
ABSTRACT
Background
Ultra-rare inherited bleeding disorders (BDs) present important challenges for generating a strong evidence foundation for optimal diagnosis and management. Without disorder-appropriate treatment, affected individuals potentially face life-threatening bleeding, delayed diagnosis, suboptimal management of invasive procedures, psychosocial distress, pain, and decreased quality-of-life.
Research design and methods
The National Hemophilia Foundation (NHF) and the American Thrombosis and Hemostasis Network identified the priorities of people with inherited BDs and their caregivers, through extensive inclusive community consultations, to inform a blueprint for future decades of research. Multidisciplinary expert Working Group (WG) 3 distilled highly feasible transformative ultra-rare inherited BD research opportunities from the community-identified priorities.
Results
WG3 identified three focus areas with the potential to advance the needs of all people with ultra-rare inherited BDs and scored the feasibility, impact, and risk of priority initiatives, including 13 in systems biology and mechanistic science; 2 in clinical research, data collection, and research infrastructure; and 5 in the regulatory process for novel therapeutics and required data collection.
Conclusions
Centralization and expansion of expertise and resources, flexible innovative research and regulatory approaches, and inclusion of all people with ultra-rare inherited BDs and their health care professionals will be essential to capitalize on the opportunities outlined herein.
Plain Language Summary
Living with an ultra-rare inherited bleeding disorder is challenging. Patients can feel alone and unsure of where to find support because their disorder is so rare. In this paper, a group of ultra-rare bleeding disorder experts, including doctors, researchers, regulators, patient advocates, and patients, identify the research that could best improve the lives of people with these disorders. They propose a national network of specialists who can help doctors, who may never have seen these disorders before, to find the right diagnosis faster. A centralized laboratory specialized in ultra-rare bleeding disorders could also improve diagnosis and do research studies. This would help us learn, for example, how symptoms change throughout a patient’s life, how effective different treatments are, and what it is like for patients to live with these disorders. A second research priority is to better understand each individual disorder so that the best treatments can be chosen or developed. A pathway showing doctors which treatment options to try, in which order, would help them help their patients. The third research priority is to make it easier to study new treatments for ultra-rare bleeding disorders. This requires designing studies with very small numbers of participants, identifying meaningful outcomes to measure, and convincing pharmaceutical companies to invest in these studies. International agreement on these requirements would allow more patients to participate and benefit from the research. These top-priority research goals should greatly improve knowledge about, and diagnosis and treatment of, ultra-rare inherited bleeding disorders.
1. Introduction
1.1. Rare versus ultra-rare disorders
Rare disorders or diseases have a prevalence of ≤4 or 5 per 10,000 people [Citation1]; the term ‘orphan disease’ refers to those occurring in <200,000 people in the total United States (US) population [Citation2–4]. The 7,000 to 8,000 rare disorders are estimated to affect approximately 400 million people globally and 30 million in the US [Citation4] with licensed therapeutics only available to treat <5% [Citation5]. Even less common, ultra-rare disorders are also estimated to impact millions of people globally [Citation6]. ‘Ultra-orphan’ diseases have a prevalence <1 per 50,000 [Citation7] and some ultra-rare genetic mutations define an N-of-1 population or affect <30 people worldwide [Citation8].
With a combined prevalence of approximately 1.5 per 10,000 [Citation9], hemophilia A and B (clotting factor VIII and IX deficiency, respectively), the best known and researched inherited bleeding disorders (BDs), are rare. While important work remains to be done [Citation10], decades of hemophilia research have yielded clinical practice guidelines [Citation11] and a variety of safe and effective treatment options [Citation12,Citation13]. Making up 3–5% of inherited BDs, the ultra-rare disorders are a heterogenous group of quantitative and/or qualitative clotting factor deficiencies and platelet disorders () [Citation14–20].
Table 1. Estimated prevalence, correlation of factor level with clinical presentation, and availability of a single clotting factor concentrate of selected ultra-rare inherited bleeding disorders.
Overall, the estimated prevalences of individual ultra-rare inherited BDs range from 1:500,000 to 1:2,000,000 globally () [Citation14,Citation20,Citation21]; some studies report considerably higher rates (e.g. [Citation22–25]) depending on the countries/populations investigated, outreach and identification approaches and capacity, and registry details [Citation15].
Generating research evidence to support accurate diagnosis, optimal management, and regulatory approval of safe and effective therapeutics poses challenges common to many ultra-rare disorders [Citation26]. We, therefore, identify herein priority initiatives to advance these key questions for all ultra-rare inherited BDs [Citation27]:
How can we better understand the biology of these ultra-rare disorders?
How can we stimulate research and optimize the regulatory process to vastly improve diagnosis and targeted treatment?
1.2. Diagnosis
Ultra-rare inherited BDs are underrecognized and underdiagnosed [Citation28]. Bleeding type, site, severity, age at onset, and duration vary enormously between disorders and individuals [Citation29]. Correlation between factor activity level and clinical presentation differs between disorders and individuals [Citation18,Citation19,Citation30]. Despite multiple valuable registry initiatives and efforts to characterize genotype–phenotype (genome-phenome) relationships in specific factor deficiencies (e.g. [Citation31,Citation32]), adequate natural history data are not uniformly available to fully characterize severities with corresponding factor levels as appropriate [Citation16–19,Citation21,Citation29,Citation33]. These foundational knowledge gaps impede the establishment of uniform diagnostic algorithms and prevalence rates.
Application of the proposed distinct clinical and laboratory severity thresholds for the more commonly encountered disorders [Citation18] remains approximate due to a lack of strong evidence. Attempts to standardize Bleeding Assessment Tools (BATs) for ultra-rare inherited BDs [Citation34–37], show promise but require further validation [Citation29]. While advances in sequencing and molecular diagnostic techniques provide powerful new tools to identify variants (formerly termed causative mutations), many are classified as variants of unknown significance (VUS) [Citation38], and symptomatic individuals may remain undiagnosed [Citation28,Citation39–41].
1.3. Treatment
Highly purified single clotting factor concentrates (CFC) are the treatment of choice for people with ultra-rare inherited BDs if available [Citation20]. In their absence, fresh frozen plasma (FFP) is often used to replace deficient procoagulants [Citation30]. FFP infusion, however, is complicated by risks of volume overload, allergic or infusion reactions, and viral transmission [Citation20]. Thromboembolic events may be associated with prothrombin complex concentrates (PCCs) which contain varying amounts of factors II, IX, X, and sometimes VII; and factor XI concentrates (no factor XI concentrates have received regulatory approval in the US) [Citation14].
Antifibrinolytic agents (e.g. tranexamic acid and aminocaproic acid) are commonly used for minor bleeding, perioperatively, and for heavy menstrual bleeding (HMB) in people with ultra-rare BDs [Citation14]. Platelet transfusions are an option for inherited platelet disorders or thrombocytopenia; however, they carry risks of viral/bacterial infection, anaphylaxis, transfusion-related acute lung injury, and platelet refractoriness [Citation20]. Desmopressin (DDAVP), potentially in combination with antifibrinolytics, may be useful to manage mild-to-moderate bleeding in some inherited platelet disorders, however the mechanism of action remains poorly understood [Citation42–44]. Clinical trials, case studies/series, and international registries will add to a growing experience with therapeutic options individually or in combination, on-demand or prophylactically; however, data for people with ultra-rare inherited BDs are still insufficient to support evidence-based clinical practice guidelines [Citation14,Citation20,Citation30,Citation41].
1.4. Research challenges and opportunities
Research into ultra-rare inherited BDs faces a number of important challenges [Citation26]. Potential research participants are few in number and diagnostic challenges diminish the likelihood of identifying them. Very few academic/clinical research centers have the finances, resources, advanced study design capabilities, and specialized laboratory services to conduct studies with only one to two participants. Longitudinal natural history and molecular characterization studies can only amass sufficient data if conducted through multicenter national/international collaborations.
