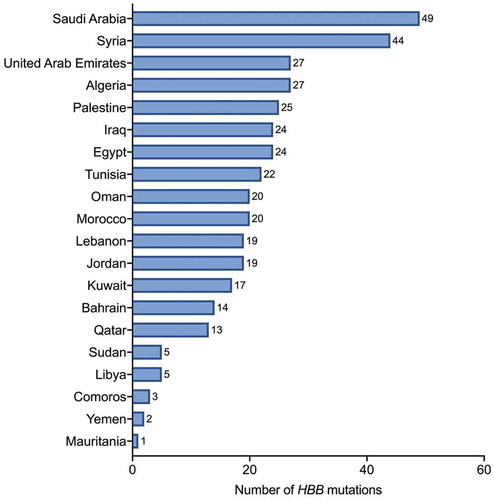
Most of the mutations were captured in Saudi Arabia (49), followed by Syria (44), Algeria, and United Arab Emirates (27, each), Palestinian territories (25), Egypt and Iraq (24, each), Tunisia (22), Morocco and Oman (20, each), Jordan and Lebanon (19, each), Kuwait (17), Bahrain (14), Qatar (13), Libya and Sudan (5, each), Comoros (3), Yemen (2), and Mauritania (1) (). Our search strategy and selection criteria yielded no β-thal-related mutations in two Arab countries (Djibouti and Somalia).
Figure 4. Number of HBB mutations identified in each Arab country
4. Discussion
This study comprehensively and systematically summarizes all the published peer-reviewed articles related to the β-thal in the Arab world. Our searching strategy captured 105 mutations in the HBB gene (, and Supplementary ), 99 of the detected β-thal mutations in Arab countries were found to be shared with the other ethnic groups, while six mutations were found to be unique to Arabs (). The Arab world covers a wide area from the Atlantic Ocean in the west to the Arabian Sea in the east, including the Middle East and North Africa. In addition to its geographic diversity and due to the historic and current immigrations, the ethnicity of the population in the Arab world is diverse, comprising of different origins, including Asian, Kurdish, Turkish, Africans, and European. This diverse ethnic demographic is reflected in the large number of β-thal mutations that were detected in Arab countries (Supplementary ).
4.1. Consanguinity and HBB gene mutations
Arab countries display some of the highest rates of consanguinity around the world, ranging from 20% to 50%, with average rates from 20% to 30%, across the 22 Arab countries [Citation44]. Consanguinity reported to confer a relatively higher risk in developing β-thal [Citation45]. In Algeria, among 60 patients the consanguinity rate was 50% [Citation46], in Iraq the rate was 52.8% among 152 patients [Citation47], 49% among 189 patients in Syria [Citation48], 55%, and 31.5% among 200 and 73 patients, respectively, in Egypt [Citation49,Citation50], while in Palestinian territories the rate was higher than 92.2% among 51 patients [Citation51]. This is reflected in the higher prevalence rate of β-thal carriers and patients in the Arab world and in similar cultures like in the Maldives where the carrier frequency of β-thal is more than 16% [Citation7,Citation52], compared to Australia, North Europe, South Africa, and America where the consanguinity rate is very low, and the carrier frequency is considerably less [Citation53].
4.2. Distribution of HBB gene mutations in the Arab world
In the present study, most of the HBB gene mutations were detected in Saudi Arabia (49) and Syria (44). Syria has a distinguished sort of population with huge diversity in ethnicity due to the historic colonization of the Roman, Ottoman, and Mongols empires in the region and because of migration [Citation54]. In Saudi Arabia, the wide spectrum of HBB mutations is thought to be due in different reasons based on the region of the country. In the eastern province of Saudi Arabia where the β-thal prevalence is high and most of the identified HBB mutations are also detected in the neighboring countries, these mutations are thought to be introduced by population migration flow [Citation55–57], whereas in Mecca, although the prevalence of β-thal is low, HBB mutations are believed to be due to the settlement of the Hajj pilgrimages from around the world in the places near the holy city [Citation58]. On the other hand, Comoros (3) Yemen (2) and Mauritania (1) recorded the lowest numbers of HBB gene mutations, which can be due to the dearth of reported data.
4.3. Unique mutations in Arabs
Although the spectrum of β-thal mutations in the Arab region is highly diversified and mostly shared with other ethnic groups, we identified six HBB gene mutations that our search strategy identified as unique to Arabs (), according to scientific literatures, and mutation databases (HGMD, ClinVar, EVS, and LOVD).
