
ABSTRACT
Introduction
Comprehensive information about atypical hemolytic uremic syndrome (aHUS) is relatively scarce outside of Europe and North America. This narrative review assembles available published data about the clinical presentation and management of aHUS in Latin America.
Areas covered
A search conducted in February 2023 of the MEDLINE (from inception), Embase (from inception), and LILACS/IBECS (1950 to 2023) databases using search terms ‘atypical hemolytic uremic syndrome’ and ‘Latin America’ and their variations retrieved 51 records (full papers and conference abstracts) published in English, Spanish, or Portuguese. After de-duplication, manual screening of titles/abstracts and addition of author-known articles, 25 articles were included of which 17 (68%) are full papers. All articles were published during the years 2013–2022. Articles include cohort studies, a registry analysis, and case reports from Argentina, Brazil, Chile and Columbia. Overall, Latin American patients with aHUS present the classic epidemiological, clinical, and genetic characteristics associated with this condition as described in other world regions. Depending on the country and time of reporting, aHUS in Latin America was treated mainly with plasma therapy and/or eculizumab. Where reported, eculizumab substantially improved aHUS-related outcomes in almost all adult and pediatric patients.
Expert opinion
Eculizumab has dramatically altered the natural course of aHUS, improving prognosis and patient outcomes. Addressing economic challenges and investing in healthcare infrastructure will be essential to implement strategies for timely detection and early treatment of aHUS in Latin America.
1. Introduction
Primary atypical hemolytic uremic syndrome (aHUS) is a rare form of thrombotic microangiopathy (TMA) characterized by microangiopathic hemolytic anemia, thrombocytopenia, and end-organ damage, primarily acute kidney injury [Citation1,Citation2]. The estimated incidence is 0.5 to 2 cases per million person-years [Citation1,Citation3]. Irrespective of etiology, the pathology of aHUS involves formation of microvascular thrombi and occlusion of glomerular capillaries [Citation1–4]. aHUS must be identified and treated promptly to avoid progression to kidney failure and multiple end-organ damage.
Primary aHUS arises from genetic or acquired abnormalities which result in uncontrolled activation of the complement pathway [Citation1–4]. Additionally, there are numerous clinical scenarios (e.g. infection, pregnancy, malignancy, autoimmune diseases, use of certain toxic medications, metabolic conditions, transplants, hematological diseases, hemodynamic alterations) that cause TMAs which can mimic or unmask aHUS. Secondary HUS is distinct from primary aHUS but with some overlap as complement dysregulation has been described in several of these settings [Citation3,Citation4].
Interest in aHUS has escalated since approval in 2011 of eculizumab, the first-in-class complement component 5 (C5) inhibitor. A Global aHUS Registry (NCT01522183) was initiated in 2012 with the aim of characterizing the natural history of aHUS and reporting treatment outcomes for a wide range of patients [Citation5]. Approximately 1900 patients, although predominantly from Europe and North America, were enrolled in the registry as of October 2020 [Citation6]. As such, comprehensive information about aHUS is scarce in other world regions, including Latin America. Based on a systematic literature search, this narrative review provides a brief overview of aHUS and summarizes available data on the clinical presentation and treatment of aHUS in Latin American countries.
2. Overview of atypical hemolytic uremic syndrome
2.1. Pathophysiology and clinical presentation
The hereditary or sporadic/acquired form of aHUS (primary aHUS) is caused by genetic mutations or acquired autoantibodies against specific complement regulatory proteins (e.g. factor H) that result in uncontrolled activation of the complement alternative pathway [Citation1,Citation2]. In brief, spontaneous hydrolysis of the complement protein C3 leads to formation of C5 convertases which split C5 into C5a, a strong chemoattractant, and C5b, a precursor of the lytic cell membrane attack complex (MAC) C5b-9 () [Citation7]. Failure to regulate complement allows for abnormal formation of MAC on endothelial cells, particularly within the renal vasculature with deleterious effect [Citation8].
Figure 1. Complement alternative pathway. CFH, CFI and MCP are complement regulatory factors. C3, CFB and CFD are complement activation factors. C3, complement component 3; CFB, complement factor B; CFD, complement factor D; CFH, complement factor H; CFI, complement factor I; MAC, membrane attack complex; MCP, membrane cofactor protein.
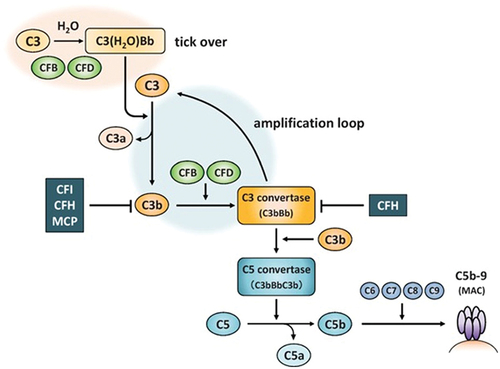
Renal manifestations of aHUS include elevated creatinine levels, decreased estimated glomerular filtration rate (eGFR), renal hypertension, proteinuria, and glomerular hematuria. Extra-renal manifestations occur in about 10% to 20% of patients and include gastrointestinal symptoms (e.g. diarrhea, bleeding, nausea, vomiting); neurological complications (e.g. headache, confusion, seizure, stroke, vision problems); pulmonary symptoms (e.g. dyspnea, pulmonary hemorrhage, pulmonary edema); cardiovascular symptoms (e.g. myocardial infarction, hypertension, cardiomyopathy); and vascular symptoms (finger necrosis) [Citation2,Citation9].
Primary aHUS can occur at any age although it is somewhat less frequent in children under 18 years of age than in adults (40 vs 60% of cases) [Citation5,Citation10]. The frequency of aHUS is approximately equal in boys and girls when onset is in childhood but there is a female preponderance in adults [Citation5].
2.2. Genetic contributions
Genes associated with primary aHUS include C3, CD46 (also known as membrane cofactor protein [MCP]), complement factor I (CFI), complement factor B (CFB), complement factor H (CFH) and its related proteins CFHR1, CFHR3, CFHR4, CFHR5, thrombomodulin (THBD), and plasminogen (PLG) [Citation1,Citation11,Citation12]. The type of genetic mutation determines disease severity, prognosis, treatment response, and need for renal transplantation in patients with aHUS [Citation4]. Although the DGKE gene has been associated with an aHUS diagnosis due to a similar clinical presentation [Citation13], it is not part of the complement pathway and hence does not confer a complement-mediated disease.
Gene mutations are present in about 60% of all aHUS cases [Citation12]. However, aHUS appears to be a ‘double whammy’ disease in that, whereas genetic alterations predispose to the disorder, a precipitating condition or event is required for the disease to manifest clinically [Citation1,Citation11].
2.3. Diagnosis
The diagnosis of primary aHUS is one of exclusion, especially in the absence of a family history. Individuals presenting TMA are investigated for thrombotic thrombocytopenic purpura (TTP) which can be excluded if serum ADAMTS13 activity is normal (i.e. >10%). Shiga toxin-producing Escherichia coli hemolytic uremic syndrome (STEC-HUS), or ‘typical HUS,’ can be excluded if fecal and serological tests for STEC are negative. In the absence of these causes, patients are evaluated for precipitating conditions or triggers of TMA which include transplantation (hematopoietic stem cell, solid organ), malignant hypertension, certain drugs, tumors, pregnancy, infections, and autoimmune diseases (e.g. systemic lupus erythematosus). These triggers can either transiently cause TMA that resolves after appropriate management or can precipitate primary aHUS in persons with a predisposed genetic background. If ‘secondary HUS’ is refractory to treatment of the underlying condition or withdrawal of the offending agent, a diagnosis of primary aHUS can be made [Citation3,Citation14,Citation15]. Secondary HUS is responsible for the majority of TMA cases (≈60% versus 15% for primary aHUS) [Citation16]. Cobalamin C deficiency is a separate condition caused by mutations in the MMACHC gene that can present systemic manifestations including TMA/hemolytic uremic syndrome (HUS) and must be ruled out in the diagnostic work-up of aHUS [Citation17].
