
ABSTRACT
Introduction
Cold agglutinin disease (CAD) is driven by IgM autoantibodies reactive at <37°C and able to fix complement. The activation of the classical complement pathway leads to C3-mediated extravascular hemolysis in the liver and to intravascular hemolytic crises in case of complement amplifying conditions. C3 positivity at direct Coombs test along with high titer agglutins are required for the diagnosis. Treatment is less standardized.
Areas covered
This review recapitulates CAD diagnosis and then focus on the evolving management of the disease. Both current approach and novel targeted drugs are discussed. Literature search was conducted in PubMed and Scopus from 2000 to 2024 using ‘CAD’ and ‘autoimmune hemolytic anemia’ as keywords.
Expert opinion
Rituximab represents the frontline approach in patients with symptomatic anemia or disabling cold-induced peripheral symptoms and is effective in 50–60% of cases. Refractory/relapsing patients are an unmet need and may now benefit from complement inhibitors, particularly the anti-C1s sutimlimab, effective in controlling hemolysis thus improving anemia in >80% of patients, but not active on cold-induced peripheral symptoms. Novel drugs include long-acting complement inhibitors, plasma cells, and B-cell targeting agents (proteasome inhibitors, anti-CD38, BTKi, PI3Ki, anti-BAFF). Combination therapy may be the future answer to CAD unmet needs.
1. Introduction
Cold agglutinin disease (CAD) is a rare form of autoimmune hemolytic anemia (prevalence about 16 per million, incidence 1 per million year), representing 20–30% of all autoimmune hemolytic anemias (AIHAs) [Citation1–4]. CAD is caused by an IgM autoantibody able to agglutinate and lyse red blood cells (RBC) at temperatures below 37°C. The IgM autoantibodies are pentamers with a well-known ability to strongly activate complement (C), causing both extravascular hemolysis C3d-mediated in the liver, and intravascular hemolysis in case of massive C activation until the final C5b-9 complex [Citation5]. The limiting factor of this pathogenetic mechanism is the temperature of reaction of cold agglutinins, which ranges from 4 to 37°C, being those closer to body temperature the most pathogenic. Clinically, this phenomenon is responsible for the circulatory symptoms (acrocyanosis, Raynaud-like phenomenon, ulcerations up to gangrene) in the acral parts of the body, where temperatures reach the optimal range for antigen–antibody reaction. Anemia is mainly due to RBC destruction, but also to the variable inability of bone marrow compensation, whose marker is reticulocytes. Other signs of hemolysis are increased unconjugated bilirubin and lactate dehydrogenase, along with reduced haptoglobin. Clinically, fatigue is particularly present in CAD, greater than expected for a given hemoglobin value, probably due to continuous complement activation and production of pro-inflammatory mediators [Citation2–5]. Importantly, an increased risk of thrombosis has been reported in this disease, as in other complement-mediated hemolytic conditions [Citation6–10]. Notably, anemia due to RBC destruction is complement-mediated, while circulatory symptoms are mainly due to IgM agglutination. In a recent international study involving 232 patients [Citation8], about 70% of patients had hemolytic anemia with no or mild peripheral circulatory symptoms, 21% hemolytic anemia with severe circulatory symptoms, and 9% circulatory symptoms with compensated hemolysis. Around half of patients required transfusions during the chronic course/acute exacerbations of the disease.
CAD is an established clonal B-cell lymphoproliferative disorder of the bone marrow, recently included into the World Health Organization classification of hematolymphoid neoplasms in the group of mature B-cell neoplasms, plasma cell neoplasia, and other diseases with paraproteins, thus distinct from other malignant lymphomas [Citation11]. Consistently, most of the cold agglutinins are monoclonal usually of the IgM class with κ light-chain restriction. For a proper diagnosis, it is important to exclude secondary forms (identified as cold agglutinin syndrome or secondary CAD), which are associated with lymphomas, neoplasms, autoimmune diseases, and infections (typically Mycoplasma pneumoniae, Epstein-Barr virus, cytomegalovirus, or recently SARS-CoV-2). The diagnosis requires an extensive work up to rule out secondary forms and is established by the direct antiglobulin test (DAT) which is strongly positive for complement (C3d) and by a cold agglutinin titer ≥ 64 at 4°C. Notably, bone marrow reexamination by a centralized expert center greatly increased the proportion of CAD diagnosis versus other indolent lymphoproliferative disorders [Citation12,Citation13]. As other AIHAs, patients with CAD often present with blood group discrepancies, and that is used as a diagnostic pointer to perform cold antibody screening, followed by titration (based on the screening result).