The mutation c.420dupT in the C-terminal region of the β-globin gene was identified in a young Kuwaiti patient with transfusion-dependent β-thal major and chromaturia (reddish/brown discoloration of the urine). This frameshift mutation in exon 3 resulted from a thymidine insertion in codons 139/140. The patient displayed clinical features, indicating early onset of the β-thal major phenotype of ineffective erythropoiesis with the request of blood transfusions. Hematopoietic stem cell transplantation was a successful treatment alternative for this severe form of β-thal major [Citation59]. Another frameshift mutation was discovered in an ethnic Qatari patient with transfusion-dependent β-thal. This frameshift mutation resulted from the deletion of 192 bp within the β-globin gene spanning exon 1, intron 1, and the first two bases of exon 2 resulting in a pre-mature appearance of a stop codon [Citation60]. Deletion in a β-globin gene is very uncommon in β-thal, where less than 10% of the 240 β-thal mutations reported involve a 150–200 bp deletion [Citation61,Citation62]. This mutation is compound heterozygous with c.92 + 5 G > T mutation, resulting in a β-thal major phenotype [Citation60]. The third frameshift mutation in exon 2, c.150delC, is homozygous and of Jordanian origin. The generation of premature a stop codon resulted in the β-thal major with severe dependency on blood transfusions [Citation63].
Another mutation is the deletion of 9.6 kb extending from exon 1 of the δ-globin gene to the IV-II sequence II of the HBB gene, which was detected in Sudanese adult males. The patient blood investigation showed microcytosis, hypochromia, elevated Hb F level, and normal Hb A2 levels, while the molecular analysis revealed a homozygous deletion of a single a-globin gene of 3.7kb, which could explain microcytosis and hypochromia, but not the elevated Hb F. Therefore, multiplex ligation-dependent probe amplification (MLPA) was used that showed this deletion. This mutation is reported in carriers of δ β-thal carriers or hereditary persistence of fetal hemoglobin (HPFH) [Citation64].
Besides frameshift mutations, a point mutation at the promoter of the HBB gene was identified in a Moroccan female (c.-240 G > A), this mutation exhibits a β-thal intermedia phenotype in compound heterozygosity with a β0 mutation or a silent phenotype in a heterozygous carrier. The patient was a 20-year-old who showed symptoms of moderate anemia, increased Hb A2, and slightly increased Hb F level. The patient was transfused initially and had an enlarged spleen by the age of 10. However, splenectomy was performed, and the patient remained asymptomatic without the need for transfusion for the rest of her life [Citation65].
Finally, c.92 + 2 T > G is a splice-site mutation of Tunisian origin [Citation66]. This mutation was later identified in Morocco [Citation67]. The mutation resulted from a severe β0 allele in homozygous form. However, no data regarding the clinical phenotype were mentioned in both studies [Citation66,Citation67].
4.4. Common mutations among Arab countries shared with other ethnic groups
Among the 99 shared mutations with other ethnic groups, the most common HBB gene mutations with the highest frequencies among the Arab countries were the following: c.93–21 G > A, c.118 C > T, c.92 + 1 G > A, c.92 + 6 T > C, c.92 + 5 G > C, c.315 + 1 G > A, and c.27dupG (). These mutations show heterogeneity in the associated β-thal phenotypes among the different Arab countries.
The c.93–21 G > A is a Mediterranean mutation found frequently in the Arab countries with different rates ranging from about 1% in Bahrain and UAE to 48% in Egypt. The c.93–21 G > A mutation is of Turkish origin, and it is thought to be spread in the Eastern Mediterranean area and North Africa during the Ottoman Empire, which controlled the region from 16th to early 20th centuries [Citation8]. Additionally, it is thought that c.93–21 G > A is introduced to the Arab world through human migration.
The mutation c.118 C > T is a Mediterranean mutation of Roman origin and is detected with various prevalent rates, highest in the North African countries where it ranges between 26%-49% of the total HBB gene mutations. It is thought that c.118 C > T got spread in the Arab countries during the Roman Empire which governed the region up to the fifth-century BC [Citation8]. An alternative suggestion is that c.118 C > T may have originated from North Africa then spread in Europe through immigration [Citation8].