2.4. Genetic testing
Although genetic testing can have diagnostic and prognostic value in aHUS, identifying a genetic variant is not essential for a diagnosis due to incomplete genetic penetrance and because some pathological variants may not yet be identified or covered by conventional panel testing [Citation1]. Genetic testing may be useful in cases of aHUS associated with pregnancy, hypertensive emergency, autoimmune diseases, and transplantation since complement dysregulation is known to have a pivotal role in these situations [Citation3,Citation18]. Evidence is insufficient at present to recommend genetic testing in other secondary HUS subtypes. In a French cohort (n = 110) with secondary HUS attributed to causes such as drugs, infections and malignancies (also autoimmune diseases in this cohort), the frequency of complement gene rare variants was found to be similar between patients (5%) and healthy controls (6%) [Citation19]. Nevertheless, rare genetic variants have also been described in many such cases [Citation3].
2.5. Therapeutic strategies
Current therapeutic strategies for aHUS are based on functional restoration of the complement system [Citation2]. Previously, aHUS was managed with plasma infusion or plasma exchange which supplemented functional complement regulatory proteins and eliminated autoantibodies against the complement system; however, response to plasma therapy varies by mutation [Citation2,Citation20,Citation21]. In a French series of 214 patients treated prior to eculizumab (2000 to 2008), prognosis with plasma therapy was poor, with end-stage renal disease (ESRD) or death occurring in approximately one-third of pediatric patients and two-thirds of adult patients within 3‒5 years of aHUS onset [Citation10]. Plasma exchange is still considered standard therapy prior to excluding deficient ADAMTS13 activity as the cause of TMA and confirming a diagnosis of aHUS, but it is no longer generally considered a first-line option for treating aHUS unless complement inhibitors are not available [Citation1,Citation2].
Advances in understanding the role of complement defects and the development of specific humanized monoclonal antibodies that bind to C5 has dramatically improved the natural course of aHUS. Eculizumab is now considered first-line therapy for initial and recurrent episodes of complement-mediated aHUS [Citation1,Citation2]. Eculizumab binds with high affinity to C5, blocking its cleavage into C5a and C5b and, hence, preventing the formation of MAC (C5b-9). Stopping uncontrolled C5 activation results in hematologic remission and restoration of renal function [Citation22]. A meta-analysis (137 patients, 4 studies) found that eculizumab reduced the need for dialysis by 70% after 26 weeks and provided a complete TMA response in 60% of aHUS patients at 26 weeks and in 65% at 2 years [Citation23]. Ravulizumab has been re-engineered from eculizumab to lengthen its half-life, extending the maintenance administration interval from every 2 weeks to every 8 weeks [Citation24]. Approved in 2019, ravulizumab has been shown to be effective and safe in treating aHUS in clinical trials in adults [Citation25] and children [Citation26,Citation27]. An indirect comparison based on clinical trial data concluded that clinical outcomes do not differ between ravulizumab and eculizumab [Citation28]. Eculizumab recipients can safely be switched to ravulizumab [Citation27].
Debate continues as to whether complement inhibitors have a role in treating secondary HUS. At present there appears to be support for their use in aHUS associated with pregnancy/postpartum, malignant hypertension, and transplantation due to the high prevalence of underlying genetic abnormalities associated with complement system regulation in these subtypes [Citation3]. In other subtypes of secondary HUS, the approach is pragmatic: treat the underlying disease or withdraw the offending agent. If there is no improvement within a reasonable timeframe, a trial of C5 inhibitor may be considered since cases of secondary HUS refractory to conventional treatment may have an underlying pathophysiology of complement dysregulation [Citation3].
3. Management of aHUS in Latin America
3.1. Search strategy
A bibliographic search was conducted to identify relevant studies of aHUS in Latin American populations reported in English, Spanish, or Portuguese. The MEDLINE (PubMed), Embase (Ovid Technologies, Inc.), and LILACS/IBECS (scientific health information from Latin America and Caribbean countries) databases were searched.
The search strategy was adapted for each database (PubMed, Embase, LILACS/IBECS) and was constructed by combining MESH/EMTREE terms (PubMed and Embase controlled vocabulary) and free-text searching in the title/abstract fields. The search strategy is available as a Supplementary File. The search terms (‘atypical hemolytic uremic syndrome’ and ‘Latin America’) and their variations were identified based on main topics, keywords, and controlled vocabularies. To ensure the comprehensiveness of the search, term variations were included in the free-text search, and truncation was used for some terms. On the LILACS/IBECS database, Spanish and Portuguese terms were included in the search strategy. Limits/filters (publication date and language) were applied as required for each search question. PubMed and Embase searches were limited by language (English, Portuguese, Spanish). The LILACS/IBECS search was limited by publication date (1950 to 2023). Following a search of all databases, results were consolidated and duplicates were removed. A preliminary manual screening of the results was carried out based on title/abstract information, and irrelevant articles were excluded.
3.2. Results
3.2.1. Literature search and selection
The literature search conducted in February 2023 retrieved 51 titles, comprising full papers and conference abstracts (). Following de-duplication and manual screening of titles and abstracts, 23 articles remained. Six of these articles were rejected: one was a review and five were abstracts of four papers published in full, leaving 17 articles. One additional article was identified in a post-February 2023 supplementary PubMed search. Seven author-known articles (all case reports) were added. A total of 25 articles are included in the narrative synthesis.
Figure 2. Results of a literature search to identify relevant studies on atypical hemolytic uremic syndrome in Latin American populations, published over the last 10 years in English, Spanish, or Portuguese.
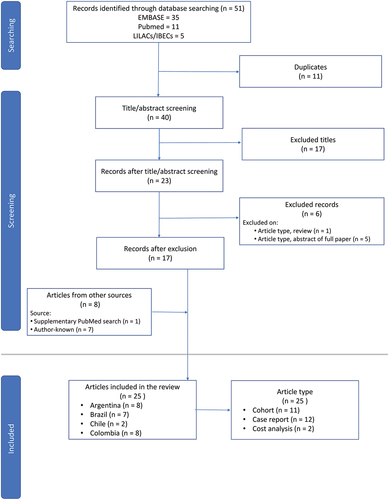
All articles were published during the years 2013–2022. Seventeen (68%) of the 25 selected articles are full papers [Citation29–45]. The remaining eight articles (32%) are conference abstracts [Citation46–53].
All selected articles report data from one of four Latin American countries: eight (32%) from Argentina [Citation38,Citation39,Citation46,Citation47,Citation49–52]; seven (28%) from Brazil [Citation30,Citation37,Citation40–43,Citation48]; two (8%) from Chile [Citation32,Citation35]; and eight (32%) from Colombia [Citation29,Citation31,Citation33,Citation34,Citation36,Citation44,Citation45,Citation53].
Eleven (44%) articles are cohort/registry analyses [Citation30,Citation39–44,Citation47,Citation48,Citation50,Citation51], 12 (48%) are case reports [Citation29,Citation31–36,Citation38,Citation45,Citation46,Citation49,Citation52] and two (8%) are cost analyses [Citation37,Citation53].
Most articles (n = 18; 72%) primarily describe the clinical presentation and outcomes of aHUS in adults and/or children [Citation29–36,Citation38–40,Citation42–46,Citation48,Citation49], with eleven of these articles being case reports [Citation29,Citation31–36,Citation38,Citation45,Citation46,Citation49]. Four articles (16%) have a primary focus on the results of genetic testing studies in Argentina [Citation47,Citation50–52], including one case report [Citation52]. The remaining three articles (12%) relate to specific issues encountered with eculizumab, including the effects of treatment interruption due to drug shortages [Citation41] and treatment cost [Citation37,Citation53].
Across all studies, data were reported for 376 patients with aHUS.
3.2.2. Clinical presentation
Four cohort studies and a registry analysis from Argentina [Citation39,Citation50], Brazil [Citation42,Citation43], and Colombia [Citation44] included from 20 to 75 patients with aHUS, and reported various data relating to clinical presentation and treatment (). Across the five studies, patients were predominantly female (50‒75%) and data were reported for 106 children (age <18 years) [Citation39,Citation42,Citation43,Citation50] and 105 adults [Citation39,Citation42–44].
Table 1. Clinical presentation, treatment, and outcomes in adults and/or children with atypical hemolytic uremic syndrome in Latin American populations.
Table 2. Genetic testing in studies of patients with atypical hemolytic uremic syndrome in Latin American populations.