Without a specific diagnosis affected individuals may face life-threatening bleeding and suboptimal management of invasive procedures. Psychosocial distress and uncertainty, pain, and decreased quality-of-life (QoL) including ability to work/attend school, participation in social activities and sports, sexual functioning, and mental health may result [Citation45–49]. Misdiagnosis can lead to inappropriate therapies or procedures such as splenectomy [Citation50] and hysterectomy [Citation51]. Affected individuals, and those who care for them, become lived experience experts (LEE) in their ultra-rare inherited BDs, amassing a wealth of experience and insight which should be shared with clinicians and researchers [Citation52]. Their involvement in the prioritization of the research that will most benefit them is essential.
1.5. Community-identified areas of priority research
To this end, in 2020, the National Hemophilia Foundation (NHF) initiated a collaboration with the American Thrombosis and Hemostasis Network (ATHN) to develop a National Research Blueprint for Inherited Bleeding Disorders rooted in the expressed needs of the community [Citation27,Citation53]. Their extensive community consultations intentionally included the representation of ultra-rare inherited BDs [Citation27,Citation54]. Improving access to specialized diagnosis and treatment for ultra-rare inherited BDs and research into new therapies for all inherited BDs were priorities shared across stakeholder groups [Citation27].
The NHF State of the Science Research Summit (SOSRS) Steering Committee (SC) charged six expert Working Groups (WG), with distilling community priorities in specific focus areas into concrete research questions [Citation27]. WG3, entitled ‘Research Priorities for Ultra-rare Inherited Bleeding Disorders,’ was charged with identifying initiatives to advance understanding of the biology of ultra-rare disorders, stimulate research, and optimize the regulatory process to vastly improve diagnosis and targeted treatment (). Community discussions at the NHF SOSRS (September, 2021) contributed to the refinement, of these goals for the National Research Blueprint for Inherited Bleeding Disorders [Citation27,Citation53,Citation55].
2. Methods
2.1. Working Group composition and workings
The SC and cochairs, Amy D. Shapiro, MD, Diane Nugent, MD, and Suchitra S. Acharya, MD recruited 21 national and international experts to WG3 (). Three LEEs with ultra-rare inherited BDs [Citation52] were full and equal WG members; supported (virtually) by an NHF staff person their expertise was proactively engaged by the cochairs. The diverse knowledge, experience, and context of each WG member, spanning the stakeholder spectrum, contributed substantially to the identification and prioritization of initiatives with the greatest potential to transform the lives of people with ultra-rare inherited BDs.
Table 2. Members of Working Group 3, by subgroup.
Rather than attempting to delineate research agendas for individual disorders, the WG sought opportunities to potentially benefit all ultra-rare inherited BDs. Subgroups focusing on three domains of community priorities () met virtually over approximately 6 months to identify current gaps/obstacles and strategize solutions, concentrating on processes and infrastructure. The full WG was reconvened to combine their outputs. The results pertinent to health care delivery networks were shared with WG5 Diversity, Equity and Inclusion; Health Services Research; and Implementation Science [Citation56], and to infrastructure and workforce with WG6 Facilitating Priority Research in the Inherited Bleeding Disorders Community [Citation57].
2.2. Feasibility–impact–risk scoring
NHF charged all WGs with scoring the feasibility, impact, and risk of prioritized initiatives against pre-defined criteria (Supplementary Table S1) [Citation27,Citation54,Citation55]. Totaling these three score areas yielded a single value to facilitate comparison between questions with different strengths and challenges. A highly feasible question that could be relatively easy to investigate (maximum score: 14, range: 0 to 14), with a significant impact on the target population (maximum score: 14, range: 0 to 14), and involving very low risk (maximum score: 0, score range: −10 to 0, greater risk scored as increasingly negative value), would achieve the maximum score (28).
Cochairs scored their subgroup’s questions/initiatives, circulated results, and resolved discrepancies through subgroup discussion. Not all initiatives easily lent themselves to feasibility-impact-risk scoring (see Limitations discussion), and some evaluations were approximate. The Diagnostics, Systems Biology, and Mechanistic Science subgroup did not score risk, reporting feasibility-impact sum scores only.
2.3. NHF State of the Science Research Summit
Feedback was solicited from >880 LEEs, physicians, researchers, multidisciplinary care team professionals, and federal and industry partners at the (virtual) NHF SOSRS, September 12–15, 2021 [Citation27]. To reduce the burden of participation on underrepresented segments of the inherited BDs community, NHF, supported by an NHLBI R13 grant (R13HL158209), offered LEEs from diverse racialized/ethnic communities, the lesbian, gay, bisexual, transgender, and queer (LGBTQ) community, the aging community, and the geographically challenged, the option of facilitated Remote Participation Groups. The facilitator’s summary of the discussion of the WG3 session by one such group (five individuals), and SOSRS live discussions [Citation27,Citation54], informed the Discussion and Conclusions of this paper.
3. Results
In seeking to identify research opportunities to advance the needs of all people with ultra-rare inherited BDs, WG3 determined that the community-identified priorities fell into three focus domains:
Diagnostics, systems biology, mechanistic science
Clinical, data collection, research infrastructure
Regulatory process for novel therapeutics and required data collection
The key questions, important elements, and modalities of addressing these three domains were determined (). Top priority actionable initiatives were identified for each, scored for feasibility, impact, and risk and these scores totaled (, ). The scoring matrix, with the evaluation of each initiative against each feasibility, impact, and risk criterion, is detailed in Supplementary Table S2. The output of the subgroup deliberations and rationale for prioritization of these initiatives are summarized below.
Figure 1. Working Group 3 research priorities for ultra-rare inherited bleeding disorders schematic of community-identified areas for priority research framework POC: point of care.
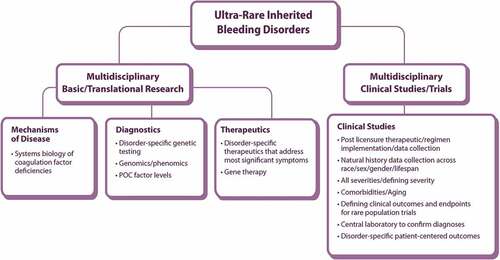
Figure 2. Questions and elements/modalities considered by the WG3 subgroup to address community priorities in diagnostics, systems biology, and mechanistic science of ultra-rare inherited bleeding disorders.

Figure 3. Questions and elements/modalities considered by WG3 to address clinical, data collection, and research infrastructure community priorities for ultra-rare inherited bleeding disorders.

Figure 4. Questions and elements/modalities considered by WG3 to address community priorities in regulatory processes for novel therapeutics and required data collection for ultra-rare inherited bleeding disorders.

Figure 5. Plot of feasibility, impact, and risk scores of the questions evaluated by the three WG3 subgroups.
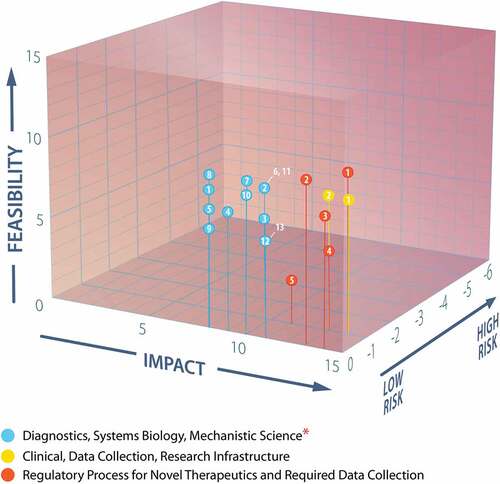
Table 3. Actionable initiatives scored for feasibility, impact, and risk in the three domains of ultra-rare inherited bleeding disorders community priorities.
