
ABSTRACT
Introduction
In β-thalassemia, imbalanced globin synthesis causes reduced red blood cell survival and ineffective erythropoiesis. Suppressed hepcidin levels increase ferroportin-mediated iron transport in enterocytes, causing increased iron absorption and potentially iron overload. Low hepcidin also stimulates ferroportin-mediated iron release from macrophages, increasing transferrin saturation (TSAT), potentially forming non-transferrin-bound iron, which can be toxic. Modulating the hepcidin–ferroportin axis is an attractive strategy to improve ineffective erythropoiesis and limit the potential tissue damage resulting from iron overload. There are no oral β-thalassemia treatments that consistently ameliorate anemia and prevent iron overload.
Areas covered
The preclinical and clinical development of vamifeport (VIT-2763), a novel ferroportin inhibitor, was reviewed. PubMed, EMBASE and ClinicalTrials.gov were searched using the search term ‘VIT-2763ʹ.
Expert opinion
Vamifeport is the first oral ferroportin inhibitor in clinical development. In healthy volunteers, vamifeport had comparable safety to placebo, was well tolerated and rapidly decreased iron levels and reduced TSAT, consistent with observations in preclinical models. Data from ongoing/planned Phase II studies are critical to define its potential in β-thalassemia and other conditions associated with iron overabsorption and/or ineffective erythropoiesis. If vamifeport potentially increases hemoglobin and reduces iron-related parameters, it could be a suitable treatment for non-transfusion-dependent and transfusion-dependent β-thalassemia.
1. Introduction
β-thalassemia is caused by the partial or complete loss of β-globin synthesis, which leads to a relative excess of α-chains of hemoglobin and, consequently, ineffective erythropoiesis. The imbalance between α- and β-chains results in formation of free α-globin-heme complexes in red blood cell (RBC) precursor membranes and precipitation of hemichromes on the RBC membrane. These promote oxidative damage, thereby inducing intra-marrow apoptosis of RBC precursors and also reducing the lifespan of circulating RBCs [Citation1,Citation2].
Clinically, β-thalassemias are classified according to dependence on blood transfusions necessitated by the degree of imbalance between the α- and β-chain production [Citation3]. Patients who need regular blood transfusions for survival are defined as having transfusion-dependent thalassemia (TDT), which includes β-thalassemia major, and severe hemoglobin E/β-thalassemia [Citation3]. In TDT, the objective of treatment is to maintain trough hemoglobin levels above 9–10 g/dL using recurrent chronic transfusion to inhibit ineffective erythropoiesis. Blood transfusions and, to a lesser extent, enhanced gastrointestinal iron absorption, result in progressive iron accumulation and, without treatment, inevitably result in iron overload, particularly in the liver, endocrine system and heart [Citation4]. Non-transferrin bound iron (NTBI) appears in the plasma of patients with systemic iron overload when the binding capacity of transferrin is exceeded [Citation5]. Aberrant uptake of NTBI with subsequent excess iron deposition as hemosiderin can cause organ toxicity, manifesting as cirrhosis in the liver, damage to the endocrine glands that impairs hormone production, and cardiac disease, the latter of which is historically the major cause of death in patients with TDT [Citation6]. Patients are usually prescribed iron chelation therapy (ICT) to treat iron overload, but iron excretion is relatively inefficient and slow and places a high treatment burden on patients [Citation7,Citation8]. Chelation treatment seeks to balance the toxic effects of excess iron with the potential toxicity of excessive iron chelation. This balance can be difficult to optimize as the side effects associated with ICT are not uncommon and can be significant. For patients who are unable to tolerate ICT, the current lack of alternative therapies is problematic. If left untreated, iron overload increases the risk of complications and can have a negative impact on survival [Citation8–10]. At present, patients in higher-income countries who receive regular transfusions and who are on regular ICT have a life expectancy beyond the fifth decade. However, even in optimally treated patients, life expectancy remains well below that of the general population [Citation11].
Patients who need occasional or intermittent transfusions are defined as having non-transfusion-dependent thalassemia (NTDT), which includes some patients with β-thalassemia intermedia, mild to moderate hemoglobin E/β-thalassemia, and hemoglobin H disease [Citation3]. Patients with NTDT comprise a wide range of phenotypes driven by the degree of globin chain imbalance, mainly due to the underlying molecular defects (either β0 or β+-thalassemia); their clinical symptoms are generally a result of ineffective erythropoiesis, anemia, and iron overload [Citation12,Citation13]. In addition to iron toxicity, complications are similar to those reported in patients with TDT, and can include hypercoagulability and thromboembolic disease, pulmonary hypertension, extramedullary hematopoiesis pseudo-tumors, leg ulcers, gallstones, pregnancy complications, and endocrine disorders such as osteoporosis and delayed puberty [Citation3,Citation14]. In patients with NTDT, iron loading occurs more slowly than in patients with TDT and is primarily caused by increased intestinal absorption secondary to chronic anemia and tissue hypoxia, rather than transfusion [Citation15]. Patients with NTDT are typically able to maintain hemoglobin levels above 7 g/dL, but they may require occasional transfusions; for example, when pregnant, when undergoing a surgical procedure, or if they have an overt infection [Citation3]. Over time, they may require more transfusions due to disease-related complications, particularly associated with progressive anemia becoming less tolerated with increasing age [Citation3]. The prognosis for patients with NTDT has historically been better than for TDT [Citation16]. However, with improvement of blood transfusion and ICT for TDT, mortality and thalassemia-related complications have substantially decreased in these patients [Citation17], while there is now greater recognition of a high level of morbidity associated with NTDT [Citation18]. Without appropriate treatment, patients with NTDT may experience frequent morbidity and a poor quality of life. Clinical management of NTDT with occasional blood transfusions can add to iron overload and lead to alloimmunization, as is observed in patients with TDT [Citation16,Citation19–21]. Other treatment options for NTDT and TDT are currently limited to splenectomy, bone marrow transplant, which can potentially be curative, and agents that are not yet approved for use specifically in NTDT, such as luspatercept (for which a clinical trial in NTDT is ongoing) and hydroxyurea () [Citation22–28]. Supportive therapies such as folic acid supplementation, antibiotic, antiviral and antifungal therapies, intensive care, and/or nutritional support can also be used.
