
ABSTRACT
Introduction: Inflammatory bowel diseases (IBD) are on the rise worldwide. This review covers the current concepts of the etiology of Crohn´s disease and ulcerative colitis by focusing on an unbalanced interaction between the intestinal microbiota and the mucosal barrier. Understanding these issues is of paramount importance for the development of targeted therapies aiming at the disease cause.
Area covered: Gut microbiota alterations and a dysfunctional intestinal mucosa are associated with IBD. Here we focus on specific defense structures of the mucosal barrier, namely antimicrobial peptides and the mucus layer, which keep the gut microbiota at a distance under healthy conditions and are defective in IBD.
Expert commentary: The microbiology of both forms of IBD is different but characterized by a reduced bacterial diversity and richness. Abundance of certain bacterial species is altered, and the compositional changes are related to disease activity. In IBD the mucus layer above the epithelium is contaminated by bacteria and the immune reaction is dominated by the antibacterial response. Human genetics suggest that many of the basic deficiencies in the mucosal response, due to Paneth cell, defensin and mucus defects, are primary. Nutrition may also be important but so far there is no therapy targeting the mucosal barrier.
1. Introduction
Inflammatory bowel diseases (IBD) are on the rise in a world-wide epidemic [Citation1]. After an initial upswing in Western societies, particularly in Nordic countries, currently many Asian countries experience a dramatic rise in the incidence of ulcerative colitis, followed by Crohn´s disease [Citation2]. This rapid epidemiological development may be viewed as a consequence of the impact of urbanization, formula feeding rather than breast-feeding as well as changes in food technology, with shifts from plant-based foods and fiber to refined sugar and animal fat and emulsifiers [Citation3]. At the same time, in Asia, there is a rapid change from traditional (for example, Chinese) medicine to Western medicine, including increased hygiene and use of antibiotics early in childhood. It is plausible that many of these factors play a major role in the IBD epidemic, since the genetic predisposition is rather stable and should not evolve that rapidly. Nevertheless, both IBD´s are archetypical examples of a genetic/environmental crosstalk ultimately resulting in health or disease. Recently, the genetic studies have pinpointed more than 200 genetic alterations in IBD with precise odds ratios, whereas the environmental factors are comparably ill-defined [Citation3,Citation4].
At any rate, a likely common denominator and target of these environmental factors is the intestinal microbiome [Citation5]. After a recalculation, it has become established that humans harbor as many ‘bugs’ as they contain human cells (3–4 × 1013) in their body, and this multitude has to be kept at a distance from our mucosal surfaces [Citation6]. The bacterial load is excessive, particularly in the distal small intestine and colon, which are the sites where IBD typically develops. Accordingly, the focus in IBD pathophysiology has recently shifted from an autoimmune concept of a hyperactive and dysregulated adaptive immune system to a defective crosstalk between innate immunity and a mucosal barrier, which is opposed to an extremely complex microbiome, including bacteria, fungi, and viruses [Citation7,Citation8]. A list of arguments for the prime (not necessarily primary) role of the microbiota in instigating disease is derived from several lines of evidence, including animal models, risk factors, surgery sequelae, immune response and barrier as well as therapeutic issues as detailed below () [Citation9–Citation14].
Table 1. Arguments for the key role of microbes in IBD.
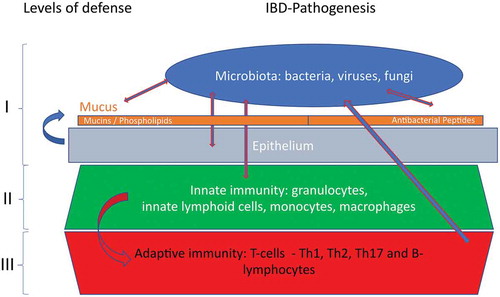
As a consequence of these complex and convincing arguments, there is currently little debate about the important role of the intestinal microbiome but, unfortunately, the hen and egg question remains unanswered. In this overview, we try to delineate the confrontation between luminal commensal bacteria and the mucosal barrier, focusing on the secreted, extracellular protection mechanisms, namely antimicrobial peptides and mucus. These secreted elements of the mucosal barrier are complemented by a sealed layer of epithelial cells, connected by critical tight junctions, and also mucosal macrophages () [Citation15,Citation16]. Undeniably, both Crohn´s disease and ulcerative colitis may be interpreted as a chronic condition where the microbiota prevails and succeeds in a slow invasion of host structures, thereby activating all defense levels of the barrier (). However, despite all these levels play a role in IBD, these cellular barriers are beyond the scope of this review.
Figure 1. The different levels of mucosal defense against invasion of the microbiota. This expert review will focus on level I.
We admit being eclectic but will focus on the human data and refer to experimental mouse data where appropriate. Several prior and excellent reviews on this topic have appeared, but none has directly addressed the microbiome in opposition to mucus and antimicrobial peptides during IBD [Citation17–Citation20]. When using the term microbiome in this review, we will focus on the bacterial microbiome, although there is now rising interest and some initial data also with respect to the gut fungome and virome [Citation21,Citation22].
2. Role of the intestinal microbiome in IBD
Based on a revolutionary technical advance from crude bacterial cultures to analysis of microbial 16S-rRNA and finally to metagenomic sequencing, the complexity of the microbiome has become established. Since most species are not culturable in vitro, only the genomic data give the full picture. A human gut microbial gene catalog was established by metagenomic sequencing and the human microbiome project consortium established the structure, function, and diversity of the healthy human microbiome at various body sites [Citation23,Citation24]. Definition of the gut microbiome variation in the population is ongoing and a new genomic blueprint has been published recently [Citation25,Citation26]. Rather than looking at ratios, it was suggested to focus on the microbial load, which is quantitated by flow cytometry [Citation27]. In parallel, the metatranscriptome was related to the metagenome of the human gut microbiome [Citation28]. This microbiota, of course, is not stable but evolves significantly over time from immediately after birth at least until the pre-adolescent phase [Citation29]. Within the bowel, there is clear spatial distribution, both along the intestine as well as transversally when comparing the crypt to the mid-lumen bacteria [Citation30].
2.1. Microbiota alterations in IBD
The first and repeatedly confirmed finding was a diminished richness and diversity of the fecal intestinal microbiome in Crohn´s disease [Citation31]. Detailed analysis revealed that among the Firmicutes 43 distinct ribotypes were identified in the healthy controls, compared with only 13 in Crohn´s disease. In this study, the Clostridium leptum group was particularly diminished, also confirmed by fluorescent in situ hybridization (FISH), whereas the Prevotella subgroup was even increased. Another study from Sweden suggested that the distance to the healthy plane of the fecal microbiome was maximal in ileal Crohn´s disease, somewhat smaller in colonic Crohn´s disease and minimal in ulcerative colitis [Citation32]. In a classic study in new onset Crohn´s patients bacterial community membership was associated independently with intestinal inflammation, antibiotic use and therapy [Citation33]. It was demonstrated convincingly that enteral nutrition therapy responders as well as anti-TNF responders normalized their microbiome whereas the non-responders did not. Yet, most early studies were limited by using exclusively fecal material, which does not necessarily represent the mucosal microbiome [Citation33]. In addition, fecal bacteria are affected by such simple variables as stool consistency, suggesting that many of the changes observed may simply be due to diarrhea secondary to IBD [Citation34].
It was established in 2002 that there is also a fundamental difference with respect to the mucosal microbiota between controls and patients with IBD. In the diseased mucosa, there were high concentrations of bacteria attached to the epithelium as a thick bacterial band whereas in controls the mucosa contained little bacteria [Citation35]. In IBD there were inclusions of polymorphic bacteria within solitary enterocytes and these bacteria were deemed to be of fecal origin. After this pioneering work the same group in Berlin followed up with a detailed study of this biofilm of bacteria in IBD, suggesting that most of it consisted of Bacteroides fragilis [Citation36]. In experimental animals this species, however, acts as an anti-inflammatory agent increasing regulatory T-cells [Citation37].
In a subsequent detailed study of colonic mucosal bacteria, the diversity was again found to be diminished [Citation38]. In the first larger study it was noticed that IBD samples were depleted of Lachnospiraceae and Bacteroidetes but enriched for Proteobacteria and Actinobacteria [Citation39]. Later, the most profound analysis was presented using a large cohort of treatment-naïve patients with new-onset pediatric Crohn´s disease [Citation33]. Although there was significant overlap in a principal component analysis between both IBD´s and controls, an axis with an increased abundance of Enterobacteriaceae, Pasteurellacaea, Veillonellaceae and Fusobacteriaceae and a decreased abundance in Erysipelotrichales, Bacteroidales and Clostridiales could be defined. Notably, this axis correlated strongly with the disease status, i.e. inflammation had a significant impact on the microbiota. It was also demonstrated that a superb differentiation could be obtained using the principal bacterial metabolic pathways that were either increased or decreased in Crohn´s disease. Thus, function may well be more important than species composition.