Co-existing conditions or triggers were reported in three studies [Citation42–44], and were identified in ≈ 60% of patients (predominantly adults) in Brazilian studies in children and adults [Citation42,Citation43], and in 100% of adult patients in the Colombian study [Citation44]. Co-existing conditions or triggers included concomitant infections, pregnancy, hypertension, autoimmune diseases, and use of TMA-inducing medications in kidney transplant patients [Citation42–44]. Common co-existing conditions/triggers were pregnancy/postpartum, infections and TMA-inducing medications in adults [Citation42–44] and infections in children [Citation42,Citation43].
Where reported, common clinical characteristics at presentation included gastrointestinal symptoms (e.g. diarrhea), hypertension, and altered neurological function, as well as low levels of serum C3, hemoglobin, and platelets, and elevated levels of creatinine [Citation42–44,Citation50]. Children had lower hemoglobin and platelet levels and higher lactate dehydrogenase (LDH) levels than adults in the Brazilian database study [Citation43].
Two case reports from Colombia reported unusual presentations in patients diagnosed with aHUS including ocular involvement in a 23-year-old male who also had renal, hematological, neurological and cardiovascular involvement [Citation29]; and association of aHUS with nephrotic syndrome in a 4-year-old child [Citation36]. Case reports from Chile described two women with catastrophic aHUS that was likely mediated by complement-amplifying factors. The first report involved a 17-year-old female patient diagnosed with aHUS at 3 years of age who had reversion and recurrence of symptoms (including after discontinuation of eculizumab) in association with intercurrent respiratory infections [Citation35]. In the second report, a 25-year-old female patient presented malignant hypertension and severe renal failure. Extensive genetic testing revealed a CFH mutation. Etiological investigation of the cause of aHUS pointed to oral contraceptive use, gastrointestinal infection and/or malignant hypertension as possible triggers in this genetically vulnerable patient [Citation32,Citation35].
3.2.3. Genetic testing
summarizes the results of two Argentinian genetic testing studies [Citation47,Citation51], as well as genetic testing results from three of the clinical presentation studies discussed above [Citation42–44] and in two studies of kidney transplant patients with aHUS [Citation30,Citation40]. In these latter five studies, 40–79% of patients underwent genetic testing [Citation30,Citation40,Citation42–44]. Where reported, a common test was the aHUS panel which comprised amplification and next-generation sequencing of entire coding regions of ADAMTS13, C3, CD46, CFB, CFHR1, CFHR2, CFHR3, CFHR5, CFI, DGKE, PIGA, THBD genes and including 10 base pairs next to exons [Citation30,Citation40,Citation42,Citation43,Citation47,Citation51]. Also described was use of multiplex ligation-dependent probe amplification (MLPA) analyses to test for deletions or duplications within or including CFH, CFHR1, CFHR2, CFHR3 and CFHR5 genes [Citation30,Citation40,Citation43,Citation51].
The proportions of aHUS patients with identified genetic abnormalities were 57% (12/21 tested patients) [Citation47] and 42% (13/31) [Citation51] in the Argentinian studies; 89% (24/27) in the Brazilian cohort study [Citation42] and 67% (22/33) in the Brazilian database study [Citation43]; and 87.5% (7/8) in the Colombian study [Citation44]. In the Brazilian studies in kidney transplant patients with aHUS, variants were identified in 67% (4/6) [Citation30] and 74% (14/19) [Citation40] of tested patients.
Mutations in CFH were the most common variant, being identified in 12.5% [Citation42], 21% [Citation43], 23% [Citation51], 24% [Citation47], 26% [Citation40] and 38% [Citation44] of tested aHUS patients. Other common variants differed between studies, which is partly due to between-study differences in the type of genetic analyses performed. For example, the CFHR1-R3 deletion was identified in 16‒33% of tested patients in three Brazilian studies [Citation30,Citation40,Citation43], but was not reported in the other studies. CD46 variants were identified in five patients (all children) in the Argentinian report, occurring in 62.5% of tested children and in 23.8% of tested patients overall [Citation47]. In other studies, one adult patient was reported to have a CD46 variant [Citation43]. CFI variants were reported in 21% [Citation40] and 25% [Citation44] of tested patients in two small studies compared with 4.8% [Citation47], 5.7% [Citation51] and 12.1% [Citation43] of tested patients in other studies. Several variants (e.g. C2, CFB, CHRH5, CHRH2, MMACHC, ADAMTS13, and THBD) were reported in only a few patients across all studies.
Variant combinations were identified across all studies. Most patients had mutations or variants associated with one other gene, although several patients had three variants.
Where reported, the proportion of variants classified as pathogenic/likely pathogenic ranged from 27.0% in the Brazilian cohort study [Citation42], to 40.0% [Citation51] and 50% [Citation47] in two Argentinian studies, to 50% in the Colombian study [Citation44], and to 42% in a Brazilian cohort study in patients with aHUS either as a primary disease or relapse after kidney transplantation [Citation40]. A further 17% [Citation44], 32% [Citation40], 46.7% [Citation51], 50.0% [Citation47], and 67.6% [Citation42] of identified variants were classified as being of unknown pathological significance. Where reported, only few variants were considered to be benign/likely benign, with two variants (13.3%) considered likely benign in the Argentinian prospective cohort study [Citation51] and two variants (5.4%) considered benign/likely benign in a Brazilian retrospective cohort study [Citation42].
Variants were reported slightly more frequently in children than in adults in the Brazilian cohort study (90.1% vs 82.3%); however, fewer children than adults (18.2% vs 62.5%) had a pathogenic/likely pathogenic variant [Citation42]. The genetic profile was reported to be similar between children and adults in the Brazilian database study [Citation43].
Genetic findings were similar in a study that examined the effects of unplanned eculizumab discontinuation [Citation41]. A complement variant was identified in 16 of 25 episodes of eculizumab discontinuation (64%), with the CFH and CFHR1/3 variants (each in 32% of episodes) and the C3 variant (20%) being most common. In a multivariate regression analysis, the presence of a CFH variant was not an independent predictor of relapse after eculizumab discontinuation, and a Kaplan Meier analysis found no difference in the cumulative risk of relapse between patients with or without the CFH variant [Citation41].
A case report presented the complex results of genetic testing in a 27-year-old Argentinian female with postpartum-associated aHUS [Citation52]. The patient had inherited a novel CFH variant and an ultra-rare THBD variant from her mother, as well as a novel CFI variant from her father. Biomarkers and complement function testing indicated that she had low levels of C3 and CFH, and low-normal levels of CFI.
A 25-year-old female patient from Chile who presented malignant hypertension and severe renal failure was found to have a previously-unreported CFH mutation (p.Tyr1177Cys) in the SCR20 domain. Mutations in this region increase the risk for primary aHUS on account of the role of the SCR20 domain on the inhibitory action of factor H in the complement alternative pathway [Citation32].
In an Argentinian cohort study [Citation50], 54 children with clinical features of aHUS were screened for the presence of autoantibodies directed against factor H, of which eight (14.8%) tested positive (defined as a mean titer of ≥100 arbitrary units [AU]). No differences in clinical or laboratory features were found between anti-CFH-positive children (mean titer 1797 AU; range 179‒4361 AU; n = 6) and anti-CFH-negative children (mean titer 3.6 AU; range 0‒27 AU; n = 29), except that the mean LDH concentration was significantly higher in the anti-CFH-positive group than in the anti-CFH-negative group (4687 vs 3131 IU/L; p = 0.05) [Citation50].
