
ABSTRACT
Introduction
Bypassing agents (BPAs) are used to treat acute bleeding episodes, manage bleeding during perioperative care, and prophylactically minimize bleed occurrence in persons with hemophilia A or B with inhibitors (PwHABI). However, the effectiveness of BPAs that have been prescribed for the last several decades can be variable, motivating the development of a new recombinant activated factor VII, eptacog beta.
Areas Covered
This review covers key eptacog beta findings from phase 1b and phase 3 (PERSEPT) clinical trials, which formed the basis for its regulatory approval to treat PwHABI ages 12 and older. Descriptions of eptacog beta structure and glycosylation profile, mechanism of action, preclinical study results, and cost analyses are also presented.
Expert Opinion
PwHABI have had only two options for bleed treatment for the past several decades. With its distinct glycosylation profile, eptacog beta offers a novel therapy aiming to improve upon BPAs currently in use, providing an option with more than one dosing regimen and a rapid response that allows most bleeds to be treated with just one dose. This has become particularly important given the use of subcutaneous medications (e.g., emicizumab) for prophylaxis of bleeding. Clinicians should consider eptacog beta as a BPA for all PwHABI.
1. Introduction
Hemophilia A (HA) and hemophilia B (HB) are inherited, X-linked bleeding disorders caused by pathogenic F8 or F9 mutations, leading to deficiencies in the essential clotting proteins factor VIII (FVIII) or factor IX (FIX), respectively. Prevalence has been estimated as 17.1 cases per 100,000 males for HA and 3.8 cases per 100,000 males for HB [Citation1]. Spontaneous bleeding into joints, muscles, or soft tissues, easy bruising, and excessive bleeding following trauma or surgery are characteristic symptoms. Hemophilia severity is classified as mild (>5–40%), moderate (1–5%), or severe (<1%) relative to normal clotting factor activity [Citation2]. Bleeding episodes (BEs) in hemophilia patients can be treated by administering recombinant or plasma-derived FVIII or FIX concentrates to replace the deficient clotting factor.
The most serious complication of treating hemophilia with replacement factor concentrates is the emergence of neutralizing antibodies (inhibitors) against FVIII or FIX. Inhibitors arise in about 30% of patients with severe HA and 5–10% of patients with severe HB [Citation3–5]. The presence of inhibitors impairs replacement therapy efforts, as factor replacement products are rendered ineffective. Compared to non-inhibitor patients, persons with hemophilia A or B with inhibitors (PwHABI) experience more BEs [Citation6] and joint pain [Citation7] and face an increased risk of intracranial hemorrhage [Citation8], range of motion limitations [Citation9], and mortality [Citation10,Citation11], as well as substantially higher treatment costs [Citation12,Citation13]. Caregivers of PwHABI report a greater burden and impact on family life than caregivers of non-inhibitor patients [Citation14]. Surgery in PwHABI is challenging given the inherent potential for bleeding in a setting where factor replacement prophylaxis is marginally effective or completely ineffective [Citation15].
Eradication of inhibitors involves immune tolerance induction (ITI), a lengthy regimen that is not always effective, particularly in HB patients [Citation16,Citation17]. Additionally, bleeding events in PwHABI can be treated with bypassing agents (BPAs) including activated prothrombin complex concentrate (aPCC, FEIBA; Takeda) [Citation18], eptacog alfa (NOVOSEVEN; Novo Nordisk), a recombinant activated factor VII (rFVIIa) product that is produced in cultured baby hamster kidney (BHK) cells [Citation19], and eptacog beta (coagulation factor VIIa [recombinant]-jncw, SEVENFACT; HEMA Biologics, LLC and LFB SA), a rFVIIa that has recently entered clinical use [Citation20]. These BPAs promote secondary hemostasis by providing clotting factors (aPCC) [Citation21] or by activating the extrinsic coagulation pathway (eptacog alfa and eptacog beta) [Citation22] to increase thrombin generation and form stable clots. Instances of BEs in HA patients with or without inhibitors are greatly reduced through prophylactic administration of emicizumab (Hemlibra; Roche), an effective, antibody-based agent [Citation23]; however, emicizumab cannot be used to treat breakthrough BEs, and BPAs are still needed for acute bleed management in persons with HA with inhibitors (PwHAI) receiving emicizumab. Similarly, concizumab has been recently approved in Canada for bleed prophylaxis in persons with HB with inhibitors (PwHBI) [Citation24], and BPAs will be needed to treat breakthrough BEs in patients receiving concizumab prophylaxis.
The hemostatic response to aPCC or eptacog alfa in PwHABI during acute bleed management can be effective, but is also unpredictable as these BPAs show variable inter-patient and intra-patient efficacy [Citation25–27]. Furthermore, some PwHABI have BEs that are refractory to either aPCC or eptacog alfa alone [Citation28]. While administering sequential combinations of these BPAs can be effective in cases where BEs cannot be controlled by a single BPA, this approach potentially carries an elevated risk of thrombotic complications [Citation27,Citation29,Citation30]. Novel BPAs that have reliable efficacy and favorable safety profiles in PwHABI have been a pressing medical need; however, the development of such BPAs has been arduous. The bioengineered rFVIIa analogs N7-GP, vatreptacog alfa, and BAY 86–6150 each advanced to human trials as candidate BPAs, but ultimately these clinical programs did not progress owing to either insufficient dose-response [Citation31] or adverse immunological events [Citation32,Citation33]. Marzeptacog alfa, another engineered rFVIIa variant, has been explored as a BPA in clinical trials [Citation34]; however, clinical development was also discontinued [Citation35].
To address the unmet medical needs of inhibitor patients, eptacog beta was developed as a BPA for the treatment of BEs in PwHABI ages 12 and older, receiving regulatory approval from the U.S. Food and Drug Administration (FDA) in 2020 [Citation20] and from the European Medicines Agency (EMA) in 2022 [Citation36]. This review will describe the results from the eptacog beta clinical program. Descriptions of eptacog beta structure and glycosylation profile, mechanism of action, preclinical characterization, and cost analyses are also presented.
2. Introduction to eptacog beta
2.1. Eptacog beta structure
Eptacog beta is expressed in the mammary glands of transgenic rabbits using rPro technology (a production platform that provides recombinant proteins with mammalian-type post-translational modifications) and is subsequently purified from collected rabbit milk [Citation37,Citation38]. Eptacog beta is produced as a zymogen and activated during purification to a form consisting of a light chain (152 amino acids) and a heavy chain (254 amino acids) linked by a disulfide bond [Citation38]. The light chain features an N-terminal γ-carboxyglutamate (Gla) domain followed by 2 epidermal growth factor (EGF)-like domains, while the heavy chain contains a catalytic serine protease domain () [Citation41]. Endogenous FVIIa and recombinant variants such as eptacog beta have several types of post-translational modifications, including γ-carboxylation at 10 potential vitamin K-dependent γ-carboxylation sites within the Gla domain and O-linked glycosylation at Ser52 and Ser60 residues within the first EGF-like domain. In addition, N-linked glycosylation is found at Asn145 (light chain) and Asn322 (heavy chain), while Asp63 represents a potential β-hydroxylation site [Citation37,Citation38,Citation42]. The immunogenic epitopes α-1,3-Gal and N-glycolyl-neuraminic acid are not present in eptacog beta [Citation37,Citation38].
Figure 1. Eptacog beta domain structure and post-translational modification sites [Citation39,Citation40]. The light and heavy chains are connected by a disulfide bond. The 10 sites for γ-carboxylation (amino acid positions 6, 7, 14, 16, 19, 20, 25, 26, 29, and 35) in the Gla domain are shown, as well as sites for O-linked glycosylation (Ser52 and Ser60), N-linked glycosylation (Asn145 and Asn322), and β-hydroxylation (Asp63). GLA, γ-carboxyglutamic acid (Gla) domain; EGF, epidermal growth factor-like domain.
![Figure 1. Eptacog beta domain structure and post-translational modification sites [Citation39,Citation40]. The light and heavy chains are connected by a disulfide bond. The 10 sites for γ-carboxylation (amino acid positions 6, 7, 14, 16, 19, 20, 25, 26, 29, and 35) in the Gla domain are shown, as well as sites for O-linked glycosylation (Ser52 and Ser60), N-linked glycosylation (Asn145 and Asn322), and β-hydroxylation (Asp63). GLA, γ-carboxyglutamic acid (Gla) domain; EGF, epidermal growth factor-like domain.](/cms/asset/1a3a9098-f8b1-4d4a-a736-b870f57b4989/ierr_a_2248385_f0001_oc.jpg)
2.2. Mechanism of action of FVIIa [activated endogenous FVIIa, eptacog alfa, eptacog beta]
2.2.1. Mechanism of action of endogenous FVIIa and of rFVIIa during pharmacotherapy
Endogenous FVIIa and its recombinant forms such as eptacog beta initiate localized hemostatic activity by binding extravascular tissue factor (TF) at the site of vascular injury (). All 4 FVIIa domains form extensive TF contacts, and O-linked glycosylation at FVIIa Ser52 and Ser60 participate in productive FVIIa-TF interactions [Citation39,Citation43]. The resulting FVIIa-TF complex, in the presence of calcium, converts the zymogen factor X (FX) to the enzyme activated factor X (FXa). FXa, together with activated factor V (FVa) converts prothrombin to thrombin (). In addition, platelets are activated by collagen from the freshly-exposed extracellular matrix and by the newly-generated thrombin [Citation44,Citation45]. Infused rFVIIa at pharmacological dosing levels in PwHABI binds these activated platelets, and in turn activates FX directly to drive a focused thrombin burst precisely at the site of injury, culminating in thrombin-mediated conversion of fibrinogen to fibrin and localized hemostasis () [Citation22,Citation46,Citation47]. rFVIIa may also facilitate hemostasis by competing with protein C (PC) for endothelial protein C receptor (EPCR) binding, thus inhibiting PC activation and downstream activated PC (APC)-dependent anticoagulation pathways that inactivate FVa () [Citation48,Citation49]. In addition, rFVIIa can induce EPCR- and protease-activated receptor-1 (PAR-1)-dependent release of extracellular vesicles (EVs) from endothelial cells, which may further promote hemostatic processes at the site of the wound () [Citation50].