3.1. Diagnostics, systems biology, mechanistic science
Examining the challenges and opportunities in this domain () revealed a need to connect primary care physicians to specialized expertise for immediate consultation and to create a network of specialized centers to facilitate diagnosis and expedite national research collaborations.
3.1.1. Connecting primary care physicians to specialized expertise
Many primary care physicians lack the training or awareness to know where to refer, or whom to consult, regarding an unexplained bleeding tendency. People with ultra-rare inherited BDs are uncommonly encountered even by hematologists at hemophilia treatment centers (HTC). A national resource (website, telephone hotline, or telehealth/e-consult) connecting those seeking assistance in complex cases with specialized clinicians and researchers would improve the rate and accuracy of diagnosis. Research centers would also benefit from samples and phenotypes with which to advance further studies.
3.1.2. Formalizing and supporting a national network for advanced diagnosis
A formal network of hemostasis researchers/centers across the country, in addition to providing an important diagnostic resource to physicians, would facilitate the synergistic advance of research into different ultra-rare inherited BDs. Such a network should:
Link centers with advanced technology and expert research teams, such that each may specialize in their focus area and benefit from one another’s capabilities
Include a national database of phenotypic and family history data linked to a national biospecimen repository (plasma, tissue, and genetic samples)
Proactively liaise HTCs with primary care physicians to facilitate referrals, information, and resource sharing
Train the HTC and referral center workforce in the optimal collection and handling of samples to minimize error and pre-analytic variables impacting assay results
3.1.3. National research collaborations
A national network of hybrid labs, with the accreditation to issue formal diagnoses as well as the expertise to carry out advanced research to identify an ultra-rare inherited BD is critically needed, especially for people with a novel genetic variant and presentation. Paradoxically, centers and individuals with some federal funding for ultra-rare inherited BD research may be prohibited from using these funds for clinical diagnostic services [Citation58]. Additionally, despite knowledgeable researchers, these labs lack the College of American Pathologists (CAP)/Clinical Laboratory Improvements Amendments (CLIA) diagnostic accreditation required by Centers for Medicare and Medicaid Services (CMS) to report results [Citation59,Citation60]. Most large centralized clinical labs and hospitals use standardized diagnostic assays or kits that are not designed to identify specific ultra-rare disorders. CAP/CLIA accredited diagnostic facilities generally lack the advanced investigational capacities and expertise of basic research labs. Thus, the workforce at the new hybrid centers must have knowledgeable directors and technicians trained to detect and characterize ultra-rare inherited BDs. An overarching research protocol with centralized institutional review board (IRB) approval linked to the national sample repository, and its testing capabilities, is also suggested.
A collaborative national approach to genetic analysis of ultra-rare inherited BDs may help address issues of cost and the evidence level required to characterize new pathogenic variants [Citation28]. ClinVar, the public archive of reports of relationships between human genetic variations and phenotypes hosted by the National Center for Biotechnology Information (NCBI) with intramural National Institutes of Health (NIH) funding [Citation61] is a resource of growing utility. Next-generation sequencing panels designed to characterize platelet and clotting factor gene sequence pathologic variants and whole-genome sequencing will be key to accelerating genomic approaches to diagnosing ultra-rare inherited BDs.
Diagnostic research teams connected through this network will benefit from resource and expertise sharing, especially as their areas of investigation align (e.g. clotting factor deficiencies or platelet disorders).
3.2. Clinical, data collection, research infrastructure
Addressing the community-prioritized clinical needs () will require important advances in diagnostic tools and data to support treatment algorithms. Data collection must be more comprehensive, rigorous, and efficient and include the outcomes most important to people with ultra-rare inherited BDs. All these initiatives require substantial financial, workforce, and infrastructure support.
3.2.1. Clinical needs
Defining severity categories of specific ultra-rare inherited BDs is important to predict outcomes and guide treatment decisions [Citation19,Citation28,Citation62]. Further validation and harmonization of BATs, particularly that developed from the Rare Bleeding Disorders (EN-RBD) registry [Citation35] and the International Society for Thrombosis and Haemostasis (ISTH)-BAT, could help advance this research [Citation62–66]. Characterizing potential hallmarks of individual disorders would benefit diagnostic pathways and accuracy.
Evidence-based guidelines akin to those of the Children’s Oncology Group [Citation67] are largely absent for the treatment of rare inherited BDs. Such algorithms are needed to guide the use of various treatment options individually or in combination, and in different clinical scenarios (e.g. acute on-demand treatment, different types of prophylaxis, surgery, etc.). Potential utility of non-factor products such as those that block tissue factor pathway inhibitor (TFPI) or antithrombin (AT) [Citation68,Citation69] should be investigated, especially in disorders without an available replacement CFC.
3.2.2. Data collection
Several initiatives (e.g. Community Counts [Citation70], ATHN10 [Citation71]) collect select data on rare factor and platelet disorders, but more rigorous and extensive data collection is needed in a wider number of affected individuals. Collecting complete natural history, treatment, and longitudinal outcome data from all people with ultra-rare inherited BDs, including those not receiving care at HTCs, into a single central database, such as the ATHNdataset [Citation72] has the potential to be impactful and increase individual participation. This database should be linked to the national biospecimen repository to advance phenotyping and severity prediction models. Its analysis would support the development of the algorithms described above. Patient-reported outcomes such as psychosocial well-being and experiences of daily living often lead the list of community-prioritized concerns [Citation27] yet these data remain largely uncaptured in these populations. The health-related QoL and health utility assessment tools in the hemophilia cohort of ATHN Transcends [Citation73] should be adapted to capture these.
3.2.3. Research infrastructure
Adequate infrastructure support is essential to all the centralized data collection, sample banking, expert network, and research collaboration initiatives identified. The important benefits of resource and expertise sharing may be most easily reaped between centers with similar foci, but are expected to extend throughout the network. A skilled workforce is needed to collect and expertly process samples, and for data entry. Centralized resources, such as a biorepository (potentially building upon the existing ATHN biorepository [Citation73]) and a quality control or reagent standardization initiative, have the potential to improve diagnostic accuracy and reduce expenses. Expertise and funding will be required to design and execute complex molecular sample analyses (genotypes, epigenetics, genetic disease modifiers) evolving with technological advances. As explored in depth by WG6, workforce cross-training in innovative approaches to informatics, data science, epidemiology, and small clinical trial design will be key [Citation57].
3.3. Regulatory process for novel therapeutics and required data collection
Established regulatory pathways optimized for more prevalent conditions impose requirements often unattainable for ultra-rare disorders with insurmountable cost, time, and complexity burdens. Addressing the community priorities () will require innovative solutions to regulatory obstacles.
3.3.1. Innovations in data collection
The centralized national, or harmonized international data collection proposed above would support accumulating the evidence base to understand a number of key factors, such as how existing therapeutics are being used (including off-label), their real-world safety and efficacy outcomes, and the need for novel therapeutics and indications. Clearly demonstrating the absence of effective treatment options for specific populations may prompt regulators to consider adjusting endpoint/outcome thresholds for provisional approval. A therapeutic currently approved for a different indication could be accepted to treat a high-need population upon demonstration of its safety and potential benefits. Under provisional approval, further information supporting use could be captured with ongoing data collection to further elucidate and confirm benefits. Adaptive clinical trial designs, as explored by WG6 [Citation57], may facilitate maximization of the impact of data generated from a limited number of participants.