Historically, treatment of CAD had been mainly based on supportive measures (cold avoidance, folic acid, and transfusions) while standard treatments of IgG-mediated warm AIHA (steroids, immunosuppressants, and splenectomy) are poorly effective or discouraged [Citation1,Citation5,Citation8]. Transfusions are generally administered in severe/symptomatic forms to obtain an appropriate hemoglobin value until specific treatments become effective. Transfusions should be administered slowly (not exceeding 1 ml/kg/h) using a blood warmer. In CAD treatment is indicated in case of symptomatic anemia, marked fatigue, or important circulatory symptoms, while a watch-and-wait approach can be adopted in mild cases [Citation5]. Treatment should be aimed at suppressing the two main pathogenic mechanisms of CAD, namely autoantibody production and complement activation. In the recent years, several B-cell target therapies have emerged, along with complement-inhibitors, providing new therapeutic options in CAD () [Citation14]. Here, we will discuss these new drugs and the possible evolving management of the disease.
Figure 1. The dual pathogenic mechanism in cold agglutinin disease (CAD): autoantibody production and complement activation and main available drugs targeting these mechanisms.
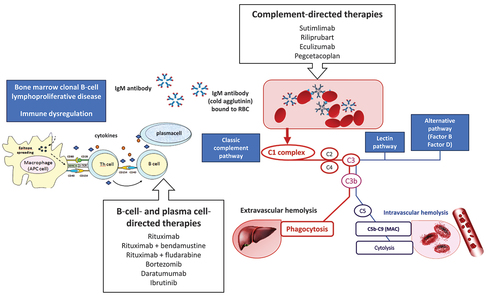
2. B-cell and plasma cell target therapies
The anti-CD20 monoclonal antibody rituximab has been the first B-cell target therapy for CAD. Evidence from several observational retrospective and prospective nonrandomized studies in the last 20 years have established that the drug at the dose of 375 mg/m2/week for 4 weeks is able to induce a response in about 50% of patients [Citation1,Citation5,Citation15–19]. However, complete responses are rare (less than 5% of patients) and the median duration of response is less than 1 year. Rituximab has a B-cell depleting effect and reduces the activity antigen-presenting cells and production of cytokines [Citation18,Citation19]. The drug is well tolerated, and retreatment is possible, particularly in case of relapse occurring at least 2 years after the initial response. However, rituximab is not worldwide available, nor indicated/reimbursed by health assurances. Moreover, a proportion of patients may experience prolonged lymphopenia/hypogammaglobulinemia and reduced response to vaccinations [Citation1,Citation5]. In 2017, Berentsen et al. [Citation20] reported that addition of fludarabine to rituximab increased response rate to 76%, with complete response in 21% of patients, and prolonged response duration to more than 5 years. The combination resulted however in a significant toxicity, mainly infective. The same Author reported that the combination of rituximab plus bendamustine resulted in similar overall response rate (78%), better complete responses (53%), and longer response duration (more than 7 years), compared with rituximab plus fludarabine [Citation21]. The new combination had a moderate and manageable toxicity. Both studies were prospective and non-randomized trials and the real word use of the two combinations (rituximab plus fludarabine and rituximab plus bendamustine) are not available so far.
Some case-reports and a small prospective study have reported the efficacy of the proteosome inhibitor bortezomib in CAD [Citation22]. The pilot study involving 21 patients by Rossi et al. described a response in 32% of patients (16% complete) with a median duration of response of 16 months and an acceptable toxicity [Citation23]. Recently, Pasquale et al. [Citation22] reviewed the available literature, and all the six adult cases reported responded (4 complete responses), receiving variable doses of the drug either IV or subcutaneous, for various courses (from 1 to 13), either single agent or in combination with rituximab-dexamethasone (N = 1) or cyclophosphamide-dexamethasone (N = 1). The good toxicity profile and the possibility to repeat cycles in case of response suggest that bortezomib is good therapeutic option in the setting of relapsed/refractory cases.
The anti-CD38 monoclonal antibody daratumumab has been used in CAD with the rationale of targeting long-lived plasma cells in the spleen and bone marrow in the relapsed/refractory setting. Most are case reports recently reviewed, showing good responses in two patients with CAD [Citation24,Citation25] however with the caveat of reporting successful treatments only. Importantly, efficacy on cold-induced symptoms has been observed. The report by Zaninoni et al. also included the investigation on immunoregulatory cytokines, showing patient’s IL-6 and IL-10 levels increased compared to controls, and TNF-α and IFN-γ reduced in the first phase of daratumumab treatment [Citation24]. Contrarily, IFN-γ levels increased after several cycles possibly reflecting the engagement of this cytokine in daratumumab-mediated cytotoxicity against plasma cells. Moreover, TGF-β was increased, further amplifying IL-17-mediated pro-inflammatory and autoimmune response and boosting cytotoxic activity against plasma cells.