Figure 2. Coagulation events that lead to localized hemostasis at the site of vessel injury take place via TF-dependent, platelet-dependent, and EPCR-dependent pathways. (a) FVIIa binds to exposed TF at the site of injury, leading to FX activation. Subsequent formation of the prothrombinase complex (FXa:FVa) leads to the generation of a small amount of thrombin on the TF-presenting cell. TPFI and AT modulate FVIIa procoagulant activity. (b) Platelets are activated by exposed extravascular collagen at the site of the wound and by the newly-generated thrombin. Binding of infused rFVIIa to activated platelets in PwHABI generates additional FXa, which binds FVa to form prothrombinase on the activated platelet surface, ultimately producing a burst of thrombin, fibrin formation, and localized hemostasis. (c) PC binds to EPCR and is converted to APC. The anticoagulant activity of APC inactivates the coagulant cascade and prevents dysregulated procoagulant activity. (d) rFVIIa can compete for PC binding to EPCR thereby reducing PC activation to APC and enhancing thrombin burst. rFVIIa can be sequestered from the circulatory system by EPCR-mediated uptake into the extravascular space. rFVIIa can stimulate release of procoagulant extracellular vesicles (EVs) from endothelial cells.
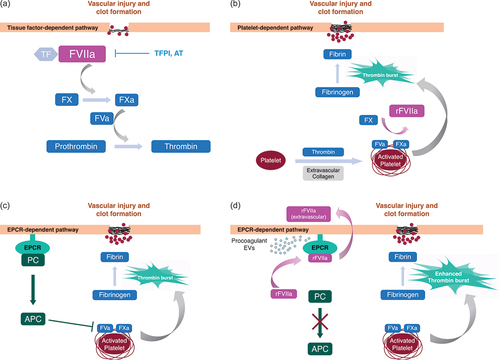
Multiple regulatory mechanisms modulate FVIIa procoagulant activity during and following clot formation (). FVIIa has low intrinsic activity in the absence of TF or activated platelets [Citation51,Citation52], diminishing the likelihood of FVIIa triggering unnecessary clotting at undamaged tissue sites. Systemic coagulation is also prevented by specific regulatory mechanisms, such as those involving tissue factor pathway inhibitor (TFPI), which inhibits the TF-FVIIa complex and prothrombinase [Citation53,Citation54], or antithrombin (AT), which inhibits thrombin and other serine protease clotting factors, including FVIIa () [Citation55,Citation56]. Elevated thrombin levels can promote the formation of the thrombin-thrombomodulin complex, which acts upon EPCR-bound PC to generate the anticoagulant APC and dampen thrombin generation () [Citation49]. Animal experiments have shown that EPCR can transport rFVIIa into extravascular tissue, storing rFVIIa in an activated form and possibly prolonging the hemostatic effect of rFVIIa () [Citation57]. Further experiments in mouse models indicate that administered eptacog beta can be sequestered in a dose-dependent manner within knee joints for several days [Citation58]. FVIIa clearance and storage mechanisms may thus provide additional processes that avoid undesired coagulation, as well as extend eptacog beta bioavailability.
2.2.2. Distinguishing molecular and functional features of eptacog beta
Different FVII glycoforms display different autoactivation rates and affinities for TF [Citation59]. Although eptacog beta and eptacog alfa have identical amino acid sequences (both encoding the human FVII protein), eptacog beta and eptacog alfa are each a distinct FVII glycoform. The major N-linked glycosylation modification presented by eptacog alfa is a bisialylated biantennary glycan with a core fucose unit and α2,3 sialic acid linkages to a galactose residue [Citation60], while a monosialylated biantennary glycan with α2,6 sialic acid linkages and lacking fucose predominates in eptacog beta [Citation37]. In addition, eptacog beta is γ-carboxylated at 9 of 10 glutamic acid residues within the Gla domain [Citation38], while eptacog alfa is partially γ-carboxylated at the final glutamic acid site (Glu35) [Citation61]. Asp63 is a potential β-hydroxylation site on FVIIa, but is unmodified in eptacog alfa and modified at low abundance (4%) in eptacog beta [Citation38,Citation61].
Along with distinct glycosylation profiles and other post-translational modification differences, eptacog beta and eptacog alfa also show functional differences. Eptacog beta demonstrates enhanced binding to activated platelets as compared with eptacog alfa [Citation62]. In vitro binding of eptacog beta to human umbilical cord vein cells (HUVEC), due in part to EPCR interactions, is 25–50% greater than observed for eptacog alfa [Citation62]. Given that eptacog beta and eptacog alfa have identical amino acid sequences, and that functional differences among various human FVIIa glycoforms have previously been observed [Citation59], the differences in EPCR or activated platelet binding between eptacog beta and eptacog alfa likely stem from post-translational modification differences.
The enhanced EPCR binding exhibited by eptacog beta may have physiological implications, as studies in cell culture and mouse model systems suggest that pharmacological levels of rFVIIa may compete with PC for EPCR binding and downregulate the APC anticoagulation pathway [Citation48,Citation63,Citation64]. In addition, rFVIIa-EPCR interactions may trigger signaling events that mediate processes beyond hemostasis including endothelial cell barrier protection [Citation64,Citation65]. The clinical impact of enhanced eptacog beta binding to EPCR or to activated platelets is currently unknown.
3. Phase 1b pharmacokinetic, pharmacodynamic, and safety studies of eptacog beta
Higher rFVIIa dosing is thought to result in an increased thrombin burst, leading to the formation of a more stable clot and more effective hemostasis [Citation22]. However, identifying optimal rFVIIa dosing for BE treatment is complicated by a lack of validated pharmacodynamic (PD) markers that reliably predict clinical efficacy. To support phase 3 clinical trials of eptacog beta in PwHABI, a phase 1b dosing escalation study was undertaken (the first report of a clinical study of eptacog beta; NCT01708564) [Citation66]. Males with congenital hemophilia (with or without inhibitors) ages 18–75 years were eligible for enrollment. Exclusion criteria were recent treatment with a FVII- or FVIIa-containing product, recent surgery (within the previous month), history of arterial or venous thromboembolic events (within past 2 years), allergy or sensitivity to rabbits, active or ongoing bleeding, a coagulation disorder other than hemophilia A or B, a low platelet count, and being immunosuppressed (as measured by CD4+ count).
Lower dosing than eptacog alfa was selected for the eptacog beta phase 1b trial based on preclinical in vitro and in vivo eptacog beta activity studies conducted by LFB SA [Citation66]. Escalating doses of 25 and 75 µg/kg, 25 and 225 µg/kg, or 75 and 225 µg/kg eptacog beta were administered to 15 subjects with hemophilia A or B with or without inhibitors and without an active bleed. Eleven subjects had HA and 4 subjects had HB, with 4 of the 15 subjects having inhibitors to either FVIII or FIX. Pharmacokinetic (PK) parameters were determined by FVIIa activity assay and subsequent noncompartmental modeling (). The maximum FVIIa concentration (Cmax) and area under the concentration-time curve (time of dosing to infinity; AUC0-inf) were dose-dependent, while eptacog beta clearance, distribution volume, and half-life changed relatively little for each dose level (). The eptacog beta plasma half-life from this study (~2 h) was similar to that described for eptacog alfa [Citation67].
Table 1. PK and PD parameters from the phase 1b study of eptacog beta [Citation66].
A number of surrogate PD markers for hemostasis in this trial showed changes at different dose levels. Peak thrombin generation increased with increasing eptacog beta dose in a platelet-rich thrombin generation assay (TGA) relative to baseline levels (), and prothrombin fragment F1 + 2 levels exhibited dose-dependent increases at 1–2 h post-eptacog beta infusion. Reductions in activated partial thromboplastin time (aPTT) and prothrombin time (PT) were also observed as eptacog beta dosing increased. Lastly, Ducore et al. reported increased clot firmness with escalated eptacog beta dosing, as measured by rotational thromboelastometry (ROTEM; ) [Citation66]. Collectively, these data are consistent with increased thrombin generation and improved clot stability associated with increased eptacog beta dose.
No hypersensitivity, allergic reaction, or other adverse immunologic events were reported during this study, and eptacog beta was well tolerated. Three adverse events occurred that were possibly treatment-related: one subject experienced two instances of mild dizziness after receiving a 25 and a 75 µg/kg eptacog beta infusion, and another subject experienced mild headache after receiving 25 µg/kg eptacog beta. Each of these three adverse events resolved without intervention. No thrombotic events were reported.
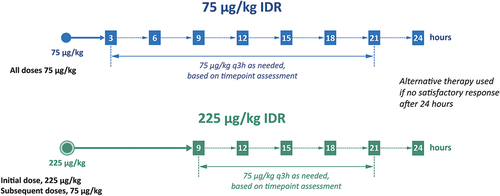
The safety, PK, and PD data from this phase 1b study supported use of 75 and 225 µg/kg dosing in phase 3 trials. The total AUC0-inf data obtained from the different doses used suggested a 3-h dosing interval between 75 µg/kg doses, and that a 9-h dose-dependent interval between an initial 225 µg/kg and subsequent doses of eptacog beta is pharmacokinetically supported [Citation66].