The U.S. Food and Drug Administration (FDA) may grant accelerated approval to a drug for a serious or life-threatening condition upon demonstration that it has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, often with a requirement for post-approval studies to verify and describe anticipated clinical benefits and support the safety profile [Citation74,Citation75]. Facilitating qualification of biomarkers, surrogate endpoints, and patient-reported outcomes for regulatory purposes would be expected to enhance successful application of the accelerated approval pathway [Citation76,Citation77].
3.3.2. Regulatory streamlining and incentives
Streamlining regulatory processes may be particularly impactful for novel therapeutics. If the gene therapy approach to correct single-base mutations continues its recent promising progress, approval may soon be sought for an increasing number of therapeutics for very limited populations with specific causative mutations [Citation78,Citation79]. Differences in regulatory requirements between national jurisdictions complicate matters as initiatives deemed sufficient in one country/region may need to be repeated with modifications to satisfy others [Citation78]. Further harmonization of requirements and coordinated streamlining of efforts between national and international regulatory agencies will benefit ultra-rare disorders [Citation3,Citation80]. Early, frequent, and close communication between those developing therapeutics and regulatory authorities should facilitate navigation of regulatory processes, especially for those without prior experience [Citation77,Citation78]. Ensuring all developers are well informed of the requirements will avoid common pitfalls through judicious design. Innovators that capitalize on all opportunities and incentives will shorten time to approval, and potential development cost, of safe and effective diagnostics and therapeutics for ultra-rare disorders [Citation81].
In the US, orphan therapeutics may be eligible for financial incentives such as market exclusivity and tax benefits, intensive development guidance, and expedited review [Citation75,Citation78,Citation82–84]. Other regulatory authorities offer similar programs and initiatives; despite this approved therapies are available for <5% of rare disorders [Citation3,Citation4,Citation82], and therefore regulators should consider further incentives.
4. Discussion
The establishment of a research agenda for the coming decades is an opportunity to direct efforts into initiatives with the greatest potential to transform the experiences of people with ultra-rare inherited BDs. The diverse stakeholders, researchers, clinicians, and LEEs of WG3 together sought to identify these opportunities in diagnostics, systems biology, and mechanistic science; to streamline clinical, data collection, and research infrastructure; and to fast track the regulatory process for novel therapeutics. In so doing, they identified recurring needs for centralization of expertise and resources, innovative solutions, and inclusion and outreach.
4.1. Limitations
Improving the experiences and outcomes of all people with ultra-rare inherited BDs requires prioritization of research themes and broad initiatives. The NHF feasibility-impact-risk scoring matrix (Suppl Table S1) may be well suited to evaluating the very focused research questions addressing aspects of individual BDs as derived by WGs 1, 2, and 4 [Citation10,Citation85,Citation86]. WG3 endeavored to apply the intent of the matrix to rank the initiatives they prioritized (), however this was very challenging for far-reaching initiatives, and some evaluations were approximate. The Diagnostics, Systems Biology and Mechanistic Science subgroup concluded that they could not apply the risk criteria to their initiatives and scored only feasibility and impact. The detailed scoring criteria, and WG3ʹs consideration of each element of feasibility, impact, and risk for each initiative, are presented in the Supplementary Table S2 interactive chart; readers are invited to explore this chart and conduct their own assessments. The nature of ultra-rare disorders renders the practicalities of implementation of all the initiatives prioritized herein challenging; some of these challenges, such as the need for a unified national approach and international harmonization, are discussed below. Achieving even a few of these initiatives will improve care for people with all rare inherited BDs, however committed champions are required to drive these initiatives forward. The NHF SOSRS sought to identify and rank the initiatives, in six key areas, with the greatest transformative potential. The ongoing National Research Blueprint multidisciplinary initiative seeks to establish the strategies and approaches necessary for the execution of this research; the operationalization and implementation of the research agenda is the aspiration of the next stages of this effort [Citation53,Citation87].
4.2. Centralization of expertise and resources
A network of hybrid expert diagnostic/research laboratories, investing in existing concentrations of expertise, accrediting them to provide formal diagnoses to complex cases, would constitute an invaluable advancement. Connecting the network to HTCs and primary care physicians would facilitate more rapid and accurate diagnoses for people with bleeding tendencies, guidance in diagnostic and treatment options for physicians, and potential participants to further research initiatives to advance knowledge and care. With very few widely dispersed individuals affected by each ultra-rare disorder and the phenotypic variability within each disorder, formal networks connecting expertise to need are essential. Infrastructure and financing will be required to fund and connect specialized centers to one another and to the larger community, as well as to accommodate the increased volume of patients and samples at the centers. The success of the My Life, Our Future national initiative in the genotypic-phenotypic characterization of variants responsible for hemophilia A and B [Citation88], inspires hope that a similar approach to ultra-rare inherited BDs might prove fruitful. A centralized reagent standardization service and assay quality control initiative would improve the accuracy of more routine diagnoses of ultra-rare inherited BDs as well.
Centralized state-of-the-art data and biospecimen collection, leveraging and supporting centers with the appropriate technology and expertise, will maximize learnings from research participants and build the evidence base. Harmonization with other networks, databases, and registries nationally [Citation89] and internationally (e.g. EN-RBD and United Kingdom Haemophilia Centre Doctors’ Organization registries [Citation16,Citation19,Citation90–92]) would amplify the power of the dataset, potentially generating cohorts large enough to meet regulatory requirements and contribute to the approval of safe and effective therapeutics. The nature of US health care delivery presents serious challenges to the design and implementation of a research infrastructure that centralizes data and expertise, nationally or beyond. The absence of a single national provider system and the wide variety of caregivers and delivery networks are significant challenges to a single unified national reporting initiative.
4.3. Innovative solutions
Poor characterization of specific disorder severities and critical factor thresholds, combined with small heterogenous patient populations and long time frames for disease progression, present challenges to achieving traditionally required clinical endpoints when demonstrating therapeutic efficacy in rare and ultra-rare disorders [Citation76,Citation83]. The FDA and other regulatory agencies are aware of these challenges and motivated to identify solutions [Citation3]. Flexibility and streamlining of approval processes and elaboration of post-approval surveillance, capitalizing on a unified centralized data collection mechanism that meets regulatory requirements, will allow more people to access potentially beneficial treatment options and build the evidence base for treatment algorithms.
Real-world data describing the health status of individuals routinely collected from diverse sources such as electronic health records, claims/billing activities, registries, generated by patients at home or on mobile devices, may contain important evidence regarding risks/benefits of therapeutics, including those prescribed off-label [Citation93]. High-quality longitudinal natural history data may serve as a comparator for single-arm treatment trials and support product submissions to regulatory agencies [Citation94,Citation95]. The 21st Century Cures Act mandating the FDA expands use of these data to support new indications for already approved drugs [Citation3] is highly relevant to inherited ultra-rare BDs research.
Developers may elect to fulfill the FDA's recommended long-term follow-up of human gene therapy products [Citation96] with stand-alone databases, not easily queried by clinicians or researchers. A combined international, lifespan database is needed to detect rare and delayed safety events in the few people receiving gene therapy for a rare or ultra-rare disorder, scattered across the globe [Citation97]. The World Federation of Hemophilia (WFH) Gene Therapy Registry collaboration with the ISTH Scientific and Standardization Committee (SSC), European Haemophilia Consortium (EHC), NHF, ATHN, industry partners, and regulatory liaisons aims to recruit 100% of eligible post-gene therapy people with hemophilia worldwide [Citation98,Citation99]. Importantly, the core dataset in this prospective, observational, longitudinal registry was developed by a multi-stakeholder steering committee, following European Medicines Agency (EMA) and FDA guidance [Citation98].