The Bruton tyrosine kinase (BTK) inhibitor ibrutinib has been investigated in a multinational retrospective study involving 15 patients with CAD (primary or secondary to chronic lymphocytic leukemia or Waldenström macroglobulinemia), all heavily pretreated, 73% transfusion dependent, and 60% with circulatory symptoms [Citation26]. Treatment induced a rapid and notable improvement of both the hemolytic anemia and acrocyanosis, regardless of underlying pathology or MYD88 mutational status. These data suggest that BTK inhibition could be particularly useful in patients failing chemo-immunotherapy or with prominent acrocyanosis, which is not considered complement mediated. reports the standard and novel B-cell and plasma cell-directed therapies for CAD.
Table 1. Standard and novel B-cell and plasma cell- directed therapies for cold agglutinin disease (CAD).
3. Complement-directed therapies
Given the key pathogenic role of complement in hemolytic anemia of CAD, several complement inhibitors have been investigated in this disease [Citation14]. The first one, the anti-C5 complement inhibitor eculizumab, was investigated about 15 years ago in a prospective open-label trial involving 13 patients [Citation27]. The study demonstrated a clear reduction of hemolysis but a minor effect on hemoglobin levels, with a fraction of patients gaining transfusion independence.
More recently, the monoclonal antibody anti-C1s sutimlimab has been extensively studied and nowadays FDA and EMA approved for CAD [Citation14,Citation28]. The first open-label study involved 24 subjects with Hb <10 g/dL who had received at least one transfusion in the previous 6 months [Citation29]. The primary end point was Hb increase ≥2.0 g/dL from baseline or attainment of Hb ≥12.0 g/dL with no transfusion. The primary endopoint was met by 54% of patients and 83% showed an Hb increase >1.0 g/dL. Efficacy was very quick, with significant improvement of Hb as soon as after 1 week of treatment, and a mean increase of 2.6 g/dL. Bilirubin and C4 levels normalized, and fatigue markedly improved in the first weeks of treatment. Participants were vaccinated against N. meningitidis, S. pneumoniae, and H. influenzae but did not receive prophylactic antibiotics. There were no meningococcal infections or other serious adverse events related to the study drug [Citation29]. The 2-years extension study confirmed the results, with normalization of Hb level in half and transfusion independence in almost the totality of patients [Citation30,Citation31]. The subsequent placebo-controlled phase 3 trial investigated the use of sutimlimab in 42 subjects not previously transfused but with Hb <10 g/dL. Again, a rapid Hb increase was observed, accompanied by bilirubin decrease and patients’ reported outcomes amelioration in sutimlimab versus placebo arm [Citation31]. In detail (73%) patients in the treatment arm met the primary end point criteria (Hb increase ≥1.5 g/dL and avoidance of transfusion, as compared with only 2 (10%) in the placebo arm. Furthermore, responses were sustained over 2 years with a good safety profile, requiring, however, continuous IV administration every 2 weeks, as disease activity reoccured after treatment interruption [Citation31]. Finally, a more comprehensive evaluation of patients’ reported outcomes confirmed the efficacy of sutimlimab versus placebo, particularly on fatigue, an important issue impacting quality of life in CAD patients [Citation32]. [Citation33].
More recently, the second-generation active C1s inhibitor riliprubart (formerly SAR445088, BIVV020), characterized by a prolonged half-life and thus a longer dosing interval than that of sutimlimab, is currently under investigation in CAD. A pilot open-label study showed that a single IV dose led to classical complement inhibition, control of hemolysis, and improvement in anemia, which was sustained for 15 weeks [Citation34].
Finally, the anti-C3 inhibitor pegcetacoplan has been studied in a Phase 2 open-label study in AIHA [NCT03226678], including 13 patients with CAD. Favorable results on hemoglobin and hemolytic parameters have been reported in seven patients (mean Hb increased from 8.9 to 11.6 g/dL) at the European Hematology Association in 2019, but final report awaits publication [Citation14,Citation35,Citation36].
reports the standard and novel complement- directed therapies for CAD.
Table 2. Standard and novel complement-directed therapies for cold agglutinin disease (CAD).
