
ABSTRACT
Introduction
The direct antiglobulin test (DAT) or Coombs test is the cornerstone of the diagnosis of autoimmune hemolytic anemia (AIHA). It can be performed by several methods with different sensitivity and specificity and enables the distinction of warm, cold, and mixed forms, which require different therapies.
Areas covered
The review describes the different DAT methods, including the tube test with monospecific antisera, microcolumn and solid phase methods that are routinely accessible in most laboratories. Additional investigations include the use of cold washes and low ionic salt solutions, the identification of auto-Ab specificity and thermal range, the study of the eluate, and the Donath-Landsteiner test, available in most reference laboratories. Experimental techniques are the dual-DAT, flow cytometry, ELISA, immuno-radiometric assay, and mitogen-stimulated DAT, which may help the diagnosis of DAT-negative AIHAs, a clinical challenge with delayed diagnosis and possible improper therapy. Further diagnostic challenges include the correct interpretation of hemolytic markers, the infectious and thrombotic complications, and the possible underlying conditions (lymphoproliferative disorders, immunodeficiencies, neoplasms, transplants, and drugs)
Expert opinion
These diagnostic challenges may be overcome by a ‘hub’ and ‘spoke’ organization among laboratories, a clinical validation of experimental techniques, and a continuous dialogue between clinicians and immune-hematologic laboratory experts.
1. Introduction
Autoimmune hemolytic anemia (AIHA) is a rare and heterogeneous disease due to premature destruction of erythrocytes by several immune effectors, mainly autoantibodies and complement. The estimated incidence and prevalence of the disease are 1–3/100,000 people per year and 17:100,000, respectively [Citation1,Citation2]. The cornerstone of the diagnosis is the direct antiglobulin test (DAT) or Coombs test, originally described in 1945, and nowadays performed by several methods with different sensitivity and specificity. The DAT allows the distinction of ‘warm’ type AIHA (wAIHA), mainly due to IgG antibodies that react at 37°C and may weakly activate complement, and ‘cold’ forms (cold agglutinin disease, CAD), due to IgM that strongly activate complement [Citation1–4]. Most cold agglutinins are monoclonal and can cause hemolysis or agglutination-mediated ischemic symptoms in the acral circulation if their thermal amplitude allows binding and complement activation in vivo. Rarer AIHAs include the mixed (wAIHA plus CAD) and atypical forms (IgA- and warm IgM-driven, and DAT-negative), and the ultra-rare paroxysmal cold hemoglobinuria (PCH) [Citation4–6]. In addition to the thermal and biochemical characteristics of the autoantibody, further complexity to the diagnostic work-up relies on the underlying disease/condition, as about 50% of all AIHAs are secondary to systemic autoimmune diseases, lymphoproliferative disorders, immunodeficiencies, neoplasms, infections, transplants, drugs, including the new check-point inhibitors and chimeric antigen receptor T-cells (CAR-T) [Citation7–15]. Finally, besides autoantibodies and complement, the pathogenic mechanisms include cellular effectors (mononuclear phagocytes, T-lymphocytes), dysregulation of regulatory cells and cytokines, and last but not least the bone marrow compensatory response [Citation3,Citation16]. The resulting clinical picture is highly heterogenous, from fully compensated to life-threatening and fulminating forms, and may even vary in the same patient during time. This review will address the diagnostic challenges in the diagnosis of primary versus secondary AIHAs, focusing on the pitfalls of the DAT, the correct interpretation of hemolytic markers, the clinical signs and symptoms that may guide the diagnosis, and the concomitant hemolytic conditions that may puzzle the diagnosis.
2. The direct antiglobulin test (DAT)
The DAT was first described in 1945 by Robin Coombs (after whom it is named), Arthur Mourant and Rob Race in Cambridge, and was done in test tubes. Since its first description, the DAT has been applied in to investigate immediate and delayed alloimmune hemolytic transfusion reactions, hemolytic disease of the newborn, and autoimmune hemolysis occurring in AIHAs [Citation17,Citation18]. The principle of the test relies on an anti-globulin reagent that binds to the Fc portions of the antibody bound to red cells, promoting the formation of a visible agglutination in a tube [Citation1,Citation17–21]. The anti-globulin reagent is indispensable to reveal IgG, IgA or complement fractions bound to the red cell since they are unable to directly agglutinate erythrocytes. At variance the anti-globulin reagent is not strictly necessary to detect IgM since they are pentamers, able to cause direct agglutination by bridging the gap between adjacent erythrocytes that normally repel each other by negative charge of sialic acid residues (). Thus, spontaneous agglutination at 20°C is a very simple and old test, able to reveal the presence of anti-erythrocyte autoantibodies of the IgM class [Citation1,Citation2,Citation17–21]. Sometimes, this agglutination may result in an alarm from the laboratory, as the coulter is unable to give proper results. Warm washes and re-testing are necessary to give a correct result, and a prompt communication between laboratory and clinicians may give important hints to guide the diagnosis [Citation1,Citation7].
Figure 1. The principle of the direct antiglobulin test (DAT).
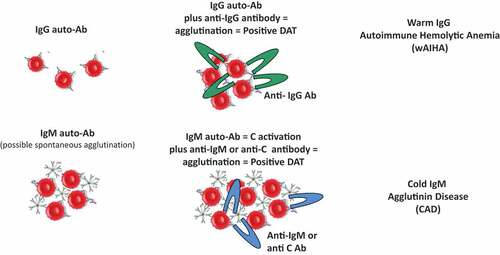
Historically, the DAT has been performed with the tube technique and broad antiglobulin reagents, containing a variable mixture of anti-IgG, -IgM, -IgA and -complement fractions. This resulted in a semi-quantitative positivity (generally expressed as 1+ to 4+ or more), without distinguishing the isotype of the autoantibody and its thermal amplitude. Moreover, the content of anti-IgG, -IgM, -IgA and -complement fractions was highly variable, generally greater for the former and lower for the latter, possibly giving false negative results. To overcome this problem, the use of monospecific antisera (anti-IgG, -IgM, -IgA and -C) are nowadays commonly used in reference laboratories to properly diagnose the various types of AIHA, as they require different treatments [Citation1–4,Citation17–21].