4. Phase 3 clinical trials of eptacog beta
4.1. PERSEPT 1: eptacog beta efficacy and safety in PwHABI ≥12 years of age
PERSEPT 1 (NCT02020369) was the pivotal phase 3 trial that led to FDA approval of eptacog beta for treatment of BEs in adults and adolescents (ages 12 and older) with hemophilia A or B and inhibitors using 75 and 225 µg/kg initial dose regimens (IDRs) [Citation20,Citation68]. This trial was a global, multicenter, open-label, prospective study of the efficacy, immunogenicity, and safety of eptacog beta, and enrolled 27 PwHABI for treatment of mild, moderate, or severe BEs. Male subjects 12–75 years of age with congenital hemophilia A or B and inhibitors could enroll, provided that inhibitor titers were ≥5 Bethesda units (BU) per mL (i.e., high-titer inhibitors) or, if inhibitor titers were <5 BU/mL, subjects had a history of either a high anamnestic response to FVIII or FIXFootnote1 or demonstrated refractory hemostatic responses to increased FVIII or FIX dosing. Experiencing at least 3 BEs in the 6 months prior to the trial was an additional inclusion requirement. PERSEPT 1 exclusion criteria were diagnosis with a coagulation disorder other than hemophilia A or B or any life-threatening disease or condition that represented a patient hazard or could skew trial results, recent surgery (within the previous month), a history of arterial or venous thromboembolic events (within the last 2 years), an allergy or sensitivity to rabbits, a low platelet count, hepatic/renal impairment, an active malignancy (non-melanoma skin cancer allowed), and immunosuppressed (as measured by CD4+ count). Use of prophylactic regimens was not among the exclusion criteria. Mild BEs included bleeds that had just started and with few symptoms, i.e., BEs with little or no pain, little or no change in the range of motion of the affected joint (if a joint BE), and mild restriction of mobility and activity. Moderate BEs included BEs that involved swelling or pain including some decrease in range of motion of an affected joint (if a joint BE) or moderate decrease in mobility and activity. Severe BEs were those that were considered life- or limb-threatening, or could lead to significant blood loss, pain, or permanent nerve damage.
Subjects received an initial dose of either 75 or 225 µg/kg eptacog beta (depending upon randomized IDR assignment) for BE treatment. Subjects were advised to treat BEs as soon as possible, and within 4 h after bleed occurrence. Subjects evaluated the efficacy and safety for mild or moderate BE treatment at 3 h and 9 h post-initial eptacog beta infusion in the 75 and 225 µg/kg IDRs, respectively, and subsequently treated with 75 µg/kg eptacog beta q3h as needed until the bleed resolved (). No additional study drug was allowed beyond the 21-h timepoint for either IDR. If bleed resolution was not achieved at 24 h, then alternative therapies could be initiated. Severe BEs were to be treated according to severe BE dosing regimens that used 2-h dosing intervals following the administration of 75 µg/kg eptacog beta, or a 225 µg/kg eptacog beta infusion, followed if necessary 6 h later with 75 µg/kg every 2 h until hemostasis is achieved [Citation68]. Severe BEs could be treated at home initially, and were subsequently required to be treated in a hospital setting. Subjects were crossed over to the alternate IDR every 3 months without a washout period for the duration of the trial (~17 months) [Citation68,Citation69]. A stringent set of criteria were used to ascertain bleed treatment success of mild or moderate BEs in PERSEPT 1. Bleed control was rated by the subject or caregiver at each evaluation timepoint according to a predefined four-point hemostasis evaluation scale of ‘excellent,’ ‘good,’ ‘moderate,’ or ‘none’ [Citation68]. Treatment of a mild or moderate BE was considered successful if all of the following criteria were met: (i) a hemostasis evaluation of ‘excellent’ or ‘good’ was obtained (‘moderate’ or ‘none’ being considered a lack of efficacy); (ii) no additional use of eptacog beta was needed within 24 h after the first ‘excellent’ or ‘good’ response”; (iii) no alternative hemostatic agent or blood product was needed; and (iv) no increase in pain was reported, as measured on a Visual Analog Scale (VAS). The primary endpoint for PERSEPT 1 was the successful treatment of a mild or moderate BE at 12 h post-initial infusion of eptacog beta based on the above criteria.
Figure 3. The 75 and 225 µg/kg eptacog beta IDRs in PERSEPT 1 and PERSEPT 2.
Subjects in PERSEPT 1 ranged in age from 12 to 54 years (median of 31 years). Twenty-five subjects were diagnosed with HA and 2 with HB. Subjects treated a total of 465 mild or moderate BEs, with 252 and 213 BEs treated using the 75 and 225 µg/kg IDRs, respectively. The majority of BEs (98%) were treated at home [Citation20] and 85% of mild or moderate BEs were treated within 1 h of symptom onset [Citation68]. Joint bleeds comprised the majority (85%) of all mild or moderate BEs in PERSEPT 1, and target joint BEs in 15 subjects made up 34% of all mild or moderate joint bleeds [Citation70,Citation71].
Subject- or caregiver-reported treatment success proportions for all mild or moderate BEs in PERSEPT 1 at 12 h post-initial infusion were 82% and 91% in the 75 and 225 µg/kg IDRs, respectively [Citation20], indicating greater efficacy with the 225 µg/kg IDR. Successful mild or moderate BE control required a median of 1 eptacog beta infusion in the 225 µg/kg IDR and 2 infusions in the 75 µg/kg IDR [Citation20]. Eptacog beta treatment success for joint BEs at 12 h (80.6% and 91.5% for 75 and 225 µg/kg IDRs, respectively) [Citation70] was congruent with observations for all BEs. Resolution of bleeding was achieved with a single 225 µg/kg infusion for 84% of all mild or moderate BEs treated while the subject was assigned to the 225 µg/kg IDR [Citation72]. Treatment success proportions of 94–100% were observed at 24 h for adults and adolescents in the 2 IDRs during PERSEPT 1 [Citation72]. A decrease in pain levels at 12 h post-initial infusion were reported in 87% and 86% of subjects assigned to 75 and 225 µg/kg IDRs, respectively [Citation68]. A low incidence of rebleeding at the same anatomical location (1/465 mild or moderate BEs; 0.2%) was observed prior to 24 h in the trial [Citation68]. Three severe BEs (a traumatic intramuscular hemorrhage, a spontaneous right hip joint bleed, and a spontaneous renal hemorrhage) received a good or excellent evaluation by 12 h following initial eptacog beta treatment. All 3 severe BEs occurred while subjects were assigned to the 225 µg/kg IDR [Citation68].
No thrombotic, hypersensitivity, allergic, or anaphylactic events were reported during PERSEPT 1 and no neutralizing antibodies were found. Two subjects experienced treatment-related treatment-emergent adverse events (TEAEs). One subject experienced 4 instances of infusion-site discomfort and 2 infusion-site hematoma events while assigned to the 75 µg/kg IDR, which were considered to be mild by the site investigator and all resolved. Another experienced increased body temperature while assigned to the 225 µg/kg IDR (), which was considered to be of moderate severity and resolved with treatment. While 1 subject experienced acute tonsillitis and subarachnoid hemorrhage that were considered serious adverse events (SAEs), neither of these SAEs were deemed to be related to eptacog beta administration (). Overall, eptacog beta was well tolerated in PERSEPT 1 [Citation68].
Table 2. Treatment-related and serious adverse events from PERSEPT 1, PERSEPT 2, and PERSEPT 3 [Citation69]. AE, adverse event. SAE, serious adverse event.
4.2. PERSEPT 2: eptacog beta efficacy and safety in PwHABI <12 years of age
PERSEPT 2 (NCT02448680) was a multicenter, open-label, prospective phase 3 trial that evaluated the efficacy and safety of eptacog beta for acute bleed treatment in pediatric subjects under 12 years of age with hemophilia A or B and inhibitors. PERSEPT 2 represents the first reported prospective study to focus solely on PwHABI <12 years for bleed treatment with a rFVIIa BPA [Citation73]. Subjects under 6 months of age must have experienced at least 3 BEs since birth to be eligible for enrollment. A known or suspected hypersensitivity to the active ingredient or any excipients was also among the exclusion criteria. Other inclusion and exclusion criteria for PERSEPT 2 were as described for PERSEPT 1 (see above). As with PERSEPT 1, the use of prophylactic measures was not among the exclusion criteria in PERSEPT 2.
The trial design for PERSEPT 2 followed that of PERSEPT 1, with subjects randomized to treat BEs using either 75 or 225 µg/kg IDRs () and crossed over to the alternate IDR every 3 months over the duration of the trial (~21 months) without a washout period. Criteria for determining bleed treatment success were identical to those used in PERSEPT 1. Bleed control and pain levels were evaluated by the caregiver together with the pediatric subject when possible (depending upon subject age and verbal capacity). The primary efficacy endpoint for PERSEPT 2 was the same as in PERSEPT 1: successful treatment of a mild or moderate BE at 12 h post-initial infusion of eptacog beta. Treatment success proportions for mild or moderate BEs at 12 h were compared to an objective performance criterion (OPC) of 55%, which was ascertained from published reports of rFVIIa treatment success in adult and pediatric PwHABI [Citation73] and chosen as the benchmark for minimal eptacog beta efficacy needed to meet the clinical endpoint of the trial. This OPC was considered the only benchmark available for comparison when PERSEPT 2 was designed, given that a prospective study of rFVIIa efficacy in PwHABI younger than 12 years had never been performed before [Citation73].
PERSEPT 2 enrolled 25 subjects (23 with HA and 2 with HB) for treatment of mild, moderate, or severe BEs. PERSEPT 2 subject ages ranged from 1 to 11 years (median of 5.0 years). Subjects received eptacog beta for 546 mild or moderate BEs, with 239 and 307 BEs treated using the 75 and 225 µg/kg IDRs, respectively. Ninety-two percent of these BEs were treated at home. Joint bleeds comprised 68% of all mild or moderate BEs in PERSEPT 2, and target joint BEs made up nearly 20% of all mild or moderate joint bleeds.