Funding these innovative resources will require innovative solutions. Collaborations between organizations such as NHF and ATHN, building upon their existing infrastructure and generating diverse funding streams, will be necessary [Citation57]. With approval from oversight agencies, revenue generated by the 340B US federal discounted prescription drug purchasing program that most HTCs participate in [Citation100], could be directed to some of these initiatives. Savings from improved diagnostic accuracy and reduced misspending on suboptimal or ineffective treatments [Citation28,Citation50] may motivate investment from insurance companies.
Pharmaceutical development decisions are usually based upon economic considerations [Citation77]; the return on investment from market approval of therapeutics for ultra-rare disorders rarely meets their thresholds [Citation3,Citation4]. Elements of expected investment cost/revenue (e.g. time to market, revenue stream duration, and competitive landscape) contribute to the financial valuation of a therapeutic under development. Components that positively influence any of these elements, such as a series of small regulatory changes accelerating/improving the likelihood of approval, pragmatic regulatory flexibility and incentives, and innovative reimbursement models, may drive development [Citation77]. Conventional reimbursement cost-effectiveness analysis is not well suited for the consideration of orphan drugs and rare disorders [Citation101]. Expanding the valuation context to better address uncertainty and equity may yield a more flexible, objective, and reproducible cost-effectiveness framework that better values therapeutics for ultra-rare disorders [Citation101].
A novel collaboration between the FDA and Foundation for NIH aims to demonstrate cost-effective approaches to the development of novel highly specialized therapeutics. Bringing together 35 private, public, and nonprofit partners, the Bespoke Gene Therapy Consortium seeks to create a platform to deliver adeno-associated virus (AAV)-mediated gene therapy for many disorders, particularly those with populations too small to be commercially viable [Citation102]. By funding research projects to improve AAV vector production and transgene expression, developing harmonized and validated sets of manufacturing and preclinical testing requirements, and creating a standardized regulatory submission package with streamlined templates, master regulatory files, and uniform manufacturing processes, the consortium aims to facilitate and accelerate progression of gene therapies for rare diseases from conception to assessment and, if safe and effective, patient access [Citation78,Citation79,Citation102,Citation103].
4.4. Inclusion and outreach
With so few individuals affected by each disorder, engaging each ultra-rare inherited BD patient and their health care professionals with research initiatives is critical. HTCs are key in connecting potential research participants with opportunities to contribute to advancing the understanding of their disorder and ultimately improve their own care. Unknown numbers of people with ultra-rare inherited BDs receive care at non-HTC institutions, remain undiagnosed, or may not have access to appropriate care at all. Advocacy organizations, such as NHF and less formal LEE networks, will be essential in reaching these individuals and ensuring they have a medical home where they feel secure and cared for and can engage with initiatives that are right for them. Access to care remains a critical determinant of who benefits from research advances; outreach efforts will be key to capitalize on the potential for research to advance health equity for all people with inherited BDs.
The majority of the world’s population, and therefore also people with ultra-rare inherited BDs, do not live in high-income countries [Citation104]. Countries facing resource constraints currently diagnose only a small proportion of people with hemophilia and even fewer with other inherited BDs [Citation105,Citation106]. Inclusion of all countries in ultra-rare inherited BDs research collaborations, expected to greatly enrich the knowledge base and improve outcomes for many, would require substantial outreach and diagnostic capacity building.
Health care professionals may not receive adequate training in the recognition and appropriate workup of ultra-rare BDs. The expertise networks proposed herein must include outreach and training initiatives to raise awareness of the hallmarks of undiagnosed bleeding tendencies. Disseminating the availability of centralized diagnostic and management expertise will be key to their successful activation. A broad awareness raising campaign must reach primary care physicians, HTC health care professionals, and hematologists/other specialists in clinics beyond the HTC network, touching everywhere a person with an ultra-rare inherited BD may seek care. Engaging providers with innovative research projects may also entice further involvement. As discussed by WG6, innovative recruitment, retainment, and training initiatives to grow the classical hematology and hemostasis research workforce are required [Citation57].
LEE perspective
As lived experience experts (LEE), we were invited to participate in all meetings and discussions as the research initiatives were debated and prioritized. We gave our perspectives as patients, family members, and patient organization leaders regarding the impact that these research goals would have on patients’ everyday lives and experiences. We also participated in the editing process and final review of the manuscript.Participating in this project was a very big honor. We gained exposure to some of the most experienced and influential people in the ultra-rare inherited BDs space, and we engaged them in direct conversation on behalf of patients. We felt that our voices were heard and that our priorities were respected in the group conversations.The final research priorities presented in this paper reflect the true priorities of people with ultra-rare inherited BDs. Highlights of our influence include emphasizing a collaborative approach to diagnosis and research; sharing information widely to help patients get a diagnosis faster, and sharing clinical experiences and samples to better draw conclusions about the natural evolution of symptoms and treatment responses. Because our patient groups are so small, collaborative sharing of information is the best way to advance knowledge and understanding of these disorders. Understanding that small populations need different clinical trials and drug approval regulations is also paramount to getting treatments to patients more quickly, yet safely. This priority was informed by LEE discussions of their experience in trials and ability to gain access to potentially life-saving treatments under development. We, the WG3 LEEs, cannot be prouder of the initiatives prioritized in this paper, and their potential to create major impacts for people with ultra-rare inherited BDs.
5. Conclusions
WG3 Research Priorities for Ultra-rare Inherited Bleeding Disorders was charged with addressing two key questions: How can we better understand the biology of these rare disorders? How can we stimulate research and optimize the regulatory process to vastly improve diagnosis and targeted treatment? While exciting advances in the understanding and treatment options of more common inherited BDs may make the headlines, the priorities outlined herein present important, highly impactful opportunities to transform the experiences of people with ultra-rare inherited BDs.
List of abbreviations
Declaration of interests
The authors are integrated members of the inherited bleeding disorders community: people with inherited bleeding disorders, their family members, health-care providers, and researchers (including physicians, nurses, physical therapists, pharmacists, social workers/psychologists, geneticists/genetic counselors, etc.), industry partners, government officials/regulators, local community organization representatives and others.