Further complexity to correctly interpret the DAT resides on the ability of autoantibodies to activate complement. As already stated, IgM are potent activators of the classical complement pathway. However, they may detach from the cell during washing procedures or even in vivo, thus giving a negative result. In this case, complement fractions (mainly C3d) remain on the erythrocyte surface, allowing a positive DAT with anti-complement antisera. As a matter of fact, IgG3, IgG1 and, to a small extent, IgG2 are also able to activate complement, resulting in a weak positivity with anti-complement antisera, but generally most of the complement positivity detected by DAT is caused by IgM autoantibodies [Citation1,Citation4,Citation20,Citation21].
The DAT detects antibodies bound to erythrocytes, thus it may be positive for alloantibodies in recently transfused patients. Their presence has been reported in 1/3 of patients with AIHA, is often masked by autoantibodies, and can cause serious hemolytic reactions in case of transfusion with incompatible blood, further aggravating the clinical picture. The identification of coexisting alloantibodies is fundamental in complex cases and when transfusions do not result in an increase of hemoglobin values. The usual immunoabsorbance tests and more recently the extended genotyping techniques are not widely performed, are time consuming and may be challenging, particularly in emergency situations making it difficult to find compatible units [Citation1,Citation4,Citation20,Citation22–24].
The DAT may be positive in the ‘passenger lymphocyte syndrome,’ a complication of both solid-organ and stem cell transplants, caused by the production of antibodies by the donor’s B lymphocyte against recipient erythrocytes. The incidence has been reported to be 9%, 29%, and 70% for kidney, liver, and heart-lung transplants, respectively, most commonly, in the setting of ABO mismatches (examples: a group B liver transplanted into a group AB recipient, group 0 lung into a group A recipient) [Citation3,Citation7].
Finally, DAT positivity should always be interpreted in a clinical context, as it may be positive after administration of various therapeutics (intravenous immunoglobulins, Rh immune globulins, anti-lymphocyte/anti-thymocyte globulins, and daratumumab), and in diseases with elevated serum globulins or paraproteins. Furthermore, the DAT may be positive in about 0.1% of healthy donors, without clinical and laboratory evidence of hemolytic disease [Citation1,Citation20,Citation21].
3. The indirect antiglobulin test and other serological investigation
The indirect antiglobulin test (IAT) reveals the presence of auto- or alloantibodies in patient’s serum [Citation1,Citation19]. The serum is incubated with standardized red blood cells panels of known antigenicity, and then anti-human globulin is added. A positive test (revealed by agglutination) identifies the antigenic specificity of the antibody. The IAT test is used in prenatal testing of pregnant women, in testing prior to a blood transfusion, and in the identification of alloantibodies in recently transfused patients. In AIHA, it may be positive, allowing the characterization of the antigen specificity (not strictly necessary for the diagnosis), usually the Rh system in wAIHA and the I/i erythrocyte antigens in CAD [Citation1]. In the latter, it is important to perform the titer of cold agglutinins in serum, since a value less than 64 is considered associated with secondary forms of cold AIHAs, while a greater titer is generally required to confirm the diagnosis of CAD [Citation2,Citation4,Citation25].
Additional serologic tests include the study of the eluates, i.e., the antibodies eluted from autologous erythrocytes. This may improve the sensitivity by concentrating the autoantibody and help the diagnosis in difficult cases of drug-induced AIHAs [Citation1,Citation18,Citation19]. Finally, the Donath-Landsteiner biphasic hemolysin (an antibody that binds in cold and hemolyses at body temperatures) is the cornerstone of the diagnosis of PCH, an ultra-rare AIHA usually seen in children after a viral infection and in the past in syphilis [Citation26,Citation27].
4. Sensitivity and specificity of routine DAT methods
In the conventional tube test, the agglutination is detected visually as clumps of erythrocytes. Although still used, this DAT method is nowadays replaced/supported by semi-automated methods such as the gel microcolumn and solid-phase tests [Citation1,Citation19–21]. In the former, patient’s erythrocytes are filtered through a gelatinous matrix mixed with anti-human reagents, in which agglutinated erythrocytes are trapped (positive test) while the non-agglutinated pass through the column (negative). In the solid-phase assay, patient’s erythrocytes are incubated into microplate wells coated with anti-human reagents, and their adherence to the wells (positive test) or not (negative) are evaluated using a spectrophotometer in an automated fashion ().
Figure 2. DAT in microcolumn (a) and solid phase (b).
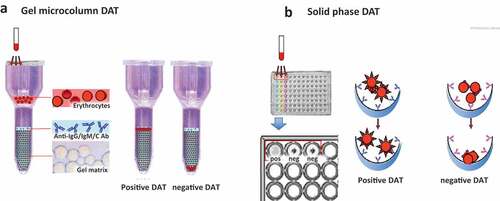
In the past years, several studies have compared the traditional tube test with more automated techniques. Dittmar et al [Citation28] found that tube agglutination and gel microcolumn DATs were concordant in 69 patient samples (positive in 49 and negative in 20 samples) but discordant in 15, and gel microcolumn was more sensitive than tube. This was confirmed in a larger study on 9,862 blood samples submitted to a reference laboratory [Citation29], where sensitivity for DATs was 100% for gel microcolumn, and 50.7% for tube, addressing the problem of DAT-tube negative AIHAs. Of note, the traditional DAT-tube with polyspecific or anti-IgG and anti-C antisera may yield false-negative results due to the prevailing presence of IgA. Although IgA are generally associated with IgG autoantibodies, it is advisable to include this reagent in routine testing. Furthermore, low-affinity autoantibodies may detach from erythrocytes during washing procedures, suggesting the use of low ionic strength solutions or cold washings in case of persistent clinical suspicion of AIHA.