The c.92 + 1 G > A is a Mediterranean mutation, although being common among Arab countries, its frequency in the Czech Republic is the highest (36%) [Citation68] while in Arab its 26% in Egypt, and lower in other countries. The c.92 + 6 T > C is a Mediterranean mutation of Portuguese origin and found common with low frequency in the Arab countries except in Palestinian territories where it counted for almost half of HBB gene mutations [Citation69]. Interestingly, the c.92 + 6 T > C mutation was reported to be localized in the mountain region of the Palestinian territories which reflects the isolation of the population of this region, which was inhabited by the Samaritans [Citation69].
The c.92 + 5 G > C is an Asian Indian mutation found with high frequency in Oman (73%), United Arab Emirate (47%), and Qatar (35%), while its frequency is much lower in the other Arab countries. This mutation is well established in Asian countries but not among the Arab countries. Most likely, the mutation was introduced to Oman, the United Arab Emirates, and Qatar by the immigration and inhibition of Baluch people who are originally localized in Iran, Afghanistan, and Pakistan.
The c.315 + 1 G > A, a mutation that was reported in various parts of the world, was found in high frequency in Iraq, while in the rest of Arab countries it was common with less frequencies [Citation8]. Interestingly, this mutation is reported to have a high frequency in Iran, suggesting that it may have introduced to Iraq during the Persian Empire [Citation8]. The last common most frequent HBB mutation observed in our study was c.27dupG, which is reported among several Arab countries, highest in Qatar (26%) compared to its frequencies in the nearby countries such Bahrain and Iran. However, this data should be reported with caution due to the limited number of samples used in the study from Qatar [Citation70].
4.5. Genotype-phenotype correlations among Arab patients with β-thal
The primary determinant of the phenotype severity is the type of β-thal allele (β0, β+, β++). This review identifies several Arab mutations affecting transcription, RNA processing, and translation. A wide variety of mutations were found to affect mRNA processing. Point mutations such as substitution affecting the invariant dinucleotides GT or AG can abolish normal mRNA splicing resulting in a β0 phenotype [Citation3]. In Tunisia, the c.92 + 2 T > G, a splice junction mutation identified in a homozygous or heterozygous state with a β0 allele showed complete absence of β-globin production [Citation66]. Such mutation as this is typical for a β-thal major phenotype since most cases are homozygous for the β0 allele (β0/β0) and some cases are compound heterozygous (β+/β0) [Citation10]. Mutations at conserved consensus sequence sites can also reduce the efficiency of splicing leading to mild to severe β-thal phenotype [Citation3]. Homozygous and compound heterozygous in association with a β0 allele status of the β+ transcriptional mutant c.-240 G > A in the promoter region manifested in β-thal media and β-thal minor, respectively. The mutation occurs in the (50–300) region of promoter elements required for regulation of gene expression [Citation65]. Several frameshift mutations in homozygous, heterozygous, and compound heterozygous states were also reported, most of the outcome is a stop codon leading to β-thal major phenotype [Citation59,Citation60,Citation65,Citation71]. A strong correlation is linked between genotype and phenotype in almost all of the unique Arab mutations identified based on the allele type (β0, β+) ().
Diagnosis of a β-thal trait is made depending on the increase in Hemoglobin A2 (HbA2) levels which is <3% in healthy individuals and >3.5% in those with β-thal trait [Citation72]. However, using HbA2 levels alone is challenging. A study from Saudi Arabia showed that HbA2 level was <3.5% in β-thal trait individuals who were confirmed to have β-thal HBB gene mutations [Citation72]. β-thal trait individuals who were carrying c.17_18het_delCT, c.25_26delAA, c.218 G > C, c.281 G > T, c.370A > C, or c.431A > T, HBB mutations had HbA2 level <3%, while those with c.[118 C > T], c.79 G > A, or c.92 þ 5 G > C had borderline HbA2 levels (3.1–3.9%) [Citation72]. Thus, based on the HbA2 level only, these β-thal trait individuals would have been identified falsely as healthy, which is very critical. If β-thal trait-married couple is diagnosed falsely as healthy, there is a 25% probability they would have a child with β-thal major [Citation2]. This observation indicates that in case of β-thal trait the phenotype does not necessarily reflects the genotype and that making diagnosis only based on phenotype can be misleading; hence, molecular tools such as sequencing the HBB gene is an important molecular diagnostic tool.
Regarding genotype and phenotype correlation in the common mutations in the Arab world, the variation or lack of information concerning the specific β-thal phenotype along with its associated clinical manifestations presented some difficulties in determining an accurate phenotype-genotype correlation within the Arab countries, and between Arab countries and the other ethnic groups. Additionally, when the information about the clinical presentation associated with some HBB gene mutations in the other ethnic groups where the mutation is reported was not available, only the data available from the Arab countries were compared. Overall, three of the most common Arab mutations are B+ mutations, these include c.92 + 5 G > C, c.93–21 G > A, and c.92 + 6 T > C. The rest are β0 mutations and include c.118 C > T, c.92 + 1 G > A, c.315 + 1 G > A, and c.27dupG.