On the other hand, approximately a third of Crohn´s patients harbor adherent-invasive E. coli in their mucosa, which survive in macrophages and are clearly pro-inflammatory [Citation40,Citation41]. This is contrasted by a protective commensal group, Faecalibacterium prausnitzii, diminished in Crohn´s disease [Citation42]. This scenario, of course, is suggestive of a required balance between more aggressive or more defensive (‘anti-inflammatory’) species at the mucosal site. Thus, it is rather the ecology that is important and not a single culprit [Citation43].
2.2. Microbiota as hen or egg in IBD?
To gain insight into the hen and egg question, i.e. the potentially primary role of the microbiome, several situations and experimental approaches may help. These relate to a) changes in the microbiome in related diseases (specificity?) and during the course of disease, b) the interaction of host genetics and of the environment with the microbiota and c) finally, the impact of changing the bacterial composition on the induction, perpetuation or improvement of inflammation. First, it should be noted that in IBD the microbiological fecal changes were similar to those with non-IBD gastrointestinal complaints, but both differed from healthy controls [Citation44]. The microbial metabolic shifts of type II diabetes are also similar to IBD [Citation45]. Along the same lines, a comparative study of the gut microbiota in several immune-mediated inflammatory diseases detected common microbial taxa with differential abundance, but some taxa were more specific for the individual diseases [Citation46]. Therefore, there seems to be some microbial differences, but only a limited specificity of the changes common in IBD.
Next, a close relationship between the inflammatory state and the gut microbiota would be suggestive of secondary effects. As mentioned above, the axis of increased and decreased bacterial taxa correlated nicely with disease activity [Citation33]. Even more convincingly, also discussed above, therapy response has a tendency to normalize the microbiome, although treatment is given also to non-responders which maintain their ‘dysbiotic’ microbiota [Citation47]. In addition, when comparing single patients in remission or exacerbations, significant differences in fecal microbial composition were observed with pronounced interindividual variations [Citation48]. In ulcerative colitis, the gut microbiota tends to normalize in cases with a long-term remission. Nevertheless, some form of dysbiosis persists even in long-term remitters with mucosal healing and microbial instability as well as low rates of Firmicutes and Faecalibacterium appear to be related to a high risk of relapse [Citation49–Citation51]. In two separate Swiss cohorts the mucosal microbiomes were stable at an intra-individual level over time, rather independent of disease activity, but out of 83 taxa analyzed, Enterobacteriaceae and Klebsiella in Crohn´s versus Ruminococcus and Prevotella in ulcerative colitis showed changes according to disease activity [Citation52]. Similarly, differences in the microbiome over time were greatest in the presence of ongoing inflammation [Citation53]. Conspicuously, the patchy distribution of ileal lesions in Crohn´s was not linked to differences in the attached local bacterial community [Citation54]. In a more recent study microbiome differences were significant with respect to endoscopic inflammation as opposed to none. However, there was no difference between matched intra-individual colonic biopsies in mucosa-associated microbiota composition comparing injured and non-injured sites, although lesions were associated with significant bacterial encroachment [Citation55]. It may be concluded that inflammation has a significant impact on the microbiota. Data in laboratory animals confirm an inflammation-associated enterotype [Citation56]. Also, for example, ileal inflammation induced by Toxoplasma gondii drives dysbiosis and bacterial invasion in the mouse [Citation57].
Furthermore, there is little doubt that the host and its genetic background shapes the microbiota. Within a population, at least three different ‘enterotypes’ coexist with predominant Bacteroides, Prevotella or Ruminococcus [Citation58]. Even if these groups are not as distinct as suggested originally but more continuous, the variation within a population with similar environment is likely to be genetically influenced [Citation59]. Accordingly, the mucosal biopsy microbiome is much more similar in monozygotic twins than in dizygotic twins [Citation60]. The fecal microbiome communities were more related between healthy twins than between twins with Crohn´s, especially when these were discordant for the disease [Citation61]. In a Swedish twin trial, it was furthermore concluded that disease phenotype outweighed genotype effects on the fecal and mucosa-associated microbiome [Citation62]. Despite this, the healthy siblings of Crohn´s-patients in another Swedish trial clearly also had a lower diversity than controls, which may reflect a predisposition [Citation63]. Comparing Crohn´s patients with their healthy relatives also emphasized the dramatic impact of the disease compared to a common genetic and environmental background [Citation64]. However, the healthy relatives also differed from healthy controls.
Looking at genetic detail, the best-known modulator of the microbiome in Crohn´s is the most important genetic link: NOD2 (nucleotide-binding oligomerization domain 2) [Citation65]. NOD2 regulates particularly ileal bacteria and ileal bacteria regulate crypt NOD2 in the mouse while NOD2 is also important for the human microbiota [Citation66,Citation67]. The autophagy gene ATG16-L1 also appears to be associated with shifts in human ileal microbial composition and the endosomal stress protein XBP1 gene as well [Citation68,Citation69]. In an interesting study on three levels, including mucosal gene regulation, mRNA splicing, and adherent microbiota signatures, their interaction was ‘uncoupled’ during any inflammation but particularly in IBD [Citation70]. Avoiding the impact of disease on the microbiome, a Dutch group recently demonstrated a significant impact of IBD risk genes on certain bacteria like Roseburia even in healthy individuals [Citation71]. However, the full picture using genome-wide analysis and microbiome studies only gradually evolves and is certainly not restricted to the gut [Citation72–Citation74].
As a matter of fact, this genetic part is complemented, and maybe sometimes antagonized, by the environment. Interestingly in the large Swiss trial mentioned above, the major factors in this regard were the body mass index (BMI) and age [Citation52]. Overall lifestyle, including sport, smoking and alcohol consumption, were also correlated with the gut microbiota [Citation52]. Probably the key factor accessible to modification is the diet, which probably is impacting on the epidemiology discussed above [Citation75]. The long-term dietary pattern determines the enterotype and the short-term change in diet from herbivorous to carnivorous modifies composition dramatically [Citation76,Citation77]. Some feel that a Westernized diet is the most ubiquitous environmental factor in IBD and this is probably mediated at least in part by the microbiome [Citation78,Citation79]. Interestingly, Chinese patients with IBD also exhibited a reduced diversity and tended to have had a Western diet in childhood [Citation80].
Finally, the microbiome would be proven to be the hen and not the egg if IBD-feces would induce IBD. But in the only instance where this could have happened with fecal material from an individual later developing Crohn´s disease, luckily none of the 31 fecal transfer recipients eventually got this disease [Citation81]. However, in some experimental models some degree of inflammation could be induced by stool transfer, although in another mouse study the NOD2-associated dysbiosis did not drive mucosal tissue and immune alterations [Citation82–Citation84]. Fecal material transferred from UC patients to gnotobiotic mice did not induce inflammation [Citation85]. Notably, the confirmed observations in several controlled trials that fecal transfer from healthy controls actually may induce remission in a subgroup of patients clearly demonstrates at least a potential protective role [Citation86–Citation88]. Here more diverse donor microbiota were superior, but ironically a sterile fecal filtrate also was effective, at least in C. difficile colitis [Citation89–Citation91]. Conspicuously, this benefit is only seen in ulcerative colitis patients, which also respond well to some probiotics, in sharp contrast to Crohn´s disease [Citation92]. Taken together, there is little controversy that invading microbes induce inflammation in IBD, and in turn inflammation affects microbial composition, but the microbiota also may be more or less aggressive during the course of disease, or even protective, if derived from a healthy host in the form of a fecal transfer. The pro and con arguments for a primary vs. secondary role are summarized in and .
Figure 2. Possible interactions of the gut microbiota with the mucosal barrier, ultimately leading to IBD. In the left scenario, the microbiota is altered, leading to barrier defects and inflammation in a susceptible host. In the right scenario, primary mucosal barrier defects lead to altered gut microbiota, and eventually an onset of inflammation.
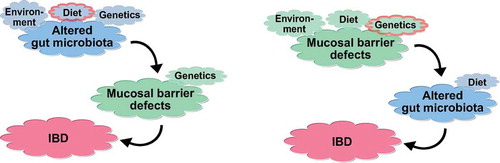
Table 2. Arguments for microbes being hen or egg in IBD.
3. Role of antimicrobial peptides in IBD
The realm of antimicrobial peptides (AMPs) as produced by essentially all epithelia potentially confronted with microbes and also by some inflammatory cells like granulocytes had been discovered and investigated in depth by the end of the last century: AMPs are virtually ubiquitous in nature, from multicellular organisms to man [Citation93]. The microbial distribution in man is not uniform but specific for given organs or regions, and the natural peptides governing their presence or absence are just as diverse. The role of AMPs in IBD has come to some attention only more recently [Citation94]. Most important in the gut epithelium are the α-defensins HD5 and HD6, produced by the small-intestinal Paneth cells, and the ß-defensins (constitutive HBD1 and inducible HBD2 and HBD3), mostly in the gastric and colonic epithelium [Citation94]. These are complemented in the Paneth cell by lysozyme and various lectins, like the REG´s, and in the colon by the cathelicidine LL-37 [Citation95]. A new kid on the block is high-mobility-group box 2, all along the gastrointestinal tract [Citation96]. Essentially all AMPs are positively charged and therefore bind to the negatively charged mucins electrostatically. This binding appears to be reversible because mucus samples are still antibacterially active [Citation97]. Thus, the function of mucus, as discussed below in detail, is both physical impenetrance and bacterial killing by incorporated AMPs.