4. Ongoing trials with B-cell targeted therapies and complement inhibitors in CAD
As shown in , several trials are ongoing in CAD, both directed at inhibiting antibody production or targeting several components of the complement cascade. Interestingly, two studies are investigating the association of bortezomib plus rituximab in difficult settings, i.e. refractory AIHAs and post-transplant cytopenias, including AIHA and ITP. However, results, even preliminary, are not available so far. The phosphoinositide 3-kinase (PI3K) inhibitor parsaclisib has been investigated in 25 patients with AIHA, of whom 9 CAD. Results, presented at European Hematology Association 2022 [Citation14,Citation37] showed a hemoglobin response in 64% of patients (32% complete responses), with Hb improvements as early as week 2 that further increased over 12 weeks of treatment, and were sustained through the extension period. Consistently, meaningful improvements in the FACIT-F subscale were observed. However, responses were higher among patients with wAIHA (75% response; 50% CR), making this option less attractive for CAD patients.
Table 3. Ongoing trials in cold agglutinin disease (CAD).
Zanubrutinib is a next-generation BTK inhibitor, more selective for BTK than ibrutinib, and with a better safety profile, currently studied in several lymphoproliferative disorders. The study in CAD is an open-label designed to assess hemoglobin response and duration of response, along with change in IgM, monoclonal-protein, hemolytic parameters, and efficacy on circulatory symptoms. The trial is not yet recruiting (NCT06067048).
Povetacicept is a dual APRIL/BAFF antagonist that modulates B lymphocyte function and production of pathogenic autoantibodies being developed for multiple autoimmune and/or inflammatory diseases. The study is an open label in patients with wAIHA, CAD, and ITP and is actively recruiting (NCT05757570), although no results are available so far.
Regarding complement inhibition, further studies are in progress with riliprubart, the second-generation active C1s inhibitor already demonstrated effective in CAD. Eligible patients include CAD either untreated or previously treated with the drug with a favorable benefit-to-risk profile (NCT04802057). Moreover, the oral Factor B inhibitor Iptacopan, which has been proven highly effective in paroxysmal nocturnal hemoglobinuria, is under investigation in a basket study assessing efficacy, safety, and pharmacokinetics in CAD and ITP (NCT05086744). The study has completed enrollment and results are awaited. Finally, based on the results of the open-label trial, the anti-C3 inhibitor pegcetacoplan has been investigated in a Phase 3 study versus placebo. However, the study has been recently suspended and results await publication (NCT05096403).
Finally, a prospective observational study on CAD and cold agglutinin syndrome (secondary CAD) is actively recruiting (NCT05791708), with the purpose to gain more information on the clinical characteristics, diagnosis, treatment, complications, and outcome of these rare diseases.
5. Therapeutic algorithm in CAD
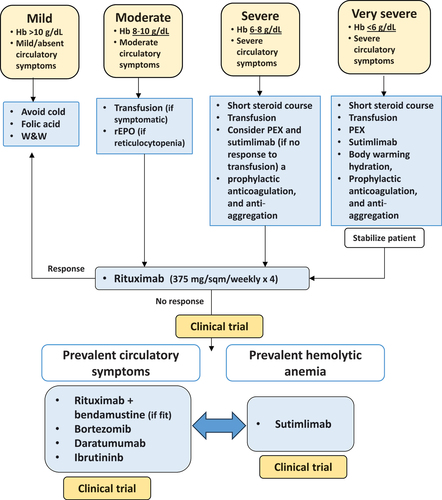
CAD is a highly heterogeneous disease, generally with a chronic course marked by acute flares due to complement-amplifying conditions (infections), cold-exposure, surgery, and other unpredictable/undefined factors [Citation1,Citation5]. Therefore, it is difficult to design a unique therapeutical algorithm and therapy should be evaluated on a case-by-case basis. Patients with mild anemia and circulatory symptoms can benefit from cold avoidance, folic support, and a watch and wait strategy, occasional blood support and recombinant erythropoietin (in case of reticulocytopenia) [Citation38–40]. Patients with symptomatic anemia, fatigue and disabling circulatory symptoms, which represent the majority of subjects, require treatment. The few very severe patients or the severe hemolytic episodes occurring in the chronic setting, with profound life-threating anemia or acral ulcers/gangrene need urgent therapeutic intervention. The latter include a short steroid course, plasma-exchange and blood transfusions, body warming, hydration, prophylactic anticoagulation, and anti-aggregation. The most attractive urgent measure is certainly complement inhibition, although evidence in this setting is poor. Sutimlimab, given the rapid action would be particularly useful, although not worldwide available and costly [Citation35]. In the experience of the Authors, even eculizumab may be beneficial in these dramatic cases. However, complement inhibition acts on one of the main pathogenic mechanisms of the disease, but has no effect on the production of the autoantibody, requiring therefore continuous administration [Citation1,Citation35]. Moreover, complement inhibition is poorly effective on circulatory symptoms, which mainly depend on IgM agglutination. Thus, a