With respect to phenotype severity, the c.92 + 5 G > C mutation showed variable clinical presentations for β-thal intermedia and transfusion-dependent β-thal major in homozygous or compound heterozygous states. The combination of the described mutations with other genetic modifiers might contribute to phenotypic severity. In Mauritius and Maldives, c.92 + 5 G > C is associated with β-thal Intermedia [Citation73,Citation74]. Similarly, in Lebanon, Iraq, Jordan the c.92 + 5 G > C is linked with β-thal Intermedia [Citation47,Citation63,Citation75], while in Qatar and Palestine it causes severe β-thal phenotype [Citation70,Citation76] and in Saudi, Algeria, and United Arab Emirates, the mutation is associated with both β-thal major and intermedia [Citation47,Citation77,Citation78]. The c.93–21 G > A and c.92 + 6 T > C mutations present as β-thal intermedia in the heterozygous state or can range in severity from β-thal intermedia to β-thal major in homozygous and compound heterozygous states. The c.93–21 G > A mutation is linked with β-thal Intermedia in Egypt, Lebanon, Iraq, Jordan, Tunis [Citation47,Citation50,Citation63,Citation71,Citation75], while in Morocco, the United Arab Emirates and Saudi the mutation is associated with β-thal major [Citation58,Citation67,Citation79], in Qatar, Libya, and Palestine it causes severe transfusion-dependent β-thal, and in Algeria the mutation is associated with both β-thal major and intermedia [Citation46]. The c.92 + 6 T > C causes β-thal Intermedia in Kuwait, Iraq, Lebanon, Libya, Egypt [Citation47,Citation50,Citation75,Citation80,Citation81], while in Saudi and United Arab Emirates it causes β-thal major [Citation77,Citation79] and it is associated with both β-thal Intermedia and major in Palestine and Morocco [Citation67,Citation69,Citation76].
For β0 mutations, c.118 C > T typically results in transfusion-dependent β-thal major in homozygous and compound heterozygous states. In Argentina, c.118 C > T is associated with β-thal major [Citation82]; similarly, in Saudi, Qatar, and Syria the mutation is associated with β-thal major [Citation70,Citation83,Citation84]. On the other hand, c.118 C > T causes β-thal intermedia in Iraq [Citation47], while in Morocco, Algeria, and Egypt the mutation is linked with both β-thal major and intermedia [Citation46,Citation50,Citation67]. c.92 + 1 G > A present as β-thal intermedia or major. In Palestine, Syria, and Iraq the mutation causes β-thal intermedia [Citation47,Citation51,Citation85], while in Saudi Arabia it causes β-thal major [Citation58] and it is associated with both β-thal intermedia and major in Algeria and Egypt [Citation46,Citation50]. Finally, c.315 + 1 G > A and c.27dupG range in clinical presentation between β-thal intermedia or major in homozygous or compound heterozygous states. The c.315 + 1 G > A is associated with both β-thal intermedia and major in Iran [Citation86], as well as in Saudi Arabia and Palestine [Citation76,Citation77], while it causes β-thal major in the United Arab Emirates, Syria, Lebanon, Algeria, and Qatar [Citation70,Citation75,Citation78,Citation79,Citation84], and it is linked with β-thal intermedia in Iraq and Kuwait [Citation47,Citation80]. The c.27dupG is associated with a β-thal major in Saudi, Syria, Qatar [Citation48,Citation70,Citation83], while in Iraq it causes β-thal intermedia and it is associated with both β-thal intermedia and major in Palestine, Egypt [Citation49,Citation69].
4.6. Limitations
We encountered some limitations in our study. First, the number of study subjects was relatively low in most of the included studies. Second, there is a huge variation among Arab patients with β-thal and their associated clinical manifestations. Third, we found a high methodological heterogeneity among the captured studies, which precluded our ability to draw firm conclusions in the context of phenotype-genotype correlation. Fourth, the death and lack of data in some Arab countries, which makes it difficult to comprehensively draw a genetic epidemiology picture in the Arab world. Finally, some published reports contained incomplete data that contain no genetic analysis for the captured patients.