Table 1. Advantages and disadvantages of current and potential therapies for NTDT
Iron homeostasis is mediated primarily via the hepcidin–ferroportin axis [Citation29]. Hepcidin is a hormone secreted by the liver in response to iron loading and inflammation, which regulates iron levels by binding and occluding the iron transporter ferroportin [Citation30], and targeting it for degradation [Citation19,Citation31]. Ferroportin is the sole cellular efflux transporter for iron, and its expression and function on iron-exporting cells is regulated post-translationally by hepcidin, which binds to ferroportin, resulting in its internalization via endocytosis and subsequent degradation in lysosomes [Citation29,Citation32]. Erythropoiesis exerts strong control over iron homeostasis [Citation33]. Anemia and hypoxia arising from ineffective erythropoiesis result in high levels of erythropoietin and erythroferrone, which respectively stimulate erythropoiesis and suppress hepcidin [Citation34–36]. The reduced level of hepcidin causes an increase in the absorption of dietary iron from upregulated ferroportin expression and release of recycled iron from macrophages, ultimately resulting in an excess of NTBI and potentially fatal iron toxicity [Citation19]. Consequently, modulation of the hepcidin–ferroportin axis has the potential to limit non-transfusional iron overload. In addition, iron overload from hepcidin insufficiency appears to exacerbate ineffective erythropoiesis [Citation37], and it has been proposed that modulation of iron homeostasis could improve ineffective erythropoiesis, thereby ameliorating anemia [Citation7]. Potential mechanisms of action to mediate the improvements in erythropoiesis include a reduction in serum iron levels with lowered transferring saturation (TSAT), an effect on monoferric transferrin levels, or an effect on macrophage iron levels. Furthermore, iron restriction decreases erythropoiesis via the TfR2-Scribble pathway that regulates the expression of erythropoietin (EPO) receptors on the cell surface of the immature erythroid precursors in mice [Citation38]. This leads to the resistance of the erythron to EPO and to a reduction in RBC production. Conversely, iron restriction also leads to an increase in heme-regulated eIF2α kinase (HRI) activity, which phosphorylates eIF2α and thereby decreases protein translation in immature erythroid precursors and also suppresses mTORC1 signaling of activated EPO receptors () [Citation39]. mTORC1 signaling preferentially leads to erythroid cell proliferation and inhibits cell differentiation. These effects would favor cell differentiation over proliferation, contributing positively to reduce ineffective erythropoiesis. Data from animal models have shown that TSAT decreases α-globin hemichrome formation, oxidative damage of the developing RBCs in the bone marrow, and in turn ameliorates erythropoiesis and increases hemoglobin levels [Citation40,Citation41].
Figure 1. Molecular pathways of iron-restricted erythropoiesis through translational control at eIF2#x3B1;P, TfR2/Scrib and mTORC1 signaling. Reproduced from Zhang S, et al. Blood 2018;131(4):450–461 [39], with permission from the American Society of Hematology. #xA9; 2018 by the American Society of Hematology. Left: Steady-state erythropoiesis in the bone marrow under iron sufficiency. In iron and thus heme abundance, HRI homodimer is inactive because of full occupancies of heme onto the 4 HRI heme-binding domains and so is unable to phosphorylate eIF2#x3B1;, thus permitting global protein synthesis, mainly globin proteins in erythroid cells. Sufficient hemoglobin production maintains oxygen-delivering capacity in blood without hypoxia. Middle: Activation of HRI-ISR under iron deficiency mitigates ineffective erythropoiesis. Under iron/heme deficiency, HRI in bone marrow erythroid precursors is activated by the dissociation of heme. HRI then induces ISR, phosphorylating eIF2#x3B1;, which inhibits globin protein synthesis and results in a decrease of hemoglobin content and consequently induction of tissue hypoxia stress. In addition, eIF2#x3B1;P selectively enhances the translation of ATF4 mRNA to alleviate ROS levels. HRI-ISR also inhibits mTORC1 signaling to mitigate ineffective erythropoiesis in the spleen. Right: Elevated mTORC1 signaling and development of ineffective erythropoiesis in mutant mice defective in HRI-ISR signaling in iron deficiency. Hypoxia induced by iron deficiency stimulates Epo production in the kidney and increases Epo in blood circulation. In the spleen, binding of Epo to its receptors in erythroid precursors induces AKT/mTORC1 signaling, thus phosphorylating 4EBP1 and S6K/S6 to increase protein synthesis, promote proliferation, and inhibit erythroid differentiation, which are the characteristics of ineffective erythropoiesis. HRI-ISR serves as feedback to inhibit mTORC1 signaling activity, inhibiting the development of ineffective erythropoiesis in iron deficiency. 4EBP1, eukaryotic translation initiation factor 4E binding protein 1; AKT, protein kinase B; ATF4, activating transcription factor 4; eIF2#x3B1;, eukaryotic translation initiation factor 2 subunit 1; Epo(R), erythropoietin (receptor); Fe, iron; HRI-ISR, heme-regulated eIF2#x3B1; kinase-integrated stress response; mRNA, messenger ribonucleic acid; mTORC1, mammalian target of rapamycin complex 1; P, phosphorylated; ROS, reactive oxygen species; Scrib, scribble; S6(K), ribosomal protein S6 (kinase); TfR2, transferrin receptor 2.
![Figure 1. Molecular pathways of iron-restricted erythropoiesis through translational control at eIF2#x3B1;P, TfR2/Scrib and mTORC1 signaling. Reproduced from Zhang S, et al. Blood 2018;131(4):450–461 [39], with permission from the American Society of Hematology. #xA9; 2018 by the American Society of Hematology. Left: Steady-state erythropoiesis in the bone marrow under iron sufficiency. In iron and thus heme abundance, HRI homodimer is inactive because of full occupancies of heme onto the 4 HRI heme-binding domains and so is unable to phosphorylate eIF2#x3B1;, thus permitting global protein synthesis, mainly globin proteins in erythroid cells. Sufficient hemoglobin production maintains oxygen-delivering capacity in blood without hypoxia. Middle: Activation of HRI-ISR under iron deficiency mitigates ineffective erythropoiesis. Under iron/heme deficiency, HRI in bone marrow erythroid precursors is activated by the dissociation of heme. HRI then induces ISR, phosphorylating eIF2#x3B1;, which inhibits globin protein synthesis and results in a decrease of hemoglobin content and consequently induction of tissue hypoxia stress. In addition, eIF2#x3B1;P selectively enhances the translation of ATF4 mRNA to alleviate ROS levels. HRI-ISR also inhibits mTORC1 signaling to mitigate ineffective erythropoiesis in the spleen. Right: Elevated mTORC1 signaling and development of ineffective erythropoiesis in mutant mice defective in HRI-ISR signaling in iron deficiency. Hypoxia induced by iron deficiency stimulates Epo production in the kidney and increases Epo in blood circulation. In the spleen, binding of Epo to its receptors in erythroid precursors induces AKT/mTORC1 signaling, thus phosphorylating 4EBP1 and S6K/S6 to increase protein synthesis, promote proliferation, and inhibit erythroid differentiation, which are the characteristics of ineffective erythropoiesis. HRI-ISR serves as feedback to inhibit mTORC1 signaling activity, inhibiting the development of ineffective erythropoiesis in iron deficiency. 4EBP1, eukaryotic translation initiation factor 4E binding protein 1; AKT, protein kinase B; ATF4, activating transcription factor 4; eIF2#x3B1;, eukaryotic translation initiation factor 2 subunit 1; Epo(R), erythropoietin (receptor); Fe, iron; HRI-ISR, heme-regulated eIF2#x3B1; kinase-integrated stress response; mRNA, messenger ribonucleic acid; mTORC1, mammalian target of rapamycin complex 1; P, phosphorylated; ROS, reactive oxygen species; Scrib, scribble; S6(K), ribosomal protein S6 (kinase); TfR2, transferrin receptor 2.](/cms/asset/cb30cd26-a37d-44bf-850f-d0f561943343/ierr_a_1935854_f0001_oc.jpg)