Most AMPs act on the bacterial cell wall directly, whereas others like HBD1 and HD6 form nets to prevent bacterial invasion [Citation98,Citation99]. Some, like HBD1 and HD6, have to be linearized through reduction of the disulfide bonds to be activated, most likely to act specifically in the periplasmic space through bacterial oxidoreductases [Citation100–Citation102]. A recent quite surprising finding was the significant antibiotic activity of a multitude of defensin fragments formed by natural proteolytic enzymes contained in intestinal secretions [Citation103]. Thus, defensins like HD5 do not only act as intact peptides but may, under physiological conditions, disintegrate and explode like a ‘bombshell’ into multiple shorter peptides with differential antibacterial activities toward several different strains. This shows that there is still a lot to learn about our natural defenses.
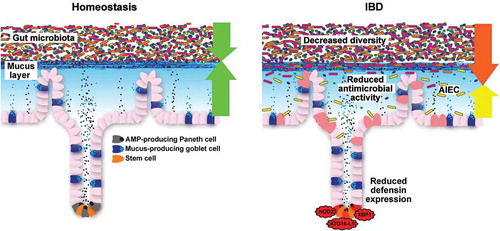
Their important contribution to IBD derives from the fact that several of the observations and arguments in may be explained by disturbances in this system. For example, the changes in the microbiome composition and the bacterial contamination of the mucosa as well as the inner layer of the mucus may well be mediated by defects in this chemical defense [Citation104]. Notably, this applies primarily to Crohn´s disease, whereas in ulcerative colitis the AMP system seems to be adequately induced, here the mucus structure and function predominate in pathophysiology (see below). With respect to Crohn´s disease, it has long been known that patients may either exhibit ileal, ileocolonic (mostly ileocecal) or isolated colonic disease and this classification was recently corroborated by genetic studies [Citation105]. This localization effect is likely be related to local differences of innate immunity (such as defensins) in the interaction with the microbes rather than of systemic adaptive immunity ().
Figure 3. Small-intestinal barrier function during homeostasis and IBD. During homeostasis, the gut microbiota is held at a distance through a mucus layer that is loaded with antimicrobial peptides (AMPs), which are predominantly produced by Paneth cells. During IBD, decreased diversity of the gut microbiota is consistently observed over most studies. Moreover, reduced antimicrobial activity and adherent-invasive E. coli (AIEC) are observed in ileal biopsies, which may be a result of defective Paneth cell function in Crohn’s disease.
Our initial interest in small-intestinal defensins was prompted by the observation that NOD2 was intensely expressed by the Paneth cell [Citation106]. In two independent cohorts in Germany and the USA Paneth cell α-defensins HD5 and HD6 mRNA and protein were diminished in small intestinal but not colonic Crohn´s disease [Citation107,Citation108]. The low HD5 was not related to inflammation but to the NOD2 genetic status. In Australian patients, low HD5 in ileal Crohn´s disease was confirmed but related to inflammation and not NOD2 [Citation109]. Low HD5 expression levels were also observed in younger (<18 years) patients [Citation110]. More recently, in an unsupervised expression analysis of American pediatric patients, again HD5 expression was reduced, related to enhanced interferon-γ and tended to be particularly low in NOD2-mutated children at an age >10 years [Citation111]. In even younger patients this was not observed. Others had found an abnormal HD5 in Japanese patients, where the Paneth cell defect is related to LRRK and not NOD2 or ATG16L1 [Citation112,Citation113]. In contrast, other Paneth cell peptides like lysozyme or sPLA2 (secretory phospholipase A2) were not diminished in the small intestine [Citation107,Citation108]. These observations were recently complemented by morphological observations of deranged Paneth cell granules and crinophagy in ileal Crohn´s [Citation114,Citation115]. These aberrations are likely to reflect functional impairment and, most interestingly were related to the established genetic links of Crohn´s disease, which are now known to be associated exclusively with ileal involvement [Citation114]. Corroborating these data in man, it was demonstrated elegantly in the mouse that mutations or deficits in NOD2, the autophagy gene ATG16L1 as well as genes of endosomal stress and even smoking negatively impact the Paneth cell and may induce ileal inflammation as well as microbiome changes [Citation116–Citation119]. Finally, a key regulator of the Paneth cell differentiation under normal conditions as well as in Crohn´s disease is the Wnt system [Citation120,Citation121].
Colonic Crohn´s disease, compared to ulcerative colitis, is characterized by low HBD1, regulated by PPARγ, and a compromised induction of HBD2 and HBD3 [Citation122,Citation123]. This was associated with a low mucosal antibacterial activity against B. vulgatus and E. faecalis but not S. aureus [Citation124]. Independent studies confirmed this defective induction in Crohn´s disease compared to ulcerative colitis. It is unclear whether this defect is related to the absence of inductor stimuli like butyrate, Muc2 or even vitamin D [Citation125,Citation126]. Genetic associations with the ß-defensin gene number are controversial. Interestingly, Paneth cells, normally rare in the proximal and absent from the distal colon, may appear in the inflamed IBD colon, and both α-defensins HD5 and HD6 as well as lysozyme and sPLA2 are expressed [Citation107,Citation108,Citation127]. Most of this appears to be due to Paneth cell metaplasia as an additional antibacterial defense in the inflamed colon but some of these peptides are also produced by non-Paneth epithelial cells. Notably, another interesting peptide called bactericidal/permeability increasing protein (BPI) was demonstrated to be linked to the disease course in ulcerative colitis [Citation128]. Lower levels were associated with a more severe disease course. It may be concluded that the role of Paneth cell defensins and Paneth cell alterations are well established in ileal Crohn´s disease whereas the colonic AMP´s and their defects require much more research [Citation129].
4. Role of the mucus layer
4.1. Structure of the mucus layer
As a first line of defense, the intestinal mucus layer protects the host epithelium from intestinal content, which includes the trillions of gut bacteria, but also undigestible food particles. Mucus is constitutively secreted from specialized secretory cells, called goblet cells, that are dispersed over the intestinal epithelium. The major structure-forming protein of mucus is mucin 2 (MUC2), a highly O-glycosylated glycoprotein of about 5100 amino acids and a molecular weight of about 2.7 MDa [Citation130,Citation131]. MUC2 monomers are linked through C-terminal disulfide bridges in the endoplasmic reticulum of goblet cells, and the formed dimers further trimerize in the Golgi [Citation132,Citation133]. Secretory vesicles then store the mucin oligomers in the absence of water, low pH and high concentration of calcium, which allows dense packaging until secretion, which is orchestrated by the NLRP6 inflammasome through the induction of autophagy [Citation134,Citation135].
Goblet cells express Toll-like receptors (TLRs) 1–5 as well as their adaptor protein MyD88, and are thus capable of bacterial recognition [Citation136]. In the small intestine, goblet cells are predominantly found in the bottom of the crypts of Lieberkühn whereas in the colon a higher density of goblet cells is detected on the mouth of the crypts. Also, colonic goblet cells express much higher levels of TLRs 1, 2, 4, and 5 while small-intestinal goblet cells express slightly higher levels of TLR3, indicating that small intestinal and colonic goblet exhibit specialized transcriptional profiles [Citation136]. Even more, a distinct subset of goblet cells has been identified in the colon: this specialized ‘sentinel’ goblet cell is located close to the crypt opening, where it safeguards the crypt against bacteria that trespassed through the mucus layer. Once this cell is activated by bacterial ligands, it releases its mucus and transmits alarm signals to adjacent goblet cells, which also secrete their mucus to repel the bacterial invaders [Citation137].
With its highly hydrated, gel-like composition, mucus lubricates the passage of stool through the intestinal channel and thereby protects the epithelium from physical damage. While this has long been considered to be the only task of intestinal mucus, the last decade or so has demonstrated that mucus is much more dynamic and complex than a simple grease. In fact, the mucus barrier varies in the longitudinal and vertical axes of the intestine, as we will discuss below. Besides the structure-forming MUC2, the mucus layer contains bacterial and dietary proteins and peptides, but also a core mucus proteome of about 50 proteins in the mouse colon [Citation138]. So far, it is not completely understood on a molecular level how the protective mucus layer is built up, but non-mucin proteins, hydrogen bonding, calcium ions and swelling of up to 100–1000 times of its initial volume are involved [Citation134,Citation139,Citation140,Citation141].
Likely dictated by the different functions of the small and large intestine, the characteristics of the mucus layer also differ between those two segments. In the small intestine, mucus is formed by a single non-attached layer that can be easily removed by aspiration [Citation142]. Remarkably, studies in germ-free mice have shown that the presence of bacteria is required for this characteristic detachment, as it takes 5 weeks of bacterial colonization until the small intestinal mucus layer can be normally removed [Citation143]. This phenomenon is likely mediated through the microbiota-dependent metalloprotease meprin β, which is shed into the luminal mucus in bacteria-colonized mice and has been demonstrated to cleave MUC2 from the goblet cell surface [Citation144].