Treatment success proportions for mild or moderate BEs at 12 h post-initial infusion were 65% and 60% in the 75 and 225 µg/kg IDRs, respectively, and were reported by the subject or caregiver. At 24 h, treatment success proportions were 97% for the 75 µg/kg IDR and 98% for the 225 µg/kg IDR [Citation73]. Successful bleed control required a median of 2 eptacog beta infusions in the 225 µg/kg IDR and 3 infusions in the 75 µg/kg IDR. Bleed recurrence (defined as bleeding at the same anatomical location within 24 h of the last ‘good’ or ‘excellent’ assessment) was observed in 8 of 546 (1.5%) mild or moderate BEs. An alternative BPA was needed for 12 of 546 BEs (2.2%), with 8 of these BEs occurring in one subject. Pain relief was seen for 93% (75 µg/kg IDR) and 91% (225 µg/kg IDR) of mild or moderate BEs at 12 h. The mean pain VAS scores decreased 71% and 65% from baseline in the 75 and 225 µg/kg IDRs, respectively.
Pipe et al. noted one subject (age 4 years) who experienced 46 of the 546 mild or moderate BEs and had an outsized effect on efficacy results [Citation73]. Treatment success proportions for this subject were 6% and 14% at 12 h in the 75 and 225 µg/kg IDRs, respectively, and were 100% for both IDRs at 24 h, indicating a delayed but effective response to eptacog beta at the 24-h timepoint. When excluding this outlier subject, treatment success proportions for mild or moderate BEs in all other subjects at 12 h increased to 70% and 65% for the 75 and 225 µg/kg IDRs, respectively.
Three severe BEs (a spontaneous renal hemorrhage, a traumatic intracranial hemorrhage [ICH], and a traumatic elbow bleed) occurred in PERSEPT 2 while subjects were assigned to the 225 µg/kg IDR. Hemostasis evaluations were not captured at 12 or 24 h for the severe elbow BE by the treating physician. While the renal hemorrhage and ICH were not controlled at 12 or 24 h, hemostatic control of the ICH was achieved following 2 days of eptacog beta treatment (as assessed by computed tomography), and control of the renal hemorrhage was achieved after approximately 4 days of eptacog beta treatment [Citation73].
While treatment success proportions at 12 h for mild or moderate BEs were not statistically different from the OPC of 55% that was established for the study [Citation73] (i.e., the clinical endpoint for the trial was not met), these results might best be viewed in light of the recognized difficulties that caregivers face in determining bleed treatment efficacy in children with hemophilia [Citation74,Citation75], particularly for younger patients having limited ability to communicate bleed status and pain levels [Citation76]. Caregiver decisions to stop or continue treating in this setting are highly subjective [Citation74,Citation75], and caregivers may opt to continue treating based on either past experience with similar BEs or out of concern for rebleeding [Citation75]. These subjective aspects of bleed management by the caregivers may potentially contribute to the longer rFVIIa treatment durations observed in children as compared with adults [Citation77]. Pipe et al. surmised that caregiver uncertainty in assessing bleed resolution and a bias toward continued treatment could have resulted in conservative estimates of bleed treatment success at the 12-h timepoint, and obscure dose-dependent differences in treatment efficacy between the two IDRs [Citation73].
No thrombotic, hypersensitivity, allergic, or anaphylactic events were reported during PERSEPT 2, and no neutralizing antibodies were found. Three SAEs (ICH, paresis, and dysentery) that resolved with treatment were recorded, and were considered unrelated to eptacog beta administration (). No treatment-related TEAEs occurred, and overall eptacog beta was well tolerated in PERSEPT 2.
4.3. PERSEPT 3: eptacog beta in PwHABI undergoing major or minor procedures
Prior to the availability of BPAs, elective major surgeries in PwHABI were often avoided given the risk of bleeding complications [Citation15]. The growth in clinical experience with BPAs in the surgical setting [Citation15,Citation78,Citation79] allows physicians to consider such procedures for their patients with increased confidence, albeit with appropriate caution and planning for the possibility of excessive bleeding. PERSEPT 3 (NCT02548143) was a multicenter, open-label, single-arm prospective phase 3 trial that evaluated the efficacy and safety of eptacog beta during major or minor elective surgeries or other invasive procedures in PwHABI [Citation80]. Male patients between ages 6 and 75 with congenital hemophilia A or B and inhibitors were eligible to participate; other inclusion and exclusion criteria were as described for PERSEPT 1 [Citation80]. Further exclusion criteria encompassed a known or suspected hypersensitivity to the active ingredient or any excipients, gastric or duodenal ulcer disease, ITI therapy, and use of aspirin or other non-steroidal anti-inflammatory drugs (NSAIDS) within 1 week of surgery and during eptacog beta treatment. Use of prophylactic measures was not among the exclusion criteria in PERSEPT 3.
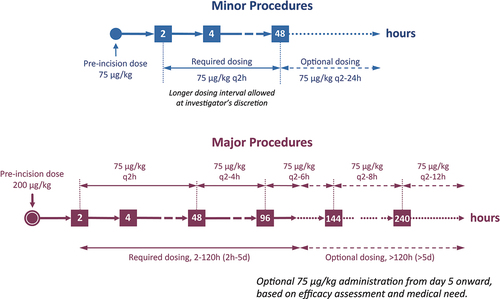
Major procedures were defined in PERSEPT 3 as those anticipated to need 5 or more days of factor replacement in hemophilia patients and involving entry into a body cavity or a similarly complex procedure. Minor procedures were those anticipated to need fewer than 5 days of factor replacement [Citation69,Citation80]. Subjects undergoing a major procedure received a bolus infusion of 200 µg/kg eptacog beta just prior to the initial incision, while subjects undergoing a minor procedure received a preoperative 75 µg/kg dose. Subsequent 75 µg/kg eptacog beta infusions were given q2h intraoperatively and postoperatively up to 48 h. Beyond 48 h, dosing intervals could be progressively increased at the discretion of the investigator (). Antifibrinolytics were allowed during PERSEPT 3. Dosing continued for at least 2 days for minor procedures and for at least 5 days for major procedures.
Figure 4. Eptacog beta dosing schedules for minor and major procedures in PERSEPT 3.
Intraoperative and postoperative efficacy of eptacog beta was recorded by the surgeon/investigator as ‘excellent,’ ‘good,’ ‘moderate,’ or ‘poor,’ based upon intraoperative and postoperative blood loss levels, transfusion requirements, and need for intensive care unit services relative to that seen for a comparable procedure in a patient without a bleeding disorder [Citation80]. ‘Excellent’ or ‘good’ hemostatic evaluations were considered a satisfactory response to eptacog beta, while ‘moderate’ or ‘poor’ evaluations were considered an insufficient response. Hemostatic evaluations were made intraoperatively, postoperatively at 24 and 48 h following procedure completion, and 24 and 48 h following the last eptacog beta infusion. The primary efficacy endpoint for PERSEPT 3 was the percentage of procedures with a ‘good’ or ‘excellent’ hemostatic evaluation at 48 h after the last eptacog beta infusion, taking into consideration the totality of all intraoperative and postoperative assessments, oozing, blood transfusions, and total amount of eptacog beta used.
Six minor and 6 major procedures were performed in 12 subjects (all with severe HA) in PERSEPT 3, with subject ages ranging from 2 to 56 years (median of 20.0 years). Minor procedures included 3 circumcisions and 3 tooth extractions. All 6 major procedures were lower extremity orthopedic surgeries (2 knee surgeries, 2 amputations, a hip replacement, and an achilloplasty). Intraoperative hemostatic assessments of ‘good’ or ‘excellent’ were reported for all 12 procedures. Mean intraoperative blood loss was estimated to be lower than the mean maximum predicted blood loss for comparable operations in a person without a bleeding disorder for both major and minor procedures.
The treatment success proportion for minor procedures at 48 h after the last eptacog beta infusion was 100% (5 successes and 1 not evaluable due to withdrawal of consent). The treatment success proportion at the primary endpoint for major procedures was 66.7%. Four procedures were completed successfully and 2 were considered failures. A hip replacement procedure was considered a failure owing to a post-procedural hematoma that led to trial discontinuation and infusion of aPCC. A total knee replacement was also considered a failure at postoperative day 7, as a BE at the surgical site led to eptacog beta efficacy being rated as poor. Eptacog beta was discontinued, and the subject subsequently received packed red blood cells, eptacog alfa, and aPCC to control the BE. The use of an alternative agent (or alternating agents) as rescue therapy to control a postoperative hemorrhage is reported in the literature [Citation81,Citation82]; furthermore, the hip and knee arthroplasty bleeding complications during PERSEPT 3, while unwelcome, were consistent with surgical experiences reported by Erturan et al. for PwHABI [Citation15]. The overall treatment success proportion at the primary endpoint for both minor and major procedures was 81.8%, as assessed by the surgeon/investigator.
No thrombotic, hypersensitivity, allergic, or anaphylactic events occurred in PERSEPT 3, and no anti-eptacog beta antibodies were reported. No treatment-related AEs or SAEs were encountered during the minor procedures. Three AEs that were considered treatment-related by the investigator were reported in 1 subject undergoing hip replacement surgery. This subject experienced a post-procedural hematoma (considered a treatment-related AE) and was discontinued from the study. The subject was subsequently given aPCC. Two days later, this subject experienced acute blood loss anemia (an SAE) from a gastrointestinal hemorrhage (an SAE) and succumbed 3 days following the hip replacement procedure. While the investigator considered the gastrointestinal hemorrhage and blood loss anemia to be probably treatment-related, the independent data monitoring committee (DMC), in considering the short half-life of eptacog beta and the switch to aPCC 2 days prior to the gastrointestinal hemorrhage, concluded that the 2 SAEs were unlikely related to eptacog beta (). This subject had previously participated in PERSEPT 1, successfully treating 25 BEs with eptacog beta without experiencing any AEs.