There is a need for new treatments that are effective and reduce the development of thalassemia-related complications, which are well tolerated, convenient, and that will improve long-term adherence and quality of life for patients. Given its central role in iron homeostasis, the hepcidin–ferroportin axis represents an attractive therapeutic target for β-thalassemia to improve ineffective erythropoiesis and associated iron overload. Since ineffective erythropoiesis and tissue hypoxia inhibit hepcidin production, resulting in iron overload, agents have been developed to reproduce the effects of hepcidin. Studies in mice indicate a potential for such agents in the treatment of NTDT [Citation40] and several have entered clinical development () [Citation42–46]. Data from preliminary human studies using injected hepcidin mimetics (PTG-300 and LJPC-401) showed that the agents were generally well tolerated and effected reductions in serum iron levels and decreased TSAT in healthy volunteers [Citation43,Citation45]; LJPC-401 also reduced serum iron levels in patients with iron overload [Citation44]. These agents aim to decrease TSAT, either to improve erythropoiesis (PTG-300) or to decrease myocardial iron uptake (LJPC-401) (for a review, see [Citation47]). However, these preparations are not orally active and would require frequent parenteral dosing. Another approach to restrict TSAT and improve erythropoiesis is by targeting expression of transmembrane protease serine 6 (TMPRSS6), a transmembrane serine protease that reduces hepcidin production. Findings from preclinical studies in hemoglobinopathy mouse models show that Tmprss6 suppression can improve anemia, ineffective erythropoiesis, and splenomegaly; data also suggest improved activity in combination with iron-chelating agents [Citation48,Citation49]. Initial safety and pharmacodynamic (PD) findings from a Phase I clinical study of IONIS TMPRSS6-LRX in healthy volunteers showed a reduction in serum iron and TSAT at Week 10 following 20 or 40 mg subcutaneous doses at Week 1, 4, 6 and 8; treatment-emergent adverse events were generally mild [Citation46]. A Phase IIa study with IONIS TMPRSS6-LRX is ongoing (NCT04059406).
Table 2. Summary of drugs in clinical development targeting the hepcidin–ferroportin axis
Direct inhibition of the ferroportin receptor represents a novel approach to targeting the hepcidin–ferroportin axis [Citation50]. Vamifeport (previously known as VIT-2763) is an oral ferroportin inhibitor that has been specifically designed to target the hepcidin–ferroportin axis and ameliorate ineffective erythropoiesis [Citation42]. In this review, we summarize the preclinical and clinical development of vamifeport, which is currently undergoing Phase IIa clinical evaluation in patients with NTDT, with Phase IIb evaluation in patients with TDT planned. As this is a historical, narrative review tracking the development of vamifeport, there was no prespecified search strategy. In line with this, PubMed, EMBASE and ClinicalTrials.gov databases were searched for all potentially relevant items using the search term ‘VIT-2763ʹ.
2. Preclinical findings
2.1. Vamifeport mechanism of action and in vitro studies
Vamifeport is a small-molecule oral inhibitor of the iron transporter ferroportin. Screening a library of small molecules for modulators of ferroportin internalization led to the identification of vamifeport, the chemical structure of which is shown in [Citation50].
Figure 2. Chemical structure and mechanism of action of vamifeport (VIT-2763)
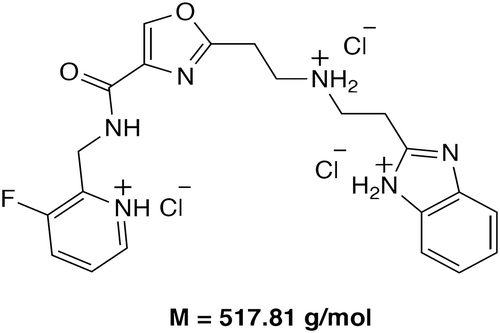
In vitro findings demonstrated that vamifeport acts as a ferroportin inhibitor, competing with hepcidin for binding to ferroportin, displacing hepcidin bound to recombinant ferroportin, and reducing cellular iron efflux with a potency similar to that of hepcidin () [Citation50–53]. In a competition assay using the mouse macrophage cell line J774, vamifeport and unlabeled synthetic hepcidin showed similar potency in competing with fluorescently labeled hepcidin for binding to ferroportin (mean IC50 ± standard deviation [SD] 9 ± 5 nM vs 13 ± 4 nM, respectively). The effect of vamifeport and hepcidin on ferroportin function was evaluated in T47D, a human breast cancer cell line endogenously expressing ferroportin, incubated with iron sulfate labeled with the stable isotope 58Fe. Vamifeport and hepcidin both demonstrated dose-dependent inhibition of cellular iron efflux, with vamifeport being slightly more potent than hepcidin (mean ± SD EC50 of 68 ± 21 nM for vamifeport vs 123 ± 46 nM for hepcidin). Immunoprecipitation experiments using J774 cells showed that vamifeport and hepcidin similarly triggered ferroportin internalization and degradation. Both vamifeport and hepcidin induced ferroportin ubiquitination within 10–20 min, followed by degradation within 1–2 h. There were slight differences in the efficacy and kinetics of vamifeport and hepcidin [Citation50].
Table 3. Key findings from preclinical studies of vamifeport (VIT-2763)
In terms of selectivity, at a single concentration of 10 μM, vamifeport showed no significant in vitro effects on any of the targets screened in a panel of 70 receptors, ion channels, and transporters, or in a selection of 27 enzyme assays. The strongest off-target effects were observed with IC50s at least 1,000-fold higher than those found for ferroportin in the J774 assay. Studies using a cell line expressing doxycycline-inducible fluorescent-labeled human ferroportin provide further evidence that ferroportin expression is needed for the inhibitory effect of vamifeport [Citation50].
2.2. In vivo studies
PD studies measuring plasma iron showed that the biological activity of vamifeport in rodents was related to reduced iron egress into the plasma. In a mouse model of acute hypoferremia (C57BL/6 mice fed a standard rodent diet), oral administration of 60 mg/kg vamifeport resulted in a rapid decrease in serum iron at 1 and 3 h post-dose that was similar to hepcidin: 40% and 58% lower, respectively, than the vehicle control [Citation50]. Similarly, in healthy rats fed a standard rodent diet, plasma iron decreased over time after oral vamifeport dosing; minimum serum iron levels were reached after 4 h, with levels recovering within 8–24 h [Citation50]. To determine the effect on intestinal iron absorption, a single 3-, 10-, or 30-mg/kg dose of vamifeport or vehicle control was administered to anemic Wistar rats (fed a low-iron diet to develop anemia) followed by an oral iron bolus. Vamifeport at 10 or 30 mg/kg, but not 3 mg/kg, significantly suppressed the serum iron increase 3 h after the oral iron bolus, compared with control () [Citation51].
