
ABSTRACT
Aim
To determine the immunogenicity, safety, and efficacy of rurioctocog alfa pegol in previously untreated patients (PUPs) with severe hemophilia A (HA).
Methods
This prospective, phase 3 study (NCT02615691) was conducted in PUPs, or patients with ≤2 exposure days (EDs) prior to screening, aged <6 years with severe HA. The primary endpoint was incidence of factor VIII (FVIII) inhibitor development. This protocol-specified interim analysis was conducted after 50 patients had completed ≥50 EDs without developing FVIII inhibitors or had developed a confirmed inhibitor at any time.
Results
Of the enrolled patients, 59/80 (73.8%) received ≥1 dose of rurioctocog alfa pegol; 54 received prophylaxis, and 35 on-demand treatment. Incidence of inhibitor development was 0.19 (10/52). Total annualized bleeding rate (95% CIs) was 3.2 (2.0–5.0) for patients receiving prophylaxis and 3.2 (1.6–6.3) for on-demand treatment. Hemostatic efficacy of most bleedings was rated as ‘excellent’ or ‘good’ after 24 hours (122/131 [93.1%]) and at resolution (161/170 [94.7%]). Five patients received ≥1 dose of rurioctocog alfa pegol for immune tolerance induction (ITI) and 1 patient was defined as having ITI success. Thirteen patients experienced 14 treatment-related adverse events, including 10 cases of FVIII inhibitor development.
Conclusion
This is the first prospective study of rurioctocog alfa pegol for the treatment of PUPs with severe HA.
Trial Registration
This trial is registered at ClinicalTrials.gov (CT.gov identifier: NCT02615691).
1. Introduction
Hemophilia A is a congenital recessive X-linked disorder characterized by deficiency or absence of clotting factor VIII (FVIII), leading to frequent, acute, and prolonged spontaneous or traumatic bleeding events [Citation1]. The management of hemophilia A includes replacement of FVIII through intravenous injections of recombinant or plasma-derived FVIII, to achieve adequate hemostasis [Citation2–5], in addition to the use of non-factor therapies such as emicizumab [Citation3]. Patients can be treated on-demand, defined as episodic replacement therapy only at the time of a clinically evident bleed, or prophylactically, defined as regular administration of therapeutic products to maintain hemostasis [Citation4]. The current standard of care for severe hemophilia A (FVIII ≤1%) includes prophylaxis with FVIII to prevent bleeds, target joint development, and hemophilic arthropathy [Citation3].
Development of an inhibitory antibody to exogenous FVIII is the most serious complication in the management of hemophilia A. Anti-FVIII neutralizing antibodies inhibit the activity of FVIII, potentially rendering FVIII concentrate treatment ineffective [Citation6,Citation7]. FVIII inhibitors develop in 20–35% of previously untreated patients (PUPs) with severe hemophilia A [Citation8]. Inhibitors typically develop soon after initial exposure to exogenous FVIII, usually within the first 50 exposure days (EDs), and particularly in the first 20 EDs [Citation7]. A recent study found a median of 11 EDs to inhibitor development [Citation9,Citation10]. Patients who develop FVIII inhibitors can have substantially increased morbidity due to higher bleeding rates [Citation11], as well as an increased risk of death [Citation12]. In addition, patients may experience decreased quality of life and an increased cost of care [Citation11,Citation13]. Various risk factors have been associated with the development of FVIII inhibitors, including disease severity, genetic factors, treatment regimen, and the type of FVIII replacement therapy used [Citation7,Citation14]. Immune tolerance induction (ITI), involving the frequent infusion of FVIII to induce FVIII antigen-specific tolerance, is the only proven method for the eradication of FVIII inhibitors [Citation15].
Rurioctocog alfa pegol is a recombinant FVIII protein covalently bound to 20 kDa polyethylene glycol (PEG) chains intended for FVIII replacement therapy. In the US, rurioctocog alfa pegol (Adynovate®; Baxalta US Inc., a Takeda company, Lexington, MA, USA) is indicated in children and adults with hemophilia A for the on-demand treatment and control of bleeding episodes, perioperative management, and routine prophylaxis to reduce the frequency of bleeding episodes. In Europe, rurioctocog alfa pegol (Adynovi™; Baxalta Innovations GmbH, a Takeda company, Vienna, Austria) is indicated for the treatment and prophylaxis of bleeding in patients ≥12 years with hemophilia A. PEGylation of FVIII prolongs its half-life, allowing clinicians to use more flexible treatment regimens. The use of extended half-life recombinant FVIII treatments, such as rurioctocog alfa pegol, gives clinicians the option to reduce the frequency of treatment administration, or maintain infusion frequency whilst increasing residual FVIII activity, without compromising therapeutic benefit.
