
ABSTRACT
Introduction
Up to one third of colorectal cancers show familial clustering and 5% are hereditary single-gene disorders. Hereditary non-polyposis colorectal cancer comprises DNA mismatch repair-deficient and -proficient subsets, represented by Lynch syndrome (LS) and familial colorectal cancer type X (FCCTX), respectively. Accurate knowledge of molecular etiology and genotype-phenotype correlations are critical for tailored cancer prevention and treatment.
Areas covered
The authors highlight advances in the molecular dissection of hereditary non-polyposis colorectal cancer, based on recent literature retrieved from PubMed. Future possibilities for novel gene discoveries are discussed.
Expert commentary
LS is molecularly well established, but new information is accumulating of the associated clinical and tumor phenotypes. FCCTX remains poorly defined, but several promising candidate genes have been discovered and share some preferential biological pathways. Multi-level characterization of specimens from large patient cohorts representing multiple populations, combined with proper bioinformatic and functional analyses, will be necessary to resolve the outstanding questions.
1. Introduction: lynch syndrome
1.1. MLH1, MSH2, MSH6, and PMS2 mutations in lynch syndrome predisposition
Lynch syndrome (LS) is an autosomal dominant disorder caused by a defect in one of the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, or PMS2 [Citation1]. It represents the MMR-deficient subgroup of hereditary non-polyposis colorectal cancer (HNPCC) (please see section 2.1. below for the evolution of nomenclature). LS is one the most prevalent hereditary cancer syndromes in man, with 1:226 individuals as the highest reported population frequency to date [Citation2]. LS accounts for 1–3% of unselected colorectal or endometrial carcinomas and ~15% of those with high-degree microsatellite instability (MSI) or absent MMR protein in tumor tissues [Citation3].
LS-associated MMR genes are conserved in evolution (). A specific clinical phenotype or syndrome exists for all known human MMR genes, except for MSH4 and MSH5 whose protein products are necessary for meiotic (and possibly mitotic) recombination but do not participate in MMR [Citation4], and PMS1 whose product forms a complex with MLH1 but no specific function has been identified for this complex (hMutLβ) to date [Citation5]. Of the over 3000 unique germline sequence variants of the four LS-associated MMR genes deposited in the InSiGHT locus-specific database, the relative shares are 40% for MLH1, 34% for MSH2, 18% for MSH6, and 8% for PMS2 [Citation6]. MLH1 and MSH2 proteins are obligatory components in all types of heterodimers that the MMR proteins form with each other [Citation5] which explains the predominant roles of MLH1 and MSH2 in LS predisposition. MSH6 is functionally redundant with MSH3 when these proteins form a complex (hMutSα and hMutSβ, respectively) with MSH2 for mismatch recognition. A MutL homolog is needed to couple mismatch recognition to the downstream events. In humans, the prevalent MutL homolog is a heterodimer of MLH1 and PMS2 (hMutLα), but MLH1 can also heterodimerize with MLH3 (hMutLγ) which is able to partially compensate for the lack of hMutLα in MMR [Citation5].
Table 1. LS-associated MMR genes in evolution and phenotypes resulting from germline mutations.
In comparison with the average population, heterozygous carriers of MMR gene mutations have significantly increased risks of cancers of the colon and rectum, endometrium, ovary, kidney, and urinary tract, upper gastrointestinal tract, and certain other organs. Compared to previous retrospective and family-based studies, recent prospective investigations have arrived at generally lower risk estimates for cancers occurring in LS, after stratification of cancer risk by the affected MMR gene, age, and gender [Citation7]. Penetrance and expression patterns strongly depend on the MMR gene involved so that germline mutations of MLH1 and MSH2 have the highest and PMS2 the lowest penetrance, whereas MSH6 mutations underlie a sex-limited trait with a high risk of gynecological cancers in females [Citation7]. Evolutionary pathways to cancer may vary (for example, colorectal cancer development in LS may or may not involve a visible polyp precursor), and differential somatic involvement of key cancer-relevant genes (such as APC and CTNNB1) may in part explain differences in (colon) tumor development and penetrance between the four LS-associated MMR genes [Citation8,Citation9].