The effects of vamifeport have been evaluated in several animal disease models (). The selection of doses tested in the preclinical models was based on dose-optimization pilot studies in the respective models. A murine model of hereditary hemochromatosis (HFE C282Y) was used to determine the impact of vamifeport on serum and organ iron concentrations. This disease is a result of mutations in genes encoding components that regulate iron homeostasis through the hepcidin–ferroportin axis, causing excessive intestinal absorption of dietary iron and pathological iron overload. Mice fed a low-iron diet were dosed with vamifeport 40 or 110 mg/kg along with stable iron isotope (58Fe) in the drinking water, with the concentration adjusted to 250 ppm iron content to correspond to the iron intake of a standard rodent diet. At the end of the study period, vamifeport had resulted in the correction of serum iron to the levels of wild-type mice and prevented iron retention in the livers of the hemochromatosis mice [Citation51,Citation53].
The effects of vamifeport on anemia, ineffective erythropoiesis, and iron overload have been studied using the Hbbth3/+ murine model of NTDT [Citation50]. Mice fed a low-iron diet were dosed orally with either 30 or 100 mg/kg vamifeport or vehicle control twice daily for 5 weeks, and between doses they had access to 58Fe-containing water, adjusted to 250 ppm iron content to correspond to the iron intake of a standard rodent diet. Findings from the study demonstrated a range of effects of vamifeport. Notably, vamifeport dosed at 30 and 100 mg/kg significantly decreased serum iron levels by 77% and 84%, respectively, compared with control, and resulted in a significant reduction in relative spleen weight by 52% and 65%, respectively. Treatment also decreased organ iron levels and improved hematologic parameters, including hemoglobin level and RBC count, demonstrating amelioration of anemia and improved erythropoiesis [Citation50]. Vamifeport reduced the percentages of early erythroid precursors in the bone marrow and spleen, and increased the percentage of mature erythrocytes, providing evidence of improved ineffective erythropoiesis [Citation50]. Importantly, vamifeport dose-dependently reduced the insoluble, membrane-bound α-globin aggregates in RBCs, compared with vehicle controls. Data also demonstrated that vamifeport reduced the percentage of reactive oxygen species (ROS)-positive RBCs and extended the lifespan of RBCs, thereby improving anemia and tissue oxygenation [Citation50]. Additionally, in a study of combination treatment with vamifeport and the iron-chelating agent deferasirox in the same preclinical model of NTDT, findings revealed no negative impact of vamifeport on the efficacy of deferasirox and vice versa [Citation52]. Vamifeport alone or in combination with deferasirox significantly increased hemoglobin, RBC counts, and hematocrit, and decreased reticulocyte numbers, reflecting improved erythropoietic efficiency. Vamifeport in the presence or absence of deferasirox decreased formation of ROS and RBC apoptosis and improved mitochondrial clearance. Vamifeport alone decreased serum iron and TSAT but did not reverse the established liver iron overload. Deferasirox had no effect on serum iron, TSAT, erythropoiesis or RBC phenotype, but decreased liver iron concentration, both alone and in the presence of vamifeport [Citation52]. Therefore, the potentiality of combining treatment with vamifeport and deferasirox relates to the drugs’ complementary effects on RBC parameters/serum iron levels and liver iron levels, respectively. Currently, three iron chelators (deferoxamine, deferiprone, and deferasirox) are available for the management of iron overload in patients with TDT [Citation54]. Deferoxamine is administered parenterally, whereas deferiprone and deferasirox are oral drugs with greater dosing convenience. Deferasirox is the only iron chelator tested in controlled clinical studies and approved in the USA and Europe for the management of iron overload in patients with NTDT [Citation55]. Combination therapy with the iron chelator deferiprone and minihepcidins or tmprss6-targeting oligonucleotides in the Hbbth3/+ thalassemia model showed no negative PD interactions [Citation40,Citation49,Citation56]. Future preclinical studies need to address potential pharmacokinetic or PD interactions of vamifeport with iron-chelating agents other than deferasirox.
Single- and repeated-dose toxicity studies have been conducted to support the clinical evaluation of vamifeport. In rodents, vamifeport was well tolerated in 14-day dosing studies, with the ‘no observed adverse effect level’ (NOAEL) being >600 mg/kg [Citation50]. In a 13-week rat study, the NOAEL was 100 mg/kg/day. Pale ears correlating with anemia and hemoglobin levels as low as 64 g/L were noted after 8 weeks at vamifeport 200 mg/kg/day, the highest dose tested, which necessitated a dosing break. After 6 days, when hemoglobin values had recovered to at least 114 g/L, dosing restarted using a 5 days on/2 days off regimen until the end of the study [Vifor, data on file (report FIT-11)]. Therefore, two scenarios need to be considered regarding long-term therapy with vamifeport; one is where iron-restricted erythropoiesis leads to therapeutic effects in β-thalassemia patients with ineffective erythropoiesis. The second scenario is where iron-deficient erythropoiesis is taking place due to an exaggerated pharmacology of vamifeport. The right balance between iron-restricted (beneficial) and iron-deficient erythropoiesis (exaggerated pharmacology) is the therapeutic target to be reached. Hemoglobin levels, hematocrit, RBC and reticulocyte counts, as well as TSAT and other iron level biomarkers, will help to define that therapeutic target ‘sweet spot’.
Outcomes from these preclinical studies show that vamifeport is a potent and well-tolerated oral ferroportin inhibitor, with a mode of action similar to hepcidin (). By blocking iron export into plasma and thereby limiting the availability of iron for erythropoiesis, vamifeport consistently improved ineffective erythropoiesis, ameliorated anemia, and prevented liver iron loading in a well-established NTDT model. These findings demonstrate the potential of vamifeport to correct a critical pathological event that occurs in NTDT and highlight the medical plausibility of vamifeport as a potential treatment for β-thalassemia. A first-in-human Phase I study was subsequently conducted in healthy adult volunteers.
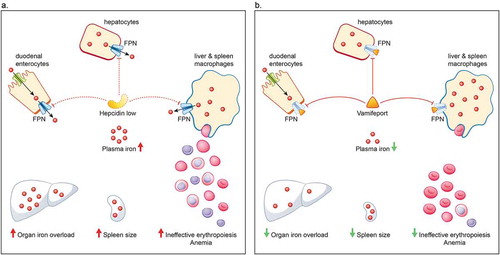
Figure 3. Hepcidin–ferroportin axis in non-transfusion-dependent thalassemia (A) and how it is affected by vamifeport (VIT-2763) (B). A. Iron homeostasis is mediated primarily via the hepcidin–ferroportin axis. Hepcidin regulates iron levels by targeting the iron transporter ferroportin for degradation. Erythropoiesis exerts a strong control over iron homeostasis, so that when erythropoiesis is impaired, iron absorption is increased, irrespective of iron stores. In patients with NTDT, anemia and hypoxia arising from ineffective erythropoiesis stimulates erythropoiesis and suppresses hepcidin. The reduced level of hepcidin causes an increase in the absorption of dietary iron and release of recycled iron from macrophages, ultimately resulting in saturation of the iron-binding capacity of transferrin and consequently generation of non-transferrin-bound iron that results in organ iron overload and further drives ineffective erythropoiesis, splenomegaly, and anemia. B. Vamifeport, a novel oral ferroportin inhibitor, acts like hepcidin to target ferroportin for degradation, thereby blocking iron export into plasma. By limiting plasma iron levels, vamifeport acts to prevent organ iron overloading, reduce spleen size, and improve erythropoiesis and anemia. FPN, ferroportin; NTDT, non-transfusion-dependent thalassemia
3. Phase I clinical study in healthy volunteers
A Phase I, randomized, double-blind, placebo-controlled study was conducted to assess the safety, tolerability, PK, and PD of single- and multiple-ascending doses of vamifeport in 72 healthy adult volunteers [Citation42]. In the single-ascending dose phase, participants received single oral doses of vamifeport (5, 15, 60, 120, or 240 mg) or matching placebo. In the multiple-ascending dose phase, participants received multiple oral doses of vamifeport (60 or 120 mg once daily or twice daily), or matching placebo for 7 days [Citation42]. Doses were given between 8 and 10 am following a 10-h overnight fast, with a further evening dose given 12 h later for the twice-daily dosing regimens.