In contrast to the small intestine, colonic mucus is formed by two distinct layers, from which the inner layer is attached to the intestinal epithelium and devoid of bacteria, while the outer layer is heavily colonized by gut bacteria and also contains dietary molecules [Citation142,Citation145]. The transition from the inner to the outer mucus layer seems to be controlled by endogenous factors, as the colon of germ-free mice displays a similar division [Citation143]. Indeed, calcium-activated chloride channel regulator 1 (CLCA1), a metalloprotease that is highly expressed in the intestine and abundant in the mucus, has recently been identified to contribute to the processing and expansion of the inner mucus layer [Citation146]. As such, CLCA1 contributes to the ‘mucus growth rate’, which has been measured to be about 240 µm/h in human and 100 µm/h mouse colon and is a characteristic feature of the inner colonic mucus layer [Citation147].
Analogous to the small intestine, full maturity of the colonic mucus is also dependent on the presence of gut microbiota. While the mucus growth rate is similar between germ-free and colonized mice, germ-free mice have a penetrable inner colonic mucus layer [Citation143]. Moreover, the intense O-glycosylation of the major mucus protein MUC2 seems to be regulated by gut microbiota-mediated expression of selective glycosyltransferases [Citation148]. Remarkably, not simply the presence of bacteria but a specific composition of gut bacteria is required to mature the colonic mucus layer: Two mouse colonies in different rooms of the same animal facility had different mucus phenotypes (i.e. penetrability) and this phenotype could be transferred to germ-free mice by microbiota transplantation [Citation149]. Yet, it is so far largely unclear how bacteria (or other community members of the gut microbiota) modulate the mucus function and which metabolites are involved.
As discussed above, mucus is also a reservoir for antimicrobial peptides [Citation150]. These host-defense molecules can kill or entrap bacteria that managed to penetrate the mucus layer and thus serve as an additional line of defense. However, despite the original assumption that such antimicrobial defense is unspecific and broad-spectrum, recent studies have shown that it is in fact quite selective. For example, Ly6/PLAUR domain containing 8 (Lypd8) protein selectively entraps Gram-negative-flagellated bacteria in the colon and thereby prevents translocation and thus the development of intestinal inflammation [Citation151]. Potentially, this could also be a mechanism how probiotic bacteria might prevent intestinal inflammation, as the probiotic Escherichia coli strain Nissle 1917 has been demonstrated to bind intestinal mucin via its flagellum and might thus occupy adherence sites for more unfavorable bacteria [Citation152]. As a companion to Gram-negative bacteria-binding Lypd8, zymogen granule protein 16 (ZG16) has been shown to selectively bind and aggregate Gram-positive bacteria in the colonic mucus [Citation153]. Through binding to peptidoglycan of the cell-wall, ZG16 prevents translocation of Gram-positive bacteria into underlying tissues, such as caudal lymph nodes and spleen.
4.2. Function of the mucus layer
Intestinal mucosal barrier defects are a hallmark of IBD and there is continuous discussion about cause and consequence [Citation154]. For patients with UC, a recent study now supports the hypothesis that a weakening of the colonic mucus barrier contributes to the development of UC pathogenesis, as a significant reduction of the structural MUC2 protein was observed in the colonic mucus of patients with active UC, independent of local inflammation [Citation155]. New-onset treatment-naïve pediatric patients with UC had compositional changes of the gut microbiome, which were associated with disease progression and severity [Citation156]. Strikingly, temporal changes in the gut microbiome were linked to the course of the disease, thus further supporting a dysfunctional host–microbe interaction in a susceptible host as a driver for UC [Citation156]. Furthermore, in the same cohort of pediatric patients, RNAseq analysis in rectal biopsies revealed reduced expression of epithelial mitochondrial genes and associated energy production pathways in active disease, some of which could be used to predict success of a corticosteroid response of the patients [Citation157]. Again, altered genes could be linked to distinct gut microbial taxa, including pathways and genes that are involved in the resolution of inflammation, which were linked to the abundance of commensal organisms, such as Lachnospiraceae, Bifidobacterium, and Ruminococcaceae [Citation157].
While a general defect of intestinal mucosal barrier function is a hallmark of IBD, specific dysfunction of the colonic mucus layer has been observed in ulcerative colitis. Various mouse models of colitis, including mice deficient in Muc2, Tlr5, Il10, with defective glycosylation, or with chemically induced dextran sodium sulfate (DSS) colitis, had bacteria penetrating into the otherwise impenetrable inner colonic mucus layer [Citation158]. Similar defects were observed in patients with active UC, and even in a subgroup of patients in remission. Yet, whether bacterial penetration into the mucus layer is the primary event and causes the disease, or whether a primary host defect allows bacteria to penetrate the mucus layer is so far not clear. However, as alterations of the gut bacteria can lead to defects of the colonic mucus layer, which are at least in part caused by increased bacterial degradation of mucus glycans, a primary role for the gut bacteria is certainly possible [Citation159–Citation161].
As altered gut microbiota composition, and thus altered production of microbial metabolites, is linked to mucus dysfunction, in return this means that specific microbes are contributing to a healthy mucus layer. Tata Element Modulatory Factor (TMF/ARA160) is a Golgi-associated protein with high abundance in colonic goblet cells and involved in the regulation of Stat3 (signal transducer and activator of transcription 3) and NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) under stress conditions [Citation162]. Remarkably, mice that lack TMF/ARA160 (TMF−/−) had decreased susceptibility to DSS-induced colitis, which was accompanied by a reduced number of bacteria colonizing the mucus layer [Citation163]. In addition, TMF−/− mice had increased expression of Muc2, which resulted in a thick, uniform colonic mucus layer that was characterized by a higher MUC2 oligomerization grade [Citation164]. Fecal gut microbiota analyses furthermore revealed that TMF−/− mice had altered gut microbiota composition with higher relative abundances of Ruminococcaceae, Roseburia, and Lactobacillus groups when compared with wild-type mice. Remarkably, when TMF−/− mice were co-housed with wild-type mice, the diminished susceptibility to induced colitis was transmissible to wild-type mice, further indicating that the gut microbiota can actively modulate mucus function, opening up a potential therapeutic avenue to block TMF/ARA160 activity [Citation164].
Since distinct gut bacteria have thus the ability to affect mucosal barrier function, modulating the gut microbiota through dietary or probiotic interventions can be used to strengthen the mucus layer. In fact, several studies have already shown a microbial effect on mucus production or function. For example, application of the probiotic strain Lactobacillus rhamnosus CNCM I-3690 to mice led to improved colonic barrier function by increasing mucus production and restoring goblet cell population [Citation165]. Similarly, two Lactobacillus reuteri strains, one human-derived and one rat-derived, reduced the inflammation markers MPO, IL-1β and IL-6 in a murine DSS colitis model, which might at least in part be due to a mucus thickness-increasing effect of the Lactobacillus strains [Citation166]. Yet, in another DSS colitis study in rats, a probiotic Lactobacillus reuteri strain has been shown to reduce DSS-associated bacterial translocation to the mesenteric lymph nodes without improving the integrity of the colonic mucus layer [Citation167]. While the first Lactobacillus reuteri study used isolated Lactobacillus strains for the treatment, the second study applied a cocktail of four different strains from different sources [Citation166,Citation167]. As immune- and mucus-modulating capacities are strain specific, and different strains might influence each other, this might explain the different results in the two murine DSS colitis models using Lactobacillus strains.
Bacteroides thetaiotaomicron (B. theta), a common intestinal inhabitant and acetate producer, increased goblet cell differentiation and thus stimulates the secretory lineage, leading to augmented mucus production in mono-colonized rats [Citation168]. Interestingly, when the rats where co-colonized with B.theta and Faecalibacterium (F.) prausnitzii, an acetate consumer and butyrate producer, the mucus-promoting effect of B.theta was abrogated. It is thus likely that in a complex intestinal community an even more complicated network of metabolite producers and consumers regulates mucus function in vivo. In addition, the anti-inflammatory commensal F. prausnitzii might play an important role in IBD, as a reduced abundance has been detected in patients with CD and oral application of the bacterium or its supernatant could reduce chemically induced colitis in mice [Citation42]. Moreover, in vitro supplementation of stool samples from CD patients with F. prausnitzii led to increased butyrate production at the costs of acetate production, which led to enhanced epithelial barrier function in a cell culture model [Citation169].