5. DAT-negative AIHA
In an attempt to increase the sensitivity of the DAT tube, flow cytometry technique has been developed, although still not routinely used in the diagnosis of AIHA. Chaudhary et al [Citation30] found that 5 out of 50 DAT-tube negative AIHA diagnosed on a clinical basis were positive by flow cytometry, even if 4 of the 5 were also positive by gel microcolumn. The Authors also addressed the question of the cutoff value of the red-blood-cell bound IgG level, which was further developed in a more recent study [Citation31]. The Authors investigated 50 clinically diagnosed DAT-tube negative AIHAs (92% positive by gel microcolumn) and found by Receiver Operating Characteristic (ROC) curve that the best cutoff values for a positive flow cytometry-DAT was 17.5% fluorescence (100% sensitivity and specificity), and 1.74 mean fluorescence intensity (76% specificity and 96% sensitivity).
Another method implemented is the immunoradiometric assay. Kamesaki et al [Citation32] studied 64 DAT-tube negative AIHA patients and demonstrated by ROC curve a 71.4% sensitivity and an 87.8% specificity. Furthermore, Fabijańska-Mitek studied 268 samples from patients with wAIHA by a quantitative enzyme-linked antiglobulin test [Citation33] and found a correlation between signs of hemolysis and amount of red cell-bound autoantibody (65.7% in those with <200 IgG molecules/red blood cell, 70.4% with 200–1000 molecules/red blood cell, and 87.9% in samples with >1000 IgG molecules/red blood cell). It is worth reminding the dual DAT [Citation34] that is useful for the detection of RBC-bound IgM warm antibodies, which fail to be revealed by standard methods, and can cause severe and fatal AIHA [Citation35].
Finally, our group [Citation36] has developed a functional and quantitative method named mitogen-stimulated (MS)-DAT, which is able to amplify by mitogen stimulation the production of autoantibodies and thus detect very low amounts of erythrocyte-bound IgG. In a comparative study among various DAT methods [Citation37], we found that DAT tube is the most specific but least sensitive test (87% and 43%, respectively); other traditional DAT methods (microcolumn/solid phase) show reduced specificity but increased sensitivity (70% and 65%, respectively), and MS-DAT is the least specific but the most sensitive test (59% and 88%, respectively), underlying that the counterpart of a greater sensitivity is a reduced specificity. Additionally, the test has been proven to be useful in the diagnosis of DAT-negative AIHAs [Citation36] and found positive in a fraction of patients with B-chronic lymphocytic leukemia [Citation38] or myelofibrosis [Citation39] without an overt diagnosis of AIHA, suggesting that in vitro mitogen stimulation could disclose a latent anti-erythrocyte autoimmunity [Citation20].
6. Hemolytic markers and their confounders
Hemolytic markers are quite known by most hematologist, and include unconjugated bilirubin, LDH haptoglobin and reticulocytes [Citation40]. They may be variably altered in the different forms of AIHAs, mostly reflecting the acuteness of the hemolytic process and degree of intravascular versus extravascular hemolysis. Symptomatic anemia usually reflects a brisk decrease in hemoglobin, typical of wAIHA plus complement activation, mixed forms, or the fulminant warm IgM AIHAs. LDH is highly elevated, as a marker of intravascular hemolysis, related to massive complement activation until the membrane attack complex (MAC); unconjugated bilirubin, is increased as well, although to a lesser extent, and reflects extravascular hemolysis in lymphoid organs, mainly in the spleen for wAIHA and in the liver for CAD. The mechanism is the so-called antibody-dependent or complement-dependent cellular cytotoxicity by macrophages or activated lymphocytes expressing Fc or complement receptors [Citation3,Citation16,Citation40,Citation41]. CAD is more frequently a chronic disease with possible flares associated with exposure to cold or infections due to concomitant massive complement activation. Regarding cold exposure, it is important to remember that agglutination and hemolysis occur only when the optimal temperature of action of the cold IgM is compatible with body temperature particularly in acral sites [Citation2,Citation41]. Haptoglobin, the scavenger of free hemoglobin is invariably consumed, and is the most sensitive hemolytic marker, often remaining measurable for months; its increase and normalization is a definitely sign of full recovery of the hemolytic process. It is worth reminding the presence of free hemoglobin (when haptoglobin scavenger activity is saturated), and hemoglobinuria/hemosiderinuria (when intravascular hemolysis is intense and prolonged). Finally, as in many chronic hemolytic conditions, ferritin may be increased, particularly for transfused patients or concomitant hemochromatosis genetic mutations, even in heterozygosity [Citation40].
Reticulocytes deserve some special consideration. They are immature erythrocytes released by the bone marrow to compensate anemia, and should be evaluated as absolute number, not percentage, which may be affected by the number/size of erythrocytes. Additionally, their number should be inversely related to hemoglobin levels/hematocrit, being the highest possible for severe anemia (good bone marrow compensation), and close to normal in mild forms. Reticulocytosis may be inadequate for a given hemoglobin level, possibly due to antibodies directed against erythroid precursors in bone marrow, or to a so-called shocked bone marrow (frequently in the context of a septic state); this occurs in about 20–40% of AIHA and is obviously associated with the most severe forms [Citation1,Citation42–45].