Vamifeport administered at single oral doses up to 240 mg or multiple oral doses up to 120 mg twice daily was well tolerated compared with placebo. There were no serious or severe adverse events (AEs) or discontinuations due to AEs [Citation42]. PK/PD data were consistent with findings from the preclinical studies. Vamifeport was rapidly absorbed, with detectable levels observed 15–30 min post-dose in most participants. In the multiple-dose group, there was no evidence of drug accumulation after 7 days of repeated dosing for both the once-daily and twice-daily dosing regimens. The elimination half-life of vamifeport was 1.9–5.3 h in the single-ascending dose phase, and 2.1–3.8 h on day 1 and 2.6–5.3 h on day 7 in the multiple-ascending dose phase.
Serum iron and TSAT are known to undergo a diurnal variation, with the highest concentrations observed in the morning/earlier part of the day, and then levels gradually decline towards the evening in healthy people [Citation57,Citation58]. This is approximately inversely related to the diurnal variation in plasma hepcidin concentrations, with noon and 8 pm values significantly higher than those at 8 am [Citation59]. In contrast, in the Phase I study of vamifeport in healthy adults [Citation42], mean serum iron and TSAT in the placebo groups appeared to be maximal at 8 h post-dose (4–6 pm) and then declined through the rest of the day. However, as there were no additional samples taken between 4 and 8 h post dose, the maximum serum iron level may not have been captured accurately in all subjects receiving placebo treatment. Following vamifeport, a rapid, temporary decrease in serum iron levels was observed with single doses ≥60 mg and all multiple doses. Multiple doses of vamifeport also temporarily decreased the mean calculated TSAT, maximally at about 4–8 h after the morning dose [Citation60], in line with the lowering of serum iron levels in all dose groups. This is an important finding because lowering TSAT is of value due to the generation of NTBI that occurs at high TSAT levels and its consequences in different pathophysiological settings [Citation60], and could also be potentially useful in the management and monitoring of iron overloaded patients with diseases like β-thalassemia. This effect of vamifeport on serum iron is consistent with the iron-lowering effects observed in healthy rodents and animal disease models. No other effects on hematologic biomarkers were observed, as expected in healthy volunteers, over the short (1-week) treatment period [Citation42]. There appeared to be small rebound increases in serum iron and TSAT up to 50% at 24 h, which were more marked following higher single doses [Citation60]. However, when dosing was given twice daily, a marked rebound was not seen, which is likely due to the fact that 36-h post-morning dose samples (i.e. 24-h post-evening dose) were not available in this study, as similar rebound effects would be expected following twice-daily dosing. These findings may suggest that the overall timing and posology of dosing may be important for optimizing benefits in serum iron and TSAT; indeed, this may become more important in β-thalassemia, where TSAT is usually already at high values and plasma iron turnover is higher than in healthy subjects.
It is interesting to note that rebound in serum iron was also observed with the use of parenteral hepcidin analogs [Citation43]. One mechanistic interpretation for rebound is that during maximum vamifeport levels, inhibition of iron release from macrophages increases labile iron within this compartment, which is subsequently released if vamifeport levels rapidly fall. The more pronounced rebound at higher doses would be explained by greater buildup of labile iron at these doses. Fortunately, such effects are unlikely to be cumulative because labile iron will be continuously incorporated into fairly kinetically stable ferritin within macrophages. However, timing between doses or dose tapering could be important in minimizing rebound of serum iron and TSAT. Also, it is currently not clear how duration of treatment over time in relation to the dose may influence the magnitude of the rebound. It will also be important to establish whether conditions associated with high iron turnover from erythrophagocytosis, such as thalassemias, are associated with greater rebound effects. Overall, the dose-dependent levels of decrease in TSAT provide further support that vamifeport, via iron restriction, has the potential to improve erythropoiesis and anemia in patients with β-thalassemia and other conditions involving ineffective erythropoiesis and disturbed blood homeostasis. On this basis, clinical trials in patients with NTDT and TDT are underway.
4. Ongoing Phase IIa study in NTDT
A Phase IIa trial (VIT-2763-THAL-201) to evaluate the safety, tolerability, PK, PD, and preliminary efficacy of multiple doses of vamifeport in subjects with NTDT is ongoing (NCT04364269; registered 28 April 2020). The primary objective of this study is to assess the safety and tolerability of vamifeport compared with placebo in adults and adolescents with NTDT over a 12-week treatment period. The primary study endpoints are AEs, serious AEs, and changes in vital signs, clinical laboratory tests, 12-lead electrocardiogram, and physical examination findings. Secondary and exploratory endpoints include assessment of hematology parameters including hemoglobin and RBC indices, iron-related markers, and biomarkers for hemopoietic/erythropoietic activity (including total serum iron, serum ferritin, serum transferrin, calculated TSAT, hepcidin, NTBI, labile plasma iron, soluble transferrin receptor [sTFR], sTFR-2, erythropoietin, and erythroferrone). Changes in iron metabolism and erythropoiesis parameters will be investigated in relation to whether vamifeport will be administered once or twice daily.
5. Planned Phase IIb study in TDT
A Phase IIb, multiple-dose, double-blind, randomized, placebo-controlled, parallel-group, multicenter trial, including adults with TDT, is being planned that will comprise a 24-week treatment phase. The primary objectives are to evaluate the efficacy of several increasing doses of vamifeport, as measured by reduction in RBC transfusion burden, and to identify the most efficacious and safe dose to take forward to Phase III studies. Secondary and exploratory endpoints include changes in hemoglobin and RBC indices, iron metabolism parameters (hepcidin, NTBI and sTFR), and markers of erythropoietic activity.