B-cell/plasma cell-directed therapy should be planned as a backbone of CAD therapy, either in the chronic or acute setting, aimed at overcoming the production of the autoantibody. Rituximab monotherapy is the most widely used therapy, although marked by low response rate and short response duration [Citation1–5,Citation35]. For patients that do not respond or relapse the choice among the several available therapies should be based on patient’s characteristics (age, comorbidity) and preferences. Rituximab plus bendamustine may be preferred in young/fit patients, particularly if bone marrow shows borderline features of lymphoproliferative disease (B-cell infiltrate > 10%) [Citation1,Citation35]. Rituximab plus fludarabine, although effective, raises important concerns about toxicity in the elderly and frail CAD population, being therefore difficult to include in a therapeutical algorithm. Bortezomib and daratumumab are both good options, particularly if circulatory symptoms are the main compliant of the patients [Citation22,Citation23]. Ibrutinib has an oral administration, avoiding repeated healthcare utilization. Whatever the choice, it should be emphasized that B-cell/plasma-cell directed therapies need time (weeks/months) to be effective. Thus, they may be planned in the acute/severe forms in addition to complement inhibitors, and a combination of both strategies may be necessary even in a fraction of ‘chronic’ patients [Citation35]. In the latter setting, sutimlimab is the best choice if hemolytic anemia is the main feature and if B-cell target therapies have failed or are contraindicated. At variance, a second-line B-cell/plasma cell-directed therapy is advisable if circulatory symptoms are prevalent, given the poor efficacy of complement inhibitors on these symptoms. The new C1s inhibitor riliprubart will probably have a more convenient schedule of administration (every 12 weeks), avoiding the burden of sutimlimab (every 2 weeks). Regarding the C3 inhibitor pegcetacoplan, the recent withdrawal of the trial would preclude any evidence-based positioning of this drug in the therapeutical algorithm of CAD, even though real-world use may provide further data in the future. Finally, an important issue to be considered is safety, as the CAD population is mainly composed by old and frail patients. Complement inhibition appears to be a safer option than chemotherapy, although direct comparative studies are not available and will be very difficult to perform. illustrates a proposed therapeutical algorithm of CAD with the existing and novel therapies sooner available.
Figure 2. Therapeutic algorithm for cold agglutinin disease (CAD) depending on the severity of clinical presentation and prevalent symptoms.
6. Conclusion
CAD is a chronic and debilitating disease, deeply impacting quality of life, and requiring treatment in most cases. The disease is highly heterogeneous, with some patients experiencing mild anemia and circulatory symptoms, few complaining of life-threatening hemolytic crisis, and the majority reporting moderate anemia with acute exacerbations and a variable degree of disabling circulatory symptoms. Treatment should be adapted to the severity of the disease, and to the acute versus chronic setting, and be aimed at suppressing the two main pathogenic mechanisms, namely autoantibody production and complement activation. Importantly, the CAD population is generally old, frail, and comorbid, thus limiting the therapeutic options. Supportive measures (cold avoidance, folic acid, and transfusions) are pivotal, and rituximab is extensively used although with limited efficacy. The combination of rituximab plus bendamustine, even though more effective, is not widely administered due to infectious concerns. In the recent years, other therapies targeting B-cells and plasma-cells have populated the therapeutic armamentarium of CAD, namely ibrutinib, bortezomib, and daratumumab. Although strong evidence is lacking, these drugs may be considered in rituximab-refractory/relapsed patients, due to their manageable toxicity. Most of the cited studies involve relatively small and homogeneous patient populations. Larger and real-life studies are needed to confirm the effectiveness and safety of new therapies across different demographics. The real novelty for CAD therapy is complement inhibition, mainly with sutimlimab, which is FDA and EMA approved. The drug is highly and rapidly effective and thus may be particularly useful in the acute/severe setting, and of course in patients relapsed/refractory or not eligible for B-cell and plasma-cell target therapies. High costs of novel therapies could be a significant barrier for the real-world application in many healthcare settings. Several trials are ongoing with new drugs, although results are still pending and, unfortunately, for some of them will never be available, preventing their release into the market.