3.2.4. Treatment and outcomes
3.2.4.1. aHUS
Three retrospective analyses described clinical outcomes with eculizumab in aHUS patients [Citation42–44] including clear benefits over plasma therapy [Citation42,Citation44]. In a nonregistry analysis of 34 Brazilian patients, 22 (65%) received plasma therapy and 31 (91%) were treated with eculizumab. Of 30 patients who were undergoing dialysis at the time of diagnosis, two discontinued dialysis after plasma therapy and 19 after eculizumab. Median time between first manifestation of aHUS and eculizumab treatment was shorter in children than in adults (30 vs 260 days) [Citation42]. A Brazilian aHUS registry analysis reported on 40 adults and 35 children who were treated before and after Brazilian Health Regulatory Agency (ANVISA) approval of eculizumab in 2017; 27.5% of patients received plasma exchange and 97.3% received eculizumab. Eculizumab use was similar irrespective of patient age: <2 years (94.1%), 2‒18 years (94.1%) or >18 years (100%). In contrast to the abovementioned analysis, the median interval between aHUS diagnosis and eculizumab treatment did not differ by age group (15 vs 30 vs 45 days in patients aged < 2, 2–18 years and >18 years, respectively) or dialysis status (16 vs 34 days in dialysis-free and dialysis patients, respectively) [Citation43]. Eculizumab was used to treat eight of 20 patients with aHUS reported in a Colombian retrospective cohort study from 2014–2018; all patients received plasma therapy. Clinical outcomes were poor among 12 patients not treated with eculizumab: two (17%) died and four (25%) progressed to ESRD. Early treatment with eculizumab led to better outcomes. Multi-organ function was restored after 4 weeks in four eculizumab recipients who began treatment within 1 week of their diagnosis, whereas three eculizumab recipients who began treatment within 3 weeks of presentation continued to have slight proteinuria [Citation44].
Neither of two retrospective cohort studies from Argentina reported use of eculizumab in aHUS patients [Citation39,Citation50]. In the 2013–2016 cohort study, adult and pediatric patients received dialysis (62%), transfusions (55%), plasmapheresis (55%), and corticosteroids (32%) [Citation39]. In the pediatric study, most children received dialysis (67% vs 55%) and/or transfusion (83% vs 69%) irrespective of their anti-CFH status, whereas use of corticosteroids (67% vs 14%) and plasmapheresis (67% vs 35%) was more common in the anti-CFH-positive than anti-CFH-negative group [Citation50]. Outcomes were not reported in either publication.
A Bothrops asper envenomation of the left foot in a 57-year-old man led to venom-induced consumption coagulopathy, which was rapidly controlled by administering Colombian polyvalent viper antivenom. The patient subsequently developed TMA and aHUS and received plasma therapy and hemodialysis. The condition resolved within 14 days [Citation45].
Several case reports of patients with aHUS described restoration of renal function and remission following treatment with eculizumab [Citation32,Citation35,Citation36,Citation46].
Two women from Chile developed terminal renal failure requiring hemodialysis, both of whom received eculizumab after an aHUS diagnosis was established [Citation32,Citation35]. In the first patient, concurrent dialysis was discontinued after 2 months. Due to a negative genetic study, eculizumab was discontinued at 6 months but restarted after the patient experienced a relapse associated with a viral respiratory infection. At 2-year follow-up she continued with eculizumab and was in hematological remission with stable renal function [Citation35]. The second patient also began eculizumab with concurrent hemodialysis three times per week. Hemodialysis was discontinued after 3 months and, at 12 months, she remained clinically stable with maintenance eculizumab dosing [Citation32,Citation35].
Two patients from Colombia who presented with unusual symptoms, including a marked loss of visual acuity (RE: 20/400 + 1; LE: 20/150 − 1) in a 23-year-old male and associated nephrotic syndrome in a 4-year-old male, received eculizumab after an aHUS diagnosis [Citation29,Citation36]. The first patient recovered his vision (20/30 in both eyes) although he remained dialysis-dependent and was ultimately diagnosed with ESRD secondary to aHUS [Citation29]. In the second patient, eculizumab controlled the disease without any further TMA events. The nephrotic syndrome resolved in 8 weeks without need for steroids or other immunosuppressants. At 2 years’ follow-up the patient remained on eculizumab with normal renal function and in remission of nephrotic syndrome. Genetic study revealed mutations in CFI, CFH and ADAMTS13 genes [Citation36].
A 2013 case report from Argentina described two children with aHUS (a 7-year-old female and a 4-month-old male) who were treated with eculizumab. Both had TMA, acute renal failure, and decreased serum C3 levels, and showed resistance to daily plasma infusion with persistent hypocomplementemia and acute renal failure. After initiation of eculizumab, renal function was completely restored without relapse in both patients [Citation46].
3.2.4.2. Kidney transplant patients with aHUS
Three studies from Brazil reported benefit with eculizumab in patients with onset of aHUS before or after kidney transplantation () [Citation30,Citation40,Citation48].
Table 3. Outcomes with eculizumab treatment in Brazilian studies of kidney transplant patients with atypical hemolytic uremic syndrome.
Seven kidney transplant patients with aHUS received eculizumab at a single center in Brazil from 2013 to 2016. Of two patients who received continuous prophylactic eculizumab because of a pre-transplant diagnosis of aHUS, one had a TMA relapse after 4 months, and the other remained asymptomatic during 16 months of follow-up. All five patients treated with continuous therapeutic eculizumab for post-transplant TMA showed improvement within 48 hours of treatment initiation. No patient experienced TMA relapse during continuous use of eculizumab (mean follow-up 21 months; range 6–42 months). One patient died at 6 months due to an Aspergillus infection [Citation30].
A retrospective multicenter cohort review of 38 kidney transplant recipients with TMA between 2007 and 2019 compared outcomes between 11 patients not treated with eculizumab, 10 patients with a pre-transplant aHUS diagnosis treated with prophylactic eculizumab and 17 patients with post-transplant TMA who received therapeutic eculizumab. The TMA graft loss rate was significantly (p < 0.001) higher in the non-treated group (91%) than in the prophylactic (10%) or therapeutic eculizumab (5.9%) treatment groups. Relative to not receiving eculizumab, the risk of TMA recurrence was reduced by 93% (hazard ratio [HR] 0.07; 95% confidence interval [CI] 0.01–0.55) with prophylactic eculizumab, and by 96% (HR 0.04; 95% CI 0.00–0.28) with therapeutic eculizumab. The non-treated group had significantly higher rates of acute rejection than the prophylactic and therapeutic eculizumab groups (67% vs 0% vs 35%; p = 0.021). The cumulative survival rate at 1000 days was lower in the non-treated group than in the prophylactic and therapeutic eculizumab groups, although the difference was not statistically significant (40% vs 100% and 70%; p = 0.131) [Citation40].
In a retrospective single-center cohort review of 306 kidney transplant recipients, 12 (3.9%) developed post-transplant TMA of whom nine were diagnosed with aHUS. Median time from kidney transplant to aHUS diagnosis was 81 days (range 35.5–134.5 days), and median time from diagnosis to eculizumab treatment was 17 days (range 4.5–134 days). Following initiation of eculizumab, five (56%) of the nine patients had improved creatinine levels and three (33%) remained on dialysis; the remaining patient died suddenly after two doses of eculizumab. Two patients with improved renal function subsequently died (one each from gastrointestinal bleeding and sepsis due to cutaneous infection) [Citation48].
Case reports from Colombia and Argentina described the benefits of eculizumab treatment in patients with post-kidney transplant aHUS [Citation31,Citation49]. A 52-year-old Colombian male developed TMA ≈5 months after transplantation and presented with neurological symptoms. After prolonged investigations, he was diagnosed with aHUS. His medical condition, which continued to worsen despite immunosuppressive treatment, improved significantly with plasma therapy, supporting the aHUS diagnosis. Treatment with eculizumab led to recovery without evidence of TMA relapse or renal deterioration [Citation31]. In the second case report, a 24-year-old female with an aHUS diagnosis from age of 2 years presented TMA ≈1 week after kidney transplantation. Renal function improved with twice-weekly plasma therapy. Eculizumab was introduced 1 month later and her eGFR improved to 50 mL/min, indicative of a mild-to-moderate loss of kidney function [Citation49].
A further case report from Argentina described the clinical course of a 19-year-old female patient after a kidney donation from her mother [Citation38]. The persistence of delayed graft function and worsening symptomatology led to a suspicion of aHUS and initiation of eculizumab. The patient continued to deteriorate and died of multiple organ failure and cardiorespiratory arrest. As the genetic study revealed heterozygous mutations (variant in THBD, deletion of CFHR1 and CFHR3), a causal relationship was uncertain. The authors suggested that factors such as reperfusion injury, transplantation and innate immune mechanisms, tacrolimus use, and infections may have had a role in the outcome [Citation38].
Case reports from Colombia describe outcomes in two patients with chronic kidney disease secondary to aHUS who underwent successful kidney transplant while receiving eculizumab, one with a factor H mutation [Citation33] and the other with negative results for all currently known mutations [Citation34]. Surgery was performed without complications and with immediate function of the kidney graft in both patients. At follow-up of 18 months and 3 years, respectively, both patients maintained their renal function without relapse of underlying disease [Citation33,Citation34].