Germline mutations in MMR genes are detectable in up to 88% of families strongly suspected of having LS based on the fulfillment of the Amsterdam criteria and presence of MSI in tumors [Citation10,Citation11]. Mutation frequencies in smaller or atypical families and families not pre-screened by MSI or immunohistochemical analyses vary between 10% and 40% depending on the method of ascertainment [Citation10,Citation12]. The detection rate of MMR gene mutations in families harboring such mutations is anticipated to increase when deep sequencing by targeted gene panels, or even whole exomes or genomes, will gradually replace single gene-based approaches. On the other hand, these new sensitive and comprehensive sequencing methods are likely to detect increasing numbers of variants of uncertain significance (VUS) which cannot be directly translated to clinical management [Citation13,Citation14]. Furthermore, panels that include predisposition genes for multiple cancer syndromes may broaden genotype-phenotype associations within the syndromes. For example, a retrospective review of clinical histories in patients who underwent multigene panel testing and were diagnosed with a germline mutation in one of the MMR genes, showed the predominance of MSH6 and PMS2 over MSH2 and MLH1 mutations and linked a hereditary breast and ovarian cancer phenotype to MSH6 and PMS2 mutations [Citation14].
1.2. Constitutional epimutations in LS predisposition
In cases suspected of having LS but with no identifiable coding region mutations, constitutional epimutations are worthy of consideration. Constitutional epimutation refers to constitutional hypermethylation at the promoter of one allele of a given (non-imprinted) gene leading to silencing of expression from that allele in all main somatic tissues. Epimutation can be induced by a genetic alteration (usually located in cis), in which case epimutation is secondary, whereas epimutation arising in the absence of any detectable genetic change is considered primary. For MLH1, primary and secondary epimutations are known, whereas constitutional epimutations of MSH2 are secondary to deletions of the 3ʹ end of the EPCAM gene (). After removal of stop codon, transcription of EPCAM reads into the adjacent, structurally normal MSH2 gene inducing its promoter methylation [Citation15]. Owing to the capability of MSH2 silencing, EPCAM is considered as a LS predisposition gene in addition to the four LS-associated MMR genes.
Table 2. Constitutional epimutations of MLH1 and MSH2 in LS predisposition.
Depending on the method of ascertainment, constitutional epimutations of MLH1 (primary or secondary) may explain 1–10% of LS-suspected cases without traditional genetic mutations and with absent MLH1 expression in tumor tissue [Citation20–Citation23]. Distinction of primary epimutations of MLH1 from those that are secondary to genetic changes is challenging, since a comprehensive screening of the MLH1 region [Citation24], and in fact, the entire genome, would be required for a reliable exclusion of a possible inducing alteration. Constitutional epimutations are rare among unselected colorectal cancer patients: in a study on one such series from Spain, no MLH1 constitutional epimutations were detected [Citation25]. On the other hand, low-level methylation may be missed by standard techniques: a sensitive method used by Damaso et al. [Citation26] was able to detect constitutional MLH1 methylation at 1% level in 1/18 (6%) patients with MLH1 hypermethylated colorectal tumors. The share of EPCAM deletion-associated constitutional epimutations of MSH2 among Lynch suspected families without conventional germline mutations and with absent MSH2 protein in tumor tissues varies between 0% and 40% depending on possible founder effects [Citation15,Citation23]. Apart from MLH1 and MSH2, no constitutional epimutations have been reported for the remaining MMR genes [Citation27].