6. Conclusions
Vamifeport has a novel mechanism of action and is the first oral ferroportin inhibitor to reach clinical development. Initial data from preclinical studies in β-thalassemia animal models showed that vamifeport ameliorated anemia, and improved ineffective erythropoiesis, while Phase I clinical studies showed that vamifeport decreased serum iron levels (in line with preclinical observations and expected PD effects) and was generally well tolerated. β-thalassemia is a lifelong disease, and improved understanding of the pathophysiology in patients with varying disease burden has helped improve management to some extent. Due to the complexity of the disease and the lack of specific therapy other than supportive care, a high unmet need for more treatment options remains. If vamifeport can potentially improve erythropoiesis markers, including increasing hemoglobin and reducing iron-related parameters such as TSAT, NTBI and ultimately storage of iron, it could be a convenient treatment option for both NTDT and TDT. Additionally, through improving erythropoiesis and the storage of iron, vamifeport could positively affect the burden of ICT in patients with NTDT and TDT. Such changes could significantly reduce morbidity and improve quality of life in patients with β-thalassemia.
7. Expert opinion
Data from preclinical and Phase I studies of vamifeport showing amelioration of ineffective erythropoiesis, a favorable safety profile, and the ability to lower serum iron levels and TSAT, supported its continued clinical evaluation [Citation42,Citation50]. Data from the ongoing Phase IIa and planned Phase IIb studies will be critical to better understanding the full potential of vamifeport. Particularly relevant will be the study endpoints relating to iron load, transfusion burden, hemoglobin level, and hemopoietic/erythropoietic activity, alongside the demonstration of an acceptable safety profile. The inclusion of adolescents in these studies may help assess the feasibility of early intervention with vamifeport, although studies in younger patients (e.g. ≤12 years of age) will also be required. In theory, starting a treatment designed to modulate iron homeostasis and decrease ineffective erythropoiesis as early as possible has the potential to reduce long-term complications such as extramedullary hematopoiesis and iron overload. Optimal dosing and intervals between doses will need to be established in patients with thalassemia. Simulating vamifeport PK following various dose administration protocols, combined with a better understanding of the underlying PK/PD relationship, should contribute to achieving this goal.
Splenomegaly (and eventually hypersplenism) can be both a consequence and cause of increased hemolysis and chronic anemia, and its development correlates with the severity of globin chain imbalance. While splenectomy, especially when there are signs of hypersplenism, may improve growth, quality of life, and hemoglobin levels for some patients, it is associated with an increased risk of sepsis, thrombosis, pulmonary hypertension, leg ulcers, and cholelithiasis [Citation20,Citation25]. In some situations, splenectomy can also lead to decreased RBC production (e.g. when markers of hemolysis are not markedly increased and the spleen is mainly a source of RBC production) [Citation61]. Data from a murine model of NTDT showed that vamifeport treatment significantly reduced spleen weight versus control, reflecting the improvement of extramedullary erythropoiesis. However, it is questionable whether such findings can be extrapolated to humans due to functional differences and differences in spleen size between species. For example, in mice, a significant proportion of erythropoiesis occurs in the spleen, but in humans the spleen does not function as an erythropoietic organ to the same extent. Nonetheless, these effects suggest that vamifeport might potentially reduce extramedullary erythropoiesis and thus splenomegaly, and eventually the need for splenectomy, thereby avoiding potentially serious complications [Citation50].
It is important to consider how vamifeport might fit into the available treatments for β-thalassemia. As these are a group of lifelong conditions requiring chronic treatment, the development of agents that are convenient and reduce hospital visits, and associated costs, is attractive. There are currently no oral treatments for β-thalassemia that consistently moderate anemia, while also reducing serum iron levels and TSAT. Hydroxyurea is orally administered and has been used for many years as an inducer of fetal hemoglobin, but results are inconsistent and most of the evidence supporting its use is retrospective or of low quality [Citation24,Citation62–71]. Controlled trials are required to examine whether there is a consistent response to hydroxyurea, irrespective of differences in thalassemia genotype, previous transfusion history, and treatment policies in different regions. As hydroxyurea and vamifeport have different modes of action, it might be possible to use these oral agents in combination to further increase hemoglobinization in β-thalassemia. This approach warrants future clinical trials. Similarly, vamifeport could be used along with ICT with the intention of improving iron distribution in iron overload conditions, by decreasing TSAT and NTBI. In the long term, improvement of ineffective erythropoiesis in thalassemia syndromes may also allow ICT dose reduction by increasing the iron loading rate from blood transfusion and/or gastrointestinal absorption. Clinical trials of vamifeport in combination with other agents, including ICT, are warranted. Compared with intravenously or subcutaneously administered agents, oral formulations may be more convenient and cost-effective, as they do not have to be administered by healthcare professionals. Oral administration may also be particularly attractive for pediatric patients. The shorter half-life may be advantageous during chronic treatment as diurnal fluctuations in pharmaceutical levels and effectiveness will better preserve non-erythropoiesis-related, iron-dependent cellular functions. Furthermore, agents that directly target ferroportin are expected to have more predictable effects than other components that stimulate or increase hepcidin levels.
Another target for therapeutic approaches in β-thalassemia is the transforming growth factor-β (TGF-β) superfamily. Luspatercept is a subcutaneously administered TGF-β ligand trap agent that is approved by the US Food and Drug Administration and the European Medicines Agency for the treatment of adults with β-thalassemia who require regular blood transfusions [Citation26,Citation72,Citation73]. Although luspatercept is not currently indicated for use in patients with NTDT [Citation26,Citation72,Citation73], it improved hemoglobin levels and transfusion requirements in patients with TDT and NTDT in a Phase II, open-label, non-randomized study [Citation28]. A Phase II, double-blind, randomized, placebo-controlled trial in patients with NTDT is ongoing (NCT03342404). Data from preclinical studies in juvenile animals have highlighted potential safety concerns about using this agent in pediatric populations [Citation26]. From first principles, however, it is not expected to decrease iron absorption or to modulate TSAT. As the mechanism of action for vamifeport differs from luspatercept, there may be potential to combine these drugs to achieve additive effects on hemoglobin levels.
Over the next 5 years, targeted treatment approaches for β-thalassemia utilizing recently approved drugs or those currently in development have the potential to change current practice, such as the use of RBC transfusions in TDT. Although several new therapeutic strategies are in clinical development, including gene therapy approaches and novel drugs that improve ineffective erythropoiesis and iron regulation, many unanswered questions about iron metabolism and homeostasis still remain.
Article highlights
The hepcidin–ferroportin axis is central to iron homeostasis and therefore represents a rational therapeutic target for β-thalassemia.
Vamifeport (VIT-2763) is a small-molecule, oral inhibitor of ferroportin that improved ineffective erythropoiesis and ameliorated anemia in preclinical models of non-transfusion-dependent thalassemia.
When administered to healthy volunteers, vamifeport had a comparable safety profile to placebo and was well tolerated. Vamifeport also rapidly decreased circulating iron levels and reduced transferrin saturation, consistent with observed effects in preclinical models.
Vamifeport is currently in clinical development for potential use in conditions with ineffective or otherwise disturbed erythropoiesis such as hemoglobinopathies (including thalassemia) and in the future may provide a novel therapeutic option for other conditions involving disturbed iron and blood homeostasis.