All the described hemolytic markers have several coexisting confounding conditions that may puzzle their interpretation [Citation40]. Underdiagnosed blood loss, vitamin/iron deficiency, renal/liver disease, and erythropoietin insufficiency may contribute to anemia. LDH is increased in several diseases that involve cellular necrosis or increased tissue turnover (myocardial infarction, heart failure, hepatitis, extreme muscular effort, and solid and hematologic tumors). Unconjugated bilirubin is typically increased in Gilbert’s syndrome, and mixed hyperbilirubinemia in liver disease. Haptoglobin, may be falsely normal/raised in inflammatory diseases, cigarette smokers, and nephritic syndrome possibly masking an underlying hemolysis; conversely, it may be reduced for liver disease, malnutrition, and congenital hypo-haptoglobinemia, without a hemolytic disease. Reticulocytes, that are not ‘truly’ hemolytic markers, may be increased in other causes of anemia requiring bone marrow compensation, such as hemorrhage, pregnancy/delivery and acclimatation. At variance a coexisting underdiagnosed bone marrow disease (myelodysplasia, aplasia, leukemia, tumors) may impair an adequate reticulocyte compensation.
7. Concomitant hemolytic conditions that may puzzle the diagnosis
Further to the latter bone marrow disease mentioned above, it is worth reminding the possible coexistence of other hemolytic conditions that may cause an overestimation of the immune-mediated hemolysis. The very long list and the diagnostic work-up is beyond the scope of this review and may be time-consuming, expensive a sometimes inconclusive. Small paroxysmal nocturnal hemoglobinuria (PNH) clones have been described in 37% of AIHA patients, and even the association of the two full diseases is reported [Citation46]. The several congenital membrane and enzyme defects, dyserythropoietic anemias, as well as hemoglobinopathies are associated with immune mediated hemolysis [Citation9]. In addition, all these chronic hemolytic anemias are frequently transfused/transfusion dependent, possibly developing acute or chronic delayed transfusion reactions of complicating the DAT determination for alloantibodies [Citation1–4]. Mechanical hemolysis (heart valve prosthesis, severe aortic stenosis, arteriovenous malformations), disseminated intravascular coagulation, hemolytic uremic syndrome, eclampsia, may challenge the differential diagnosis. Moreover, some serious infections (malaria, Clostridium sepsis, Babesia, Bartonella), poisons (snakes, spiders, acute copper poisoning, Wilson’s disease) may present with hemolytic features. Finally, the list of drug-associated hemolytic anemia is continuously adding new therapeuticals and should always be considered in the differential diagnosis or as a coexisting condition [Citation1–4,Citation47].
8. Clinical signs and symptoms that may guide or complicate the diagnosis
Several common/nonspecific symptoms are present in chronic hemolytic anemia, such as fatigue, pallor, dizziness, jaundice, gallstones, hepatosplenomegaly. Acute/severe hemolysis is marked by dyspnea, tachycardia, angina, hearth failure, and dark urine [Citation1–4,Citation48]. In CAD, typical signs/symptoms involve the circulatory district, including acrocyanosis, Raynaud-like symptoms, gangrene or ulcerations. These symptoms may mislead the diagnostic pathway, addressing consultations with angiologists, immunologists, rheumatologist with several investigations searching for vasculitides (systemic scleroderma, systemic lupus erythematosus) or even high-affinity hemoglobins/methemoglobinaemia for cyanosis [Citation25,Citation49]. Importantly, AIHAs have an increased risk of venous and arterial thromboses in various organs with consequent heterogeneous signs/symptoms. These complications occurring in up to 20% of cases are reported in the most severe cases, in those with prominent intravascular hemolysis, and in splenectomized patients, further complicating the diagnosis [Citation50–53]. Recently, low-molecular weight heparin prophylaxis has been recommended in these cases to prevent thrombotic events in lung, heart, brain, and other vital districts [Citation53]. Infections, either therapy-related or associated with an underlying underestimated congenital immunodeficiency, may further complicate the clinical picture, fueling hemolysis and impairing bone marrow compensation [Citation54]. Finally, it is worth reminding Evans Syndrome, the concomitant or subsequent association of AIHA with another autoimmune cytopenia, namely immune thrombocytopenia (ITP) or immune neutropenia, with consequent bleeding and infections that again complicate the clinical picture [Citation55].
9. Further investigation in primary versus secondary forms
Beyond hemolytic markers, vitamins and iron status, and liver and kidney tests, a plethora of examinations are useful to distinguish primary AIHAs, from those secondary to associated conditions. The latter may in fact require ad hoc therapeutic approach (i.e., substitutive IVIG in primary immunodeficiencies, anti-lymphoma agents in lymphoproliferative disorders, etc.) [Citation7,Citation16]. lists additional investigations and their indication throughout the diagnosis and management of AIHA. Serum electrophoresis, total IgG, IgA, and IgM levels, and immune fixation are useful to identify patients with associated plasma cell dyscrasias as well as primary immunodeficiencies. As possible confounders, CAD patients usually exhibit monoclonal IgM gammopathy, and treatment with rituximab or other immunosuppressants may lead to hypogammaglobulinemia. Organ and non-organ specific autoantibodies (ANA, ENA, anti-DNA, anti-thyroid ones, etc.), are suggested in patients displaying suggestive clinical features, including serositis, arthralgia, myalgia, photosensitivity, hypothyroidism, etc. These tests may aid to identify connective tissue diseases or autoimmune diseases associated with AIHA, that may require different treatments including cytotoxic immunosuppressants and specific biologic drugs. C3 and C4 complement fractions may be decreased in primary forms, particularly in CAD and in wAIHA with complement activation, but also in those associated with systemic autoimmune diseases as hallmark of broader immune activation [Citation1,Citation2,Citation7].
Table 1. Further investigation for the evaluation of primary versus secondary autoimmune hemolytic anemia (AIHA) cases. IVIG intravenous immunoglobulin; CAD cold agglutinin disease; wAIHA warm type AIHA.