4.4. Collective safety results from the PERSEPT program
In all, 60 subjects (56 with HA and 4 with HB) participated in PERSEPT 1 (27 subjects), PERSEPT 2 (25 subjects), and PERSEPT 3 (12 subjects). Two subjects from PERSEPT 1 and 2 subjects from PERSEPT 2 also participated in PERSEPT 3. None of the study subjects received emicizumab prophylaxis. The 60 subjects received a total of 3,388 infusions of eptacog beta for 1,087 acute BEs, invasive procedures, postoperative treatments, and PK measurements [Citation69]. Subjects experienced 133 TEAEs, of which 7 were serious (SAEs) and 10 were considered treatment-related (6 in a single PERSEPT 1 subject; ). None of the SAEs were considered treatment-related, and the occurrences of treatment-related AEs showed no apparent correlation with IDR assignment [Citation69]. One subject in PERSEPT 3 discontinued the trial due to a post-procedural hematoma that was considered a treatment-related TEAE (). No subjects were discontinued from PERSEPT 1 or PERSEPT 2 as the result of an adverse event. As noted above, the DMC considered the death that occurred during PERSEPT 3 to be unlikely related to eptacog beta administration.
A history of thrombotic complications was a common exclusion criterion from the PERSEPT trials, and no thrombotic events occurred during these clinical investigations. No allergic, hypersensitivity, or anaphylactic reactions were reported, and no neutralizing antibodies were detected during the PERSEPT program [Citation69].
5. Post-marketing surveillance
Emicizumab has been increasingly adopted as a prophylactic agent in individuals with HA with or without inhibitors. As noted above, BPAs such as eptacog beta are still needed to control any breakthrough bleeds in PwHAI using emicizumab. Co-administration of emicizumab and aPCC during the HAVEN clinical program revealed a potential safety issue where some PwHAI who received >100 U/kg/24 hours aPCC for ≥24 hours to treat breakthrough bleeds (either alone or in combination with eptacog alfa) experienced thrombotic microangiopathy (TMA) and thrombotic complications [Citation23,Citation83]. These findings prompted concerns that concomitant use of emicizumab and high-dose aPCC for extended periods could lead to increased incidence of thrombotic events. No TMA or thromboembolic events were associated with eptacog alfa monotherapy during the HAVEN trials [Citation23,Citation83]. A recent examination of the EudraVigilance database revealed 6 instances of thrombotic adverse drug reactions associated with concomitant administration of emicizumab and eptacog alfa [Citation84]. These observations underscore the need for continued safety monitoring of new treatments in various clinical settings.
The safety of eptacog beta in hemophilia patients with inhibitors (ages 12 and older) with or without concurrent emicizumab prophylaxis is currently being studied in the phase 4 ATHN-16 trial (NCT04647227; recruiting participants). In vitro experiments with clinically-relevant concentrations of eptacog beta and emicizumab in plasma samples from HA subjects (with and without inhibitors) showed that thrombin generation remained below that found in plasma from non-hemophilia subjects [Citation85], suggesting that eptacog beta could be safely used to control breakthrough bleeding events in patients receiving emicizumab prophylaxis without promoting thrombotic complications. The Medical and Scientific Advisory Council (MASAC) for the National Hemophilia Foundation has included the use of 75 µg/kg IDR eptacog beta among its recommendations for treating breakthrough bleeds in PwHAI receiving emicizumab [Citation86]. As additional safety data is needed to characterize the thrombotic risk of using 225 µg/kg IDR eptacog beta in PwHAI receiving emicizumb, the findings from the ongoing ATHN-16 trial (once completed) are of high interest. Aside from its use with emicizumab, continued eptacog beta utilization by physicians to treat PwHABI might lead to new data and accumulated clinical experience that could potentially support an expanded set of indications (e.g., prophylactic use outside of a perioperative setting, or acute bleed treatment during ITI therapy).
6. Regulatory affairs
Eptacog beta has been approved in the U.S. and Mexico (under the brand name SEVENFACT), as well as in the EU and the U.K. (brand name CEVENFACTA) for BE treatment in adult and adolescent hemophilia A or B patients with inhibitors (≥12 years of age) [Citation20,Citation36]. Eptacog beta is also approved in the EU for prevention of bleeding during surgery in PwHABI ages 12 and older [Citation36]. Eptacog beta is not indicated for the treatment of congenital factor VII deficiency [Citation20].
7. Cost
In addition to anticipated BPA efficacy, cost considerations may also influence BPA choice for bleed treatment in PwHABI. Ciolek et al. have compared the cost per day for treating a severe BE in an 80-kg patient using aPCC (200 U/kg/day), eptacog alfa (90 µg/kg q2h), or eptacog beta (225 µg/kg followed by 75 µg/kg 6 h later and 75 µg/kg q2h thereafter) [Citation87]. Based on an average wholesale price of $2.87/unit, $2,880/mg and $2,748/mg for aPCC, eptacog alfa and eptacog beta, respectively, the daily cost for severe bleed treatment was estimated to be $45,920 for aPCC, $241,920 using eptacog alfa, and $197,856 using eptacog beta [Citation87].
Alexander et al. developed a decision analytic model to compare utilization costs between eptacog beta and eptacog alfa for treatment of acute mild or moderate BEs in PwHABI over a 1-year timeframe [Citation88]. While head-to-head comparisons of the two BPAs in clinical trials were not available, the analysis drew upon BPA usage and reported efficacy outcomes from PERSEPT 1 and the eptacog alfa treatment arm of the adept™ 2 trial [Citation32,Citation68]. The cost model took into consideration HA and HB prevalence, the proportion of PwHABI requiring BPAs, dosing regimens of 90 µg/kg q3h for eptacog alfa and the 75 and 225 µg/kg IDRs for eptacog beta, and the wholesale acquisition unit costs for each product (priced by the microgram). Additional model inputs included expenditures for emergency department visits and hospital admissions associated with treatment failures and rebleeding [Citation88].
The model predicted annual product utilization reductions of 48% and 33% for 75 and 225 µg/kg eptacog beta IDRs, respectively, over that seen for 90 µg/kg q3h eptacog alfa. The corresponding annual cost reduction was 86% for the 75 µg/kg eptacog beta IDR and 77% for the 225 µg/kg eptacog beta IDR over the cost of eptacog alfa usage. Lower product acquisition expenditures for eptacog beta over eptacog alfa also contributed to the predicted annual cost savings [Citation88]. In accord with the study by Ciolek et al. [Citation87], this analysis suggests that eptacog beta might be preferred over eptacog alfa from a cost standpoint where a rFVIIa is indicated for bleed treatment.
In the real-world setting of eptacog beta use, Youkhana et al. calculated total micrograms utilized and cost of product in comparing the utilization of eptacog alfa and then eptacog beta during 6 months of prophylaxis, and reported that eptacog beta demonstrated favorable clinical outcome and cost reduction by 42% [Citation89].
While 270 µg/kg eptacog alfa is approved for use in the EU, none of the studies described above estimated bleed treatment costs between 270 µg/kg eptacog alfa and either 75 or 225 µg/kg eptacog beta.
8. Stability
Eptacog beta is supplied as a room temperature-stable lyophilized powder in 1 or 5 mg single-use vials and reconstituted with sterile water from prefilled syringes. Prior to reconstitution, eptacog beta can be stored between 2°C and 30°C (36–86°F). Following reconstitution, eptacog beta can be stored between 2°C and 30°C (36–86°F) for up to 4 h [Citation20].
9. Conclusion
Inhibitor development in hemophilia patients represents a serious treatment challenge. While bleed treatment using aPCC or eptacog alfa in PwHABI can be effective, outcomes are unpredictable owing to the variable inter-patient and intra-patient efficacy for each of these agents [Citation27]. Given the differential response to aPCC and eptacog alfa experienced by PwHABI, eptacog beta was developed to give PwHABI and their physicians an additional treatment option. Eptacog beta was FDA-approved in 2020 for bleed treatment in PwHABI 12 years and older [Citation20], ending a 20-year gap in regulatory approvals for new BPAs to treat PwHABI. Eptacog beta is produced in transgenic rabbits and enriched in N-linked monosialylated biantennary glycans with α2,6 sialic acid linkages, a molecular feature that may play a role in the observed improvements in eptacog beta binding to EPCR and activated platelets, relative to eptacog alfa [Citation37,Citation38,Citation62].
The treatment success proportions for the 75 and 225 µg/kg IDRs in PERSEPT 1 at 12 hours for mild or moderate BEs (91% for the 225 µg/kg IDR and 82% for 75 µg/kg IDR) [Citation20] suggest greater efficacy with the 225 µg/kg IDR. These findings, together with the TGA, ROTEM, and other PD data from the eptacog beta phase 1b study [Citation66], support the concept that higher eptacog beta dosing increases the rate of thrombin generation to better promote formation of a stable hemostatic clot and resolution of BEs. Most BEs treated with the 225 µg/kg IDR during PERSEPT 1 (84%) were successfully controlled with a single dose [Citation72], indicating that the 225 µg/kg IDR potentially offers PwHABI the relief and convenience of fewer infusions to achieve hemostasis. Nearly all mild or moderate BEs were resolved at 24 h in PERSEPT 1 and PERSEPT 2, and bleed recurrence prior to 24 h was low (0.2% of mild or moderate BEs in PERSEPT 1 and 1.5% in PERSEPT 2) [Citation68,Citation73]. A majority of the procedures in PERSEPT 3 were successfully completed [Citation80].
Subjects in the PERSEPT program experienced no thrombotic events or allergic, hypersensitivity, or anaphylactic reactions, and no neutralizing antibodies were found [Citation69]. The results establish a favorable safety profile for eptacog beta in the treatment of bleeding and perioperative management of PwHABI. This favorable safety profile is likely due to FVIIa mechanisms that mediate localized activity at the site of vessel injury [Citation51,Citation52], rapid circulatory clearance [Citation57,Citation58], and other mechanisms that dampen the likelihood of systemic coagulation [Citation49,Citation53–56]. With the efficacy and safety demonstrated during the PERSEPT trials, eptacog beta offers clinicians and PwHABI an important option for BE treatment and perioperative care.