5. Conclusion
This systemic review highlighted the up-to-date mutational spectrum of β-thal across the Arab world. Our research strategy identified 105 β-thal genetic variants, of which we were able to identify six unique β-thal mutations in six Arab countries. The high allelic heterogeneity of β-thal mutations in Arabs resulted from human migration. A high level of consanguinity marriage contributed to the increased carrier frequency among Arabs. Additionally, we observed a meaningful genotype-phenotype correlation in most of the studied mutations. Although 20 Arab countries reported genetic data on Arab patients with β-thal, the actual prevalence might be higher; therefore, well though genetic epidemiological studies are needed to determine the actual picture of the β-thal in the Arab world.
6. Expert commentary
Although beta-thalassemia (β-thal) is a common genetic disease in the Arab world, there is no current update on the most common and unique mutations that are circulated among Arab patients with β-thal. These data are needed on a large scale for improving the healthcare of Arab patients with β-thal, including genetic counseling and genetic screening. More data are needed to accurately estimate the frequency of β-thal carriers in the Arab world.
Author contributions
HZ conceptualized and designed the study, AMK and AMA wrote the initial draft of the article and performed data analysis. SY contributed in responses to the reviewers’ comments, MY and HZ critically reviewed the manuscript. All authors read and approved the final version of the report before submission.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Open Access funding provided by the Qatar National Library.
Additional information
Funding
References
- Fibach E, Rachmilewitz EA. Pathophysiology and treatment of patients with beta-thalassemia - an update. F1000Research. 6(2156–):2017.
- Galanello R, R OR. Beta-thalassemia. Orphanet J Rare Dis. 2010;5(1):11.
- Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013;3(5):a011700–a.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609–619.
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487.
- Cappellini MD, Porter JB, Viprakasit V, et al. A paradigm shift on beta-thalassaemia treatment: how will we manage this old disease with new therapies? Blood Rev. 2018;32(4):300–311. .
- De Sanctis V, Kattamis C, Canatan D, et al. β-Thalassemia distribution in the old world: an Ancient disease seen from a historical standpoint. Mediterr J Hematol Infect Dis. 2017;9(1):e2017018–e. .
- Hamamy HA, Al-Allawi NAS. Epidemiological profile of common haemoglobinopathies in Arab countries. J Community Genet. 2013;4(2):147–167.
- Higgs DR, Engel JD, G S. Thalassaemia. Lancet. 2012;379(9813):373–383.
- Chen W, Zhang X, Shang X, et al. The molecular basis of beta-thalassemia intermedia in southern China: genotypic heterogeneity and phenotypic diversity. BMC Med Genet. 2010;11(1):31. .
- Hajjej A, Almawi WY, Arnaiz-Villena A, et al. The genetic heterogeneity of Arab populations as inferred from HLA genes. PloS One. 2018;13(3):e0192269–e.
- Ibrahim Z, Makhlouf S. Linguistics in an age of globalization: Perspectives on Arabic language and teaching. American University in Cairo Press. 2008. ISBN: 9789774161490
- Panigrahi I, Agarwal S. Genetic determinants of phenotype in beta-thalassemia. Hematology. 2008;13(4):247–252.
- Zayed H. The Arab genome: health and wealth. Gene. 2016;592(2):239–243.
- Zayed H. The Qatar genome project: translation of whole-genome sequencing into clinical practice. Int J Clin Pract. 2016;70(10):832–834.
- Al-Ali M, Osman W, Tay GK, et al. A 1000 Arab genome project to study the Emirati population. J Hum Genet. 2018. DOI:10.1038/s10038-017-0402-y.
- L Z. The Spectrum of beta-Thalassemia Mutations in the Arab Populations. J Biomed Biotechnol. 2001;1(3):129–132.
- El-Hazmi MA. al-Swailem AR, Warsy AS. Molecular defects in beta-thalassaemias in the population of Saudi Arabia. Hum Hered. 1995;45(5):278–285.
- Alotibi RS, Alharbi E, Aljuhani B, et al. The frequency and spectrum of HBB gene mutation in β-Thalassemia patients in Saudi Arabia. Journal of Natural Science, Biology and Medicine. 2019;10(1):97–10.
- Daar S, Hussein HM, Merghoub T, et al. Spectrum of beta-thalassemia mutations in Oman. Ann N Y Acad Sci. 1998;850:404–406.
- Hassan SM, Hamza N, Jaffer Al-Lawatiya F, et al. Extended molecular spectrum of beta- and alpha-thalassemia in Oman. Hemoglobin. 2010;34(2):127–134. .