As mentioned before, bone marrow compensation, as highlighted by reticulocytosis, is a major determinant of AIHA severity. The evaluation of endogenous erythropoietin levels is a useful test in patients with inadequate reticulocytosis and may prompt the administration of recombinant erythropoietin agents that may improve anemia in >70% of cases [Citation56,Citation57]. Bone marrow evaluation (morphology, cytometry, cytogenetics, and biopsy) has been recently advised in CAD at diagnosis and in wAIHA relapsed after or refractory to first-line therapy with steroids [Citation4,Citation25,Citation58]. Along with whole body CT scan, it is pivotal to diagnose AIHA associated with lymphoproliferative diseases, as well as to myelodysplasia or bone marrow failure syndromes. Moreover, the type of lymphocyte infiltrate (T or B) may be helpful to choose a better target-therapy [Citation59,Citation60]. CAD deserves a specific mention, since a clonal, low-grade lymphoproliferation distinct from lymphoplasmacytic lymphoma and marginal zone lymphoma, is almost invariably present [Citation25,Citation49,Citation52]. It has been currently included in the World Health Organization (WHO) classification of hematolymphoid tumors [Citation61]. Molecular analysis may be useful, since the MYD88 L265P mutation, typical of lymphoplasmacytic lymphoma/Waldenström’s macroglobulinemia, is usually negative in CAD, whilst KMT2D and CARD11 mutations have been described [Citation62,Citation63]. Further molecular analysis and next-generation sequencing would help to confirm associated conditions in selected cases (congenital anemias, immunodeficiencies, myelo- and lympho-proliferative disorders) [Citation64].
Finally, anti-phospholipid antibodies testing (lupus anticoagulant, anti-cardiolipin and anti-beta2 glycoprotein 1 antibodies) is advised in all patients, particularly in those with history of thrombosis/miscarriages since may reveal associated anti-phospholipid syndrome (APS) and/or prompt primary anti-thrombotic prophylaxis. At variance, full thrombophilia screening should not be routinely performed and limited to selected cases (i.e., recurrence of thrombosis, unusual sites, etc.) and after consultation with thrombosis experts [Citation4,Citation53]. Serologies for major and minor hepatitis viruses and quantiferon for TBC are indicated at diagnosis, since they may disclose associated chronic/acute infections and prompt the establishment of a specific anti-infectious prophylaxis before starting immunosuppression [Citation54]. summarizes the most common conditions associated with AIHA.
Table 2. Conditions associated with autoimmune hemolytic anemia (AIHA).
10. Conclusions
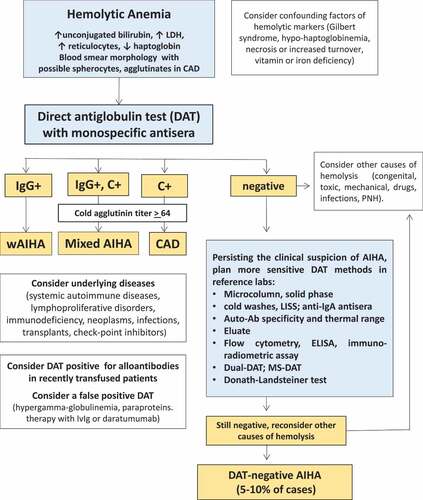
AIHA is a clinically heterogeneous disease posing several diagnostic challenges including the variable alteration of hemolytic markers and their several confounders; the latter may be overcome only by putting the disease in the clinical context and thanks to extensive laboratory testing (vitamins and iron status, and liver and kidney tests). The positivity of the direct Coombs test remains the gold standard for the diagnosis, however several methods are available and none with a 100% sensitivity/specificity. The use of tube test with monospecific anti-sera is recommended for the distinction of AIHA subtypes and is widely available. Possible falsely positive DAT should be considered in patients with plasma cell dyscrasias, and in those recently treated with IVIG or with daratumumab. For negative cases, if the clinical suspicion persists, more sensitive tests are microcolumn and solid phase DAT, cold washes, LISS, test for auto-Ab specificity and thermal range, and study of the eluate, which are available in most reference laboratories. More sophisticated techniques, including the Donath-Landsteiner test and the dual DAT should be considered under specific clinical suspect of PCH or warm IgM-driven AIHA, as well as flow cytometry, ELISA, immuno-radiometric assay, and MS-DAT that remain ‘experimental’ techniques. It should be however kept in mind that in DAT negative cases, the exclusion of other hemolytic conditions (i.e., congenital RBC membrane or enzyme defects, PNH, mechanical, microangiopathies, etc.) and the response to ex juvantibus steroids are required diagnostic criteria. In fact, more sensitive techniques show a progressive increase in sensitivity but a decrease in specificity being positive even in conditions other than AIHA, such as bone marrow failure syndromes. A comprehensive diagnostic flow-chart is shown in . The presence of alloantibodies by IAT should be evaluated since they may be difficult to diagnose and may cause life-threatening transfusion reactions. Finally, several additional tests, including serum electrophoresis, immune-fixation, organ and non-organ specific autoantibodies, anti-phospholipid antibodies, hepatitis viruses and TBC tests, bone marrow evaluation, and CT scan are useful to identify conditions associated with AIHA (i.e. immunodeficiencies, hematologic and solid tumors, autoimmune disorders, etc.) that may require ad hoc therapeutic approach or anti-thrombotic/anti-infectious prophylaxis.
Figure 3. Diagnostic algorithm of AIHA.