Post-translational modifications, such as PEGylation, can influence a protein’s biological properties, which may have an impact on immunogenicity [Citation16]. In addition, although free PEG molecules have been shown to be non-immunogenic, the conjugation of PEG in PEGylated drugs could induce an immune response. The presence of anti-PEG immunoglobulin (Ig)G and IgM may also impact efficacy due to drug clearance [Citation7,Citation17].
Identifying and understanding the immunogenicity of FVIII products has become an extremely important research question in the field of hematology in recent decades. This ongoing study aims to assess the immunogenicity, safety, and efficacy of rurioctocog alfa pegol in PUPs with severe hemophilia A and to evaluate the efficacy and safety of ITI with rurioctocog alfa pegol in patients who develop FVIII inhibitors. This analysis reports data from a prespecified interim analysis (IA) conducted after 50 patients had completed ≥50 EDs without developing an inhibitor or had developed a confirmed inhibitor at any time.
2. Patients and methods
2.1. Study design and patient population
This is an ongoing phase 3, prospective, uncontrolled, open-label, multicenter study (NCT02615691) investigating the immunogenicity, safety, and efficacy of rurioctocog alfa pegol in ≥100 evaluable PUPs aged <6 years with severe hemophilia A (baseline FVIII level <1%). During this ongoing study, patients receive on-demand and/or prophylactic therapy with rurioctocog alfa pegol for ≥100 EDs (defined as the unique calendar days that the patient received treatment), or until they develop a confirmed FVIII inhibitor. Study enrollment commenced in November 2015 and active recruitment has now completed. The overall primary completion date for the study is estimated for October 2024.
To qualify for enrollment, patients must have had ≤2 EDs to octocog alfa (parent molecule), rurioctocog alfa, or plasma transfusion (fresh frozen plasma) and no detectable, or history of, FVIII inhibitory antibodies (≥0.6 BU/mL using the Nijmegen modification of the Bethesda assay [Citation18]). These patients were considered as PUPs for the purpose of this study. Patients with previous exposure to any other commercially available FVIII concentrate were not eligible for participation. Other exclusion criteria were the diagnosis of any inherited/acquired hemostatic disorder other than hemophilia A; known hypersensitivity toward mouse or hamster proteins, PEG, or Tween 80; current or recent (<30 days) use of other PEGylated drugs prior to study participation; concomitant treatment with systemic immunomodulating drugs; severe renal impairment; and current participation in other interventional clinical studies or ≤30 days before enrollment. Patients enrolled in the study complete study visits to undergo safety, immunogenicity, and hemostatic efficacy assessments at baseline, after every 5 ± 1 EDs until 20 ± 2 EDs (Visits 1–4), after a further 10 ± 3 EDs (Visits 5 and 6) and at 50–55 EDs (Visit 7), 75 ± 5 EDs (Visit 8); and the Study Completion/Termination Visit for follow-up at 100–110 EDs. The planned duration of this first part of the study is 5 years (enrollment approximately 3 years, treatment approximately 2 years, depending on the type of treatment).
Patients who develop either high-titer FVIII inhibitors (>5.0 BU/mL) or low-titer inhibitors (≥0.6 to ≤5.0 BU/mL) plus poorly controlled bleeding despite increasing FVIII doses and/or bypassing agents during the study are eligible for ITI. In the ITI part of this study, patients complete a baseline visit, Visit 1 at Week 2 ± 2 days, Visit 2 at Week 4 ± 2 days, and subsequent visits every month ±1 week. The planned duration of patient participation in the investigation of ITI is up to 3.5 years (until immune tolerance success, failure, or a maximum of 33 months, whichever occurs first). In case of success, this includes 5–6 additional months for transitioning to twice-weekly prophylaxis, including a 3-month follow-up period.
The study is conducted in compliance with the International Council for Harmonisation Good Clinical Practice guidelines and the Declaration of Helsinki, as well as other applicable national and local ethical and legal requirements. Parents/legally authorized representative provided written assent before patients entered the study. The protocol, final approved consent/assent document, relevant supporting material, and all patient recruitment information were reviewed and approved by the relevant institutional review boards before study initiation.
2.2. Study treatment
Rurioctocog alfa pegol was administered at baseline and at all study visits except for Visit 1 (5 ± 1 EDs before visit), Visit 3 (15 ± 1 EDs before visit), and Visit 5 (30 ± 3 EDs before visit), during which administration was optional. Patients received intravenous rurioctocog alfa pegol as prophylaxis (25–50 IU/kg, up to 80 IU/kg ≥1× weekly) and/or on-demand therapy (10–50 IU/kg, up to 80 IU/kg depending on bleed severity). The frequency and dosing of treatment depended on each patient’s clinical situation and were at the investigator’s discretion. Patients who started the study receiving on-demand rurioctocog alfa pegol eventually switched to prophylactic treatment during the study. Prophylaxis had to be initiated before 3 years of age or after ≤2 joint bleeds, whichever occurred first.