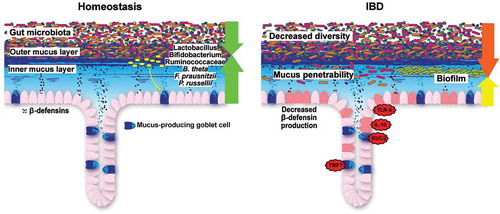
Although an increasing number of bacteria is identified that either positively or negatively modulate mucus function, the molecular details often remain elusive. However, to therapeutically utilize the microbe-mucus axis in IBD a better molecular understanding is required. How this can be achieved was demonstrated in a recent study, in which the mucin degradation capability of the bacterium Peptostreptococcus russellii was first identified through computational prediction and subsequently confirmed in vitro [Citation170]. This bacterium was able to colonize the mucus layer and mediate an increase in goblet cell number and to reduce susceptibility to DSS-induced colitis in mice, likely by producing the tryptophan metabolite indoleacrylic acid (IA). In a system of LPS-stimulated co-cultured bone marrow-derived macrophages and colonic spheroids, IA could enhance IL-10 production and increased Muc2 expression through activation of the transcription factor NRF2, which promoted anti-oxidant and anti-inflammatory immune responses [Citation170]. Finally, the authors could show that patients with UC and CD had lower fecal abundance of bacteria that can degrade terminal fucose residues from mucin glycans and to metabolize tryptophan, which correlated with lower capacity to produce IA. Consequently, facilitating the colonization of the mucus layer by health-promoting bacteria such as P. russellii could be a novel strategy to target the mucus barrier defects in patients with IBD ().
Figure 4. Colonic barrier function during homeostasis and IBD. During homeostasis, gut microbiota colonization is restricted to the outer mucus layer. Beneficial commensal bacteria, such as Lactobacillus, Bifidobacterium, Ruminococcaceae, Bacteroides thetaiotaomicron (B. theta), Faecalibacterium prausnitzii (F. prausnitzii) and Peptostreptococcus russellii (P. russellii) stimulate and maintain active mucosal barrier function. During IBD, bacterial diversity is decreased, the inner colonic mucus layer is penetrable to bacteria (ulcerative colitis) and a biofilm, mainly consisting of Bacteroides fragilis, can be observed at the mucosa (Crohn’s disease). In mice, several genetic defects recapitulate the clinical signs of ulcerative colitis.
5. Expert opinion
There is increasing evidence that the gut microbiota can modulate intestinal mucosal barrier function and that disturbances in this modulation might be a strong contributor to inflammatory bowel diseases. We believe that this opens possibilities to exploit this interaction for therapeutic interventions. Instead of dampening the inflammation, as most current treatments do, targeting the gut bacteria would strike already a stage earlier and in the best case prevent the development of chronic inflammation. While the use of specific probiotics like E. coli Nissle 1917 is already recommended for UC, ongoing research is expected to identify additional strains with specific modulatory and/or anti-inflammatory functions. Likely candidates to enter clinical practice are beneficial bacteria like F. prausnitzii or improved probiotic cocktails, as well as the above-mentioned crude fecal transfer, optimized by selecting the ‘golden donor’. However, although mechanistic data are accumulating, a remaining challenge for a successful clinical application is the large-scale cultivation and preservation of anaerobic bacteria. Yet, we are optimistic that it is only a matter of time until these challenges will be resolved.
Parallel to the development of novel probiotics, a different approach with a similar outcome is to modulate the modulator, i.e. to shift the gut microbiota with selected dietary substrates. As such, prebiotics, are known to improve host health by enriching beneficial bacteria [Citation171]. For IBD, only a few trials have been conducted and yielded mixed results, depending on clinical entity and the selected prebiotic [Citation172–Citation176]. Thus, to apply this concept for IBD requires further research, as it is not entirely clear yet which bacteria should be enriched and which dietary substances would facilitate a selective enrichment of the to-be-identified bacteria.
Recently, a novel tool to screen potential drug candidates has been described, in which a micro-engineered ‘human gut inflammation-on-a-chip’ was tested to recapitulate the barrier dysfunction mediated by intercellular host–microbiome cross-talk in vitro [Citation177]. This system demonstrated that DSS treatment strongly impaired the epithelial barrier, including mucus production, without being cytotoxic to epithelial cells. Moreover, the set-up revealed that probiotic treatment, in this case VSL#3, required an undamaged epithelial barrier to be effective, and was ineffective when the barrier was already damaged by DSS treatment [Citation177]. We think that in the future, such a chip could be used to test whether potential therapeutic compounds, including probiotic bacteria, can effectively ameliorate intestinal inflammation when the barrier is already damaged, or if they would rather be effective to maintain remission.
Modulating the second part of the equation, the antibacterial peptides, is also possible. The above-mentioned E. coli Nissle, and some other probiotics have been demonstrated to enhance inducible ß-defensin formation [Citation178]. The direct oral administration of defensins or of their fragments is also feasible and has been shown in preliminary work to be beneficial in experimental colitis [Citation179]. An alternative would be to administer the known natural inducers mentioned above, including vitamin D, or alternatively target defensin-mRNA to Paneth cells.
Finally, targeting the mucus should not only be possible using microbes, as mentioned above, but also chemicals or drugs like simple lecithin, which is known to be diminished in UC mucus. Accordingly, application of delayed-release lecithin led to an improvement of ulcerative colitis [Citation180]. Yet, simultaneous application of lecithin and the established drug for ulcerative colitis, mesalazine, did not result in the expected efficiency, probably because mesalazine removed mucin-bound lecithin [Citation181]. Other ways to reduce penetrability through chemical interactions of UC-mucus are conceivable. Another elegant way to stabilize the barrier might be achieved by enhancing Paneth cell and goblet cell differentiation through Wnt and other differentiation factors [Citation94,Citation144,Citation182].
There are multiple efforts to identify microbial signatures as diagnostic markers for IBD and changes in fecal gut microbiota composition have been identified in several cohorts of IBD patients, yet without common conclusive results [Citation32,Citation183–Citation185]. This discrepancy is likely due to different geographic origin of the cohorts, dietary habits and methodological differences in sample preparation. However, analysis of mucosa-associated bacteria has been proven to be a better predictor of Crohn’s disease than the analysis of fecal samples [Citation33]. While obtaining mucosal samples is somewhat more complicated than collecting fecal samples, it might still be a more promising approach to diagnose and/or predict IBD in treatment-naïve patients. Nevertheless, even crude stool metagenomes helped to predict the disease course in vedolizumab-treated patients [Citation186]. Considering the dropping prices in high-throughput microbiota sequencing costs, we envision that a personalized therapy, based on microbiota composition, might be possible already in the next decade or so. In such a scenario, a mucosal microbiota analysis would identify missing or depleted microbes, which could then be supplemented or enriched through probiotic or prebiotic therapy. Without doubt, before this option becomes reality, further understanding of the barrier-modifying abilities of individual gut microbiota is required.
In conclusion, accumulating evidence suggests that the barrier is of prime and maybe primary importance in IBD, but that gut microbiota are not just innocent bystanders given the chance to penetrate the mucus and mucosa by the defective host. Modulating the gut microbiota and/or the mucus by targeted interventions, either through diet (prebiotic), microbial (probiotic) or drug approaches is becoming a more and more promising option and might help to reduce the burden of IBD in the future by finally arriving at a more ‘causal’ treatment.
Article highlights
A dysfunctional interaction between the gut microbiota and the intestinal mucosal barrier is a hallmark of IBD.
Changes in gut microbiota composition during IBD are inconsistent between studies.
Are gut microbiota alterations cause (hen) or consequence (egg) of IBD?
Antimicrobial peptides and intestinal mucus are key elements of intestinal barrier function that are defective during IBD.
Targeting the gut microbiota to strengthen intestinal barrier function may become a successful strategy to treat IBD in the future.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The authors thank Stephanie Bügler-Mietens for administrative assistance with the manuscript.
Additional information
Funding
References
- Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1): 46–54.e42. quiz e30.
- Kaplan GG, Ng SC. Globalisation of inflammatory bowel disease: perspectives from the evolution of inflammatory bowel disease in the UK and China. Lancet Gastroenterol Hepatol. 2016;1(4):307–316.
- Piovani D, Danese S, Peyrin-Biroulet L, et al. Environmental risk factors for inflammatory bowel diseases: an umbrella review of meta-analyses. Gastroenterology. 2019;157(3):647–659.e4.
- The International IBD Genetics Consortium, Momozawa Y, Dmitrieva J, Théâtre E, et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat Commun. 2018;9(1):2427.
- Asakura H, Suzuki K, Kitahora T, et al. Is there a link between food and intestinal microbes and the occurrence of Crohn’s disease and ulcerative colitis? J Gastroenterol Hepatol. 2008;23(12):1794–1801.
- Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164(3):337–340.
- Antoni L. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2014;20(5):1165.
- Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. 2017;152(2):327–339.e4.
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134(2):577–594.
- Hildebrand H, Malmborg P, Askling J, et al. Early-life exposures associated with antibiotic use and risk of subsequent Crohn’s disease. Scand J Gastroenterol. 2008;43(8):961–966.
- Rutgeerts P, Goboes K, Peeters M, et al. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet. 1991;338(8770):771–774.
- Duchmann R, Kaiser I, Hermann E, et al. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol. 1995;102(3):448–455.