- Molecular BE. Basis of β-Thalassemia in the United Arab Emirates. Hemoglobin. 2011;35:581–588.
- Sirdah MM, Sievertsen J, Al-Yazji MS, et al. The spectrum of β-thalassemia mutations in Gaza Strip, Palestine. Blood Cells Mol Dis. 2013;50(4):247–251. .
- Jassim N, Arrayed S, Al-Mukharraq H, et al. Spectrum of β-thalassaemia mutations in Bahrain. 2000;22(1):8–12.
- Abuamer S, Shome DK, Jaradat A, et al. Frequencies and phenotypic consequences of association of α- and β-thalassemia alleles with sickle-cell disease in Bahrain. Int J Lab Hematol. 2017;39(1):76–83. .
- Adekile A, Al-Sherida S, Marouf R, et al. The Sub-Phenotypes of sickle cell disease in Kuwait. Hemoglobin. 2019;43(2):1–5. .
- Kountouris P, Kousiappa I, Papasavva T, et al. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: a 20-year retrospective study. Sci Rep. 2016;6(1):26371. .
- Boussiou M, Karababa P, Sinopoulou K, et al. The molecular heterogeneity of beta-thalassemia in Greece. Blood Cells Mol Dis. 2008;40(3):317–319. .
- Genc A, Tastemir Korkmaz D, Buyukleyla M, et al. Prevalence and molecular analysis of β-thalassemia in Adiyaman, Turkey. Hemoglobin. 2012;36(2):131–138. .
- Agouti I, Badens C, Abouyoub A, et al. Molecular basis of beta-thalassemia in Morocco: possible origins of the molecular heterogeneity. Genet Test. 2008;12(4):563–568. .
- Badens C, Martinez Di Montemuros F, Thuret I, et al. Molecular basis of haemoglobinopathies and G6PD deficiency in the Comorian population. Hematol J. 2000;1(4):264–268.
- R E. Identification and Molecular Characterization of the Most Common Types of Beta Thalassemia Mutations in Sudanese Patients. Am. J. Biomed. Sci. 2019;6: 237–242.
- MAF E-H, Warsy AS, Al-Swailem AR. The Frequency of 14 β-Thalassemia Mutations in the Arab Populations. Hemoglobin. 1995;19(6):353–360.
- Rosatelli MC, Tuveri T, Scalas MT, et al. Molecular screening and fetal diagnosis of beta-thalassemia in the Italian population. Hum Genet. 1992;89(6):585–589.
- Adekile AD, Gu LH, Baysal E, et al. Molecular characterization of alpha-thalassemia determinants, beta-thalassemia alleles, and beta S haplotypes among Kuwaiti Arabs. Acta Haematol. 1994;92(4):176–181. .
- Villegas A, Ropero P, González FA, et al. The thalassemia syndromes: molecular characterization in the Spanish population. Hemoglobin. 2001;25(3):273–283. .
- Scerri CA, Abela W, Galdies R, et al. The beta + IVS, I-NT no. 6 (T –> C) thalassaemia in heterozygotes with an associated Hb Valletta or Hb S heterozygosity in homozygotes from Malta. Br J Haematol. 1993;83(4):669–671. .
- Faustino P, Pacheco P, Loureiro P, et al. The geographic pattern of beta-thalassaemia mutations in the Portuguese population. Br J Haematol. 1999;107(4):903–904. .
- Adekile A, Haider M, Kutlar F. Mutations associated with beta-thalassemia intermedia in Kuwait. Med Princ Pract. 2005;14(Suppl 1):69–72.
- Daar S, Gravell D. Diagnosis of Beta-thalassaemia carriers in the sultanate of oman. Sultan Qaboos Univ Med J. 2006;6(1):27–31.
- Pessar S. Evaluation of twenty four discriminant indices for differentiating beta-thalassemia trait from iron deficiency anemia in Egyptians. Iranian Journal of Pediatric Hematology & Oncology. 2019;9(3):135-146.
- Old JM, Khan SN, Verma I, et al. A multi-center study in order to further define the molecular basis of β-thalassemia in Thailand, Pakistan, Sri Lanka, Mauritius, Syria, and India, and to develop a simple molecular diagnostic strategy by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2001;25(4):397–407.
- Henderson S, Timbs A, McCarthy J, et al. Incidence of haemoglobinopathies in various populations - the impact of immigration. Clin Biochem. 2009;42(18):1745–1756. .