Patients who developed FVIII inhibitors and subsequently enrolled in ITI received either a high-dose regimen of 100–200 IU/kg intravenous rurioctocog alfa pegol daily, or a low-dose regimen of 50 IU/kg 3× weekly at the discretion of the investigator. Patients who received treatment with any FVIII concentrate other than rurioctocog alfa pegol were discontinued from the study.
2.3. Outcome measures
The primary objective is to determine the safety, in terms of the immunogenicity, of rurioctocog alfa pegol based on the incidence of inhibitor development to FVIII (≥0.6 BU/mL using the Nijmegen modification of the Bethesda assay [Citation18]). Development of FVIII inhibitors was assessed at all study visits and was determined by a central laboratory and confirmed by a second blood sample drawn within 2 weeks of site notification of an inhibitor. However, to ensure timely availability of FVIII inhibitor results, the clinical management of the patient for ITI could be based on results generated at the local laboratory.
Secondary endpoints include the efficacy of prophylactic treatment with rurioctocog alfa pegol and on-demand treatment for the control of bleeds. Annualized bleeding rate (ABR) was assessed based on individual bleeds. Bleeds were categorized as spontaneous (definitely not trauma-related) or injury-related (definitely due to injury/trauma). Overall hemostatic efficacy rating at 24 hours after treatment initiation and at resolution of bleed were also assessed, with the patient or caregiver rating severity of bleeds (minor, moderate, or major/life-threatening) and overall treatment response using a 4-point efficacy scale. Efficacy was rated as: Excellent (full relief of pain and cessation of objective signs of bleeding after a single infusion with no additional infusion required for the control of bleeding), good (definite pain relief and/or improvement in signs of bleeding after a single infusion. Possibly requires >1 infusion for complete resolution), fair (probable and/or slight relief of pain and slight improvement in signs of bleeding after a single infusion. Required >1 infusion for complete resolution), and none (no improvement or condition worsens). The number of infusions required for treatment of bleeds, weight-adjusted consumption of rurioctocog alfa pegol, and efficacy of rurioctocog alfa pegol for perioperative management were also investigated. Safety outcomes include the development of binding IgG and IgM antibodies to FVIII, PEG-FVIII, and PEG, assessed at all study visits [Citation19,Citation20]. Adverse events (AEs) and serious AEs (SAEs) were recorded for all patients receiving rurioctocog alfa pegol throughout the study. Incremental recovery (IR) of rurioctocog alfa pegol was assessed at baseline and throughout the study by non-compartmental analyses using Pharsight WinNonlin 6.3 (Pharsight Corporation [2012]: Pharsight WinNonLin 6.3, St. Louis, MO, USA). FVIII IR was determined at baseline and at all study visits except for Visit 1 (5 ± 1 EDs before visit), Visit 3 (15 ± 1 EDs before visit), and Visit 5 (30 ± 3 EDs before visit), when IR determination was optional. The FVIII assays used were the 1-stage clotting FVIII activity and FVIII chromogenic activity.
Additional outcome measures for patients who received ITI with rurioctocog alfa pegol include the success rate of ITI. The categories were considered as success (inhibitor titer persistently <0.6 BU/mL, FVIII IR ≥66% of baseline following 84- to 96-hour washout, and FVIII half-life ≥6 hours), partial success (two of the aforementioned criteria must be met after 33 months of ITI), or failure (failure to meet any criteria within 33 months of ITI therapy or <20% reduction in inhibitor titer relative to peak inhibitor titer over any 6-month period after the first 3 months of treatment) [Citation21]. Success factors varied based on the specific protocol version. To comply with European Medicine Agency regulatory requirements, FVIII half-life of ≥6 hours was not evaluated in patients enrolled from the European Union. The rate of partial success and failure of ITI, ABR, and weight-adjusted consumption of rurioctocog alfa pegol in the ITI population were also assessed.
2.4. Statistical analysis
This prespecified IA was conducted after 50 patients had completed ≥50 EDs without developing an inhibitor or had developed a confirmed inhibitor at any time; the ED cutoff was selected because FVIII inhibitors generally develop within this time [Citation9,Citation10,Citation22,Citation23]. Demographic and baseline characteristics were summarized for the safety analysis set (SAS), comprising all patients who received ≥1 dose of rurioctocog alfa pegol. Continuous variables were reported as mean and standard deviation (SD) and categorical variables as number and percentage. The incidence of FVIII inhibitor development was assessed by computing the Clopper-Pearson exact 95% confidence interval (CI) for the proportion of patients who developed inhibitors during the study. ABR was analyzed by point and interval estimates derived by SAS procedure GLIMMIX (SAS Institute Inc. 2020. SAS/STAT® 15.2 User’s Guide. Cary, NC, USA: SAS Institute Inc.) from a negative binomial model with treatment regimen as a covariate and the logarithm of the duration of the regimen as an offset. Descriptive statistics were given for weight-adjusted consumption of rurioctocog alfa pegol per month, per year, and per bleeding/surgery event and the number of infusions per month and per year. Missing data were not imputed, and all analyses were performed using non-missing data.