Declaration of interest
J Porter has served on advisory boards for Vifor, Silence Therapeutics, Celgene, Agios, Bristol Myers Squibb, Protagonist, and Bluebird Bio. A Taher reports consultancy for Agios, Bristol Myers Squibb, Ionis, Novartis, and Vifor; research funding from Bristol Myers Squibb, Novartis and Vifor; and honoraria from Novartis. V Viprakasit reports research funding from the Research Excellence Development (RED) Program, Faculty of Medicine Siriraj Hospital, Mahidol University, Biorad, Celgene, GPO (Thailand), La Jolla, Novartis, Protagonist, Roche Diagnostics, Sebia, Silence Therapeutics, and Vifor. A Kattamis reports consultancy/advisory board membership for Bristol Myers Squibb, Novartis, Chiesi, Crisp/Vertex, Agios, Ionis, Silence Therapeutics, and Vifor; and research funding from Bristol Myers Squibb and Novartis. TD Coates is an advisory board member for Celgene/Bristol Myers Squib and Vifor; a steering committee member for Vifor; and advisor for Bluebird Bio, Chiesi/Apopharma and Sangamo. M Garbowski has a consultancy agreement with Imara and is a steering committee member for Vifor. MD Cappellini is an advisory board member for Celgene, CRISPR, Novartis, Sanofi/Genzyme, Novo, Nordisk, and Vifor. F Dürrenberger, V Manolova, F Richard are all employees of Vifor.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosure
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Medical writing support (including the development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact-checking and referencing) was provided by Julianna Solomons PhD, Aspire Scientific (Bollington, UK), and funded by Vifor Pharma AG, Glattbrugg, Switzerland. John B. Porter would like to acknowledge financial (salary) support from the UCL Biomedical Research Centre, Cardiometabolic Programme. The authors would also like to thank Nadja Stadelmann (Emmenbrücke, Switzerland) for her illustration assistance in producing .
Additional information
Funding
References
- Camaschella C. Iron and hepcidin: a story of recycling and balance. Hematology Am Soc Hematol Educ Program. 2013;2013:1–8.
- Tavazzi D, Duca L, Graziadei G, et al. Membrane-bound iron contributes to oxidative damage of beta-thalassaemia intermedia erythrocytes. Br J Haematol. 2001;112(1):48–50.
- Viprakasit V, Ekwattanakit S. Clinical classification, screening and diagnosis for thalassemia. Hematol Oncol Clin North Am. 2018;32(2):193–211.
- Jaing TH. Is the benefit-risk ratio for patients with transfusion-dependent thalassemia treated by unrelated cord blood transplantation favorable? Int J Mol Sci. 2017;18(11):2472.
- Cabantchik ZI, Breuer W, Zanninelli G, et al. LPI-labile plasma iron in iron overload. Best Pract Res Clin Haematol. 2005;18(2):277–287.
- Borgna-Pignatti C, Marsella M. Iron chelation in thalassemia major. Clin Ther. 2015;37(12):2866–2877.
- Ginzburg Y, Rivella S. β-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118(16):4321–4330.
- Payne KA, Rofail D, Baladi JF, et al. Iron chelation therapy: clinical effectiveness, economic burden and quality of life in patients with iron overload. Adv Ther. 2008;25(8):725–742.
- Trachtenberg FL, Mednick L, Kwiatkowski JL, et al. Beliefs about chelation among thalassemia patients. Health Qual Life Outcomes. 2012;10(1):148.
- Uygun V, Kurtoglu E. Iron-chelation therapy with oral chelators in patients with thalassemia major. Hematology. 2013;18(1):50–55.
- Ladis V, Chouliaras G, Berdoukas V, et al. Survival in a large cohort of Greek patients with transfusion-dependent beta thalassaemia and mortality ratios compared to the general population. Eur J Haematol. 2011;86(4):332–338.
- Shander A, Cappellini MD, Goodnough LT. Iron overload and toxicity: the hidden risk of multiple blood transfusions. Vox Sang. 2009;97(3):185–197.
- Taher AT, Saliba AN. Iron overload in thalassemia: different organs at different rates. Hematology Am Soc Hematol Educ Program. 2017;2017(1):265–271.
- Musallam KM, Taher AT, Rachmilewitz EA. β-thalassemia intermedia: a clinical perspective. Cold Spring Harb Perspect Med. 2012;2(7):a013482.
- Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–588.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11.
- Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187–1193.
- Vichinsky E. Non-transfusion-dependent thalassemia and thalassemia intermedia: epidemiology, complications, and management. Curr Med Res Opin. 2016;32(1):191–204.
- Taher AT, Musallam KM, Cappellini MD. Thalassaemia intermedia: an update. Mediterr J Hematol Infect Dis. 2009;1(1):e2009004.
- Cappellini MD, Motta I. New therapeutic targets in transfusion-dependent and -independent thalassemia. Hematology Am Soc Hematol Educ Program. 2017;2017(1):278–283.
- Franchini M, Forni GL, Marano G, et al. Red blood cell alloimmunisation in transfusion-dependent thalassaemia: a systematic review. Blood Transfus. 2019;17(1):4–15.
- Taher A, Musallam K, Cappellini MD, eds. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT). Thalassaemia International Federation: Nicosia, Cyprus. 2nd edition. 2017. [cited 2021 Apr 27]. Available from: https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-clinical-management-of-non-transfusion-dependent-thalassaemias-updated-version/
- Rigano P, Pecoraro A, Calzolari R, et al. Desensitization to hydroxycarbamide following long-term treatment of thalassaemia intermedia as observed in vivo and in primary erythroid cultures from treated patients. Br J Haematol. 2010;151(5):509–515.
- Foong WC, Ho JJ, Loh CK, et al. Hydroxyurea for reducing blood transfusion in non-transfusion dependent beta thalassaemias. Cochrane Database Syst Rev. 2016;10(10):CD011579.
- Cappellini MD, Porter JB, Viprakasit V, et al. A paradigm shift on beta-thalassaemia treatment: how will we manage this old disease with new therapies? Blood Rev. 2018;32(4):300–311.
- United States Food and Drug Administration. Reblozyl (luspatercept-aamt) Prescribing Information (2019) [updated November 2019. [cited 2021 Apr 27]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761136lbl.pdf
- Bou-Fakhredin R, Bazarbachi AH, Chaya B, et al. Iron overload and chelation therapy in non-transfusion dependent thalassemia. Int J Mol Sci. 2017;18(12):2778.
- Piga A, Perrotta S, Gamberini MR, et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with beta-thalassemia. Blood. 2019;133(12):1279–1289.
- Leecharoenkiat K, Lithanatudom P, Sornjai W, et al. Iron dysregulation in beta-thalassemia. Asian Pac J Trop Med. 2016;9(11):1035–1043.
- Aschemeyer S, Qiao B, Stefanova D, et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood. 2018;131(8):899–910.
- Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093.
- Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab. 2005;1(3):155–157.
- Papanikolaou G, Pantopoulos K. Systemic iron homeostasis and erythropoiesis. IUBMB Life. 2017;69(6):399–413.
