
1. Introduction
β-hemoglobinopathies, which include β-thalassemia and sickle cell disease (SCD), comprise a heterogeneous group of disorders of hemoglobin (Hb) synthesis and structure [Citation1]. Around 7% of the world population are carriers of a α- and/or β-globin gene disorders [http://globin.bx.psu.edu/hbvar; Citation2].
In addition to supportive therapy, hematopoietic stem cell transplantation and several different forms of gene therapy are under study. Fetal Hb (HbF) inducing drug treatment with hydroxyurea (HU) is the standard of care in SCD and had been used, albeit less successfully, in β-thalassemia. The response to this drug is highly variable [Citation3,Citation4]. Approximately, 25% of patients with β-hemoglobinopathies do not respond to HU [Citation4,Citation5], suggesting that genomic variants could play role in modulating this variable HbF response. Understanding the pharmacogenomics (PGx), the genetic basis of the interindividual differences in HbF response to HU therapy, could therefore be beneficial in terms of pharmacological treatment for this group of disorders.
2. Existing evidence to individualize HU treatment response
There are few studies focused on PGx of HbF response to HU in SCD and even fewer in β-thalassemia [Citation4,Citation5]. Genome-wide association studies (GWAS) have revealed strong associations between the quantitative trait loci (QTL) Xmnl-HBG2 within the human β-globin gene cluster and the HBS1L-MYB and BCL11A loci and HbF levels [Citation4]. In a candidate gene-based association study, where the increment in HbF at 2 years after institution of HU in SCD was the outcome measure, Ma and coworkers [Citation6] showed significant association of variants in the linkage peaks of 6q22.3–23.2 representing the HBS1L-MYB QTL and 8q11-q12, which had previous been associated with HbF levels. Genomic variants in the MAP3K5, TOX, NOS1, NOS2, ARG2, and FLT genes were also associated with HbF response. An association between SAR1 gene promoter variants and response to HU was also demonstrated [Citation7]. In 2012, Borg and coworkers [Citation8] showed, using whole-transcriptome analysis, that genomic variants in the KLF10 gene also modulated HbF levels in response to HU treatment, a finding that has been confirmed in two subsequent retrospective studies [Citation9,Citation10]. The latter study also showed that genomic variants within the MAP3K5, NOS2A, ARG2 genes could be also correlated with HU treatment response, confirming previous findings by Ma and coworkers.
These results, although confirmed in different populations and types of hemoglobinopathies, must be considered preliminary. Nonetheless, they provide hope that useful PGx biomarkers for HbF response to HU, HbF inducers yet to be approved and even for newly approved drugs for β-hemoglobinopathies like voxelotor, crizanlizumab, and luspatercept could be discovered and validated in a prospective study.
3. A call for prospective PGx studies for β-hemoglobinopathies
Application of PGx is a rapidly evolving and highly promising approach but is still in its early stage of development for β-hemoglobinopathies. As previously mentioned, apart from HU, there are no PGx data for other drugs used for treating these diseases, while the vast molecular diversity of β-hemoglobinopathies poses yet another challenge for the identification of PGx biomarkers. Also, the effect of hydroxyurea on mortality and morbidity in adult SCD, including the risks and benefits have been well studied and reported previously [Citation11,Citation12].
To proceed beyond this early stage and in order to obtain some meaningful and clinically impactful results to individualize HU treatment for β-hemoglobinopathies, a concerted and well-orchestrated effort among different clinical and research centers in different countries needs to be organized (). In particular, β-hemoglobinopathy patients need to be actively recruited in large numbers and well-characterized at the molecular level, using next-generation sequencing of both the β-globin gene cluster and known modifier genes. To incentivize clinicians to participate in this effort, the microattribution approach [Citation13] could be pursued further and expanded, similar to the CFTR2 Consortium (https://cftr2.org) that has been successfully implemented in a clinical setting [Citation14], in which a large number of clinical centers and clinicians have participated. In particular, microattribution, that was conceptually perceived and first implemented using the globin gene and related, to the hemoglobinopathies phenotypes, loci as a paradigm, aims to incentivize deposition of genomic and associated phenotypic data into the public domain, particularly in stable, comprehensively curated and internationally renowned data repositories. Strong commitment from multiple clinical centers is needed in order to collect a large number of patients and cover a wide range of clinical and genetic variability, building on and expanding beyond the published retrospective studies that included a limited number of patients.