3. Results
3.1. Patient disposition
As of the data cutoff date (30 August 2019), 80 patients were enrolled from 44 study sites in 15 countries. Of these patients, 59 received ≥1 dose of rurioctocog alfa pegol and were included in the SAS for this prespecified IA (); their baseline demographics and clinical characteristics are shown in . A hemophilia-associated gene mutation was identified in 54/59 patients (91.5%). Large deletions, inversions (introns 1 and 22), and substitution nonsense mutations were present in 29/59 patients (49.2%); small deletions, small duplications, and substitution-missense mutations were present in 21/59 patients (35.6%). Inversion of intron 22 (22/59; 37.3%), small duplication, and substitution-missense mutations (8/59; 13.6% each) were the most common gene mutations leading to severe hemophilia A ().
Figure 1. Patient disposition. Of the 18 patients who did not meet the eligibility criteria, 1 was rescreened and entered with a different patient ID. Therefore, this patient was counted twice. FVIII, factor VIII; ITI, immune tolerance induction.
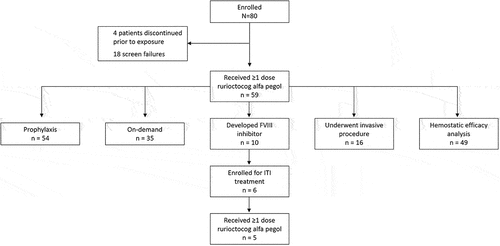
Table 1. Patient demographics and baseline characteristics.
3.2. Immunogenicity
Of the 59 patients included in the SAS, 52 patients qualified for evaluation of the primary endpoint in this IA. Positive anti-FVIII inhibitor titers were reported for 10 of these patients, resulting in an incidence (95% CI) of inhibitor development of 0.19 (0.10–0.33). Of these 10 patients, 4 (40.0%) were receiving on-demand treatment and 6 (60.0%) were receiving prophylaxis. Three of the 10 patients who developed FVIII inhibitors had 2 prior EDs to octocog alfa and 1 patient had 1 ED to fresh frozen plasma before entering the study. Mean (SD) EDs until confirmed inhibitor development was 8.0 (4.0) days. High-titer inhibitors were present in 5 patients; 5 had low-titer inhibitors. The highest inhibitor titer in patients with high-titer inhibitors ranged from 7.3 to 2004.1 BU/mL, and from 1.1 to 4.0 BU/mL in patients with low-titer inhibitors.
summarizes patients who developed ≥1 positive post-baseline binding antibody during the study. The number of positive results for FVIII, PEG-FVIII, and PEG binding antibodies increased in the first year following rurioctocog alfa pegol exposure and then declined by the second year.
Table 2. Patients receiving rurioctocog alfa pegol with at least 1 positive post-baseline binding IgG and IgM antibody to FVIII, PEG-FVIII, and PEG.
3.3. Efficacy
The 59 patients included in the SAS had 4543 EDs over a total observation period of 82.2 years. The mean (SD) total EDs to rurioctocog alfa pegol was 76.2 (42.4) days. Of these patients, 54 received prophylaxis with rurioctocog alfa pegol and these patients had 3789 EDs over a total observation period of 77.2 years, with mean (SD) EDs for rurioctocog alfa pegol prophylaxis of 70.2 (35.1) days. There were 35 patients who received on-demand treatment and received rurioctocog alfa pegol to treat bleeds (including patients who subsequently switched to prophylaxis).
During the first part of the study (prior to FVIII inhibitor detection and commencement of ITI therapy), 412 bleeds occurred; 269 of these bleeds (occurring in 47 patients) were treated with rurioctocog alfa pegol. The remaining 143 bleeds were not treated as they were mostly superficial skin bleeds (108 minor, 24 moderate, 2 major, 9 not reported). Of the total treated bleeds, 59/269 (21.9%) occurring in 18 patients were spontaneous, 172/269 (63.9%) in 41 patients were injury-related, and 37/269 (13.8%) in 15 patients were of unknown causality. The categorization for 1 bleed was not reported. Altogether, 59 joint bleeds were experienced by 22 patients. Of the treated bleeds, 139 were of minor severity, 107 moderate severity, and 22 major severity. Patients receiving prophylaxis with rurioctocog alfa pegol experienced 247 bleeds and 165 bleeds occurred in patients treated on-demand.