- Gardenghi S, Grady RW, Rivella S. Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematol Oncol Clin North Am. 2010;24(6):1089–1107.
- Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684.
- Coffey R, Ganz T. Erythroferrone: an erythroid regulator of hepcidin and iron metabolism. HemaSphere. 2018;2(2):e35.
- Gardenghi S, Ramos P, Marongiu MF, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120(12):4466–4477.
- Khalil S, Delehanty L, Grado S, et al. Iron modulation of erythropoiesis is associated with Scribble-mediated control of the erythropoietin receptor. J Exp Med. 2018;215(2):661–679.
- Zhang S, Macias-Garcia A, Velazquez J, et al. HRI coordinates translation by eIF2αP and mTORC1 to mitigate ineffective erythropoiesis in mice during iron deficiency. Blood. 2018;131(4):450–461.
- Casu C, Oikonomidou PR, Chen H, et al. Minihepcidin peptides as disease modifiers in mice affected by beta-thalassemia and polycythemia vera. Blood. 2016;128(2):265–276.
- Schmidt PJ, Toudjarska I, Sendamarai AK, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121(7):1200–1208.
- Richard F, van Lier JJ, Roubert B, et al. Oral ferroportin inhibitor VIT-2763: first-in-human, Phase 1 study in healthy volunteers. Am J Hematol. 2020;95(1):68–77.
- Porter J, Kowdley K, Taher A, et al. Effect of Ljpc-401 (synthetic human hepcidin) on iron parameters in healthy adults. Blood. 2018;132(Suppl 1):2336.
- Lal A, Piga A, Viprakasit V, et al. A phase 1, open-label study to determine the safety, tolerability, and pharmacokinetics of escalating doses of LJPC-401 (synthetic human hepcidin) in patients with iron overload. HemaSphere. 2018;2(Suppl 1):396–397.
- Nicholls A, Lickliter J, Tozzi L, et al. Hepcidin mimetic PTG-300 induces dose-related and sustained reductions in serum iron and transferrin saturation in healthy subjects. HemaSphere. 2018;2(Suppl 1):397.
- McCaleb M, Lickliter J, Dibble A, et al. Transmembrane protease, serine 6 (TMPRSS6) antisense oligonucleotide (IONIS-TMPRSS6-LRX) reduces plasma iron levels of healthy volunteers in a phase 1 clinical study. Blood. 2018;132(Suppl 1):3634.
- Porter J. Beyond transfusion therapy: new therapies in thalassemia including drugs, alternate donor transplant, and gene therapy. Hematology Am Soc Hematol Educ Program. 2018;2018(1):361–370.
- Nai A, Pagani A, Mandelli G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119(21):5021–5029.
- Casu C, Aghajan M, Oikonomidou PR, et al. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica. 2016;101(1):e8–e11.
- Manolova V, Nyffenegger N, Flace A, et al. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of beta-thalassemia. J Clin Invest. 2020;130(1):491–506.
- Nyffenegger N, Flace A, Canclini C, et al. Ferroportin inhibitors prevent iron loading in a mouse model of hereditary hemochromatosis. Am J Hematol. 2017;92(8): E471.
- Nyffenegger N, Flace A, Doucerain C, et al. The oral ferroportin inhibitor VIT-2763 improves erythropoiesis without interfering with iron chelation therapy in a mouse model of beta-thalassemia. Int J Mol Sci. 2021;22(2):873.
- Nyffenegger N, Flace A, Varol A, et al. Oral ferroportin inhibitor prevents iron overload in the HFE C282Y mouse model of hereditary hemochromatosis. Oral presentation at the European Iron Club Annual Meeting 2018, Zurich, Switzerland. [cited 2021 Apr 27]. Available from: https://ethz.ch/content/dam/ethz/special-interest/conference-websites-dam/european-iron-club-2018-dam/documents/EIC2018_Abstracts_Book.pdf
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167.
- Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92(11):1485–1493.
- Schmidt PJ, Racie T, Westerman M, et al. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of β-thalassemia intermedia. Am J Hematol. 2015;90(4):310–313.
- Sinniah R, Doggart JR, Neill DW. Diurnal variations of the serum iron in normal subjects and in patients with haemochromatosis. Br J Haematol. 1969;17(4):351–358.
- Guillygomarc’h A, Christian J, Romain M, et al. Circadian variations of transferrin saturation levels in iron-overloaded patients: implications for the screening of C282Y-linked haemochromatosis. Br J Haematol. 2003;120(2):359–363.
- Ganz T, Olbina G, Girelli D, et al. Immunoassay for human serum hepcidin. Blood. 2008;112(10):4292–4297.
- Garbowski MW, Ma Y, Fucharoen S, et al. Clinical and methodological factors affecting non-transferrin-bound iron values using a novel fluorescent bead assay. Transl Res. 2016;177:19–30.e5.
- Erslev AJ, McKenna PJ. Effect of splenectomy on red cell production. Ann Intern Med. 1967;67(5):990–997.
- Algiraigri AH, Wright NAM, Paolucci EO, et al. Hydroxyurea for nontransfusion-dependent beta-thalassemia: a systematic review and meta-analysis. Hematol Oncol Stem Cell Ther. 2017;10(3):116–125.
- Ansari SH, Lassi ZS, Khowaja SM, et al. Hydroxyurea (hydroxycarbamide) for transfusion-dependent beta-thalassaemia. Cochrane Database Syst Rev. 2019;3(3):CD012064.
- Loukopoulos D, Voskaridou E, Stamoulakatou A, et al. Hydroxyurea therapy in thalassemia. Ann N Y Acad Sci. 1998;850:120–128.
- Koren A, Levin C, Dgany O, et al. Response to hydroxyurea therapy in beta-thalassemia. Am J Hematol. 2008;83(5):366–370.
- Dixit A, Chatterjee TC, Mishra P, et al. Hydroxyurea in thalassemia intermedia–a promising therapy. Ann Hematol. 2005;84(7):441–446.
- Fucharoen S, Siritanaratkul N, Winichagoon P, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood. 1996;87(3):887–892.
- Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892.
- Bradai M, Pissard S, Abad MT, et al. Decreased transfusion needs associated with hydroxyurea therapy in Algerian patients with thalassemia major or intermedia. Transfusion. 2007;47(10):1830–1836.
- Karimi M, Darzi H, Yavarian M. Hematologic and clinical responses of thalassemia intermedia patients to hydroxyurea during 6 years of therapy in Iran. J Pediatr Hematol Oncol. 2005;27(7):380–385.
- Algiraigri AH, Wright NAM, Paolucci EO, et al. Hydroxyurea for lifelong transfusion-dependent beta-thalassemia: a meta-analysis. Pediatr Hematol Oncol. 2017;34(8):435–448.
- Markham A. Luspatercept: first approval. Drugs. 2020;80(1):85–90.
- European Medicines Agency. Reblozyl (luspatercept) summary of product characteristics (2020) [updated 11 September 2020]. [cited 2021 Apr 27]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/reblozyl#product-information-section