10. Expert opinion
In the past, patients with hemophilia had limited options for both prophylaxis and treatment of BEs [Citation90]. From the 1990s to the early 2010s, the choices for prophylaxis and treatment of bleeding were limited to clotting factor concentrates (CFCs), predominantly recombinant versions, that were quite similar to one another in their biochemical properties [Citation90,Citation91]. Since the 2010s, there has been the addition of extended half-life CFCs [Citation92], emicizumab [Citation23], and recently gene therapy (for HB) [Citation93] and a next-generation FVIII CFC with a substantially longer half-life (efanesoctocog alfa) [Citation94]. Furthermore, this menu of options is set to increase further as soon as this year and will continue to expand. Unfortunately, for PwHABI, the options have remained limited. While aPCC and eptacog alfa are approved in a limited number of countries for prophylaxis, their efficacy and treatment burden leave a lot to be desired. For patients with HA, emicizumab has certainly revolutionized the care for PwHAI and has become the standard of prophylactic care for such patients [Citation95]. Importantly however, emicizumab is only approved for HA patients and as effective as emicizumab has been shown to be, patients still experience occasional bleeding episodes requiring BPA, as well as need BPA for surgery and trauma [Citation95]. As for bleed treatment and surgical prophylaxis, the options for PwHABI have been limited to aPCC and eptacog alfa. Published studies comparing these two agents have shown variable responses between patients, and even variable responses within patients from one bleed type to another [Citation25,Citation27]. Notably, between 1999 and 2020 when eptacog beta was licensed, no new bleed treatment medications were approved for PwHABI.
Therefore, the question really is: what does eptacog beta offer that the previously available BPAs do not? For starters, having a second rFVIIa option simply offers more choice for patients – something that non-inhibitor patients with both hemophilia A and B have in abundance. Nevertheless, more choice alone is not enough to justify its use. As described above, eptacog beta is not merely a biosimilar: eptacog beta is structurally similar to eptacog alfa, but has post-translational glycan modifications that appear to be driving pharmacologic functional differences with respect to receptor affinities that could be impacting its mechanism of action [Citation37,Citation38,Citation62]. Most notable is the substantial increase in affinity for platelets as well as a higher affinity for EPCR on endothelial cells at therapeutic plasma concentrations [Citation62], which may impact its clinical efficacy. More practically, eptacog beta offers 2 IDRs that have been evaluated in the clinical development program and both doses are licensed, which is in contradistinction to eptacog alfa where only one dose is licensed in the U.S [Citation20,Citation67]. Furthermore, the PERSEPT clinical trials demonstrated a rapid treatment response, especially to the high dose IDR, with the ability to treat most bleeds with just one infusion [Citation72]. This property has gained increased importance in the emicizumab era since many patients are more challenged with venous access, given that emicizumab is subcutaneously administered and some patients either never acquired venipuncture skills (relying on central venous catheters) or have had those skills lost or diminished. Therefore, a single infusion for bleed management at home or in an outpatient setting has gained additional importance.
Lastly, what is to be made of the fact that eptacog beta did not meet its clinical endpoint (the OPC) in the pediatric trial, and is thus not approved for use in children <12 years of age? In the opinion of the authors, this is both unfortunate and unwarranted. As described above, the OPC was set based on the limited previously available data drawn primarily from adult studies. Second, if not for one subject who experienced a disproportionate number of bleeds and who appeared to have a lesser (if not absent) response to eptacog beta, the OPC would have been met. While we must be cautious not to cherry-pick the data we like while making clinical decisions, we can and often do take a more practical approach to evaluating clinical trial data. Given the earlier discussion, we do believe that eptacog beta is effective for bleed treatment in children <12 years of age and feel comfortable using it in that patient population. Although more data will be welcome to provide additional support for its use in this population, such data may be nearly impossible to collect given that bleeding rates and BPA use have plummeted since emicizumab was licensed, and relying on only HB patients with inhibitors to conduct a study for this purpose would likely be futile.
Thus, to summarize, eptacog beta offers an additional option for bleed treatment and surgical bleed prevention in PwHABI and has been demonstrated to be effective and safe in patients of all ages with both hemophilia A and B. Eptacog beta is in its first years of approval and use, and we anticipate that real-world experience will show that eptacog beta can be used successfully in all settings where eptacog alfa has been used. Clinicians should consider eptacog beta as a treatment option with advantages of safety over aPCC in hemophilia patients on emicizumab and clinical trial data supporting rapid and durable treatment response with as little as one infusion for the majority of acute bleeding events.
Article highlights
Bypassing agents are needed for bleed management in PwHABI, as factor replacement therapy becomes ineffective in these patients.
Eptacog beta is a recently-approved recombinant activated human factor VII bypassing agent for treating bleeding episodes in PwHABI 12 years of age and older.
The pivotal phase 3 PERSEPT 1 trial demonstrated the efficacy and safety of two initial dose regimens (IDRs) of 75 and 225 µg/kg eptacog beta in adult and adolescent PwHABI. Treatment success proportions at 12 h post-initial infusion were 82% for the 75 µg/kg IDR and 91% for the 225 µg/kg IDR.
Eighty-four percent of mild or moderate bleeds treated with the 225 µg/kg IDR resolved with a single 225 µg/kg dose in PERSEPT 1, and bleed recurrence prior to 24 h was low (0.2%).
PERSEPT 2 was a phase 3 trial of eptacog beta for acute bleed treatment in pediatric PwHABI younger than 12 years of age. A majority of mild or moderate bleeding episodes were successfully treated with eptacog beta treatment by 12 h, and nearly all bleeding episodes resolved by 24 h.
A majority of minor and major procedures were successfully completed using eptacog beta to manage bleeding during perioperative care in PwHAI during the PERSEPT 3 trial.
A favorable safety profile for eptacog beta was established during the PERSEPT clinical program.
Eptacog beta should be considered as a treatment option for acute bleed treatment and surgical bleed prevention in all PwHABI.
Declaration of interest
SW Pipe has received consulting fees from Apcintex, ASC Therapeutics, Bayer, BioMarin, CSL Behring, Equilibra Bioscience, GenVentiv, HEMA Biologics, Freeline, LFB, Novo Nordisk, Pfizer, Regeneron/Intellia, Roche/Genentech, Sanofi, Takeda, Spark Therapeutics, and UniQure.
AL Dunn has received consulting fees from Genentech, Kedrion, CSL Behring, BioMarin, uniQure, Sanofi, Novo Nordisk, and HEMA Biologics; has received research funding from Takeda, BioMarin, Freeline, Novo Nordisk, Sanofi, Pfizer, Genentech, and the American Society of Thrombosis and Hemostasis; is on the board of the World Federation of Hemophilia U.S.A.; and is Chair of MASAC of the National Hemophilia Foundation.
G Young has received consulting fees from Apcintex, BioMarin, CSL Behring, Genentech/Roche, HEMA Biologics/LFB, Novo Nordisk, Octapharma, Pfizer, Sanofi Genzyme, Spark, and Takeda, and funds for research support from Genentech/Roche and Takeda.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
One peer reviewer has received speaker fees from Bayer, NovoNordisk, Takeda, and Roche; and grants for scientific events from Bayer, NovoNordisk, Takeda, and Roche. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Scientific accuracy review
HEMA Biologics provided a scientific accuracy review at the request of the journal editor and advised that SEVENFACT has not been approved or cleared by FDA as safe and effective for the uses discussed in this article outside of BE treatment in adult and adolescent hemophilia A or B patients with inhibitors (≥12 years of age).
Acknowledgments
The authors would like to thank Sonia Nasr, PhD and Thomas Wilkinson, PhD from GLOVAL LLC for medical writing assistance during the preparation of this manuscript.
Notes
1. A ‘high anamnestic response’ is interpreted as the subject having a history of increased inhibitor titer in the high titer range (≥5 BU/mL) upon exposure to factor replacement product.
References
- Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019 Oct 15;171(8):540–546. doi: 10.7326/M19-1208
- Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020 Aug;26(Suppl 6):1–158.
- Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003 Jul;9(4):418–435. doi: 10.1046/j.1365-2516.2003.00780.x
- DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007 Aug;138(3):305–315. doi: 10.1111/j.1365-2141.2007.06657.x
- Male C, Andersson NG, Rafowicz A, et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2021 Jan 9;106(1):123–129.
- Oladapo AO, Lu M, Walsh S, et al. Inhibitor clinical burden of disease: a comparative analysis of the CHESS data. Orphanet J Rare Dis. 2018 Nov 9;13(1):198. doi: 10.1186/s13023-018-0929-9
- Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007 Sep;13(5):606–612. doi: 10.1111/j.1365-2516.2007.01518.x
- Nuss R, Soucie JM, Evatt B, et al. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol. 2001 Sep;68(1):37–42. doi: 10.1002/ajh.1146
- Soucie JM, Cianfrini C, Janco RL, et al. Joint range-of-motion limitations among young males with hemophilia: prevalence and risk factors. Blood. 2004 Apr 1;103(7):2467–2473.
- Walsh CE, Soucie JM, Miller CH, et al. Impact of inhibitors on hemophilia A mortality in the United States. Am J Hematol. 2015 May;90(5):400–405. doi: 10.1002/ajh.23957
- Eckhardt CL, Loomans JI, van Velzen AS, et al. Inhibitor development and mortality in non-severe hemophilia A. J Thromb Haemost. 2015 Jul;13(7):1217–1225. doi: 10.1111/jth.12990
- Gringeri A, Mantovani LG, Scalone L, et al. Cost of care and quality of life for patients with hemophilia complicated by inhibitors: the COCIS study group. Blood. 2003 Oct 1;102(7):2358–2363. doi: 10.1182/blood-2003-03-0941
- Guh S, Grosse SD, McAlister S, et al. Healthcare expenditures for males with haemophilia and employer-sponsored insurance in the United States, 2008. Haemophilia. 2012 Mar;18(2):268–275. doi: 10.1111/j.1365-2516.2011.02692.x
- Lindvall K, von Mackensen S, Elmstahl S, et al. Increased burden on caregivers of having a child with haemophilia complicated by inhibitors. Pediatr Blood Cancer. 2014 Apr;61(4):706–711. doi: 10.1002/pbc.24856
- Erturan G, Guevel B, Alvand A, et al. Over two decades of orthopaedic surgery in patients with inhibitors-quantifying the complication of bleeding. Haemophilia. 2019 Jan;25(1):21–32. doi: 10.1111/hae.13647
- Rocino A, Franchini M, Coppola A. Treatment and prevention of bleeds in haemophilia patients with inhibitors to factor VIII/IX. J Clin Med. 2017 Apr 17;6(4):46. doi: 10.3390/jcm6040046
- Young G. How I treat children with haemophilia and inhibitors. Br J Haematol. 2019 Aug;186(3):400–408. doi: 10.1111/bjh.15942
- Négrier C, Gomperts ED, Oldenburg J. The history of FEIBA: a lifetime of success in the treatment of haemophilia complicated by an inhibitor. Haemophilia. 2006;12(s5):4–13. doi: 10.1111/j.1365-2516.2006.01379.x
- Hedner U. Recombinant activated factor VII: 30 years of research and innovation. Blood Rev. 2015 Jun;29(Suppl 1):S4–8.