3.2.4.3. Discontinuation of maintenance eculizumab
Due to shortages of eculizumab in Brazil, maintenance treatment had to be interrupted in some aHUS patients, which can result in relapse [Citation41,Citation42].
A retrospective multicenter cohort review assessed the effects of eculizumab interruption during 2016–2019 [Citation41]. Episodes of exposure to risk of relapse were defined as an unplanned interruption of ≥30 days. In all, 25 episodes were identified in 24 patients. Eculizumab had been used for >365 days in 22 episodes (88%), and for 150, 182, and 350 days in the remaining three episodes. The episode population included both children and adults (median age 33 years; range 6–53 years). Eighteen patients (72%) were female, nine (36%) had post-kidney-transplant aHUS, and five (20%) were undergoing dialysis. Eleven instances of aHUS relapse after eculizumab withdrawal were reported. Following eculizumab interruption, the cumulative risk of aHUS relapse was 34% at 114 days, which increased to 44.5% at 150 days and 58% at 397 days. No correlation was found between baseline variables (age, sex, current dialysis, functioning kidney transplant, CFH variant) and increased risk of aHUS relapse. In view of the high relapse rate, the authors deemed that eculizumab interruption is ‘not safe’ [Citation41].
A Brazilian retrospective cohort study reported outcomes collected until 31 December 2018 in 34 patients with aHUS, 31 of whom received eculizumab. Of 12 patients who had to interrupt eculizumab due to drug shortages, four relapsed (three within 40 days of stopping, one not reported) for an overall relapse rate of 30% [Citation42].
3.2.5. Access to eculizumab
Access to eculizumab in both Brazil [Citation37] and Colombia [Citation53] was difficult due to economic hurdles. Market approval of eculizumab in Brazil prompted a cost reduction [Citation37].
4. Discussion
Based on available data, cases of aHUS in Latin America present the classical clinical and epidemiological characteristics of this condition as described in other world regions. Overall, there was female preponderance, and 40–50% of populations were aged <18 years in studies that included both children and adults, consistent with the age distribution reported elsewhere [Citation5,Citation10]. Frequent co-existing triggers/conditions included concomitant infections, pregnancy, hypertension, autoimmune diseases, and use of TMA-inducing medications after kidney transplantation. In addition to renal involvement, other common clinical characteristics at presentation were gastrointestinal symptoms, hypertension, and altered neurological function. Common laboratory findings were low levels of serum C3, hemoglobin, and platelets, and elevated levels of creatinine levels.
Genetic testing is an increasingly important component of aHUS management since it provides important prognostic information that can guide clinical decision making. The results of genetic testing varied across studies, depending on which genetic panels were analyzed. Identifiable predisposing variants in disease-associated genes were identified in 41–89% of tested patients in the Latin American studies, which aligns broadly with the ≈ 60% figure reported in the literature [Citation12]. Consistent with other reports [Citation1,Citation11,Citation12], mutations in the CFH gene were the most common variants in Latin American populations.
Eculizumab and ravulizumab are the only drugs approved at present to treat aHUS [Citation54,Citation55]. According to available Latin America data, depending on the country and timeframe, aHUS was treated most commonly with plasma therapy and/or eculizumab, with no mention of ravulizumab in any study. Most eculizumab data came from Brazil where eculizumab has been in use since US Food & Drug Agency approval in 2011. Across studies, eculizumab substantially improved aHUS-related outcomes in almost all adult and pediatric patients, consistent with its known effectiveness [Citation23]. Aligning with data from other regions supporting the use of eculizumab in kidney transplantation [Citation56,Citation57], prophylactic or therapeutic use of eculizumab in a Brazilian cohort of aHUS patients was associated with superior outcomes compared with no eculizumab treatment [Citation40].
Delays in diagnosis of aHUS and treatment initiationcan lead to poorer outcomes. The interval between aHUS diagnosis and initiation of eculizumab varied widely in available Latin American studies, from within the first week of diagnosis to >20 years in patients diagnosed before the drug was available, although beneficial outcomes were reported irrespective of the interval [Citation42]. The benefits of early treatment are supported by a pooled analysis of four phase 2, open-label, single-arm, prospective clinical studies of eculizumab in patients with aHUS which found that renal recovery was better in those treated ≤7 days than >7 days from aHUS manifestation [Citation58]. Although the sample size was small, a Colombian cohort study reported superior outcomes for patients who began eculizumab within 1 week versus 3 weeks of their aHUS diagnosis and for all seven early starters of eculizumab compared with one patient who began treatment 1 month post-diagnosis [Citation44]. The lack of correlation between genetic characteristics and relapse after eculizumab discontinuation [Citation41,Citation42] suggests that treatment delay after aHUS diagnosis contributes more than case severity toward relapse.
Some uncertainties with eculizumab use in patients with aHUS relate to treatment duration and the feasibility of safe discontinuation. In patients with unplanned interruption of eculizumab for ≥30 days due to drug shortages, relapse rates were at least 30% [Citation41,Citation42] and even higher after 1 year without access (68%) [Citation41]. Studies from other regions have reported broadly similar TMA relapse rates of up to 40% after eculizumab discontinuation [Citation59–63]. Nevertheless, while indefinite treatment with eculizumab may be necessary in high-risk patients with detected pathogenic variants, depending on the genetic profile, there are select groups of patients (e.g. those with coexisting or trigger conditions) who can safely stop treatment after achieving stable hematological remission. Close clinical and laboratory monitoring is recommended after eculizumab discontinuation and any decision to stop treatment must factor in the ease of restarting in the event of TMA relapse [Citation15,Citation59].
Limited data indicate that eculizumab is not easily accessible in Latin America [Citation37,Citation53] particularly in Mexico and Argentina among the five representative countries. Although pharmacoeconomic analyses should consider both the costs and benefits of treatment, cost-utility analyses of eculizumab in the treatment of aHUS are lacking overall. Certain strategies may help reduce the costs associated with use of complement inhibitors. For example, early initiation of treatment may be cost saving. A modeled analysis of hospitalization costs based on retrospective data in 222 patients with aHUS in the U.S.A. found that per-patient total hospitalization costs were 17% lower (p = 0.002) in patients who initiated eculizumab within 7 days than in those who initiated it on day 8 or later [Citation64]. Individualized modification of eculizumab therapy based on pharmacokinetic, complement, and/or genetic studies may prove to be safe and cost-effective [Citation65]. Although outside the current approved label [Citation54], potential cost-saving measures include extending the interval between dosage administration in some individuals by monitoring eculizumab and complement levels [Citation66–68] and/or suspending treatment in selected patients with a low risk of relapse, with immediate reinstatement of therapy if aHUS recurs [Citation1,Citation60–63,Citation69–71]. The choice of complement inhibitor may also affect overall costs. In a modeled cost-minimization study that took into account lost productivity costs associated with administration of eculizumab and ravulizumab in the U.S.A., overall discounted annual treatment costs (direct treatment costs plus lost productivity costs) were estimated to be 33% lower with ravulizumab than eculizumab in adults and 40% lower in children, due to less frequent administration of ravulizumab (once every 4–8 weeks) than eculizumab (once every 2 weeks) [Citation72]. As of mid-2023, ravulizumab has been approved to treat aHUS in some Latin American countries (e.g. Chile). Similar to other world regions, ravulizumab might be expected to simplify and improve the management of patients with aHUS in Latin America due to its much less frequent dosing schedule [Citation73]. A suggestion to improve access to C5 inhibitors in Latin America is to base pricing on the gross domestic product of individual countries [Citation74].
The review clearly highlights the scarcity of high-quality data about aHUS management in Latin America. This limitation stems in part from the inherent rarity of aHUS and also for reasons specific to Latin America such as restricted access to complement inhibitors and limited financial support for researchers to conduct studies and publish their work. The failure of the original search to retrieve all available Latin American articles exposes certain limitations of systematic searches which are unable to identify articles published in non-indexed local journals and may miss others due to indexing failures. Despite these irregularities in data retrieval, our familiarity with the literature gives us confidence that no articles of material significance were missed. Lastly and importantly, we acknowledge that available evidence from Latin America may not accurately reflect the contemporary management of aHUS in the region; at minimum; however, it provides a glimpse which has hitherto been absent.