The total ABR using the negative binomial model (95% CIs) for patients receiving prophylaxis with rurioctocog alfa pegol was 3.2 (2.0–5.0) compared with 3.2 (1.6–6.3) for patients receiving on-demand treatment. The annualized joint bleeding rate was 0.3 (0.2–0.6), the spontaneous bleeding rate was 1.0 (0.4–2.7), and the injury-related bleeding rate was 2.1 (1.5–2.9) for patients receiving prophylaxis compared with 0.8 (0.3–2.1), 3.1 (1.0–10.0), and 1.6 (1.1–2.5) for those receiving on-demand treatment, respectively.
Hemostatic efficacy of treatment as rated by patients, or their caregivers is shown in . Where efficacy was reported, it was mostly rated as ‘excellent’ or ‘good’ after 24 hours (122/131 [93.1%]) and at resolution (161/170 [94.7%]) for all bleeds. Patients did not report an efficacy rating at 24 hours after first infusion for 88/269 (32.7%) bleeds, and for 99/269 (36.8%) bleeds at bleed resolution. In addition, 50 bleeds were not rated at any point. Most of these unrated bleeds occurred in a small number of patients. A further 50 bleeds that resolved within 24 hours after infusion were only rated at bleed resolution.
Table 3. Hemostatic efficacy of rurioctocog alfa pegol in prophylactic treatment and control of bleeds.
3.4. Consumption
Most bleeding events were treated with a single infusion of rurioctocog alfa pegol (209/269 [77.7%]). Two infusions were required for 38 bleeds (14.1%), 3 for 16 bleeds (5.9%), 4 for 3 bleeds (1.1%), and >4 for 2 bleeds (0.7%). The mean (SD) average dose per infusion to treat a bleed was 45.7 (14.6) IU/kg (SAS; n = 59).
The weight-adjusted exposure data for the patients who received a prophylaxis regimen are presented in .
Table 4. Weight-adjusted consumption and number of infusions for patients receiving rurioctocog alfa pegol prophylaxis.
3.5. Immune tolerance induction
Of the 10 patients who developed FVIII inhibitors to rurioctocog alfa pegol during the study, 6 were enrolled to receive ITI. Of the 4 remaining patients not undergoing ITI, 2 with high-titer inhibitors left the study and were considered as having completed the study per protocol. One patient continued receiving prophylaxis with rurioctocog alfa pegol and completed the study with a negative inhibitor titer; 1 patient was receiving on-demand treatment as of data cutoff.
Of the 6 patients enrolled to receive ITI, 5 received ≥1 dose of rurioctocog alfa pegol as ITI. The remaining 1 patient only developed inhibitors immediately prior to data cutoff and therefore never received ITI treatment and left the study. Low-dose ITI was used in 3 patients (1 patient with high-titer and 2 with low-titer inhibitors), and 2 patients received a high dose (1 patient with high-titer and 1 with low-titer inhibitors). The mean (SD) number of FVIII infusions during ITI was 3.1 (1.3) infusions per week and 13.4 (5.5) infusions per month. The mean (SD) treatment dose per month was 866.3 (524.0) IU/kg (564.3 [112.8] IU/kg in the 50 IU/kg 3× weekly group and 1319.3 [623.6] IU/kg in the 100–200 IU/kg daily group). Following ITI treatment, 1 patient was defined as a complete success. This patient had high-titer FVIII inhibitors and received the high dose of rurioctocog alfa pegol (100–200 IU/kg daily). The remaining 4 patients were still undergoing ITI at the time of this IA.
3.6. Safety
A total of 283 AEs (13 considered treatment-related; 9 SAEs of FVIII inhibitor development, 2 cases of drug hypersensitivity, 1 rash, 1 increase in alkaline phosphatase) were reported in 52/59 patients who received ≥1 dose of rurioctocog alfa pegol during the first part of the study (). A further 17 AEs (1 considered treatment-related) were reported in 4/5 patients who developed FVIII inhibitors and received ≥1 dose of rurioctocog alfa pegol for ITI (). Two patients experienced drug hypersensitivity during the first part of the study, but no patients experienced drug hypersensitivity when treated with rurioctocog alfa pegol for ITI, and no thrombotic events were reported at any point during the study.
Table 5. Adverse events occurring during or after first dose of rurioctocog alfa pegol in the safety analysis set and patients who were subsequently enrolled to receive ITI.