- Duchmann R, May E, Heike M, et al. T cell specificity and cross reactivity towards enterobacteria, Bacteroides, Bifidobacterium, and antigens from resident intestinal flora in humans. Gut. 1999;44(6):812–818.
- Furrie E. Systemic antibodies towards mucosal bacteria in ulcerative colitis and Crohn’s disease differentially activate the innate immune response. Gut. 2004;53(1):91–98.
- Curciarello R, Canziani KE, Docena GH, et al. Contribution of non-immune cells to activation and modulation of the intestinal inflammation. Front Immunol. 2019;10:647.
- Rubio CA, Schmidt PT. Severe defects in the macrophage barrier to gut microflora in inflammatory bowel disease and Colon Cancer. Anticancer Res. 2018;38(7):3811–3815.
- Buttó LF, Haller D. Dysbiosis in intestinal inflammation: cause or consequence. Int J Med Microbiol. 2016;306(5):302–309.
- Marchesi JR, Adams DH, Fava F, et al. The gut microbiota and host health: a new clinical frontier. Gut. 2016;65(2):330–339.
- Dalal SR, Chang EB. The microbial basis of inflammatory bowel diseases. J Clin Invest. 2014;124(10):4190–4196.
- McIlroy J, Ianiro G, Mukhopadhya I, et al. Review article: the gut microbiome in inflammatory bowel disease-avenues for microbial management. Aliment Pharmacol Ther. 2018;47(1):26–42.
- Richard ML, Sokol H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol. 2019;16(6):331–345.
- Norman JM, Handley S, Baldridge M, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160(3):447–460.
- MetaHIT Consortium, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65.
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
- Falony G, Joossens M, Vieira-Silva S, et al. Population-level analysis of gut microbiome variation. Science. 2016;352(6285):560–564.
- Almeida A, Mitchell AL, Boland M, et al. A new genomic blueprint of the human gut microbiota. Nature. 2019;568(7753):499–504.
- Vandeputte D, Kathagen G, D'hoe K, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551(7681):507–511.
- Franzosa EA, Morgan XC, Segata N, et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci. 2014;111(22):E2329–38.
- Hollister EB, Riehle K, Luna RA, et al. Structure and function of the healthy pre-adolescent pediatric gut microbiome. Microbiome. 2015;3(1):36.
- Tropini C, Earle KA, Huang KC, et al. The Gut Microbiome: connecting spatial organization to function. Cell Host Microbe. 2017;21(4):433–442.
- Manichanh C. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55(2):205–211.
- Halfvarson J, Brislawn CJ, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol. 2017;2:17004.
- Gevers D, Kugathasan S, Denson L, et al. The treatment-naive microbiome in new-Onset Crohn’s Disease. Cell Host Microbe. 2014;15(3):382–392.
- Vandeputte D, Falony G, Vieira-Silva S, et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65(1):57–62.
- Swidsinski A, Ladhoff A, Pernthaler A, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122(1):44–54.
- Swidsinski A, Weber J, Loening-Baucke V, et al. Spatial organization and composition of the Mucosal Flora in patients with Inflammatory Bowel Disease. J Clin Microbiol. 2005;43(7):3380–3389.
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A. 2010;107(27):12204–12209.
- Ott SJ. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53(5):685–693.
- Frank DN, Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci. 2007;104(34):13780–13785.
- Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127(2):412–421.
- Lapaquette P, Glasser A-L, Huett A, et al. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12(1):99–113.
- Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–16737.
- Huttenhower C, Kostic AD, Xavier RJ. Inflammatory Bowel Disease as a model for translating the Microbiome. Immunity. 2014;40(6):843–854.
- Olbjørn C, Cvancarova Småstuen M, Thiis-Evensen E, et al. Fecal microbiota profiles in treatment-naïve pediatric inflammatory bowel disease – associations with disease phenotype, treatment, and outcome. Clin Exp Gastroenterol. 2019;12:37–49.
- Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60.
- Forbes JD, Chen CY, Knox NC, et al. A comparative study of the gut microbiota in immune-mediated inflammatory diseases—does a common dysbiosis exist? Microbiome. 2018;6(1):221.
- Lewis JD, Chen E, Baldassano R, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s Disease. Cell Host Microbe. 2015;18(4):489–500.
- Wills ES, Jonkers DMAE, Savelkoul PH, et al. Fecal microbial composition of ulcerative Colitis and Crohn’s Disease patients in remission and subsequent exacerbation. PLoS ONE. 2014;9(3):e90981.
- Rajca S, Grondin V, Louis E, et al. Alterations in the intestinal microbiome (Dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohnʼs Disease. Inflamm Bowel Dis. 2014;20(6):978–986.
- Braun T, Di Segni A, BenShoshan M, et al. Individualized dynamics in the gut microbiota precede Crohnʼs Disease Flares:. Am J Gastroenterol. 2019;114(7):1142–1151.
- Wright EK, Kamm MA, Wagner J, et al. Microbial factors associated with postoperative Crohn’s Disease Recurrence. J Crohns Colitis. 2017;11(2):191–203.
- Swiss IBD Cohort Investigators, Yilmaz B, Juillerat P, Øyås O, et al. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat Med. 2019;25(2):323–336.
- Kiely CJ, Pavli P, O’Brien CL. The role of inflammation in temporal shifts in the inflammatory bowel disease mucosal microbiome. Gut Microbes. 2018;1–9.
- Vasquez N, Mangin I, Lepage P, et al. Patchy distribution of mucosal lesions in ileal Crohnʼs disease is not linked to differences in the dominant mucosa-associated bacteria: a study using fluorescence in situ hybridization and temporal temperature gradient gel electrophoresis. Inflamm Bowel Dis. 2007;13(6):684–692.
- Libertucci J, Dutta U, Kaur S, et al. Inflammation-related differences in mucosa-associated microbiota and intestinal barrier function in colonic Crohn’s disease. Am J Physiol-Gastrointest Liver Physiol. 2018;315(3):G420–31.
- Hildebrand F, Nguyen TLA, Brinkman B, et al. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013;14(1):R4.
- Craven M, Egan CE, Dowd SE, et al. Inflammation drives dysbiosis and bacterial invasion in murine models of Ileal Crohn’s Disease. PLoS ONE. 2012;7(7):e41594.
- MetaHIT Consortium (additional members), et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180.
- Knights D, Clare S, Goulding D, et al. Rethinking ‘Enterotypes,’. Cell Host Microbe. 2014;16(4):433–437.
- Lepage P, Häsler R, Spehlmann ME, et al. Twin study indicates loss of interaction between Microbiota and Mucosa of patients with Ulcerative Colitis. Gastroenterology. 2011;141(1):227–236.
- Dicksved J, Halfvarson J, Rosenquist M, et al. Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. Isme J. 2008;2(7):716–727.
- Willing BP, Dicksved J, Halfvarson J, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139(6):1844–1854.e1.
- Hedin CR, van der Gast CJ, Stagg AJ, et al. The gut microbiota of siblings offers insights into microbial pathogenesis of inflammatory bowel disease. Gut Microbes. 2017;8(4):359–365.
- Joossens M, Huys G, Cnockaert M, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637.
- Sidiq T, Yoshihama S, Downs I, et al. Nod2: a critical regulator of Ileal Microbiota and Crohn’s Disease. Front Immunol. 2016;7.
- Petnicki-Ocwieja T, Hrncir T, Liu Y-J, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci. 2009;106(37):15813–15818.
- Knights D, Silverberg MS, Weersma RK, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6(12).
- Zhang T, DeSimone RA, Jiao X, et al. Host genes related to paneth cells and xenobiotic metabolism are associated with shifts in human Ileum-Associated Microbial Composition. PLoS ONE. 2012;7(6):e30044.
- Li E, Zhang Y, Tian X, et al. Influence of Crohn’s disease related polymorphisms in innate immune function on ileal microbiome. Plos One. 2019;14(2):e0213108.
- Häsler R, Sheibani-Tezerji R, Sinha A, et al. Uncoupling of mucosal gene regulation, mRNA splicing and adherent microbiota signatures in inflammatory bowel disease. Gut. 2017;66(12):2087–2097.
- Imhann F, Vich Vila A, Bonder MJ, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67(1):108–119.
- Bonder MJ, Kurilshikov A, Tigchelaar EF, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48(11):1407–1412.
- Kurilshikov A, Wijmenga C, Fu J, et al. Host genetics and Gut Microbiome: challenges and perspectives. Trends Immunol. 2017;38(9):633–647.
- Blekhman R, Goodrich JK, Huang K, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015;16(1):191.
- Rapozo DCM, Bernardazzi C, de Souza HSP. Diet and microbiota in inflammatory bowel disease: the gut in disharmony. World J Gastroenterol. 2017;23(12):2124.
- Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108.
- David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563.
- Chiba M. Westernized diet is the most ubiquitous environmental factor in inflammatory Bowel Disease. Perm J. 2019;23:18–107.
- Kau AL, Ahern PP, Griffin NW, et al. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474(7351):327–336.