The clinical phenotype of constitutional epimutation carriers is indistinguishable from that of conventional LS mutation carriers. For occasional carriers of MLH1 epimutation, atypical phenotypes, such as colonic polyposis, have been reported [Citation28]. EPCAM deletion carriers have a high risk of colorectal cancer, whereas only deletions extending close to the MSH2 promoter are associated with increased endometrial cancer risk [Citation29]. In analogy to conventional LS mutations, tumor tissues show MSI and absent MMR protein(s) following inactivation of the wild-type allele by a genetic or epigenetic mechanism [Citation15,Citation20,Citation30–Citation32].
Since epigenetic changes are erased on passage through the germline, they are not regularly transmitted from parent to child. Therefore, primary constitutional epimutations segregate in a non-Mendelian fashion and are seldom associated with any remarkable family history of cancer, which makes a clear distinction relative to LS arising from genetic mutations. Mosaic epigenetic inheritance and reversion of the methylated allele to normal active state have been observed in families of primary epimutation carriers, and occasionally apparent heritability is seen [Citation21,Citation33–Citation35]. In contrast, secondary epimutations of MLH1 or MSH2 give rise to classical LS families and co-segregate with their cis-acting genetic changes as dominant Mendelian traits [Citation15,Citation24]. However, there is evidence that the basic mechanism of transmission differs from that of a genetic mutation: a secondary epimutation of MLH1 was found to undergo a complete but transient reversion in the germline, being erased in spermatozoa but reinstated in somatic cells of the next generation [Citation36].
1.3. Lynch-like ‘syndrome’ – a LS mimic
MLH1 promoter methylation as an acquired (somatic) alteration is by far the most frequent cause of MMR deficiency in various carcinomas, attributable to 10–20% of all colorectal carcinomas and 30% of endometrial carcinomas [Citation37,Citation38]. Germline mutations of MMR genes indicative of LS are detected in 1–3% of unselected colorectal or endometrial carcinomas [Citation3]. MMR-deficient cancers not explained by either of the two mechanisms mentioned above represent Lynch-like syndrome (LLS). The share of all colorectal carcinomas that LLS is responsible for (2–3%) [Citation37,Citation39] is comparable to LS. Two somatic mutations of the MMR genes MLH1, MSH2, MSH6, or PMS2 are detectable in more than half of LLS tumors [Citation2,Citation37,Citation40–Citation42]; this subset of LLS tumors is referred to as double somatic cases.
A higher average age at onset compared to LS is a feature of LLS [Citation37,Citation43], but on the level of an individual patient, LLS cannot be reliably distinguished from LS on clinical or histopathological grounds [Citation39,Citation44]. Some LLS cases are young and a (minor) proportion shows a positive family history [Citation39,Citation45], raising the possibility of a hereditary entity (even in the proven double somatic subset). Rare likely pathogenic germline variants in repair genes and other genes that maintain genome integrity were detected in LLS by Vargas-Parra et al. [Citation46] (BUB1, SETD2, FAN1, and heterozygous variant of MUTYH) and Xicola et al. [Citation47] (WRN, MCPH1, BARD1, and REV3 L). Additionally, tumors from variant carriers exhibited a mutational signature different from that seen in tumors from LLS patients without such variants, suggesting a possibly distinct etiology in the former group [Citation47]. On the other hand, Pico et al. [Citation45] found that LLS patients had homogeneous clinical and pathology characteristics regardless of family history of colorectal cancer, arguing against the existence of ‘sporadic’ and ‘hereditary’ subgroups within LLS. Future studies are necessary to resolve this question.
1.4. Homozygosity for MMR gene mutations – a shift from non-polypotic colorectal cancer to polyposis
To date, 100–200 patients have been reported to have homozygous or compound heterozygous germline mutations in any one of the four LS-associated MMR genes. These patients represent the constitutional mismatch repair deficiency (CMMRD) syndrome [Citation48–Citation50]. The syndrome is characterized by childhood cancers such as hematological malignancies and brain tumors, signs of neurofibromatosis type 1 (café-au-lait spots), and early-onset colorectal cancer. The distinct tumor spectrum may reflect sensitivity of neural and hematological progenitor cells to deficient MMR through mutational inactivation of certain key genes such as NF1 [Citation51]. Since both alleles of the involved MMR gene are mutant in the germline, the respective protein is absent in normal tissues in addition to tumors. On the other hand, microsatellite length variants induced by defective MMR occur at such low levels in nonneoplastic tissues that their detection requires sensitive techniques [Citation50,Citation52,Citation53].