AD Shapiro discloses: grants or contracts from any entity: Plasminogen IV study 18G, Compassionate use of Plasminogen eye drop, Compassionate Care Intermediate size patient population, Development of International Registry on Plasminogen Deficiency; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Genentech/Roche, Novo Nordisk, Pfizer, Sigilon; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Novo Nordisk Hemophilia Foundation, IHTC Board of Directors. SS Acharya discloses: participation on a Data Safety Monitoring Board or Advisory Board: Sanofi. AS Wolberg discloses: grants or contracts from any entity: Takeda; Consulting fees: Takeda; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: U Kansas; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: ISTH, HTRS, IFRS. B Sørensen discloses: stock or stock options: Hemab Therapeutics. C Bedrosian discloses: leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Board member for two companies working in Rare diseases (non- hematological) - Crinetics and Rhythm; other financial or non-financial interests: Member, Visiting Committee for Department of Biology, MIT. CPM Hayward discloses: grants or contracts from any entity: Canadian Institutes for Health Research Operating Grant 201603PJT-364832; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Stago; support for attending meetings and/or travel: Hamilton Health Sciences, McMaster University; participation on a Data Safety Monitoring Board or Advisory Board: Stago, Werfen; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: International Society for Laboratory Hematology, THSNA. D Corzo discloses: all support for the present manuscript: Sigilon Therapeutics, uniQure Inc.; support for attending meetings and/or travel: Sigilon Therapeutics; participation on a Data Safety Monitoring Board or Advisory Board: Sigilon Therapeutics; stock or stock options: Sigilon Therapeutics. K Nammacher discloses: participation on a Data Safety Monitoring Board or Advisory Board: BioMarin Ad Board 4/13/22, employee of NHF. KJ Baumann discloses: all support for the present manuscript: NHF-Fiona Robinson; consulting fees: Sanofi Genzyme, Optum; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Sanofi Genzyme, Optum; support for attending meetings and/or travel: Sanofi Genzyme; participation on a Data Safety Monitoring Board or Advisory Board: Novo Nordisk. M Tarantino discloses: consulting fees: Pfizer, Sanofi, Sobi-Swedish Orphan Biovitrum, Medexus, Takeda, Amgen, Inc., Octapharma, Genentech, Roche, BioMarin, UCB Biosciences, Dova Pharmaceuticals, Novo Nordisk; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Doptelet; participation on a Data Safety Monitoring Board or Advisory Board: Adult ITP Ad Board. M Recht discloses: grants or contracts from any entity: Bayer, BioMarin, CSL Behring, Genentech, Grifols, Hema Biologics, LFB, Novo Nordisk, Octapharma, Pfizer, Sanofi, Spark, Takeda, uniQure; consulting fees: Catalyst Biosciences, CSL Behring, Genentech, Hema Biologics, Kedrion, NovoNordisk, Pfizer, Sanofi, Takeda, uniQure; leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Foundation for Women and Girls with Blood Disorders; Partners in Bleeding Disorders, Thrombosis and Hemostasis Societies of North America. RK Pruthi discloses: consulting fees: Merck; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: CSL Behring, Genentech Inc, Bayer Healthcare AG, HEMA Biologics, Instrumentation Laboratory. R Bialas discloses: leadership or fiduciary role in other board, society, committee or advocacy group, paid or unpaid: Plasminogen Deficiency Foundation. R Palla discloses: payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Novo Nordisk; support for attending meetings/travel: Kedrion. S Peltier discloses: payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Novo Nordisk. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
AD Shapiro and D Nugent led the working group recruitment, organization, and subdivision into subgroups. AD Shapiro, D Nugent, and SS Acharya led subgroup deliberations and analysis. All authors contributed to analysis and deliberations. AD Shapiro, D Nugent, and SS Acharya led manuscript preparation, all authors offered input and contributed to revisions of the manuscript, approved the final version to be published, and agree to be accountable for all aspects of the work. A Haidar, R Bialas, and S Peltier led writing of the Plain Language Summary and LEE Perspective sections.
Presentation of content
D Nugent, SS Acharya, and AD Shapiro presented highlights of the deliberations of Working Group 3 at the National Hemophilia Foundation State of the Science (virtual) Research Summit (SOSRS), September12–15, 2021. Summit discussions informed the Discussion and Conclusions of this paper.
Supplemental Material
Download Zip (43.4 KB)Acknowledgments
The Executive Committee of the National Hemophilia Foundation (NHF) National Research Blueprint initiative were actively engaged in the conception, design, preparation, and oversight of each of the State of the Science manuscripts in this supplement. Maria E. Santaella actively engaged with the lived experience expert (LEE) WG members throughout the process, empowering their inclusion and participation. The Executive Committee consisted of: Kevin Mills, Michael Recht, Michelle L. Witkop, Maria E. Santaella, Donna DiMichele, Keri L. Norris, Esmeralda Vázquez and Brett Spitale. The authors thank Donna DiMichele, MD for her review of the manuscript. Fiona Robinson, PhD provided professional medical writing support during manuscript development; medical illustrations were created by Matt Evans; both paid by NHF.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/17474086.2023.2175661
Additional information
Funding
References
- Richter T, Nestler-Parr S, Babela R, et al. Rare disease terminology and definitions-A systematic global review: report of the ISPOR rare disease special interest group. Value Health. 2015;18(6):906–914.
- Orphan Drug Act. Public Law 97–414 (1983).
- Epps C, Bax R, Croker A, et al. Global regulatory and public health initiatives to advance pediatric drug development for rare diseases. Ther Innov Regul Sci. 2022;56:964–975.
- Kaufmann P, Pariser AR, Austin C. From scientific discovery to treatments for rare diseases - the view from the national center for advancing translational sciences - office of rare diseases research. Orphanet J Rare Dis. 2018;13(1):196.
- Bax BE. Biomarkers in rare diseases. Int J Mol Sci. 2021;22:2.
- Crooke ST. Addressing the needs of patients with ultra-rare mutations one patient at a time: the n-lorem approach. Nucleic Acid Ther. 2022;32(2):95–100.
- Regulation (EU) No. 536/2014 of the European parliament and of the council of April 16, 2014 on clinical trials on medicinal products for human use, and repealing directive 2001/20/EC, (2014).
- Crooke ST. A call to arms against ultra-rare diseases. Nat Biotechnol. 2021;39(6):671–677.
- Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019;171(8):540–546.
- Tran DQ, Benson CC, Boice JA, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities to transform the care of people with hemophilia. Expert Rev Hematol. 2023;16(S1):19-37. DOI: 10.1080/17474086.2023.2171981.
- Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(Suppl 6):1–158. 3rd
- Croteau SE, Wang M, Wheeler AP. clinical trials update: innovations in hemophilia therapy. Am J Hematol. 2021;96(1):128–144.
- Mahlangu JN, Blanchette V, Klamroth R. Redefining prophylaxis in the modern era. Haemophilia. 2021;27(Suppl 3):21–27.
- Batsuli G, Kouides P. Rare coagulation factor deficiencies (factors VII, X, V, and II). Hematol Oncol Clin North Am. 2021;35(6):1181–1196.
- Gupta S, Acharya S, Roberson C, et al. Potential of the Community Counts registry to characterize rare bleeding disorders. Haemophilia. 2019;25(6):1045–1050.
- Mumford AD, Ackroyd S, Alikhan R, et al. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British committee for standards in haematology. Br J Haematol. 2014;167(3):304–326.
- Peyvandi F. Epidemiology and treatment of congenital fibrinogen deficiency. Thromb Res. 2012;130(Suppl 2):S7–11.
- Peyvandi F, Di Michele D, Bolton-Maggs PH, et al. Classification of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012;10(9):1938–1943.
- Peyvandi F, Palla R, Menegatti M, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European network of rare bleeding disorders. J Thromb Haemost. 2012;10(4):615–621.
- Shapiro A. The use of prophylaxis in the treatment of rare bleeding disorders. Thromb Res. 2020;196:590–602.
- Peyvandi F, Menegatti M. Treatment of rare factor deficiencies in 2016. Hematology Am Soc Hematol Educ Program. 2016;2016(1):663–669.
- Blechman S, Fruchtman Y, Perry ZH, et al. Clinical and laboratory findings in Jewish and Bedouin patients in Southern Israel who were diagnosed with factor VII deficiency. Isr Med Assoc J. 2019;21(5):318–321.
- Dorgalaleh A, Alavi SE, Tabibian S, et al. Diagnosis, clinical manifestations and management of rare bleeding disorders in Iran. Hematology. 2017;22(4):224–230.
- Rodrigues DN, Siqueira LH, Galizoni AM, et al. Prevalence of factor VII deficiency and molecular characterization of the F7 gene in Brazilian patients. Blood Coagul Fibrinolysis. 2003;14(3):289–292.
- Zhang X, Lewandowska M, Aldridge M, et al. Global epidemiology of factor XI deficiency: a targeted review of the literature and foundation reports. Haemophilia. 2022. 10.1111/hae.14687.
- Koerper MA, Frick N, Kessler CM. MASAC Consensus Conference: impediments to conducting clinical research in persons with haemophilia, von Willebrand’s disease and rare bleeding disorders. Haemophilia. 2013;19(2):188–193.
- Valentino LA, Witkop ML, Santaella ME, et al. Building the blueprint: formulating a community-generated national plan for future research in inherited bleeding disorders. Haemophilia. 2022;28(5):760–768.