- Prideaux L, Kang S, Wagner J, et al. Impact of ethnicity, geography, and disease on the microbiota in health and Inflammatory Bowel Disease. Inflamm Bowel Dis. 2013;19(13):2906–2918.
- Fischer M, Bittar M, Papa E, et al. Can you cause inflammatory bowel disease with fecal transplantation? A 31-patient case-series of fecal transplantation using stool from a donor who later developed Crohn’s disease. Gut Microbes. 2017;8(3):205–207.
- Couturier-Maillard A, Secher T, Rehman A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700–711.
- Schaubeck M, Clavel T, Calasan J, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65(2):225–237.
- Al Nabhani Z, Lepage P, Mauny P, et al. Nod2 deficiency leads to a specific and transmissible mucosa-associated microbial dysbiosis which is independent of the mucosal barrier defect. J Crohn’s Colitis. 2016;10(12):1428–1436.
- Nakamoto N, Sasaki N, Aoki R, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol. 2019;4(3):492–503.
- Rossen NG, Fuentes S, van der Spek MJ, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology. 2015;149(1):110–118.e4.
- Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149(1):102–109.e6.
- Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389(10075):1218–1228.
- Vermeire S, Joossens M, Verbeke K, et al. Donor species richness determines faecal microbiota transplantation success in inflammatory Bowel Disease. J Crohns Colitis. 2016;10(4):387–394.
- Kump P, Wurm P, Gröchenig HP, et al. The taxonomic composition of the donor intestinal microbiota is a major factor influencing the efficacy of faecal microbiota transplantation in therapy refractory ulcerative colitis. Aliment Pharmacol Ther. 2018;47(1):67–77.
- Ott SJ, Waetzig GH, Rehman A, et al. Efficacy of sterile fecal filtrate transfer for treating patients with clostridium difficile infection. Gastroenterology. 2017;152(4):799–811.e7.
- Kruis W, Fric P, Pokrotnieks J, et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut. 2004;53(11):1617–1623.
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–395.
- Ostaff MJ, Stange EF, Wehkamp J. Antimicrobial peptides and gut microbiota in homeostasis and pathology: homeostasis in the gut. EMBO Mol Med. 2013;5(10):1465–1483.
- Schauber J, Rieger D, Weiler F, et al. Heterogeneous expression of human cathelicidin hCAP18/LL-37 in inflammatory bowel diseases. Eur J Gastroenterol Hepatol. 2006;18(6):615–621.
- Küchler R, Schroeder BO, Jaeger SU, et al. Antimicrobial activity of High-Mobility-Group Box 2: a new function to a well-known protein. Antimicrob Agents Chemother. 2013;57(10):4782–4793.
- Antoni L, Nuding S, Weller D, et al. Human colonic mucus is a reservoir for antimicrobial peptides. J Crohns Colitis. 2013;7(12):e652–664.
- Chu H, Pazgier M, Jung G, et al. Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets,”. Science. 2012;337(6093):477–481.
- Raschig J, Mailänder-Sánchez D, Berscheid A, et al. Ubiquitously expressed Human Beta Defensin 1 (hBD1) forms bacteria-entrapping nets in a redox dependent mode of action. PLoS Pathog. 2017;13(3):e1006261.
- Schroeder BO, Wu Z, Nuding S, et al. Reduction of disulphide bonds unmasks potent antimicrobial activity of human β-defensin 1. Nature. 2011;469(7330):419–423.
- Schroeder BO, Ehmann D, Precht JC, et al. Paneth cell α-defensin 6 (HD-6) is an antimicrobial peptide. Mucosal Immunol. 2015;8(3):661–671.
- Wendler J, Ehmann D, Courth L, et al. Bacterial periplasmic oxidoreductases control the activity of oxidized human antimicrobial β-Defensin 1. Infect Immun. 2018;86(4):e00875–17.
- Ehmann D, Wendler J, Koeninger L, et al. Paneth cell α-defensins HD-5 and HD-6 display differential degradation into active antimicrobial fragments. Proc Natl Acad Sci. 2019;116(9):3746–3751.
- Salzman NH, Hung K, Haribhai D, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11(1):76–83.
- Cleynen I, Boucher G, Jostins L, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387(10014):156–167.
- Ogura Y, Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut. 2003;52(11):1591–1597.
- Wehkamp J. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal -defensin expression. Gut. 2004;53(11):1658–1664.
- Wehkamp J, Salzman NH, Porter E, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A. 2005;102(50):18129–18134.
- Simms LA, Doecke JD, Walsh MD, et al. Reduced -defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut. 2008;57(7):903–910.
- Perminow G, Beisner J, Koslowski M, et al. Defective Paneth Cell—mediated host defense in pediatric Ileal Crohnʼs Disease. Am J Gastroenterol. 2010;105(2):452–459.
- Haberman Y, Schirmer M, Dexheimer PJ, et al. Age-of-diagnosis dependent ileal immune intensification and reduced alpha-defensin in older versus younger pediatric Crohn Disease patients despite already established dysbiosis. Mucosal Immunol. 2019;12(2):491–502.
- Tanabe H, Ayabe T, Maemoto A, et al. Denatured human alpha-defensin attenuates the bactericidal activity and the stability against enzymatic digestion. Biochem Biophys Res Commun. 2007;358(1):349–355.
- Liu T-C, Naito T, Liu Z, et al. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight. 2017;2(6):e91917.
- VanDussen KL, Liu T-C, Li D, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s Disease. Gastroenterology. 2014;146(1):200–209.
- Thachil E, Hugot J-P, Arbeille B, et al. Abnormal activation of autophagy-induced crinophagy in paneth cells from patients with Crohn’s Disease. Gastroenterology. 2012;142(5):1097–1099.e4.
- Liu T-C, Gurram B, Baldridge MT, et al. Paneth cell defects in Crohn’s disease patients promote dysbiosis. JCI Insight. 2016;1(8).
- Liu T-C, Kern JT, VanDussen KL, et al. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. J Clin Invest. 2018;128(11):5110–5122.
- Stappenbeck TS, McGovern DPB. Paneth Cell Alterations in the Development and Phenotype of Crohn’s Disease. Gastroenterology. 2017;152(2):322–326.
- Tschurtschenthaler M, Adolph TE, Ashcroft JW, et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J Exp Med. 2017;214(2):401–422.
- Armbruster NS, Stange EF, Wehkamp J. In the Wnt of paneth cells: immune-epithelial crosstalk in small intestinal Crohn’s Disease. Front Immunol. 2017;8:1204.
- Courth LF, Ostaff MJ, Mailänder-Sánchez D, et al. Crohn’s disease-derived monocytes fail to induce Paneth cell defensins. Proc Natl Acad Sci. 2015;112(45):14000–14005.
- Peyrin-Biroulet L, Beisner J, Wang G, et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc Natl Acad Sci U S A. 2010;107(19):8772–8777.
- Wehkamp J, Harder J, Weichenthal M, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2003;9(4):215–223.
- Nuding S, Fellermann K, Wehkamp J, et al. Reduced mucosal antimicrobial activity in Crohn’s disease of the colon. Gut. 2007;56(9):1240–1247.
- Campbell Y, Fantacone ML, Gombart AF. Regulation of antimicrobial peptide gene expression by nutrients and by-products of microbial metabolism. Eur J Nutr. 2012;51(8):899–907.
- Cobo ER, Kissoon-Singh V, Moreau F, et al. Colonic MUC2 mucin regulates the expression and antimicrobial activity of β-defensin 2. Mucosal Immunol. 2015;8(6):1360–1372.
- Rubio CA. The natural antimicrobial enzyme lysozyme is up-regulated in gastrointestinal inflammatory conditions. Pathog Basel Switz. 2014;3(1):73–92.
- Magnusson MK, Strid H, Isaksson S, et al. The mucosal antibacterial response profile and fecal microbiota composition are linked to the disease course in patients with newly diagnosed ulcerative colitis. Inflamm Bowel Dis. 2017;23(6):956–966.
- Wehkamp J, Stange EF. Paneth’s disease. J Crohns Colitis. 2010;4(5):523–531.
- Allen A, Hutton DA, Pearson JP. The MUC2 gene product: a human intestinal mucin. Int J Biochem Cell Biol. 1998;30(7):797–801.
- Axelsson MAB, Asker N, Hansson GC. O-Glycosylated MUC2 Monomer and Dimer from LS 174T Cells Are Water-soluble, whereas Larger MUC2 species formed early during biosynthesis are insoluble and contain nonreducible intermolecular bonds. J Biol Chem. 1998;273(30):18864–18870.
- Asker N, Axelsson MAB, Olofsson S-O, et al. Dimerization of the human MUC2 mucin in the endoplasmic reticulum is followed by a N-Glycosylation-dependent transfer of the Mono- and Dimers to the Golgi Apparatus. J Biol Chem. 1998;273(30):18857–18863.
- Godl K, Johansson MEV, Lidell ME, et al. The N Terminus of the MUC2 mucin forms trimers that are held together within a Trypsin-resistant core fragment. J Biol Chem. 2002;277(49):47248–47256.