11. Expert opinion
Due to its rarity and heterogeneity, the diagnosis of AIHA may be challenging, particularly in relapsed/refractory cases for therapy-related confounders or in secondary forms for concomitant diseases. In this regard, the availability of new specific diagnostic tools and particularly molecular testing may be of great utility in identifying underlying conditions or complex cases with associated hemolytic disorders (congenital or acquired) that may puzzle the diagnosis. In fact, NGS panels are progressively implemented and will be further expanded in the next future, allowing the diagnosis of several congenital hemolytic anemias, such as hemoglobinopathies, erythrocyte membrane or enzyme defects, and congenital dyserythropoietic anemias. All these conditions, although rarely, may coexist with AIHAs, and/or may challenge the diagnosis. Additionally, NGS analysis will be extensively used for somatic mutations that contribute to define lymphoproliferative diseases, as well as bone marrow failure syndromes and myeloid neoplasms. Again, all these conditions may coexist with AIHAs and/or complicate the diagnosis, particularly in transfused patients. Finally, the increasing number of AIHAs observed after bone marrow transplant, or new therapies with CAR-T and checkpoint inhibitors, will reinforce the need for a comprehensive diagnostic package along with a deep interpretative knowledge of the results.
The truly unmet need is DAT-negative AIHA where a reference laboratory with a comprehensive diagnostic workup is fundamental to unravel the diagnosis, at least after the exclusion, as well as possible, of the other causes of hemolysis. In fact, while first line ex-adiuvantibus therapy with steroids is generally administered without particular concern, second-line therapy (splenectomy, rituximab, cytotoxic drugs) may be troublesome in DAT-negative AIHAs. In these cases, any further non-routinely investigations (flow-cytometry, ELISA, MS-DAT) may be of great value in avoiding delayed diagnosis and inappropriate therapies and should be strongly pursued in reference laboratories. In this context, a ‘hub’ and ‘spoke’ organization is advisable, as experimental techniques require validation and implementation to ameliorate their sensitivity and, more importantly, their specificity. Thanks to these new methods, the percentage of DAT-negative AIHAs has decreased in recent years, although difficult cases are still a challenge for reference laboratories. Overall, a tight collaboration and continuous dialogue between clinicians and immune-hematologic laboratory experts is warranted to offer the best diagnosis and therapy for atypical cases of AIHA. The latter, such as mixed forms, PCH, and DAT negative AIHAs, represent an unmet need for new therapies under investigation in clinical trials that are specifically designed for wAIHA or CAD. In the former, several drugs are actively investigated in relapsed forms, and include spleen tyrosine kinase inhibitors (fostamatinib, sovleplenib), Bruton tyrosine kinase inhibitors (ibrutinib, rilzabrutinib) phosphatidylinositol 3-kinase delta inhibitor (parsaclisib), neonatal Fc-receptor inhibitors (nipocalimab, batoclimab), and several others (obexelimab, ianalumab). In CAD complement inhibitors represent a new therapeutic option, and encompass sutimlimab, pegcetacoplan, Iptacopan, and SAR445088.
It is difficult to assess how the advances/research discussed here may impact real-world outcomes in terms of diagnosis, drug utilization and their effectiveness, implementation of treatment guidelines, and economic impact on health care resource utilization. Certainly, patients will benefit from a proper diagnosis, but therapies for difficult/atypical cases are still lacking, along with dedicated guidelines. The rarity of these forms is undoubtedly a critical factor to collect information and design ad hoc trials. Moreover, the identification of ‘hub’ reference laboratories is not easy, requires specialized and dedicated personnel and strong commitment on the study and clinical follow up of these rare diseases. The availability of new drugs in the next future, and more importantly, their clinical use in the real-world clinical practice will certainly increase the interest of clinicians for AIHAs, with a consequent increase of economic resources for dedicated diagnostic and research laboratories. Finally, continuous education of medical students and clinicians is fundamental to raise awareness about the need of a proper diagnosis of AIHAs.
Article highlights
The direct antiglobulin test (DAT) or Coombs is the cornerstone of the diagnosis of autoimmune hemolytic anemia (AIHA) and can be performed by several methods with different sensitivity and specificity.
The tube test with monospecific antisera, microcolumn and solid phase methods are routinely accessible in most laboratories. Additional investigation should be strongly pursued in reference laboratories to diagnose DAT-negative AIHAs that represent a clinical challenge.
The correct interpretation of hemolytic markers, the infectious and thrombotic complications of AIHAs, and the possible underlying conditions should be carefully considered.
A continuous dialogue between clinicians and immune-hematologic laboratory experts is fundamental to offer the best diagnosis and therapy for AIHAs.
Declaration of interest
W Barcellini has received consultancy and speaker bureau honoraria from Agios, Alexion, Novartis, Sanofi, and Sobi. B Fattizzo has received consultancy and speaker bureau honoraria from Amgen, Alexion, Annexon, Novartis, and Sobi.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Petz LD, Garratty G. Immune hemolytic anemias. 2nd ed. Philadelphia: Churchill Livingstone; 2004.
- Berentsen S, Barcellini W, Longo DL. Autoimmune Hemolytic Anemias. N Engl J Med. 2021 Oct 7;385(15):1407–1419. DOI:10.1056/NEJMra2033982
- Barcellini W, Zaninoni A, Giannotta JA, et al. New insights in autoimmune hemolytic anemia: from pathogenesis to therapy stage 1. J Clin Med. 2020;9(12):3859. DOI:10.3390/jcm9123859
- Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev. 2020;41:100648.
- Shulman IA, Branch DR, Nelson JM, et al. Autoimmune hemolytic anemia with both cold and warm autoantibodies. JAMA. 1985;253(12):1746–1748. DOI:10.1001/jama.1985.03350360072021
- Sokol RJ, Booker DJ, Stamps R, et al. Direct coombs test-negative autoimmune hemolytic anemia and low-affinity IgG class antibodies. Immunohematology. 1997;13(4):115–118. DOI:10.21307/immunohematology-2019-725
- Barcellini W, Giannotta J, Fattizzo B. Autoimmune hemolytic anemia in adults: primary risk factors and diagnostic procedures. Expert Rev Hematol. 2020;13(6):585–597.