- Tadmouri GO, Nair P, Obeid T, et al. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6(1):17. .
- Steensma DP, Hoyer JD, Fairbanks VF. Hereditary red blood cell disorders in middle eastern patients. Mayo Clin Proc. 2001;76(3):285–293.
- Abdaoui W, Benouareth DE, Djenouni A, et al. Genetic background of β-Thalassemia in Northeast Algeria with assessment of the Thalassemia severity score and description of a new β(0)-Thalassemia frameshift mutation (HBB: c.374dup; p.Pro126Thrfs*15). Hemoglobin. 2019;43(4–5):223–228. .
- Amin SS, Jalal SD, Ali KM, et al. Beta-Thalassemia Intermedia: A Single Thalassemia center experience from Northeastern Iraq. Biomed Res Int. 2020;1:1-11.
- Jarjour RA, Murad H, Moasses F, et al. Molecular update of β-thalassemia mutations in the Syrian population: identification of rare β-thalassemia mutations. Hemoglobin. 2014;38(4):272–276.
- El-Shanshory M, Hagag A, Shebl S, et al. Spectrum of Beta Globin Gene mutations in Egyptian Children with β-Thalassemia. Mediterr J Hematol Infect Dis. 2014;6(1):e2014071–e.
- Hassan TH, Salam MMA, Zakaria M, et al. Impact of Genotype of Beta Globin Gene on hepatic and myocardial iron content in Egyptian Patients with Beta Thalassemia. Indian J Hematol Blood Transfus. 2019;35(2):284–291.
- Faraon R, Daraghmah M, Samarah F, et al. Molecular characterization of β-thalassemia intermedia in the West Bank, Palestine. BMC Hematol. 2019;19(1):4.
- Mustafa I, Firdous N, Shebl FM, et al. Genetic epidemiology of beta-thalassemia in the Maldives: 23 years of a beta-thalassemia screening program. Gene. 2020;741:144544.
- Saeed U, Piracha ZZ. Thalassemia: impact of consanguineous marriages on most prevalent monogenic disorders of humans. Asian Pacific J Trop Disease. 2016;6(10):837–840.
- Murad H, Moasses F, Dabboul A, et al. Geographical distribution of β-globin gene mutations in Syria. Hematology. 2018;23(9):697–704.
- Mashi A, Khogeer H, khyatte A, et al. Molecular patterns of β-thalassemia mutations of Saudi patients referred to King Faisal Specialist Hospital and Research Center. Journal of Applied Hematology. 2017;8(3):99.
- Alaithan MA, AbdulAzeez S, Borgio JF. A comprehensive review of the prevalence of beta globin gene variations and the co-inheritance of related gene variants in Saudi Arabians with beta-thalassemia. Saudi Med J. 2018;39(4):329–335.
- Al-Ali AK, Al-Ateeq S, Imamwerdi BW, et al. Molecular bases of beta-thalassemia in the Eastern Province of Saudi Arabia. J Biomed Biotechnol. 2005;4:322–325.
- Abuzenadah AM, Hussein IM, Damanhouri GA, et al. Molecular basis of β-thalassemia in the western province of Saudi Arabia: identification of rare β-thalassemia mutations. Hemoglobin. 2011;35(4):346–357. .
- Croteau SE, Luo HY, Lehmann LE, et al. Novel dominant β-thalassemia: hb Boston-Kuwait [codon 139/140(+T)]. Pediatr Blood Cancer. 2013;60(10):E131–4. .
- Al-Obaidli A, Gerard N, Al Zadjali S, et al. A novel deletional beta-thalassemic variant in an ethnic Qatari patient. Hemoglobin. 2009;33(3):214–219. .
- Hardison RC, Chui DH, Giardine B, et al. HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum Mutat. 2002;19(3):225–233. .
- Patrinos GP, Giardine B, Riemer C, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 2004;32(Database issue):D537–D41. .
- Sadiq MF, Eigel A, Horst J. Spectrum of beta-thalassemia in Jordan: identification of two novel mutations. Am J Hematol. 2001;68(1):16–22.
- Waye J, Eng B, Got T, et al. Sudanese (δβ)0-Thalassemia: identification and Characterization of a Novel 9.6 kb Deletion. Hemoglobin. 2015;39. DOI:10.3109/03630269.2015.1057736.
- Agouti I, Bennani M, Nezri M, et al. Beta-thalassemia intermedia due to two novel mutations in the promoter region of the beta-globin gene. Eur J Haematol. 2008;80(4):346–350. .