- Ambort D, Johansson MEV, Gustafsson JK, et al. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin. Proc Natl Acad Sci. 2012;109(15):5645–5650.
- Wlodarska M, Thaiss C, Nowarski R, et al. NLRP6 Inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156(5):1045–1059.
- Knoop KA, McDonald KG, McCrate S, et al. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. 2015;8(1):198–210.
- Birchenough GMH, Nyström EEL, Johansson MEV, et al. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science. 2016;352(6293):1535–1542.
- Johansson MEV, Thomsson KA, Hansson GC. Proteomic analyses of the two mucus layers of the colon barrier reveal that their main component, the Muc2 Mucin, is strongly bound to the Fcgbp protein. J Proteome Res. 2009;8(7):3549–3557.
- McGuckin MA, Lindén SK, Sutton P, et al. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9(4):265–278.
- Meldrum OW, Yakubov GE, Bonilla MR, et al. Mucin gel assembly is controlled by a collective action of non-mucin proteins, disulfide bridges, Ca 2+ -mediated links, and hydrogen bonding. Sci Rep. 2018;8(1):5802.
- Nowarski R, Jackson R, Gagliani N, et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell. 2015;163(6):1444–1456.
- Ermund A, Schütte A, Johansson MEV, et al. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer’s patches. Am J Physiol-Gastrointest Liver Physiol. 2013;305(5):G341–7.
- Johansson MEV, Jakobsson H, Holmén-Larsson J, et al. Normalization of Host Intestinal Mucus Layers Requires Long-Term Microbial Colonization. Cell Host Microbe. 2015;18(5):582–592.
- Schütte A, Ermund A, Becker-Pauly C, et al. Microbial-induced meprin β cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc Natl Acad Sci. 2014;111(34):12396–12401.
- Johansson MEV, Phillipson M, Petersson J, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci. 2008;105(39):15064–15069.
- Nyström EEL, Birchenough GMH, van der Post S, et al. Calcium-activated chloride channel regulator 1 (CLCA1) controls mucus expansion in colon by proteolytic activity. EBioMedicine. 2018;33:134–143.
- Gustafsson JK, Ermund A, Johansson MEV, et al. An ex vivo method for studying mucus formation, properties, and thickness in human colonic biopsies and mouse small and large intestinal explants. Am J Physiol-Gastrointest Liver Physiol. 2011;302(4):G430–8.
- Arike L, Holmén-Larsson J, Hansson GC. Intestinal Muc2 mucin O-glycosylation is affected by microbiota and regulated by differential expression of glycosyltranferases. Glycobiology. 2017;27(4):318–328.
- Jakobsson HE, Rodríguez‐Piñeiro AM, Schütte A, et al. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015;16(2):164–177.
- Meyer-Hoffert U, Hornef MW, Henriques-Normark B, et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut. 2008;57(6):764–771.
- Okumura R, Kurakawa T, Nakano T, et al. Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature. 2016;532(7597):117–121.
- Troge A, Scheppach W, Schroeder BO, et al. More than a marine propeller – the flagellum of the probiotic Escherichia coli strain Nissle 1917 is the major adhesin mediating binding to human mucus. Int J Med Microbiol. 2012;302(7):304–314.
- Bergström JH, Birchenough GMH, Katona G, et al. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc Natl Acad Sci. 2016;113(48):13833–13838.
- Fritsch J, Abreu MT. The Microbiota and the Immune Response: what Is the Chicken and What Is the Egg? Gastrointest Endosc Clin N Am. 2019;29(3):381–393.
- van der Post S, Jabbar KS, Birchenough G, et al. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut. Published Online First: 26 March 2019. doi: 10.1136/gutjnl-2018-317571.
- Schirmer M, Denson L, Vlamakis H, et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe. 2018;24(4):600–610.e4.
- Haberman Y, Karns R, Dexheimer PJ, et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun. 2019;10(1):38.
- Johansson MEV, Gustafsson JK, Holmén-Larsson J, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63(2):281–291.
- Schroeder BO, Birchenough GMH, Ståhlman M, et al. Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe. 2018;23(1):27–40.e7.
- Desai MS, Seekatz AM, Koropatkin NM, et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016;167(5):1339–1353.e21.
- Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4(5):447–457.
- Abrham G, Volpe M, Shpungin S, et al. TMF/ARA160 downregulates proangiogenic genes and attenuates the progression of PC3 xenografts. Int J Cancer. 2009;125(1):43–53.
- Bel S, Elkis Y, Lerer-Goldstein T, et al. Loss of TMF/ARA160 protein renders colonic mucus refractory to bacterial colonization and diminishes intestinal susceptibility to Acute Colitis. J Biol Chem. 2012;287(30):25631–25639.
- Bel S, Elkis Y, Elifantz H, et al. Reprogrammed and transmissible intestinal microbiota confer diminished susceptibility to induced colitis in TMF−/− mice. Proc Natl Acad Sci. 2014;111(13):4964–4969.
- Martín R, Chamignon C, Mhedbi-Hajri N, et al. The potential probiotic Lactobacillus rhamnosus CNCM I-3690 strain protects the intestinal barrier by stimulating both mucus production and cytoprotective response. Sci Rep. 2019;9(1):5398.
- Ahl D, Liu H, Schreiber O, et al. Lactobacillus reuteri increases mucus thickness and ameliorates dextran sulphate sodium-induced colitis in mice. Acta Physiol. 2016;217(4):300–310.
- Dicksved J, Schreiber O, Willing B, et al. Lactobacillus reuteri Maintains a Functional Mucosal Barrier during DSS Treatment Despite Mucus Layer Dysfunction,”. Plos One. 2012;7(9):e46399.
- Wrzosek L, Miquel S, Noordine ML, et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013;11(1):61.
- Geirnaert A, Calatayud M, Grootaert C, et al. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci Rep. 2017;7(1):11450.
- Wlodarska M, Luo C, Kolde R, et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe. 2017;22(1):25–37.e6.
- Hutkins RW, Krumbeck JA, Bindels LB, et al. Prebiotics: why definitions matter. Curr Opin Biotechnol. 2016;37:1–7.
- Hafer A, Krämer S, Duncker S, et al. Effect of oral lactulose on clinical and immunohistochemical parameters in patients with inflammatory bowel disease: a pilot study. BMC Gastroenterol. 2007;7(1):36.
- Benjamin JL, Hedin CR, Koutsoumpas A, et al. Randomised, double-blind, placebo-controlled trial of fructo-oligosaccharides in active Crohn’s disease. Gut. 2011;60(7):923–929.
- Chermesh I, Tamir A, Reshef R, et al. Failure of Synbiotic 2000 to Prevent Postoperative Recurrence of Crohn’s Disease. Dig Dis Sci. 2007;52(2):385–389.
- Fujimori S, Gudis K, Mitsui K, et al. A randomized controlled trial on the efficacy of synbiotic versus probiotic or prebiotic treatment to improve the quality of life in patients with ulcerative colitis. Nutrition. 2009;25(5):520–525.
- Currò D, Ianiro G, Pecere S, et al. Probiotics, fibre and herbal medicinal products for functional and inflammatory bowel disorders. Br J Pharmacol. 2017;174(11):1426–1449.
- Shin W, Kim HJ. Intestinal barrier dysfunction orchestrates the onset of inflammatory host–microbiome cross-talk in a human gut inflammation-on-a-chip. Proc Natl Acad Sci. 2018;115(45):E10539–47.
- Wehkamp J, Harder J, Wehkamp K, et al. NF-kappaB- and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic bacterium. Infect Immun. 2004;72(10):5750–5758.
- Wendler J, Schroeder BO, Ehmann D, et al. Proteolytic Degradation of reduced Human Beta Defensin 1 generates a Novel Antibiotic Octapeptide. Sci Rep. 2019;9(1):3640.
- Stremmel W, Merle U, Zahn A, et al. Retarded release phosphatidylcholine benefits patients with chronic active ulcerative colitis. Gut. 2005;54(7):966–971.
- Stremmel W, Staffer S, Gehrke S. The Detergent Effect of Mesalazine Interferes with Phosphatidylcholine Binding to Mucin 2. Inflamm Intest Dis. 2018;3(3):107–115.
- Becker S, Schott C, Wolff C, et al. Bacteria regulate intestinal epithelial cell differentiation factors both in vitro and in vivo. PloS One. 2013;8(2):e55620.
- Tamboli CP, Neut C, Desreumaux P, et al. Dysbiosis in inflammatory bowel disease. Gut. 2004;53(1):1–4.
- Zuo T, Kamm MA, Colombel J-F, et al. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2018;15(7):440–452.
- Hall AB, Yassour M, Sauk J, et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9(1):103.
- Ananthakrishnan AN, Luo C, Yajnik V, et al. Gut microbiome function predicts response to anti-integrin biologic therapy in Inflammatory Bowel Diseases. Cell Host Microbe. 2017;21(5):603–610.e3.