5. Conclusion
Available data about aHUS in Latin American countries indicate that disease characteristics and management approaches are consistent with those reported in the rest of the world. Similar to elsewhere, mutations in CFH are the most common variants in Latin American populations. Despite access issues, eculizumab has substantially improved outcomes in patients in aHUS in Latin America. In shifting toward use of second-generation ravulizumab, patients with aHUS in Latin America might expect to achieve outcomes equivalent to those with eculizumab but with considerably less administration burden and cost.
6. Expert opinion
To our knowledge, this is the first article to assemble evidence about aHUS in Latin America, providing valuable insights into clinical characteristics, genetic profiles, treatment approaches, and regional challenges. The findings have the potential to impact positively on real-world outcomes by altering how we observe, analyze, identify, and treat our patients with aHUS. For instance, the identification of classical clinical characteristics, such as female preponderance, common triggers, and associated conditions, will aid in early and accurate diagnosis. The findings highlight the evolution in aHUS management with eculizumab, a drug that is not available on a large scale in many Latin America countries and is often difficult to access, resulting in treatment delays. Despite these hurdles, patient outcomes have been vastly superior to plasma therapy.
Addressing economic challenges and investing in healthcare infrastructure will be essential to implement strategies for timely detection and early treatment of aHUS. This approach is likely to be cost-effective given the decrease in serious complications and prevention of death with complement inhibitors. Understanding the diagnostic and therapeutic reality of aHUS in Latin America may inform health policy decisions and institutional protocols. Not least, the findings identify avenues for clinical research and challenge medical specialists to establish appropriate routes to approach this pathology. Formulating local guidelines, gathering statistical data by country and by region, integrating genetic testing into clinical practice, and optimizing the therapeutic response with complement inhibitors are aspects to be deepened. At a practical level, more research is required to identify bottlenecks and implement solutions (clinical, biomarkers, genetic) to facilitate a timely diagnosis and prompt initiation of treatment. Such advances will ultimately favor patients with aHUS by allowing for greater diagnostic capacity, more timely treatment, and the opportunity to receive effective therapy that changes the catastrophic evolution of the disease.
Other key areas for improvement in aHUS management center around education and detection. Health teams must be educated about these rare pathologies in order to increase diagnostic suspicion. Physicians in critical areas such as intensive care and coronary care units must appreciate the importance of referral to nephrologists with the expertise to exclude alternative differential diagnoses, recognize the ‘predisposing – triggering’ binomial and mediate the performance of kidney biopsy which is often the only manifestation of the disease. Collaboration between nephrology pathologists and geneticists is crucial for early recognition. The absence of a specific serological diagnostic test for aHUS is a major limitation in clinical practice. Tests that measure complement fractions offer no assurance that the alternative pathway is overactivated and is responsible for the disease. Moreover, the genetic study is not exhaustive and results can be inconclusive. Technical challenges related to genetic sequencing and personalized medicine expertise must be addressed through training and resource allocation. However, managing complementary molecular biology and genetic studies is a challenge in Latin America given that there are few resources to carry them out and few centers are trained to develop genetic studies.
The future of aHUS research lies in a collaborative multidisciplinary approach that combines clinical, genetic, pharmacoeconomic data and patient-reported outcomes. We anticipate many areas of opportunity in the complement system and its pathophysiological consequences. Understanding the mechanisms involved in complement system activation associated with genetic diversity, including alterations that exist in the absence of gene mutations, would make it possible to recognize different immunophenotypes and deliver tailored treatment. Understanding the link between the complement and coagulation systems could improve the diagnosis and treatment of pathologies such as transplant rejection, IgA nephropathy, membranous nephropathy, C3 glomerulonephritis, autoimmune diseases, and others. Some secondary causes of aHUS have demonstrated an interrelation between both pathways that warrants further investigation. Future study of aHUS should also focus on identifying biomarkers that allow for a faster and more accurate diagnosis; factors that clarify which patients to treat with complement inhibitors, optimal treatment duration; candidates for treatment discontinuation; and factors that predict relapse.
Over the next five to ten years, we can expect to gain greater insight into the true epidemiology of aHUS in Latin America and benefit from improved accessibility to advanced diagnostics, genetic testing, and targeted therapies. As technology progresses, genetic testing costs may decrease, making it attainable for a broader population. To counter the ‘data explosion’ in complement system disease, artificial intelligence may have a role in differential diagnosis and contribute to knowledge about the timing and effectiveness of current and new therapies. The availability of newer therapeutic options, including ravulizumab, will expand treatment choices. Numerous other therapies in development will make it possible to inhibit specific steps of complement activation depending on the predominant pathogenic phenomenon. Collaborative efforts among healthcare providers, governments, and pharmaceutical companies alongside investments in healthcare infrastructure are essential to gain meaningful improvements in patient care. The Latin American medical community welcomes the challenges as we strive to improve outcomes for our patients with aHUS.
Article highlights
Atypical hemolytic uremic syndrome (aHUS) is a rare form of thrombotic microangiopathy.
Comprehensive information about aHUS, as provided by the Global aHUS Registry, involves patients mainly from Europe and North America.
This narrative review, based on a systematic search, presents available published data on clinical characteristics, treatment approaches and outcomes in patients with aHUS in Latin America.
The search identified 51 titles/abstracts of which 25 articles (full papers and conference abstracts) are included in the review: 8 from Argentina, 7 from Brazil, 2 from Chile, and 8 from Colombia.
Articles include cohort studies, a registry analysis, and case reports. Seventeen articles (68%) are full papers.
Latin American patients present the classic epidemiological, clinical, and genetic characteristics associated with aHUS as reported in populations outside the region. Complement factor H (CFH) mutations are the most common variant.
In publications reporting use of eculizumab, aHUS-related outcomes were substantially improved in almost all adult and pediatric patients, including in the kidney transplant setting.
Complement component 5 (C5) inhibitors have dramatically altered the course of aHUS, improving prognosis and patient outcomes, although their use is not without challenges.
Similar to other world regions, a priority in Latin America is to optimize use of this important class of drugs.
Declaration of interest
RA Sepúlveda Palamara has participated as a speaker and received funding from AstraZeneca, Boehringer Ingelheim, and Tecnofarma. LG Modelli de Andrade has received funding from Alexion, Sanofi, and Takeda. RM Fortunato has participated as a speaker for AstraZeneca, and Boehringer Ingelheim Argentina. B Gómez has received honoraria as speaker from Alexion, Asofarma, AstraZeneca, and Roche. JF Nieto-Ríos has presented at conferences on thrombotic microangiopathies in academic events sponsored by Alexion Pharma, but declares no direct relationship with Alexion Pharma and is not an employee of that laboratory. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Medical writing support was provided by Katherine Lyseng-Williamson and Kerry Dechant, ISMPP CMPP™, on behalf of Content Ed Net, with funding from Alexion, AstraZeneca Rare Disease.
Additional information
Funding
References
- Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508–530. doi: 10.1016/j.semnephrol.2013.08.003
- Raina R, Krishnappa V, Blaha T, et al. Atypical hemolytic-uremic syndrome: an update on pathophysiology, diagnosis, and treatment. Ther Apher Dial. 2019;23(1):4–21. doi: 10.1111/1744-9987.12763
- Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “kidney disease: improving global outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539–551. doi: 10.1016/j.kint.2016.10.005
- Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847–2856. doi: 10.1182/blood-2016-11-709865
- Licht C, Ardissino G, Ariceta G, et al. The global aHUS registry: methodology and initial patient characteristics. BMC Nephrol. 2015;16:207. doi: 10.1186/s12882-015-0195-1
- Halimi JM, Al-Dakkak I, Anokhina K, et al. Clinical characteristics and outcomes of a patient population with atypical hemolytic uremic syndrome and malignant hypertension: analysis from the Global aHUS registry. J Nephrol. 2023;36(3):817–828. doi: 10.1007/s40620-022-01465-z
- Yoshida Y, Kato H, Ikeda Y, et al. Pathogenesis of atypical hemolytic uremic syndrome. J Atheroscler Thromb. 2019;26(2):99–110. doi: 10.5551/jat.RV17026
- Wada T, Nangaku M. Novel roles of complement in renal diseases and their therapeutic consequences. Kidney Int. 2013;84(3):441–450. doi: 10.1038/ki.2013.134
- Malina M, Gulati A, Bagga A, et al. Peripheral gangrene in children with atypical hemolytic uremic syndrome. Pediatrics. 2013;131(1):e331–e335. doi: 10.1542/peds.2012-0903
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554–562. doi: 10.2215/CJN.04760512
- Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844–1859. doi: 10.2215/CJN.02210310
- Noris M, Bresin E, Mele C, et al. Genetic Atypical hemolytic-uremic syndrome. In: Adam M, Mirzaa G, Pagon R, Wallace S, Bean L, Gripp K Amemia A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2007 Nov 16 [cited 2021 Sep 23]. p. 1993–2023.