- Feuille EJ, Anooshiravani N, Sullivan KE, et al. Autoimmune cytopenias and associated conditions in CVID: a report from the USIDNET registry. J Clin Immunol. 2018;38(1):28–34. DOI:10.1007/s10875-017-0456-9
- Motta I, Giannotta J, Ferraresi M, et al. Autoimmune hemolytic anemia as a complication of congenital anemias. a case series and review of the literature. J Clin Med. 2021;10(15):3439. DOI:10.3390/jcm10153439
- Gormezano NW, Kern D, Pereira OL, et al. Autoimmune hemolytic anemia in systemic lupus erythematosus at diagnosis: differences between pediatric and adult patients. Lupus. 2017;26(4):426–430. DOI:10.1177/0961203316676379
- Nenova IS, Valcheva MY, Beleva EA, et al. Autoimmune phenomena in patients with solid tumors. Folia Med (Plovdiv). 2016;58(3):195–199. DOI:10.1515/folmed-2016-0026
- Barcellini W, Giannotta JA, Fattizzo B. Autoimmune complications in hematologic neoplasms. Cancers (Basel). 2021;13(7):1532.
- Stein B, DeCredico N, Hillman L. Evaluation of the Direct Antiglobulin Test (DAT) in the setting of mycoplasma pneumoniae Infection. JAMA. 2018;319(13):1377–1378.
- Tanios GE, Doley PB, Munker R. Autoimmune hemolytic anemia associated with the use of immune checkpoint inhibitors for cancer: 68 cases from the food and drug administration database and review. Eur J Haematol. 2019;102(2):157–162.
- Azizi G, Tavakol M, Rafiemanesh H, et al. Autoimmunity in a cohort of 471 patients with primary antibody deficiencies. Expert Rev Clin Immunol. 2017;13(11):1099–1106. DOI:10.1080/1744666X.2017.1384312
- Fattizzo B, Barcellini W. Autoimmune hemolytic anemia: causes and consequences. Expert Rev Clin Immunol. 2022 Jul;18(7):731–745.
- Coombs RR, Mourant AE, Race RR. A new test for the detection of weak and incomplete Rh agglutinins. Br J Exp Pathol. 1945;26:255–266.
- LE S, Cunningham MJ. Autoimmune hemolytic anemias. In: Hillyer C, LE S, Ness P, Anderson K Roback J, editors. Blood banking and transfusion medicine: basic principles and practice. 2nd ed. Philadelphia: Churchill Livingstone; 2007. p. 557–570.
- Roback JD, Combs MR, Grossman BJ, et al. AABB Technical Manual. 16th ed. Bethesda: AABB; 2008.
- Barcellini W. Pitfalls in the diagnosis of autoimmune haemolytic anaemia. Blood Transfus. 2015;13(1):3–5.
- Parker V, Tormey CA. The direct antiglobulin test: indications, interpretation, and pitfalls. Arch Pathol Lab Med. 2017;141(2):305–310.
- Dubarry M, Charron C, Habibi B, et al. Quantitation of immunoglobulin classes and subclasses of autoantibodies bound to red cells in patients with and without hemolysis. Transfusion. 1993;33(6):466–471.
- Branch DR, Petz LD. Detecting alloantibodies in patients with autoantibodies. PMID: 9920160. Transfusion. 1999 Jan;39:6–10
- Hannon JL. Management of blood donors and blood donations from individuals found to have a positive direct antiglobulin test. Transfus Med Rev. 2012;26(2):142–152.
- Berentsen S. New insights in the pathogenesis and therapy of cold agglutinin-mediated autoimmune hemolytic anemia. Front Immunol. 2020;11:590.
- Donath J, Landsteiner K. Ueber Paroxysmale Hämoglobinurie. Münchener medizinische Wochenschrift. 1904;36:1590–1593.
- Kilty M, Ipe TS. Donath-Landsteiner test. Immunohematology. 2019;35(1):3–6.
- Dittmar K, Procter JL, Cipolone K, et al. Comparison of DATs using traditional tube agglutination to gel column and affinity column procedures. Transfusion. 2001;41(10):1258–1262.
- Novaretti MC, Jens E, Pagliarini T, et al. Comparison of conventional tube test technique and gel microcolumn assay for direct antiglobulin test: a large study. J Clin Lab Anal. 2004;18(5):255–258.
- Chaudhary R, Das SS, Gupta R, et al. Application of flow cytometry in detection of red-cell-bound IgG in Coombs-negative AIHA. Hematology. 2006;11(4):295–300.
- Fayek MH, Saad AA, Eissa DG, et al. Role of gel test and flow cytometry in diagnosis of Coombs’ negative autoimmune haemolytic anaemia. Int J Lab Hematol. 2012;34(3):311–319.
- Kamesaki T, Oyamada T, Omine M, et al. Cut-off value of red-blood-cell-bound IgG for the diagnosis of Coombs-negative autoimmune hemolytic anemia. Am J Hematol. 2009;84(2):98–101.
- Fabijańska-Mitek J, Pogłod R, Adamowicz-Salach A, et al. Quantitation of red cell-bound IgG by an enzyme-linked antiglobulin test in the patients with warm-type autoimmune haemolytic anaemia. Clin Lab Haematol. 2006;28(4):241–244.
- Bartolmäs T, Salama A. A dual antiglobulin test for the detection of weak or nonagglutinating immunoglobulin M warm autoantibodies. Transfusion. 2010;50(5):1131–1134.
- Arndt PA, Leger RM, Garratty G. Serologic findings in autoimmune hemolytic anemia associated with immunoglobulin M warm autoantibodies. Transfusion. 2009;49(2):235–242.