- Meijer K, van Heerde W, Gomez K. Diagnosis of rare bleeding disorders. Haemophilia. 2022;28(Suppl 4):119–124.
- Menegatti M, Palla R. Clinical and laboratory diagnosis of rare coagulation disorders (RCDs). Thromb Res. 2020;196:603–608.
- Peyvandi F, Garagiola I, Biguzzi E. Advances in the treatment of bleeding disorders. J Thromb Haemost. 2016;14(11):2095–2106.
- Bernardi F, Mariani G. Biochemical, molecular and clinical aspects of coagulation factor VII and its role in hemostasis and thrombosis. Haematologica. 2021;106(2):351–362.
- Napolitano M, Siragusa S, Mariani G. Factor VII deficiency: clinical phenotype, genotype and therapy. J Clin Med. 2017;6:4.
- Peyvandi F, Menegatti M, Palla R. Rare bleeding disorders: worldwide efforts for classification, diagnosis, and management. Semin Thromb Hemost. 2013;39(6):579–584.
- Kulkarni A, Lee CA, Griffeon A, et al. Disorders of menstruation and their effect on the quality of life in women with congenital factor VII deficiency. Haemophilia. 2006;12(3):248–252.
- Palla R, Siboni SM, Menegatti M, et al. Establishment of a bleeding score as a diagnostic tool for patients with rare bleeding disorders. Thromb Res. 2016;148:128–134.
- Shapiro SE, Phillips E, Manning RA, et al. Clinical phenotype, laboratory features and genotype of 35 patients with heritable dysfibrinogenaemia. Br J Haematol. 2013;160(2):220–227.
- Siboni SM, Spreafico M, Calo L, et al. Gynaecological and obstetrical problems in women with different bleeding disorders. Haemophilia. 2009;15(6):1291–1299.
- Postel MD, Culver JO, Ricker C, et al. Transcriptome analysis provides critical answers to the “variants of uncertain significance” conundrum. Hum Mutat. 2022;43:1590–1608.
- Bastida JM, Benito R, Lozano ML, et al. Molecular diagnosis of inherited coagulation and bleeding disorders. Semin Thromb Hemost. 2019;45(7):695–707.
- Nurden P, Stritt S, Favier R, et al. Inherited platelet diseases with normal platelet count: phenotypes, genotypes and diagnostic strategy. Haematologica. 2021;106(2):337–350.
- Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood. 2015;125(13):2052–2061.
- Coppola A, Di Minno G. Desmopressin in inherited disorders of platelet function. Haemophilia. 2008;14(Suppl 1):31–39.
- Desborough MJ, Oakland KA, Landoni G, et al. Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost. 2017;15(2):263–272.
- Persyn M, Athanase N, Trossaert M, et al. Effect of DDAVP on platelet activation and platelet-derived microparticle generation. Hamostaseologie. 2022;42(3):185–192.
- Kadir RA, Sabin CA, Pollard D, et al. Quality of life during menstruation in patients with inherited bleeding disorders. Haemophilia. 1998;4(6):836–841.
- Liu Z, Doan QV, Blumenthal P, et al. A systematic review evaluating health-related quality of life, work impairment, and health-care costs and utilization in abnormal uterine bleeding. Value Health. 2007;10(3):183–194.
- McGrath M, Quint EH, Weyand AC. Depression in adolescents and young adults with heavy menstrual bleeding in a referral clinic setting. Am J Hematol. 2021;96(4):E105–E8.
- van Hoorn ES, Houwing ME, Al Arashi W, et al. Patient-reported outcomes in autosomal inherited bleeding disorders: a systematic literature review. Haemophilia. 2022;28(2):197–214.
- Weyand AC, Fitzgerald KD, McGrath M, et al. Depression in female adolescents with heavy menstrual bleeding. J Pediatr. 2022;240:171–176.
- Arnold DM, Nazy I, Clare R, et al. Misdiagnosis of primary immune thrombocytopenia and frequency of bleeding: lessons from the McMaster ITP Registry. Blood Adv. 2017;1(25):2414–2420.
- Kirtava A, Drews C, Lally C, et al. Medical, reproductive and psychosocial experiences of women diagnosed with von Willebrand’s disease receiving care in haemophilia treatment centres: a case-control study. Haemophilia. 2003;9(3):292–297.
- Vázquez E, Kim M, Santaella ME. Lived experience experts: a name created by us for us, Expert Rev Hematol, 2023;16(S1):7–11 DOI: 10.1080/17474086.2023.2178410.
- Valentino LA, Witkop ML, Santaella ME, et al. The National Hemophilia Foundation State of the Science Research Summit initiative: executive summary. Expert Rev Hematol. 2023;16(S1):129-134 DOI: 10.1080/17474086.2023.2181782.
- Rauch A, Valentino LA, Mills K, et al. Big picture initiatives in bleeding disorders. Haemophilia. 2022;28(Suppl 4):53–60.
- Valentino LA, Witkop ML, Santaella ME, et al. The National Hemophilia Foundation’s State of the Science Research Summit initiative: the foundation of an inherited bleeding disorders national research blueprint. Expert Rev Hematol. 2023;16(S1):129–134. DOI: 10.1080/17474086.2023.2181782.
- Byams VR, Baker JR, Bailey C, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities in health services; diversity, equity, and inclusion; and implementation science. Expert Rev Hematol. 2023;16(S1):87-106. DOI: 10.1080/17474086.2023.2183836.
- Ragni MV, Young G, Batsuli G, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: Facilitating research through infrastructure, workforce, resources and funding. Expert Rev Hematol. 2023;16(S1):107-127 DOI: 10.1080/17474086.2023.2181781.
- National Institutes of Health. Definition of human subjects research [cited 2022 Jun 16]. Available from: https://grants.nih.gov/policy/humansubjects/research.htm.
- U.S. Food and Drug Administration. Clinical Laboratory Improvement Amendments (CLIA) [cited 2022 Apr 7]. Available from: https://www.fda.gov/medical-devices/ivd-regulatory-assistance/clinical-laboratory-improvement-amendments-clia.
- College of American Pathologists. Why CAP accreditation [cited 2022 Apr 7]. Available from: https://www.cap.org/laboratory-improvement/accreditation/why-cap-accreditation.
- Clinical Genome Resource. ClinVar [cited 2022 Apr 7]. Available from: https://www.clinicalgenome.org/data-sharing/clinvar/.
- Saes JL, Verhagen MJA, Meijer K, et al. Bleeding severity in patients with rare bleeding disorders: real-life data from the RBiN study. Blood Adv. 2020;4(20):5025–5034.
- Gresele P, Falcinelli E, Bury L, et al. The ISTH bleeding assessment tool as predictor of bleeding events in inherited platelet disorders: communication from the ISTH SSC subcommittee on platelet physiology. J Thromb Haemost. 2021;19(5):1364–1371.
- Gresele P, Orsini S, Noris P, et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: a communication from the Platelet Physiology SSC. J Thromb Haemost. 2020;18(3):732–739.
- James PD, Mahlangu J, Bidlingmaier C, et al. Evaluation of the utility of the ISTH-BAT in haemophilia carriers: a multinational study. Haemophilia. 2016;22(6):912–918.
- Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065.
- Children’s Oncology Group. The world’s childhood cancer experts [cited 2022 Jun 14]. Available from: https://childrensoncologygroup.org/.
- Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. 2018;9:135–140.
- Mahlangu JN. Progress in the development of anti-tissue factor pathway inhibitors for haemophilia management. Front Med (Lausanne). 2021;8:670526.