- Chibani J, Vidaud M, Duquesnoy P, et al. The peculiar spectrum of beta-thalassemia genes in Tunisia. Hum Genet. 1988;78(2):190–192. .
- Lemsaddek W, Picanço I, Seuanes F, et al. The beta-thalassemia mutation/haplotype distribution in the moroccan population. Hemoglobin. 2004;28:25–37.
- Divoka M, Partschova M, Kucerova J, et al. Molecular Characterization of β-Thalassemia in the Czech and Slovak Populations: mediterranean, Asian and Unique Mutations. Hemoglobin. 2016;40(3):156–162. .
- El-Latif MA, Filon D, Rund D, et al. The beta+-IVS-I-6 (T–>C) mutation accounts for half of the thalassemia chromosomes in the Palestinian populations of the mountain regions. Hemoglobin. 2002;26(1):33–40. .
- Al-Obaidli A, Hamodat M, Fawzi Z, et al. Molecular basis of thalassemia in Qatar. Hemoglobin. 2007;31(2):121–127. .
- Fattoum S, Messaoud T, Bibi A. Molecular basis of beta-thalassemia in the population of Tunisia. Hemoglobin. 2004;28(3):177–187.
- Al-Amodi AM, Ghanem NZ, Aldakeel SA, et al. Hemoglobin A(2) (HbA(2)) has a measure of unreliability in diagnosing β-thalassemia trait (β-TT. Curr Med Res Opin. 2018;34(5):945–951. .
- Verma IC, Kleanthous M, Saxena R, et al. Multicenter Study of the Molecular Basis of Thalassemia Intermedia in Different Ethnic Populations. Hemoglobin. 2007;31(4):439–452. .
- Furuumi H, Firdous N, Inoue T, et al. Molecular basis of beta-thalassemia in the Maldives. Hemoglobin. 1998;22(2):141–151. .
- Makhoul NJ, Wells RS, Kaspar H, et al. Genetic heterogeneity of Beta thalassemia in Lebanon reflects historic and recent population migration. Ann Hum Genet. 2005;69(Pt 1):55–66. .
- Darwish H, El-Khatib F, Ayesh S. Spectrum of β-globin gene mutations among thalassemia patients in the West Bank Region of Palestine. Hemoglobin. 2005;29:119–132.
- Al-Ali AK, Al-Ateeq S, Imamwerdi BW, et al. Molecular bases of beta-thalassemia in the Eastern Province of Saudi Arabia. J Biomed Biotechnol. 2005;4:322–325.
- Boudrahem-Addour N, Zidani N, Carion N, et al. Molecular heterogeneity of beta-thalassemia in Algeria: how to face up to a major health problem. Hemoglobin. 2009;33(1):24–36. .
- Baysal E. Molecular heterogeneity of beta-thalassemia in the United Arab Emirates. Community Genet. 2005;8(1):35–39.
- Adekile AD, Haider MZ, Dimovski A, et al. Mutations associated with β-thalassemia intermedia in Kuwait. Blood. 2000;96(11 PART II): 22B-22B.
- Marwan MM, Scerri CA, Zarroag SO, et al. Comparative In Vivo Expression of β+-Thalassemia Alleles. Hemoglobin. 1999;23(3):221–229. .
- Rossetti LC, Targovnik HM, Varela V. The molecular basis of beta-thalassemia in Argentina. Influence of the pattern of immigration from the Mediterranean Basin. Haematologica. 2004;89(6):746.
- El-Harth EH, Kühnau W, Schmidtke J, et al. Identification and clinical presentation of beta thalassaemia mutations in the eastern region of Saudi Arabia. J Med Genet. 1999;36(12):935–937.
- Çevirici H, Acıpayam C, Dündar Yenilmez E, et al. Investigation of beta globin gene mutations in Syrian refugee patients with thalassemia major. Turk JBiochem. 2019;44(2):126–129. .
- Murad H, Moassas F, Ali B, et al. A compound heterozygous −29 A>G and IVS-I-1 G>A mutation of HBB gene leading to β-thalassemia intermedia in a Syrian patient: A case repot. Cogent Med. 2019;6. DOI:10.1080/2331205X.2019.1581448.
- Maryami F, Azarkeivan A, Fallah MS, et al. A Large Cohort Study of Genotype and Phenotype Correlations of Beta- Thalassemia in Iranian Population. Int J Hematol Oncol Stem Cell Res. 2015;9(4):198–202.