- Barcellini W, Clerici G, Montesano R, et al. In vitro quantification of anti-red blood cell antibody production in idiopathic autoimmune haemolytic anaemia: effect of mitogen and cytokine stimulation. Br J Haematol. 2000;111(2):452–460. DOI:10.1046/j.1365-2141.2000.02380.x
- Barcellini W, Revelli N, Imperiali FG, et al. Comparison of traditional methods and mitogen-stimulated direct antiglobulin test for detection of anti-red blood cell autoimmunity. Int J Hematol. 2010;91(5):762–769. DOI:10.1007/s12185-010-0578-9
- Barcellini W, Montesano R, Clerici G, et al. In vitro production of anti-RBC antibodies and cytokines in chronic lymphocytic leukemia. Am J Hematol. 2002;71(3):177–183. DOI:10.1002/ajh.10210
- Barcellini W, Iurlo A, Radice T, et al. Increased prevalence of autoimmune phenomena in myelofibrosis: relationship with clinical and morphological characteristics, and with immunoregulatory cytokine patterns. Leuk Res. 2013;37(11):1509–1515. DOI:10.1016/j.leukres.2013.09.001
- Barcellini W, Fattizzo B. Clinical applications of hemolytic markers in the differential diagnosis and management of hemolytic anemia. Dis Markers. 2015;2015:635670.
- Barcellini W, Fattizzo B. How I treat warm autoimmune hemolytic anemia. Blood. 2021;137(10):1283–1294.
- Van De Loosdrecht AA, Hendriks DW, Blom NR, et al. Excessive apoptosis of bone marrow erythroblasts in a patient with autoimmune haemolytic anaemia with reticulocytopenia. Br J Haematol. 2000;108(2):313–315.
- Hauke G, Fauser AA, Weber S, et al. Reticulocytopenia in severe autoimmune hemolytic anemia (AIHA) of the warm antibody type. Blut. 1983;46(6):321–327.
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014;124(19):2930–2936. DOI:10.1182/blood-2014-06-583021
- Barcellini W, Zaninoni A, Fattizzo B, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol. 2018;93(9):E243–246. DOI:10.1002/ajh.25212
- Fattizzo B, Giannotta J, Zaninoni A, et al. Small paroxysmal nocturnal hemoglobinuria clones in autoimmune hemolytic anemia: clinical implications and different cytokine patterns in positive and negative patients. Front Immunol. 2020;11:1006.
- Hill QA, Stamps R, Massey E, et al. British society for haematology guidelines. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017;177(2):208–220.
- Fattizzo B, Giannotta JA, Serpenti F, et al. Difficult cases of autoimmune hemolytic anemia: a challenge for the internal medicine specialist. J Clin Med. 2020;9(12):3858.
- Berentsen S. How I treat cold agglutinin disease. Blood. 2021;137(10):1295–1303.
- Audia S, Bach B, Samson M, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. PLoS ONE. 2018;13(11):e0207218. DOI:10.1371/journal.pone.0207218
- Broome CM, Cunningham JM, Mullins M, et al. Increased risk of thrombotic events in cold agglutinin disease: a 10-year retrospective analysis. Res Pract Thromb Haemost. 2020;4(4):628–635. DOI:10.1002/rth2.12333
- Berentsen S, Barcellini W, D’Sa S, et al. Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood. 2020;136(4):480–488. DOI:10.1182/blood.2020005674
- Fattizzo B, Bortolotti M, Giannotta JA, et al. Intravascular hemolysis and multitreatment predict thrombosis in patients with autoimmune hemolytic anemia. J Thromb Haemost. 2022;20(8):1852–1858. DOI:10.1111/jth.15757
- Giannotta JA, Fattizzo B, Cavallaro F, et al. Infectious complications in autoimmune hemolytic anemia. J Clin Med. 2021;10(1):164. DOI:10.3390/jcm10010164
- Fattizzo B, Michel M, Giannotta JA, et al. Evans syndrome in adults: an observational multicenter study. Blood Adv. 2021;5(24):5468–5478. DOI:10.1182/bloodadvances.2021005610
- Fattizzo B, Michel M, Zaninoni A, et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: a multicenter international study. Haematologica. 2021;106(2):622–625. DOI:10.3324/haematol.2020.250522
- Salama A, Hartnack D, Lindemann HW, et al. The effect of erythropoiesis-stimulating agents in patients with therapy-refractory autoimmune hemolytic anemia. Transfus Med Hemother. 2014;41(6):462–468.
- Barcellini W, Fattizzo B. The changing landscape of autoimmune hemolytic anemia. Front Immunol. 2020;11:946.
- Fattizzo B, Zaninoni A, Gianelli U, et al. Prognostic impact of bone marrow fibrosis and dyserythropoiesis in autoimmune hemolytic anemia. Am J Hematol. 2018 Aug;93(4):E88–91.
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical evolution of autoimmune cytopenias to idiopathic cytopenias/dysplasias of uncertain significance (ICUS/IDUS) and bone marrow failure syndromes. Am J Hematol. 2017;92(3):E26–9. DOI:10.1002/ajh.24618
- Campo E, Jaffe ES, Cook JR, et al. The international consensus classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood. 2022;140(11):1229–1253. DOI:10.1182/blood.2022015851
- Randen U, Trøen G, Tierens A, et al. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica. 2014;99(3):497–504. DOI:10.3324/haematol.2013.091702
- Małecka A, Trøen G, Tierens A, et al. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. 2018;183(5):838–842. DOI:10.1111/bjh.15063
- Fattizzo B, Barcellini W. Autoimmune cytopenias in chronic lymphocytic leukemia: focus on molecular aspects. Front Oncol. 2020;9:1435.