- SEVENFACT® [ Package Insert]. Louisville, KY: HEMA Biologics, LLC; 2022.
- Turecek PL, Varadi K, Gritsch H, et al. FEIBA: mode of action. Haemophilia. 2004 Sep;10(Suppl 2):3–9.
- Abshire T, Kenet G. Recombinant factor VIIa: review of efficacy, dosing regimens and safety in patients with congenital and acquired factor VIII or IX inhibitors. J Thromb Haemost. 2004 Jun;2(6):899–909. doi: 10.1111/j.1538-7836.2004.00759.x
- Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017 Aug 31;377(9):809–818. doi: 10.1056/NEJMoa1703068
- Wexler M Concizumab approved in Canada for hemophilia B with inhibitors. Hemophilia News Today 2023 Apr 19 [cited Jul 20, 2023]; Available from: https://hemophilianewstoday.com/news/hemophilia-b-therapy-concizumab-be-sold-canada-alhemo/
- Astermark J, Donfield SM, DiMichele DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven Comparative (FENOC) study. Blood. 2007 Jan 15;109(2):546–551. doi: 10.1182/blood-2006-04-017988
- Gomperts ED, Astermark J, Gringeri A, et al. From theory to practice: applying current clinical knowledge and treatment strategies to the care of hemophilia A patients with inhibitors. Blood Rev. 2008 Feb;22(Suppl 1):S1–11.
- Berntorp E. Differential response to bypassing agents complicates treatment in patients with haemophilia and inhibitors. Haemophilia. 2009 Jan;15(1):3–10. doi: 10.1111/j.1365-2516.2008.01931.x
- Schneiderman J, Nugent DJ, Young G. Sequential therapy with activated prothrombin complex concentrate and recombinant factor VIIa in patients with severe haemophilia and inhibitors. Haemophilia. 2004;10(4):347–351. doi: 10.1111/j.1365-2516.2004.00912.x
- Gringeri A, Fischer K, Karafoulidou A, et al. Sequential combined bypassing therapy is safe and effective in the treatment of unresponsive bleeding in adults and children with haemophilia and inhibitors. Haemophilia. 2011 Jul;17(4):630–635. doi: 10.1111/j.1365-2516.2010.02467.x
- Teitel J, Berntorp E, Collins P, et al. A systematic approach to controlling problem bleeds in patients with severe congenital haemophilia A and high-titre inhibitors. Haemophilia. 2007 May;13(3):256–263. doi: 10.1111/j.1365-2516.2007.01449.x
- Ljung R, Karim FA, Saxena K, et al. 40K glycoPEGylated, recombinant FVIIa: 3-month, double-blind, randomized trial of safety, pharmacokinetics and preliminary efficacy in hemophilia patients with inhibitors. J Thromb Haemost. 2013 Jul;11(7):1260–1268. doi: 10.1111/jth.12237
- Lentz SR, Ehrenforth S, Karim FA, et al. Recombinant factor VIIa analog in the management of hemophilia with inhibitors: results from a multicenter, randomized, controlled trial of vatreptacog alfa. J Thromb Haemost. 2014 Aug;12(8):1244–1253. doi: 10.1111/jth.12634
- Mahlangu J, Paz P, Hardtke M, et al. TRUST trial: BAY 86-6150 use in haemophilia with inhibitors and assessment for immunogenicity. Haemophilia. 2016 Nov;22(6):873–879. doi: 10.1111/hae.12994
- Faraj A, Knudsen T, Desai S, et al. Phase III dose selection of marzeptacog alfa (activated) informed by population pharmacokinetic modeling: a novel hemostatic drug. CPT Pharmacometrics Syst Pharmacol. 2022 Dec;11(12):1628–1637. doi: 10.1002/psp4.12872
- Catalyst Biosciences, Inc. Catalyst Biosciences announces change in corporate strategy [Press Release]. 2021 Nov 12 [cited 2023 Jul 12]; Available from: https://ir.catalystbiosciences.com/news-releases/news-release-details/catalyst-biosciences-announces-change-corporate-strategy
- European Medicines Agency. CEVENFACTA® summary of product characteristics; 2022 [cited May 22, 2023]; Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/cevenfacta
- Chevreux G, Faid V, Scohyers JM, et al. N-/O-glycosylation analysis of human FVIIa produced in the milk of transgenic rabbits. Glycobiology. 2013 Dec;23(12):1531–1546. doi: 10.1093/glycob/cwt085
- Chevreux G, Tilly N, Leblanc Y, et al. Biochemical characterization of LR769, a new recombinant factor VIIa bypassing agent produced in the milk of transgenic rabbits. Haemophilia. 2017 Jul;23(4):e324–e334. doi: 10.1111/hae.13253
- Vadivel K, Bajaj SP. Structural biology of factor VIIa/tissue factor initiated coagulation. Front Biosci (Landmark Ed). 2012 Jun 1;17(7):2476–2494.
- Jurlander B, Thim L, Klausen NK, et al. Recombinant activated factor VII (rFVIIa): characterization, manufacturing, and clinical development. Semin Thromb Hemost. 2001 Aug;27(4):373–384. doi: 10.1055/s-2001-16890
- Bernardi F, Mariani G. Biochemical, molecular and clinical aspects of coagulation factor VII and its role in hemostasis and thrombosis. Haematologica. 2021 Feb 1;106(2):351–362. doi: 10.3324/haematol.2020.248542
- Hansson K, Stenflo J. Post-translational modifications in proteins involved in blood coagulation. J Thromb Haemost. 2005 Dec;3(12):2633–2648. doi: 10.1111/j.1538-7836.2005.01478.x
- Iino M, Foster DC, Kisiel W. Functional consequences of mutations in Ser-52 and Ser-60 in human blood coagulation factor VII. Arch Biochem Biophys. 1998 Apr 15;352(2):182–192. doi: 10.1006/abbi.1998.0595
- Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemost. 2005 Oct;3(10):2185–2192.
- Riedl J, Ay C, Pabinger I. Platelets and hemophilia: a review of the literature. Thromb Res. 2017 Jul;155:131–139. doi: 10.1016/j.thromres.2017.05.013
- Hoffman M, Monroe DM 3rd, Roberts HR. Activated factor VII activates factors IX and X on the surface of activated platelets: thoughts on the mechanism of action of high-dose activated factor VII. Blood Coagul Fibrinolysis. 1998 Mar;9(Suppl 1):S61–5.
- Monroe DM. Further understanding of recombinant activated factor VII mode of action. Semin Hematol. 2008 Apr;45(2 Suppl 1):S7–S11.
- Keshava S, Sundaram J, Rajulapati A, et al. Factor VIIa interaction with EPCR modulates the hemostatic effect of rFVIIa in hemophilia therapy: mode of its action. Blood Adv. 2017 Jun 27;1(15):1206–1214.
- Dahlbäck B, Villoutreix BO. The anticoagulant protein C pathway. FEBS Lett. 2005 Jun 13;579(15):3310–3316. doi: 10.1016/j.febslet.2005.03.001
- Das K, Keshava S, Ansari SA, et al. Factor VIIa induces extracellular vesicles from the endothelium: a potential mechanism for its hemostatic effect. Blood. 2021 Jun 17;137(24):3428–3442. doi: 10.1182/blood.2020008417
- Monroe DM, Hoffman M, Oliver JA, et al. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol. 1997 Dec;99(3):542–547. doi: 10.1046/j.1365-2141.1997.4463256.x
- Ruf W, Rehemtulla A, Morrissey JH, et al. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem. 1991 Aug 25;266(24):16256. doi: 10.1016/S0021-9258(18)98544-3
- Maroney SA, Haberichter SL, Friese P, et al. Active tissue factor pathway inhibitor is expressed on the surface of coated platelets. Blood. 2007 Mar 1;109(5):1931–1937. doi: 10.1182/blood-2006-07-037283
- Mast AE. Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol. 2016 Jan;36(1):9–14. doi: 10.1161/ATVBAHA.115.305996
- Vatsyayan R, Kothari H, Mackman N, et al. Inactivation of factor VIIa by antithrombin in vitro, ex vivo and in vivo: role of tissue factor and endothelial cell protein C receptor. Plos One. 2014;9(8):e103505. doi: 10.1371/journal.pone.0103505
- Olson ST, Richard B, Izaguirre G, et al. Molecular mechanisms of antithrombin-heparin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie. 2010 Nov;92(11):1587–1596.
- Clark CA, Vatsyayan R, Hedner U, et al. Endothelial cell protein C receptor-mediated redistribution and tissue-level accumulation of factor VIIa. J Thromb Haemost. 2012 Nov;10(11):2383–2391. doi: 10.1111/j.1538-7836.2012.04917.x
- Magisetty J, Pendurthi UR, Madhunapantula SV, et al. Increased accumulation and retention of rhFVIIa (eptacog beta) in knee joints of hemophilia A mice compared to wild-type mice. Thromb Haemost. 2019 Aug;119(8):1283–1294. doi: 10.1055/s-0039-1688907
- Sutkeviciute I, Mistiniene E, Sereikaite J, et al. The influence of different glycosylation patterns on factor VII biological activity. Biochimie. 2009 Sep;91(9):1123–1130. doi: 10.1016/j.biochi.2009.05.015
- Klausen NK, Bayne S, Palm L. Analysis of the site-specific asparagine-linked glycosylation of recombinant human coagulation factor VIIa by glycosidase digestions, liquid chromatography, and mass spectrometry. Mol Biotechnol. 1998 Jun;9(3):195–204.