- Brocklebank V, Wood KM, Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. 2018;13(2):300–317. doi: 10.2215/CJN.00620117
- Laurence J, Haller H, Mannucci PM, et al. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol. 2016;14 Suppl 11(11):2–15.
- Fakhouri F, Schwotzer N, Frémeaux-Bacchi V. How I diagnose and treat atypical hemolytic uremic syndrome. Blood. 2023;141(9):984–995. doi: 10.1182/blood.2022017860
- Werion A, Storms P, Zizi Y, et al. Epidemiology, outcomes, and complement gene variants in secondary thrombotic microangiopathies. Clin J Am Soc Nephrol. 2023;18(7):881–891. doi: 10.2215/CJN.0000000000000182
- Beck BB, van Spronsen F, Diepstra A, et al. Renal thrombotic microangiopathy in patients with cblC defect: review of an under-recognized entity. Pediatr Nephrol. 2017;32(5):733–741. doi: 10.1007/s00467-016-3399-0
- Leon J, LeStang MB, Sberro-Soussan R, et al. Complement-driven hemolytic uremic syndrome. Am J Hematol. 2023;98(Suppl 4):S44–S56. doi: 10.1002/ajh.26854
- Le Clech A, Simon-Tillaux N, Provôt F, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019;95(6):1443–1452. doi: 10.1016/j.kint.2019.01.023
- Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421–447. English, Spanish. doi: 10.1016/j.nefro.2015.07.005
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and if mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267–1279. doi: 10.1182/blood-2005-10-007252
- Cofiell R, Kukreja A, Bedard K, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood. 2015;125(21):3253–3262. doi: 10.1182/blood-2014-09-600411
- Pugh D, O’Sullivan ED, Duthie FA, et al. Interventions for atypical haemolytic uraemic syndrome. Cochrane Database Syst Rev. 2021;3(3): CD012862. doi: 10.1002/14651858.CD012862.pub2
- Syed YY. Ravulizumab: A review in atypical haemolytic uraemic syndrome. Drugs. 2021;81(5):587–594. doi: 10.1007/s40265-021-01481-6 Erratum in: Drugs. 2021;81(6):737. Erratum in: Drugs. 2021;81(11):1363–1364.
- Rondeau E, Scully M, Ariceta G, et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287–1296. doi: 10.1016/j.kint.2020.01.035 Erratum in: Kidney Int. 2020;98(6):1621. Erratum in: Kidney Int. 2021;99(5):1244.
- Ariceta G, Dixon BP, Kim SH, et al. The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2021;100(1):225–237. doi: 10.1016/j.kint.2020.10.046
- Tanaka K, Adams B, Aris AM, et al. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr Nephrol. 2021;36(4):889–898. doi: 10.1007/s00467-020-04774-2 Erratum in: Pediatr Nephrol. 2020 Dec 9.
- Tomazos I, Hatswell AJ, Cataland S, et al. Comparative efficacy of ravulizumab and eculizumab in the treatment of atypical hemolytic uremic syndrome: an indirect comparison using clinical trial data. Clin Nephrol. 2022;97(5):261–272. doi: 10.5414/CN110516
- Nieto-Ríos JF, Serna-Higuita LM, Calle-Botero E, et al. Atypical haemolytic-uraemic syndrome in a young patient with renal, neurological, ocular and cardiovascular involvement. [Síndrome hemolítico urémico atípico en un paciente joven con compromiso renal, neurológico, ocular y cardiovascular]. Nefrología. 2016;36(1):82–85. doi: 10.1016/j.nefro.2015.07.008
- de Andrade LGM, Contti MM, Nga HS, et al. Long-term outcomes of the atypical hemolytic uremic syndrome after kidney transplantation treated with eculizumab as first choice. PLOS ONE. 2017;12(11):e0188155. doi: 10.1371/journal.pone.0188155
- Ramírez Quintero JI, Gómez ED, Aldana Campos MC, et al. Hemolytic uremic syndrome atypical after kidney transplantation: a case report and literature review. Rev Colomb Nefrol. 2017;4(1):74–84. doi: 10.22265/acnef.4.1.267
- Sepúlveda RA, Tagle R, Jara A. Novel complement factor H mutation, a case report of atypical hemolytic uremic syndrome. Case Rep In Internal Med. 2017;4(2):13–17. doi: 10.5430/crim.v4n2p13
- Nieto-Ríos JF, Zuluaga-Quintero M, Bello-Márquez DC, et al. Successful kidney transplant with eculizumab, thymoglobulin and belatacept therapy in a highly-sensitised patient with atypical haemolytic uraemic syndrome due to factor H mutation. Nefrología (English Edition). 2018;38(4):433–437. English, Spanish. doi: 10.1016/j.nefroe.2018.04.008
- Nieto-Ríos JF, Zuluaga-Quintero M, Bello-Márquez DC, et al. Successful kidney transplantation with a protocol of eculizumab, thymoglobulin, tacrolimus, mycophenolate, and steroids in a patient with atypical hemolytic uraemic syndrome without an identified mutation. [Trasplante renal exitoso con protocol de eculizumab, timoglobulina, tacrolimus, micofenolato y esteroides en paciente con syndrome hemolítico urémico atípico sin mutación identificada]. NefroPlus. 2018;10(2):63–67.
- Tagle R, Rivera G, Walbaum B, et al. Síndrome hemolítico urémico atípico en tratamiento con eculizumab. Casos clínicos [Atypical hemolytic uremic syndrome. Report of two cases treated with eculizumab]. Rev Med Chil. 2018 Feb;146(2):254–259. Spanish. doi: 10.4067/s0034-98872018000200254
- Bello-Marquez DC, Nieto-Rios JF, Serna-Higuita LM, et al. Nephrotic syndrome associated with primary atypical hemolytic uremic syndrome. J Bras Nefrol. 2021;43(3):440–444. doi: 10.1590/2175-8239-JBN-2020-0050
- Caetano R, Rodrigues PHA, Corrêa MCV, et al. The case of eculizumab: litigation and purchases by the Brazilian Ministry of Health. Rev Saude Publica. 2020;54:22. doi: 10.11606/s1518-8787.2020054001693
- Gutiérrez R, Fortunato RM, Vigliano C, et al. Microangiopatía trombótica sistémica asociado a mutaciones del complemento en trasplante renal donante vivo. Reporte de Caso. Rev Nefrol Dial Traspl. 2020;40(2):139–145.