7. Expert opinion
There is increasing interest in AIHAs, with several new drugs at various stages of development in both CAD and warm IgG forms, which represent the majority of cases. This attention has underlined the importance of the correct diagnosis, since therapy of the two conditions is quite different. In fact, standard treatments of warm AIHA (steroids, immunosuppressants, splenectomy) are poorly effective or discouraged in CAD. On the other hand, CAD is a well-recognized lymphoproliferative disorder, with monoclonal cold agglutinins and a pivotal pathogenic role of complement activation. The two conditions are generally considered as separate, however, from an immunological point of view, warm AIHA and CAD share a common autoimmune etiology based on the dysregulation of self-tolerance, whose causes are not understood so far. Additionally, warm AIHA and CAD are not always clearly distinct, and there is a gray zone in between, supported by several evidence. First, about 5–10% of all AIHAs are the so-called mixed forms [Citation1], where both IgG and IgM autoantibodies are present. In these cases, often severe and refractory, the therapeutic choice is very difficult and not supported by clinical studies. Thus, therapy may be empirically chosen from that of warm AIHA or CAD depending on the prevalent clinical features and on a case-by-case basis. Second, complement activation is found in about 30% of warm AIHAs (DAT positive with anti-IgG and anti-complement antisera), leading to a more severe clinical picture [Citation6]. However, no clinical trials are investigating the possible therapeutic use of complement inhibitors in this setting. Third, the thermal amplitude of the autoantibody is poorly considered: while IgM that react with RBC around 4°C would generally have little clinical consequences, those that have a thermal amplitude close to body temperatures (warm IgM) are responsible for the most difficult-to-diagnose, catastrophic, and fatal cases. Again, only few cases are reported in the literature, and no trials are planned in these patients due to their rarity.
Another issue deserving discussion is the assessment of secondary forms, again generally excluded from the trials. AIHAs and CAD may be secondary to infections, lymphoproliferative disorders, tumors, connective tissue diseases, inborn errors of immunity, other autoimmune cytopenias (thrombocytopenia, neutropenia in the Evans syndrome), transplants (both solid organ and hematopoietic), chimeric antigen T-cells, and countless drugs, including novel checkpoint inhibitors [Citation2,Citation41]. The diagnostic work-up is undoubtedly challenging, and most of these difficult cases require a long follow-up to unravel the underlying disease. Therapy of these ‘secondary’ AIHAs, besides that of the associated disease when identified, is again based on medical experience and on clinical course in a case-by-case fashion.
Regardless the unmet need of mixed, warm IgG+C and warm IgM AIHAs, the novel drugs described here represent an important therapeutic advancement in the management CAD. These drugs are effective on the two main pathogenic mechanisms of the disease, i.e. production of the autoantibody by B-cells and plasma cells and complement activation. The existing B-cell/plasma cell directed therapies have the advantage of a time-limited treatment aimed at curing the disease, both the hemolytic anemia and the circulatory symptoms. Their disadvantages include the low rate and duration of response of rituximab monotherapy, and the toxicity of chemotherapy in the elderly and frail CAD patients. Complement inhibitors, particularly the extensively studied sutimlimab, have evident benefits given the quick efficacy in terms of Hb improvement, the high rate of responses and the good safety profile. However, they are not effective on peripheral cold-induced symptoms, and will probably require long-term fortnightly intravenous infusions, thus impacting on patient’ convenience. Given the high cost of complement inhibitors and their indefinite administration, this therapy will raise sustainability issues in healthcare systems, especially in low- to middle-income countries. The comprehensive armamentarium here described may adapt to patient’s age, comorbidity, and preferences, as well as fit with disease characteristics and clinical course. Real-world experience in complex cases currently excluded from trials would certainly add knowledge to CAD management and ameliorate the therapeutical algorithm.
Article highlights
CAD patients with symptomatic anemia or disabling cold-induced peripheral symptoms need treatment, while cold avoidance and folic acid supplement are indicated for asymptomatic/mild symptomatic patients.
Transfusions are indicated for anemia symptoms, considering comorbidities, recombinant erythropoietin in case of reticulocytopenia, and plasma exchange for severe and very-severe anemia.
Among B-cell targeting therapies aimed at reducing the production of the cold agglutinin, rituximab represents the frontline approach. However, responses are observed in 50–60% of cases and are not durable. Combination with bendamustine or fludarabine increases response rates but also toxicity.
The anti-C1s complement inhibitor sutimlimab is effective in improving anemia and controlling hemolysis in > 80% of patients but is not active on cold-induced peripheral symptoms.