4. Discussion
This ongoing prospective study is the first and only to investigate the immunogenicity, safety, and efficacy of the PEGylated, extended half-life, recombinant FVIII replacement therapy, rurioctocog alfa pegol for the treatment of PUPs. In addition, these are the first data assessing the use of rurioctocog alfa pegol for ITI in children. The availability of this interim data is extremely important given that rurioctocog alfa pegol is currently indicated for routine prophylaxis in children and adults with hemophilia A in the US. Rurioctocog alfa pegol is a recombinant FVIII protein with covalently bound PEG chains that is produced in a Chinese hamster ovary cell line. The aim of PEGylation is to retain the functionality of the molecule, improve its pharmacokinetic properties (decrease clearance and thereby increase half-life), and maintain treatment effectiveness while reducing the need for frequent injections [Citation24]. However, assessing inhibitor development and confirming the safety profile in PUPs, who have no (or limited) previous exposure to rurioctocog alfa pegol is essential, especially given that post-translational modification can impact the immunogenicity of therapeutics [Citation16].
The incidence (95% CI) of inhibitor development in this IA (19% [10.0%–33.0%]) was lower than reported in other studies in PUPs [Citation10,Citation25–27]. The SIPPET study, a large, prospective study in which patients were randomized to receive either plasma-derived FVIII or recombinant FVIII, found a cumulative incidence (95% CIs) for all inhibitors of 26.8% (18.4%–35.2%) in patients treated with plasma-derived FVIII and 44.5% (34.7%–54.3%) in patients treated with recombinant FVIII [Citation28]. In addition, recent data investigating the use of turoctocog alfa pegol in PUPs found an inhibitor incidence of 29.9% [Citation29]. However, it is important to note that there are several differences between the patients included in these studies and this is an interim analysis of an ongoing study. The incidence of high-risk FVIII mutations and family history of FVIII inhibitors was lower in this study compared with these previously published trials, indicating that the patients in this current study may have had a lower overall risk of inhibitor development. In addition, this manuscript reports an IA rather than a full patient follow-up. As a result, any comparisons should be made with caution. The time to inhibitor development was consistent with previous studies of recombinant FVIII products in PUPs with hemophilia A [Citation10,Citation26,Citation30].
The ABR reported for the SAS in this IA was similar for patients receiving prophylaxis and those receiving on-demand treatment. This similarity in ABR between treatment regimens could be explained by the fact that these are PUPs, and the median patient age is <12 months, meaning that most patients will start with on-demand treatment to avoid over exposure to FVIII and patients may only switch to prophylactic treatment in response to increased bleeding rates. This creates a bias, with patients experiencing higher bleeding rates more likely to be those receiving prophylaxis, and those with a lower bleeding rate more likely to be receiving on-demand treatment. In addition, owing to the age of the patients, most were given a once weekly prophylaxis regimen, rather than the label recommended regimen of twice weekly. Patients receiving on-demand treatment experienced numerically higher spontaneous ABRs (3.1) and numerically lower injury-related ABR (1.6) than those on prophylaxis (1.0 and 2.1, respectively), as expected in this patient population. Where a hemostatic efficacy rating after 24 hours or following bleed resolution was provided, it was mostly reported as ‘excellent’ or ‘good.’ A factor that may have contributed to the high rate of unrated bleeds is that the patients are PUPs, mostly <1 year of age. Therefore, noncompliance to the requested process for rating bleed severity is not unexpected given that the caregivers had likely not previously participated in such a process.
No thromboembolic AEs were reported, and no patients experienced drug hypersensitivity when treated with rurioctocog alfa pegol for ITI, even when the high-dose regimen was used. These data indicate that rurioctocog alfa pegol has a safety and efficacy profile in PUPs consistent with that reported in previously treated pediatric, adolescent, and adult patients with hemophilia A for the treatment and prevention of bleeds [Citation31–33].
The data reported here are limited by the fact that this was a prespecified IA and included only 59 patients, 10 of whom developed FVIII inhibitors. Calculations of incidence of inhibitor development may also be biased toward higher rates owing to the inclusion/exclusion criteria for this IA. However, including patients with ≤2 prior EDs might have reduced inhibitor risk estimates by excluding patients who are highly susceptible to inhibitor formation. An analysis assessing the rate for risk factors will be performed once all patients have completed the study. In addition, as of the data cutoff, only 1 patient had completed ITI. Therefore, all results should be interpreted with caution.
A key strength of this study is that it was conducted in a diverse patient population compared with previous PUP studies, which typically has a population with >70% White patients [Citation10,Citation26], making the results more generalizable to the wider population. This is especially important given that the risk of inhibitor formation is impacted by patients’ race [Citation34].
5. Conclusion
Interim data show that rurioctocog alfa pegol was efficacious at maintaining hemostasis and treating bleeds in PUPs with severe hemophilia A, with comparable immunogenicity to that demonstrated for other rFVIII products and a similar safety profile to studies in previously treated patients. These results complement previously published data. Further data from the final analysis are needed to draw conclusions on the use of rurioctocog alfa pegol for ITI.