- Dos Santos C, Paiva J, Romero ML, et al. Thrombotic microangiopathies: First report of 294 cases from a single institution experience in Argentina. eJHaem. 2021;2(2):149–156. doi: 10.1002/jha2.154
- Nga HS, Palma LMP, Ernandes Neto M, et al. Thrombotic microangiopathy after kidney transplantation: Analysis of the Brazilian atypical hemolytic uremic syndrome cohort. PLOS ONE. 2021;16(11):e0258319. doi: 10.1371/journal.pone.0258319
- Neto ME, de Moraes Soler L, Vasconcelos HVG, et al. Eculizumab interruption in atypical hemolytic uremic syndrome due to shortage: analysis of a Brazilian cohort. J Nephrol. 2021;34(4):1373–1380. doi: 10.1007/s40620-020-00920-z
- Palma LMP, Eick RG, Dantas GC, et al. Atypical hemolytic uremic syndrome in Brazil: clinical presentation, genetic findings and outcomes of a case series in adults and children treated with eculizumab. Clin Kidney J. 2020;14(4):1126–1135. doi: 10.1093/ckj/sfaa062
- Vaisbich MH, de Andrade LGM, de Menezes Neves PDM, et al. Baseline characteristics and evolution of Brazilian patients with atypical hemolytic uremic syndrome: first report of the Brazilian aHUS registry. Clin Kidney J. 2022;15(8):1601–1611. doi: 10.1093/ckj/sfac097
- Cabarcas-Barbosa O, Aroca-Martínez G, Musso CG, et al. Atypical hemolytic uremic syndrome in the Colombian Caribbean: its particular characteristics. Int Urol Nephrol. 2022;54(6):1323–1330. doi: 10.1007/s11255-021-03011-5
- CañCañAs CA, Vecino MJ, Posso-Osorio I. Atypical hemolytic uremic syndrome in a patient with Bothrops asper envenomation. Wilderness Environ Med. 2022;33(1):109–115. doi: 10.1016/j.wem.2021.08.010
- Lopez LC, Adragna MS, Briones LM. Preservation of renal function in atypical haemolytic uremic syndrome (aHUS) by eculizumab in two Argentinean children [abstract no. P-SUN108]. Pediatr Nephrol. 2013;28(8):1570. doi: 10.1007/s00467-013-2523-7
- Bonany P, Olavarrieta L, Perez-Perez J. Complement-aHUS/PTT in Argentina, five years experience of genetic international laboratory [abstract no. PO-341]. Pediatr Nephrol. 2016;31(10):1873–1874. doi: 10.1007/s00467-016-3467-5
- Miranda SMC, Souza PAM, Pereira GM, et al. Evolution of post-transplant atypical hemolytic uremic syndrome (aHUS) [abstract no. SA-PO526]. J Am Soc Nephrol. 2017;28:815.
- Vazquez MC, Alonso V, Costantini MS, et al. Atypical hemolytic uremic syndrome (AHUS) post kidney transplant (KT) [abstract no. 204]. Transplant Int. 2017;30:445. doi: 10.1111/tri.13053
- Dos Santos C, Mogollon Molina N, Alberto MF, et al. Anti-factor H autoantibody-associated hemolytic uremic syndrome in an Argentinean pediatric cohort [abstract no. PB611]. Res Pract Thromb Haemost. 2018;2:296–297. doi: 10.1002/rth2.12125
- Dos Santos C, Ghiringhelli Borsa N, Taylor A, et al. Complement abnormalities in a cohort of Argentinean patients with atypical hemolytic uremic syndrome [abstract no. PB1599 Res Pract Thromb Haemost. 2019;3:747. doi: 10.1002/rth2.12229
- Taylor A, Dos Santos C, Ghiringhelli Borsa N, et al. Comprehensive complement analysis of atypical hemolytic uremic syndrome in Argentina: a case study [abstract no. 10]. Mol Immunol. 2019;114:413–414. doi: 10.1016/j.molimm.2019.08.016
- Alvis Zakzuk J, Gamero K, Fernandez Mercado JC, et al. Costs of treatment of atypical hemolytic uremic syndrome in poor population of Colombia [abstract no. PRO11]. Value Health. 2020;23:S330–S331. doi: 10.1016/j.jval.2020.04.1242
- European Medicines Agency. Solaris. Summary of Product Characteristics. [cited 2023 Aug 18]. Available from: https://www.ema.europa.eu/en/documents/product-information/soliris-epar-product-information_en.pdf
- European Medicines Agency. Ultomiris. Summary of Product Characteristics. [cited 2023 Aug 18]. Available from: https://www.ema.europa.eu/en/documents/product-information/ultomiris-epar-product-information_en.pdf
- Siedlecki AM, Isbel N, Vande Walle J, et al. Eculizumab use for kidney transplantation in patients with a diagnosis of atypical hemolytic uremic syndrome. Kidney Int Rep. 2018;4(3):434–446. doi: 10.1016/j.ekir.2018.11.010
- Glover EK, Smith-Jackson K, Brocklebank V, et al. Assessing the impact of prophylactic eculizumab on renal graft survival in atypical hemolytic uremic syndrome. Transplantation. 2023;107(4):994–1003. doi: 10.1097/TP.0000000000004355
- Vande Walle JV, Delmas Y, Ardissino G, et al. Improved renal recovery in patients with atypical hemolytic uremic syndrome following rapid initiation of eculizumab treatment. J Nephrol. 2017;30(1):127–134. doi: 10.1007/s40620-016-0288-3
- Olson SR, Lu E, Sulpizio E, et al. When to stop eculizumab in complement-mediated thrombotic microangiopathies. Am J Nephrol. 2018;48(2):96–107. doi: 10.1159/000492033
- Duineveld C, Bouwmeester R, van der Heijden JW, et al. Outcome of atypical haemolytic uraemic syndrome relapse after eculizumab withdrawal. Clin Kidney J. 2020;14(8):1939–1945. doi: 10.1093/ckj/sfaa241
- Ariceta G, Fakhouri F, Sartz L, et al. Eculizumab discontinuation in atypical haemolytic uraemic syndrome: TMA recurrence risk and renal outcomes. Clin Kidney J. 2021;14(9):2075–2084. doi: 10.1093/ckj/sfab005
- AlZabali S, AlBatati S, Rahim K, et al. A multicenter study evaluating the discontinuation of eculizumab therapy in children with atypical hemolytic uremic syndrome. Children (Basel). 2022;9(11):1734. doi: 10.3390/children9111734
- Baskin E, Fidan K, Gulhan B, et al. Eculizumab treatment and discontinuation in pediatric patients with atypical hemolytic uremic syndrome: a multicentric retrospective study. J Nephrol. 2022;35(4):1213–1222. doi: 10.1007/s40620-021-01212-w Erratum in: J Nephrol. 2022 Feb 7.
- Ryan M, Donato BMK, Irish W, et al. Economic impact of early-in-hospital diagnosis and initiation of eculizumab in atypical haemolytic uraemic syndrome. Pharmacoeconomics. 2020;38(3):307–313. doi: 10.1007/s40273-019-00862-w
- Avila Bernabeu AI, Cavero Escribano T, Cao Vilarino M. Atypical hemolytic uremic syndrome: new challenges in the complement blockage era. Nephron. 2020;144(11):537–549. doi: 10.1159/000508920
- Gatault P, Brachet G, Ternant D, et al. Therapeutic drug monitoring of eculizumab: Rationale for an individualized dosing schedule. MAbs. 2015;7(6):1205–1211. doi: 10.1080/19420862.2015.1086049
- Volokhina E, Wijnsma K, van der Molen R, et al. Eculizumab dosing regimen in atypical HUS: possibilities for individualized treatment. Clin Pharmacol Ther. 2017;102(4):671–678. doi: 10.1002/cpt.686
- Sridharan M, Go RS, Willrich MAV. Clinical utility and potential cost savings of pharmacologic monitoring of eculizumab for complement-mediated thrombotic microangiopathy. Mayo Clin Proc Innov Qual Outcomes. 2022;6(5):458–464. doi: 10.1016/j.mayocpiqo.2022.03.005
- Menne J, Delmas Y, Fakhouri F, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019;20(1):125. doi: 10.1186/s12882-019-1314-1
- Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438–2449. doi: 10.1182/blood.2020009280
- Bouwmeester RN, Duineveld C, Wijnsma KL, et al. Early eculizumab withdrawal in patients with atypical hemolytic uremic syndrome in native kidneys is safe and cost-effective: results of the CUREiHUS Study. Kidney Int Rep. 2022;8(1):91–102. doi: 10.1016/j.ekir.2022.10.013
- Levy AR, Chen P, Johnston K, et al. Quantifying the economic effects of ravulizumab versus eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Med Econ. 2022;25(1):249–259. doi: 10.1080/13696998.2022.2027706
- Ter Avest M, Langemeijer SMC, Blijlevens NMA, et al. Dose optimalization of subcutaneous ravulizumab is predicted to yield significant savings and to improve patient friendliness. Br J Clin Pharmacol. 2023;89(3):1211–1215. doi: 10.1111/bcp.15602
- Nga HS, Palma LMP, Ernandes Neto M, et al. Eculizumab in low-middle income countries: how much does a life cost? J Nephrol. 2022;35(4):1255–1257. doi: 10.1007/s40620-022-01282-4