Novel drugs include plasma cell and B-cell targeting agents (bortezomib, daratumumab, parsaclisib, ibrutinib, zanubrutinib, and povetacicept) and complement inhibitors (riliprubart, pegcetacoplan, and iptacopan).
Declaration of interest
W Barcellini has received consultancy and speaker’s bureau from Alexion, Agios, Novartis, Sanofi, and Sobi. B Fattizzo has received consultancy and speaker’s bureau from Alexion, Novartis, Roche, Sanofi, and Sobi.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
W Barcellini designed and wrote the article. B Fattizzo wrote the article and revised it for important intellectual content. All authors approved the article.
Additional information
Funding
References
- Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev. 2020;41:100648. doi: 10.1016/j.blre.2019.100648
- Barcellini W, Giannotta J, Fattizzo B. Autoimmune hemolytic anemia in adults: primary risk factors and diagnostic procedures. Expert Rev Hematol. 2020;13(6):585–597. doi: 10.1080/17474086.2020.1754791
- Berentsen S, Barcellini W, Longo DL. Autoimmune Hemolytic Anemias. N Engl J Med. 2021;385(15):1407–1419. doi: 10.1056/NEJMra2033982
- Barcellini W, Zaninoni A, Giannotta JA, et al. New insights in autoimmune hemolytic anemia: from pathogenesis to therapy stage 1. J Clin Med. 2020;9(12):3859. doi: 10.3390/jcm9123859
- Berentsen S. How I treat cold agglutinin disease. Blood. 2021;137(10):1295–1303. doi: 10.1182/blood.2019003809
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014;124(19):2930–2936. doi: 10.1182/blood-2014-06-583021
- Barcellini W, Zaninoni A, Fattizzo B, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol. 2018;93(9):E243–E246. doi: 10.1002/ajh.25212
- Berentsen S, Barcellini W, D’Sa S, et al. Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood. 2020;136(4):480–488. doi: 10.1182/blood.2020005674
- Broome CM, Cunningham JM, Mullins M, et al. Increased risk of thrombotic events in cold agglutinin disease: a 10-year retrospective analysis. Res Pract Thromb Haemost. 2020;4(4):628–635. doi: 10.1002/rth2.12333
- Fattizzo B, Bortolotti M, Giannotta JA, et al. Intravascular hemolysis and multitreatment predict thrombosis in patients with autoimmune hemolytic anemia. J Thromb Haemost. 2022;20(8):1852–1858. doi: 10.1111/jth.15757
- Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720–1748. doi: 10.1038/s41375-022-01620-2
- Fattizzo B, Barcellini W. Autoimmune hemolytic anemia: causes and consequences. Expert Rev Clin Immunol. 2022;18(7):731–745. doi: 10.1080/1744666X.2022.2089115
- Barcellini W, Fattizzo B. Strategies to overcome the diagnostic challenges of autoimmune hemolytic anemias. Expert Rev Hematol. 2023;16(7):515–524. doi: 10.1080/17474086.2023.2216930
- Bortolotti M, Barcellini W, Fattizzo B. Molecular pharmacology in complement-mediated hemolytic disorders. Eur J Haematol. 2023;111(3):326–336. doi: 10.1111/ejh.14026
- Hill QA, Stamps R, Massey E, et al. British society for haematology. The diagnosis and management of primary autoimmune haemolytic anaemia. Br J Haematol. 2017;176(3):395–411. doi: 10.1111/bjh.14478
- Berentsen S, Ulvestad E, Gjertsen BT, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. 2004;103(8):2925–2928. doi: 10.1182/blood-2003-10-3597
- Schöllkopf C, Kjeldsen L, Bjerrum OW, et al. Rituximab in chronic cold agglutinin disease: a prospective study of 20 patients. Leuk Lymphoma. 2006;47(2):253–260. doi: 10.1080/10428190500286481
- Barcellini W, Zaja F, Zaninoni A, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. 2012;119(16):3691–3697. doi: 10.1182/blood-2011-06-363556
- Barcellini W, Zaja F, Zaninoni A, et al. Sustained response to low-dose rituximab in idiopathic autoimmune hemolytic anemia. Eur J Haematol. 2013;91(6):546–551. doi: 10.1111/ejh.12199
- Berentsen S, Randen U, Vågan AM, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. 2010;116(17):3180–3184. doi: 10.1182/blood-2010-06-288647
- Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017;130(4):537–541. doi: 10.1182/blood-2017-04-778175