Declaration of interest
RF Sidonio has acted as a paid consultant to Bayer, Takeda, Octapharma, Genentech, Sanofi, Sobi, Novo Nordisk, Tremeau Pharmaceuticals, Guardian Therapeutics, Uniqure, BioMarin, and Spark. He has received investigator-initiated research funding from Takeda, Octapharma, and Genentech.
AA Thompson has acted as a paid consultant to Agios, Beam, bluebird bio, and Celgene/Bristol Myers Squibb. She has received institutional funding for research from Baxalta, BioMarin, bluebird bio, Celgene, CRISP/Vertex, Editas, Graphite Bio, and Novartis. Her current affiliation is the Division of Hematology, Children’s Hospital of Philadelphia.
F Peyvandi has received honoraria as a speaker at educational symposia from Grifols, Roche, Sanofi, Sobi, and Takeda; member of the advisory board of BioMarin, Roche, Sanofi, Sobi, and Takeda.
SL Yeoh has received honoraria as a speaker from Pfizer, Novo Nordisk, Roche, Grifols, Octapharma, and Takeda.
AB Antmen has acted as a paid consultant for Takeda, Pfizer, Roche, and Novo Nordisk.
W Engl is a previous employee of Baxalta Innovations GmbH, a Takeda company.
S Tangada is an employee and stockholder of Takeda.
B Ewenstein is a previous employee and stockholder of Takeda.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A peer reviewer on this manuscript has received speaker fees from Bayer, Novo Nordisk, Hoffman-La Roche, and Takeda, consultancy fees from Hoffman-La Roche and Takeda, and scientific event grants from Bayer, Novo Nordisk, Hoffman-La Roche, and Takeda. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Author contributions
W Engl, S Tangada and B Ewenstein were involved in the conception, design, study conduct, analysis, and interpretation of the clinical trial. RF Sidonio, AA Thompson, F Peyvandi, O Stasyshyn, SL Yeoh, D Sosothikul, and AB Antmen were involved in study conduct and interpretation of the clinical trial. C Maggiore was involved in analysis and interpretation of the clinical trial. All authors revised the manuscript critically for intellectual content and gave their final approval for it to be published.
Acknowledgments
The authors would like to thank Christine Knoll (Center for Cancer and Blood Disorders, Phoenix Children’s Hospital, Phoenix, AZ, USA) for her contributions to the study conduct and interpretation of the clinical data. Under the direction of the authors, medical writing and editorial support was provided by Laura Harrison PhD of Excel Medical Affairs (Fairfield, CT, USA) and was funded by Takeda Development Center Americas, Inc., Lexington, MA, USA.
Data availability statement
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participants data supporting the results of the study, will be made available after the publication of study results within 3 months from initial request to researchers who provide a methodologically sound proposal. The data will be provided after its de-identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
Additional information
Funding
References
- Mannucci PM, Tuddenham EG. The hemophilias–from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. doi: 10.1056/NEJM200106073442307
- Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003;361(9371):1801–1809. doi: 10.1016/S0140-6736(03)13405-8
- Srivastava A, Santagostino E, Dougall A, et al. WFH Guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1–158. doi: 10.1111/hae.14046
- Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388(10040):187–197. doi: 10.1016/S0140-6736(15)01123-X
- Ljung R. Aspects of prophylactic treatment of hemophilia. Thromb J. 2016;14(Suppl 1):30. doi: 10.1186/s12959-016-0103-3
- Witmer C, Young G. Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther Adv Hematol. 2013;4(1):59–72. doi: 10.1177/2040620712464509
- Prezotti ANL, Frade-Guanaes JO, Yamaguti-Hayakawa GG, et al. Immunogenicity of current and new therapies for hemophilia A. Pharmaceuticals. 2022;15(8):911. doi: 10.3390/ph15080911
- Jardim LL, Chaves DG, Rezende SM. Development of inhibitors in hemophilia A: An illustrated review. Res Pract Thromb Haemost. 2020;4(5):752–760. doi: 10.1002/rth2.12335
- Bray G. Inhibitor questions: plasma-derived factor VIII and recombinant factor VIII. Ann Hematol. 1994;68(Suppl 3):S29–34. doi: 10.1007/BF01774527
- Liesner RJ, Abraham A, Altisent C, et al. Simoctocog alfa (Nuwiq) in previously untreated patients with severe haemophilia A: Final results of the NuProtect study. Thromb Haemost. 2021;121(11):1400–1408. doi: 10.1055/s-0040-1722623
- Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007;13(5):606–612. doi: 10.1111/j.1365-2516.2007.01518.x
- Walsh CE, Soucie JM, Miler CH, et al. Impact of inhibitors on hemophilia A mortality in the United States. Am J Hematol. 2015;90(5):400–405. doi: 10.1002/ajh.23957
- Nerich V, Tissot E, Faradji A, et al. Cost-of-illness study of severe haemophilia A and B in five French haemophilia treatment centres. Pharm World Sci. 2008;30(3):287–292. doi: 10.1007/s11096-007-9181-4
- Iorio A, Halimeh S, Holzhauer S, et al. Rate of inhibitor development in previously untreated hemophilia A patients treated with plasma-derived or recombinant factor VIII concentrates: a systematic review. J Thromb Haemost. 2010;8(6):1256–1265. doi: 10.1111/j.1538-7836.2010.03823.x
- Dimichele D. Immune tolerance therapy for factor VIII inhibitors: moving from empiricism to an evidence-based approach. J Thromb Haemost. 2007;5(Suppl 1):143–150. doi: 10.1111/j.1538-7836.2007.02474.x
- Jefferis R. Posttranslational Modifications and the Immunogenicity of Biotherapeutics. J Immunol Res. 2016;2016:5358272. doi: 10.1155/2016/5358272
- Kozma GT, Shimizu T, Ishida T, et al. Anti-PEG antibodies: properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv Drug Deliv Rev. 2020;154-155:163–175. doi: 10.1016/j.addr.2020.07.024
- Verbruggen B, Novakova I, Wessels H, et al. The Nijmegen modification of the Bethesda assay for factor VIII: C inhibitors: improved specificity and reliability. Thromb Haemost. 1995;73(2):247–251. doi: 10.1055/s-0038-1653759
- Whelan SFJ, Hofbauer CJ, Horling FM, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121(6):1039–1048. doi: 10.1182/blood-2012-07-444877
- Lubich C, Allacher P, de la Rosa M, et al. The mystery of antibodies against polyethylene glycol (PEG) – what do we know? Pharm Res. 2016;33(9):2239–2249. doi: 10.1007/s11095-016-1961-x
- Hay CRM, DiMichele D. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119(6):1335–1344. doi: 10.1182/blood-2011-08-369132
- Gouw SC, van der Bom JG, Auerswald G, et al. Recombinant versus plasma-derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood. 2007;109(11):4693–4697. doi: 10.1182/blood-2006-11-056317
- Kempton CL, White GC. How we treat a hemophilia A patient with a factor VIII inhibitor. Blood. 2009;113(1):11–17. doi: 10.1182/blood-2008-06-160432
- Stidl R, Fuchs S, Bossard M, et al. Safety of PEGylated recombinant human full-length coagulation factor VIII (BAX 855) in the overall context of PEG and PEG conjugates. Haemophilia. 2016;22(1):54–64. doi: 10.1111/hae.12762
- Liesner RJ, Abashidze M, Aleinikova O, et al. Immunogenicity, efficacy and safety of Nuwiq® (human-cl rhFVIII) in previously untreated patients with severe haemophilia A-Interim results from the NuProtect study. Haemophilia. 2018;24(2):211–220. doi: 10.1111/hae.13320
- Yaish H, Matsushita T, Belhani M, et al. Safety and efficacy of turoctocog alfa in the prevention and treatment of bleeds in previously untreated paediatric patients with severe haemophilia A: Results from the guardian 4 multinational clinical trial. Haemophilia. 2020;26(1):64–72. doi: 10.1111/hae.13883
- Collins PW, Palmer BP, Chalmers EA, et al. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000-2011. Blood. 2014;124(23):3389–3397. doi: 10.1182/blood-2014-07-580498
- Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and Neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054–2064. doi: 10.1056/NEJMoa1516437
- Male C, Konigs C, Dey S, et al. The safety and efficacy of N8-GP (turoctocog alfa pegol) in previously untreated pediatric patients with hemophilia A. Blood Adv. 2022;7:620–629. doi: 10.1182/bloodadvances.2022007529
- Auerswald G, Thompson AA, Recht M, et al. Experience of Advate rAHF-PFM in previously untreated patients and minimally treated patients with haemophilia A. Thromb Haemost. 2012;107(6):1072–1082. doi: 10.1160/TH11-09-0642
- Klamroth R, Windyga J, Radulescu V, et al. Rurioctocog alfa pegol PK-guided prophylaxis in hemophilia A: results from the phase 3 PROPEL study. Blood. 2021;137(13):1818–1827. doi: 10.1182/blood.2020005673
- Mullins ES, Stasyshyn O, Alvarez-Roman MT, et al. Extended half-life pegylated, full-length recombinant factor VIII for prophylaxis in children with severe haemophilia A. Haemophilia. 2017;23(2):238–246. doi: 10.1111/hae.13119
- Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126(9):1078–1085. doi: 10.1182/blood-2015-03-630897
- Miller CH, Benson J, Ellingsen D, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18(3):375–382. doi: 10.1111/j.1365-2516.2011.02700.x