- Centers for Disease Control and Prevention. About community counts [cited 2022 Jun 16]. Available from: https://www.cdc.gov/ncbddd/hemophilia/communitycounts/about.html.
- American Thrombosis and Hemostasis Network. ATHN 10: leveraging the ATHNdataset to document the state of rare coagulation disorders in the United States [cited 2022 Apr 12]. Available from: https://www.athn.org/what-we-do/national-projects/athn10.html.
- American Thrombosis and Hemostasis Network. Advancing research with better data through the ATHNdataset [cited 2022 Apr 12]. Available from: https://sysadmin.athn.org/what-we-do/national-projects/athndataset.html.
- American Thrombosis and Hemostasis Network. ATHN Projects and Research Studies—an Overview [cited 2022 Apr 12]. Available from: https://www.athn.org/what-we-do/national-projects/athn-projects-and-research-studies.html.
- U.S. Food and Drug Administration. Expedited programs for regenerative medicine therapies for serious conditions - guidance for industry [cited 2022 Jun 19]. Available from: https://www.fda.gov/media/120267/download.
- U.S. Food and Drug Administration. Guidance for industry: expedited programs for serious conditions – drugs and biologics [cited 2022 Jun 23]. Available from: https://www.fda.gov/media/86377/download.
- Kakkis ED, O’Donovan M, Cox G, et al. Recommendations for the development of rare disease drugs using the accelerated approval pathway and for qualifying biomarkers as primary endpoints. Orphanet J Rare Dis. 2015;10:16.
- European Federation of Pharmaceutical Industries and Associations. Challenges and facilitators in the development of orphan and paediatric medicines [cited 2022 Sept 28]. Available from: https://www.efpia.eu/publications/downloads/access-to-medicines/challenges-and-facilitators-in-the-development-of-orphan-and-paediatric-medicines/.
- Marks P. Enhancing gene therapy regulatory interactions. Expert Opin Biol Ther. 2022;22(9):1073–1074.
- Marks P, Witten C. Toward a new framework for the development of individualized therapies. Gene Ther. 2021;28(10–11):615–617.
- World Health Organization. WHO considerations on regulatory convergence of cell and gene therapy products [cited 2022 Jun 18]. Available from: https://cdn.who.int/media/docs/default-source/biologicals/ecbs/who-public-consultation_cgtp-white-paper_16_dec_2021.pdf?sfvrsn=18f6c549_5.
- U.S. Food and Drug Administration. INTERACT meetings [cited 2022 Jun 23]. Available from: https://www.fda.gov/vaccines-blood-biologics/industry-biologics/interact-meetings.
- Chan AYL, Chan VKY, Olsson S, et al. Access and unmet needs of orphan drugs in 194 countries and 6 areas: a global policy review with content analysis. Value Health. 2020;23(12):1580–1591.
- Kempf L, Goldsmith JC, Temple R. Challenges of developing and conducting clinical trials in rare disorders. Am J Med Genet A. 2018;176(4):773–783.
- U.S. Food and Drug Administration. Fast Track [cited 2022 Jun 23]. Available from: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/fast-track.
- Sidonio RF Jr., Bryant PC, Di Paola J, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities for mucocutaneous bleeding disorders. Expert Rev Hematol. 2023;16(S1):39-54. DOI: 10.1080/17474086.2023.2171983.
- Baldwin MK, Ahmadzia HK, Bartlett DL, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research to advance the health of people with inherited bleeding disorders with the potential to menstruate. Expert Rev Hematol. 2023;16(S1):71-86 DOI: 10.1080/17474086.2023.2175660.
- National Hemophilia Foundation. National Research Blueprint - A world without bleeding disorders starts with research [cited 2022 Jan 23]. Available from: https://www.hemophilia.org/research/national-research-blueprint.
- Johnsen JM, Fletcher SN, Dove A, et al. Results of genetic analysis of 11 341 participants enrolled in the my life, our future hemophilia genotyping initiative in the United States. J Thromb Haemost. 2022;20(9):2022–2034.
- Acharya SS, Coughlin A, Dimichele DM. North American rare bleeding disorder study g. rare bleeding disorder registry: deficiencies of factors II, V, VII, X, XIII, fibrinogen and dysfibrinogenemias. J Thromb Haemost. 2004;2(2):248–256.
- Rare Bleeding Disorders Network. RRBD rare bleeding disorders database - Studies [cited 2022 Jun 17]. Available from: https://rbddorg.serversicuro.it/014/index.php?option=com_content&view=category&id=11&Itemid=104.
- Bolton-Maggs PH, Perry DJ, Chalmers EA, et al. The rare coagulation disorders–review with guidelines for management from the United Kingdom Haemophilia Centre Doctors’ Organisation. Haemophilia. 2004;10(5):593–628.
- Viswabandya A, Baidya S, Nair SC, et al. Correlating clinical manifestations with factor levels in rare bleeding disorders: a report from Southern India. Haemophilia. 2012;18(3):e195–200.
- U.S. Food and Drug Administration. Real-world evidence [cited 2022 Jun 19]. Available from: https://www.fda.gov/science-research/science-and-research-special-topics/real-world-evidence.
- Gross AM. Using real world data to support regulatory approval of drugs in rare diseases: a review of opportunities, limitations & a case example. Curr Probl Cancer. 2021;45(4):100769.
- Izem R, Buenconsejo J, Davi R, et al. Real-world data as external controls: practical experience from notable marketing applications of new therapies. Ther Innov Regul Sci. 2022;56:704–716.
- U.S. Food and Drug Administration. Long term follow-up after administration of human gene therapy products - guidance for industry [cited 2022 Jun 19]. Available from: https://www.fda.gov/media/113768/download.
- Konkle BA, Coffin D, Pierce GF, et al. World Federation of Hemophilia gene therapy registry. Haemophilia. 2020;26(4):563–564.
- Konkle B, Pierce G, Coffin D, et al. Core data set on safety, efficacy, and durability of hemophilia gene therapy for a global registry: communication from the SSC of the ISTH. J Thromb Haemost. 2020;18(11):3074–3077.
- World Federation of Hemophilia. WFH gene therapy registry: now live! [cited 2023 Jan 23]. Available from: https://wfh.org/article/wfh-gene-therapy-registry-now-live/.
- Health Resources & Services A. 340B drug pricing program [cited 2022 Jun 19]. Available from: https://www.hrsa.gov/opa/index.html.
- Postma MJ, Noone D, Rozenbaum MH, et al. Assessing the value of orphan drugs using conventional cost-effectiveness analysis: is it fit for purpose? Orphanet J Rare Dis. 2022;17(1):157.
- The Foundation for the National Institutes of Health. Accelerating medicines partnership® bespoke gene therapy consortium (AMP® BGTC) (poster) [cited 2022 Jun 19]. Available from: https://fnih.org/sites/default/files/2022-05/FNIH%20BGTC_Overview%20Fnl.pdf.
- The Foundation for the National Institutes of Health. Accelerating medicines partnership®bespoke gene therapy consortium (AMP® BGTC) (webpage) [cited 2022 Jun 19]. Available from: https://fnih.org/our-programs/AMP/BGTC.
- Our World In Data. Population by income level, 1960 to 2020 Accessed 20 Jan 2023. . Available from https://ourworldindata.org/grapher/population-by-income-level
- World Federation of Hemophilia. World Federation of Hemophilia Report on the Annual Global Survey 2021. Montréal Canada: WFH; 2022 Accessed 20 Jan 2023. Available from: https://www1.wfh.org/publications/files/pdf-2324.pdf.
- Witkop ML, Robinson F, DiMichele D. Soliciting international perspectives on an American national research agenda for inherited bleeding disorders, Exp Rev Hem, 2023;16(S1):13–17. DOI: 10.1080/17474086.2023.2178411.