- Pasquale R, Giannotta JA, Barcellini W, et al. Bortezomib in autoimmune hemolytic anemia and beyond. Ther Adv Hematol. 2021;12:20406207211046428. doi:10.1177/20406207211046428
- Rossi G, Gramegna D, Paoloni F, et al. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018;132(5):547–550. doi: 10.1182/blood-2018-03-835413
- Zaninoni A, Giannotta JA, Gallì A, et al. The immunomodulatory effect and clinical efficacy of daratumumab in a patient with cold Agglutinin disease. Front Immunol. 2021;12:649441. doi: 10.3389/fimmu.2021.649441
- Mohamed A, Alkhatib M, Alshurafa A, et al. Refractory cold agglutinin disease successfully treated with daratumumab. A case report and review of literature. Hematology. 2023;28(1):2252651. doi: 10.1080/16078454.2023.2252651
- Jalink M, Berentsen S, Castillo JJ, et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood. 2021;138(20):2002–2005. doi: 10.1182/blood.2021012039
- Röth A, Bommer M, Hüttmann A, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. 2018;2(19):2543–2549. doi: 10.1182/bloodadvances.2018024190
- Cavallaro F, Barcellini W, Fattizzo B. Antibody based therapeutics for autoimmune hemolytic anemia. Expert Opin Biol Ther. 2023;23(12):1227–1237. doi: 10.1080/14712598.2023.2274912
- Röth A, Barcellini W, D’Sa S, et al. Sutimlimab in cold agglutinin disease. N Engl J Med. 2021;384(14):1323–1334. doi: 10.1056/NEJMoa2027760
- Röth A, Broome CM, Barcellini W, et al. Long-term sutimlimab improves quality of life for patients with cold agglutinin disease: CARDINAL 2-year follow-up. Blood Adv. 2023;7(19):5890–5897. doi: 10.1182/bloodadvances.2022009318
- Röth A, Barcellini W, D’Sa S, et al. Sustained inhibition of complement C1s with sutimlimab over 2 years in patients with cold agglutinin disease. Am J Hematol. 2023;98(8):1246–1253. doi: 10.1002/ajh.26965
- Röth A, Berentsen S, Barcellini W, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood. 2022;140(9):980–991. doi: 10.1182/blood.2021014955
- Röth A, Broome CM, Barcellini W, et al. Sutimlimab provides clinically meaningful improvements in patient-reported outcomes in patients with cold agglutinin disease: results from the randomised, placebo-controlled, phase 3 CADENZA study. Eur J Haematol. 2023;110(3):280–288. doi: 10.1111/ejh.13903
- D’Sa S, Vos JMI, Barcellini W, et al. Safety, tolerability, and activity of the active C1s antibody riliprubart in cold agglutinin disease: a phase 1b study. Blood. 2024;143(8):713–720. doi: 10.1182/blood.2023022153
- Berentsen S, Fattizzo B, Barcellini W. The choice of new treatments in autoimmune hemolytic anemia: how to pick from the basket? Front Immunol. 2023;14:1180509. doi:10.3389/fimmu.2023.1180509
- Gertz M, Roman E, Fattizzo B, et al. Inhibition of C3 with APL-2 controls haemolysis and increases haemoglobin levels in subjects with autoimmune haemolytic anaemia (AIHA). EHA Library. 2019;3(S1):S899. doi: 10.1097/01.HS9.0000561876.96057.48
- Barcellini W, Murakhovskaya I, Terriou L, et al. Long-term efficacy and safety results from an ongoing open-label phase 2 study of parsaclisib for the treatment of autoimmune hemolytic anemia (AIHA). Hemasphere. 2022;6(Supplement 3):186–187. doi: 10.1097/01.HS9.0000844036.02998.15
- Fattizzo B, Michel M, Zaninoni A, et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: a multicenter international study. Haematologica. 2021;106(2):622–625. doi: 10.3324/haematol.2020.250522
- Fattizzo B, Pedone GL, Brambilla C, et al. Recombinant erythropoietin in autoimmune hemolytic anemia with inadequate bone marrow response: a prospective analysis. Blood Adv. 2023;8(5):1322–1327. doi: 10.1182/bloodadvances.2023011798
- Versino F, Revelli N, Villa S, et al. Transfusions in autoimmune hemolytic anemias: frequency and clinical significance of alloimmunization. J Intern Med. 2023 Nov 27. doi: 10.1111/joim.13753
- Hill QA, Stamps R, Massey E, et al. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017;177(2):208–220. doi: 10.1111/bjh.14654