- Thim L, Bjoern S, Christensen M, et al. Amino acid sequence and posttranslational modifications of human factor VIIa from plasma and transfected baby hamster kidney cells. Biochemistry. 1988 Oct 4;27(20):7785–7793. doi: 10.1021/bi00420a030
- Grandoni J, Perret G, Forier C. Kinetic analysis and binding studies of a new recombinant human factor VIIa for treatment of haemophilia. Haemophilia. 2017 Mar;23(2):300–308. doi: 10.1111/hae.13110
- Ghosh S, Pendurthi UR, Steinoe A, et al. Endothelial cell protein C receptor acts as a cellular receptor for factor VIIa on endothelium. J Biol Chem. 2007 Apr 20;282(16):11849–11857. doi: 10.1074/jbc.M609283200
- Rao LVM, Esmon CT, Pendurthi UR. Endothelial cell protein C receptor: a multiliganded and multifunctional receptor. Blood. 2014 Sep 4;124(10):1553–1562. doi: 10.1182/blood-2014-05-578328
- Sen P, Gopalakrishnan R, Kothari H, et al. Factor VIIa bound to endothelial cell protein C receptor activates protease activated receptor-1 and mediates cell signaling and barrier protection. Blood. 2011 Mar 17;117(11):3199–3208. doi: 10.1182/blood-2010-09-310706
- Ducore J, Lawrence JB, Simpson M, et al. Safety and dose-dependency of eptacog beta (activated) in a dose escalation study of non-bleeding congenital haemophilia A or B patients, with or without inhibitors. Haemophilia. 2017 Nov;23(6):844–851. doi: 10.1111/hae.13357
- NOVOSEVEN® RT [Package Insert]. Bagsværd, Denmark: Novo Nordisk A/S; 2020.
- Wang M, Lawrence JB, Quon DV, et al. PERSEPT 1: a phase 3 trial of activated eptacog beta for on-demand treatment of haemophilia inhibitor-related bleeding. Haemophilia. 2017 Nov;23(6):832–843. doi: 10.1111/hae.13301
- Escobar M, Castaman G, Boix SB, et al. The safety of activated eptacog beta in the management of bleeding episodes and perioperative haemostasis in adult and paediatric haemophilia patients with inhibitors. Haemophilia. 2021 Nov;27(6):921–931. doi: 10.1111/hae.14419
- Journeycake JM, Hermans C, Ducore JM, et al. Single 225µg/kg dose treatment with eptacog beta (factor VIIa, recombinant) results in rapid hemostasis in joint bleeds for persons with hemophilia A or B with inhibitors: a PERSEPT1 subset analysis [abstract]. Blood. 2020;136(Supplement 1):2–3.
- Reding MT, Mancuso ME, Acharya S, et al. Eptacog beta efficacy in treating mild or moderate bleeds in target joints of individuals with hemophilia A or B and inhibitors in PERSEPT 1 [abstract]. Blood. 2022;140(Supplement 1):5597–5599. doi: 10.1182/blood-2022-156110
- Boggio L, Mancuso ME, Acharya S, et al. Eptacog beta efficacy at 24 hours post-infustion for mild or moderate bleeds in individuals with hemophilia A or B and inhibitors [abstract]. Am J Hematol. 2023 Feb;98:E13.
- Pipe SW, Hermans C, Chitlur M, et al. Eptacog beta efficacy and safety in the treatment and control of bleeding in paediatric subjects (<12 years) with haemophilia A or B with inhibitors. Haemophilia. 2022;28:548–556. doi: 10.1111/hae.14563
- Young G, Shapiro AD, Walsh CE, et al. Patient/caregiver-reported recombinant factor VIIa (rFVIIa) dosing: home treatment of acute bleeds in the Dosing Observational Study In Hemophilia (DOSE). Haemophilia. 2012 May;18(3):392–399. doi: 10.1111/j.1365-2516.2011.02704.x
- Valentino LA, Walsh CE, Reding MT, et al. Patient- and caregiver-reported bleeding symptoms and reasons for starting and stopping treatment with recombinant factor VIIa: analysis of the Dosing Observational Study in Haemophilia (DOSE). Haemophilia. 2012 Jul;18(4):554–560. doi: 10.1111/j.1365-2516.2012.02762.x
- Amby LK, Seremetis S, Obergfell A, et al. Challenges of defining reliable clinical surrogate end points in haemophilia trials: a critical review. Blood Coagul Fibrinolysis. 2009 Oct;20(7):488–493. doi: 10.1097/MBC.0b013e32832c8803
- Gruppo RA, Kessler CM, Neufeld EJ, et al. Assessment of individual dose utilization vs. physician prescribing recommendations for recombinant activated factor VII (rFVIIa) in paediatric and adult patients with congenital haemophilia and alloantibody inhibitors (CHwI): the Dosing Observational Study In Hemophilia (DOSE). Haemophilia. 2013 Jul;19(4):524–532. doi: 10.1111/hae.12113
- Coppola A, Windyga J, Tufano A, et al. Treatment for preventing bleeding in people with haemophilia or other congenital bleeding disorders undergoing surgery. Cochrane Database Syst Rev. 2015 Feb;9(2):CD009961. doi: 10.1002/14651858.CD009961.pub2
- Mingot-Castellano ME, Alvarez-Roman MT, Lopez-Fernandez MF, et al. Spanish consensus guidelines on prophylaxis with bypassing agents for surgery in patients with haemophilia and inhibitors. Eur J Haematol. 2016 May;96(5):461–474. doi: 10.1111/ejh.12730
- Escobar M, Luck J, Averianov Y, et al. PERSEPT 3: a phase 3 clinical trial to evaluate the haemostatic efficacy of eptacog beta (recombinant human FVIIa) in perioperative care in subjects with haemophilia A or B with inhibitors. Haemophilia. 2021 Nov;27(6):911–920. doi: 10.1111/hae.14418
- Ju HY, Jang HL, Park YS. The efficacy of bypassing agents in surgery of hemophilia patients with inhibitors. Blood Res. 2015 Sep;50(3):173–178. doi: 10.5045/br.2015.50.3.173
- Bossard D, Carrillon Y, Stieltjes N, et al. Management of haemophilic arthropathy. Haemophilia. 2008;14(s4):11–19. doi: 10.1111/j.1365-2516.2008.01734.x
- Levy GG, Asikanius E, Kuebler P, et al. Safety analysis of rFVIIa with emicizumab dosing in congenital hemophilia A with inhibitors: experience from the HAVEN clinical program. J Thromb Haemost. 2019 May 24;17(9):1470–1477. doi: 10.1111/jth.14491
- Maria A, Alessandro C, Declan N, et al. Hemorrhagic and thrombotic adverse events associated with emicizumab and extended half-life factor VIII replacement drugs: EudraVigilance data of 2021. J Thromb Haemost. 2023 Mar;21(3):546–552.
- Grandoni J, Duretz V, Bonzo D, et al. Exploratory in vitro evaluation of thrombin generation of eptacog beta (recombinant human fviia) and emicizumab in congenital haemophilia A plasma. Haemophilia. 2021 Mar;27(2):321–328. doi: 10.1111/hae.14253
- Medical and Scientific Advisory Council (MASAC). MASAC recommendations concerning products licensed for the treatment of hemophilia and other bleeding disorders. MASAC Document #272; (NY), NY: National Hemophilia Foundation; 2022.
- Ciolek AM, Arnall J, Moore DC, et al. Eptacog beta for bleeding treatment and prevention in congenital hemophilia A and B with inhibitors: a review of clinical data and implications for clinical practice. Ann Pharmacother. 2022 Jul;56(7):831–838. doi: 10.1177/10600280211049394
- Alexander WA, Jensen I, Hathway J, et al. Bleeding in patients with hemophilia who have inhibitors: modeling US medical system utilization and cost avoidance between recombinant factor VIIa products with different clinical dosing requirements. J Manag Care Spec Pharm. 2022 May;28(5):518–527. doi: 10.18553/jmcp.2022.21197
- Youkhana K, Abajas Y. Successful use of eptacog beta prophylaxis in an adolescent with severe hemophilia B with high-titer inhibitor resulting in decreased bleeding episodes and cost of treatment [abstract]. Am J Hematol. 2023 Feb;98:E122.
- Berntorp E, Shapiro AD. Modern haemophilia care. Lancet. 2012 Apr 14;379(9824):1447–1456. doi: 10.1016/S0140-6736(11)61139-2
- Pipe SW. The promise and challenges of bioengineered recombinant clotting factors. J Thromb Haemost. 2005 Aug;3(8):1692–1701. doi: 10.1111/j.1538-7836.2005.01367.x
- Berntorp E, Fischer K, Hart DP, et al. Haemophilia. Nat Rev Dis Primers. 2021 Jun 24;7(1):45. doi: 10.1038/s41572-021-00278-x
- Pipe SW, Leebeek FWG, Recht M, et al. Gene therapy with etranacogene dezaparvovec for hemophilia B. N Engl J Med. 2023 Feb 23;388(8):706–718. doi: 10.1056/NEJMoa2211644
- von Drygalski A, Chowdary P, Kulkarni R, et al. Efanesoctocog alfa prophylaxis for patients with severe hemophilia A. N Engl J Med. 2023 Jan 26;388(4):310–318.
- Linari S, Castaman G. Concomitant use of rFVIIa and emicizumab in people with hemophilia A with inhibitors: current perspectives and emerging clinical evidence. Ther Clin Risk Manag. 2020;16:461–469.