Figure 1. Overview of the stepwise approach to identify, validate and possibly incorporate PGx biomarkers into routine clinical practice based on individualized HU drug treatment response
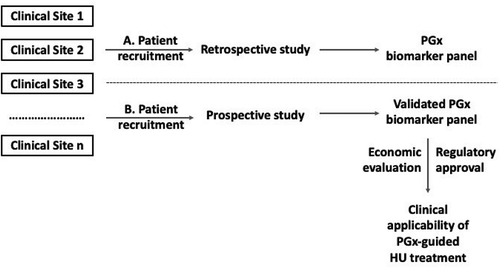
This coupled with the development of gene-specific next-generation sequencing (NGS) panels [Citation15] will allow fine mapping of the genomic variants that contribute not only to the disease molecular heterogeneity but also to HU drug response, which will allow not only proceeding agnostically in the discovery phase but also to establish statistically significant associations with a large study sample. NGS could be performed in a centralized manner (a single or few laboratories) to ensure data homogeneity and consistency.
The on-going whole-genome sequencing and phenotyping consortia such as Trans Omics for Precision Medicine [TOPMed; Citation16] and the UK BioBank [Citation17] are focused on phenotypes such as commonly measured blood cell parameters. Our proposed PGx consortium potentially could leverage these rich repositories of genotype and phenotype data to improve fine mapping of genomic variants with drug responses.
Creation of a strong consortium of motivated clinicians and laboratory scientists will allow the launch of a multi-site prospective PGx clinical study to confirm part or ideally all of the preliminary findings from the discovery phase and also give the evidence required by regulatory bodies to establish a panel of PGx biomarkers that could be eventually used by clinicians to rationalize HU treatment response. Establishment of such a multicenter effort is needed in order to recruit substantial number of patients required to reach a sound genotype-phenotype association of genome-guided HU treatment response, given the vast clinical heterogeneity of these patients, associated with the underlying globin gene variants. In parallel, a dedicated team of economists would engage to perform economic evaluation of the above therapeutic options to assist decision makers to define whether they can be adopted in healthcare systems to contribute towards reducing the burden of the annual healthcare expenditures [Citation18] and possibly decide on the reimbursement of those interventions by the payers [Citation19]. This approach would eventually enable integration of such a PGx biomarker panel for individualizing HU treatment response among β-hemoglobinopathies patients ().
Declaration of interests
GP Patrinos is full Member and National Representative at the European Medicines Agency, Committee for Human Medicinal Products (CHMP) – Pharmacogenomics Working Party, Amsterdam, the Netherlands. Support for RC Hardison is from the National Institutes of Health grants R24DK106766, R01CA178393, and R01DK054937. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Patrinos GP, Antonarakis SE. Human hemoglobin. In: Vogel and Motulsky’s human genetics: problems and approaches. Fourth Edition ed. Berlin Heidelberg: Springer-Verlag; 2011. p. 365–401.
- Giardine BM, Joly P, Pissard S, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2021;49:D1192–D1196.
- Patrinos GP, Grosveld FG. Pharmacogenomics and therapeutics of hemoglobinopathies. Hemoglobin. 2008;32:229–236.
- Karamperis K, Tsoumpeli MT, Kounelis F, et al. Genome-based therapeutic interventions for β-type hemoglobinopathies. Hum Genomics. 2021;15:32.
- Gravia A, Chondrou V, Sgourou A, et al. Individualizing fetal hemoglobin augmenting therapy for β-type hemoglobinopathies patients. Pharmacogenomics 2014;15:1355–1364.
- Ma Q, Wyszynski DF, Farrell JJ, et al. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea. Pharmacogenomics J. 2007;7:386–394.
- Kumkhaek C, Taylor JG 6th, Zhu J, et al. Fetal haemoglobin response to hydroxycarbamide treatment and sar1a promoter polymorphisms in sickle cell anaemia. Br J Haematol. 2008;141:254–259.
- Borg J, Phylactides M, Bartsakoulia M, et al. KLF10 gene expression is associated with high fetal hemoglobin levels and with response to hydroxyurea treatment in β-hemoglobinopathy patients. Pharmacogenomics 2012;13:1487–1500.
- Elalfy MS, El Sherif NH, Kamal TM, et al. Klf10 Gene, a secondary modifier and a pharmacogenomic biomarker of hydroxyurea treatment among patients with hemoglobinopathies. J Pediatr Hematol Oncol. 2017;39:e155–e162.
- Kolliopoulou A, Siamoglou S, John A, et al. Role of genomic biomarkers in increasing fetal hemoglobin levels upon hydroxyurea therapy and in β-thalassemia intermedia: a validation cohort study. Hemoglobin. 2019;43:27–33.
- Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651.
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85:403–408.
- Giardine B, Borg J, Higgs DR, et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat Genet. 2011;43:295–302.
- Sosnay PR, Siklosi KR, Van Goor F, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013;45:1160–1167.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159.
- Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590:290–299.
- Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209.
- Patrinos GP, Mitropoulou C. Measuring the value of pharmacogenomics evidence. Clin Pharmacol Ther. 2017;102:739–741.
- Simeonidis S, Koutsilieri S, Vozikis A, et al. Application of economic evaluation to assess feasibility for reimbursement of genomic testing as part of personalized medicine interventions. Front Pharmacol. 2019;2:10.