The predominant genes underlying CMMRD are PMS2 and MSH6 [Citation50]. When heterozygous, PMS2 mutations cause predisposition to LS with low penetrance [Citation7,Citation54], whereas homozygosity or compound heterozygosity for PMS2 mutations gives rise to CMMRD with full penetrance [Citation50]. Adenomatous polyposis ranging from a few polyps to familial adenomatous polyposis-like phenotype is a regular feature of CMMRD [Citation55,Citation56]. In addition to CMMRD due to biallelic germline mutations of the four LS-associated MMR genes (especially PMS2), polyposis is the leading phenotypic feature in the rare patients with biallelic germline mutations of MSH3 [Citation57] and MLH3 [Citation58]. In heterozygous states, MSH3 mutations are not known to be associated with any apparent disease phenotype [Citation57], and MLH3 mutations may act as low-penetrance susceptibility factors for colorectal cancer [Citation59,Citation60]. In CMMRD, germline inactivation of both alleles of the MMR gene in question, together with somatic polymerase exonuclease domain mutations, was shown to result in an ‘ultra-hypermutated’ tumor phenotype (over 100 mutations/Mb) in brain tumors, whereas gastrointestinal polyps revealed no increased mutational load compared with adult polyps [Citation61]. Moreover, no microsatellite instability (at dinucleotide or trinucleotide repeats) was observed in a polyp from a homozygous carrier of a nonsense mutation in PSM2 [Citation62]. Tumor tissues from biallelic MSH3 mutation carriers showed a distinct form of microsatellite instability called EMAST (Elevated Microsatellite Alterations at Selected Tetranucleotide Repeats) [Citation57]. No instability at any type of microsatellite repeats was found in polyps or cancer tissues from biallelic MLH3 mutation carriers [Citation58]. Absent or low-degree MSI may reflect partial redundancy between the different hMutS and hMutL dimers (see above).
At present, no definitive explanation is available for polyp predisposition in biallelic carriers of MMR gene mutations. At least one somatic APC mutation was detected in each of three PMS2-associated polyps examined by Siraj et al. [Citation62], implying that inactivation of APC, a gatekeeper of cellular proliferation, is important for tumor development. In MSH3-associated tumors, the observed inflammatory infiltration and the spectrum of somatic APC mutations (small deletions at repeat sequences) pointed to the significance of MMR deficiency in tumorigenesis [Citation57]. Finally, in MLH3-associated tumors, multiple loci revealed allelic imbalance, suggesting the possible involvement of chromosomal segregation or recombination-related mechanisms [Citation58].
2. Familial colorectal cancer type x
2.1. Division of hereditary non-polyposis colorectal cancer into two main entities
MMR-proficient families meeting the Amsterdam criteria [Citation63,Citation64] are referred to as familial colorectal cancer ‘type X’ (FCCTX); ‘type X’ implies that the genetic basis is largely unknown [Citation65,Citation66]. Prior to the discovery of the LS-associated MMR genes in the 1990 s, the Amsterdam Criteria were used to identify families with a presumably inherited form of colorectal carcinoma termed HNPCC. Molecular analyses have revealed that LS accounts for approximately half of such families, whereas, in the remaining families (FCCTX), no MSI or absent MMR protein in tumor tissue or germline MMR gene defect is detectable [Citation65]. FCCTX families show site-specific (mainly distal) colorectal cancer, as opposed to colonic (mainly proximal) and extracolonic cancers seen in LS (). The mean age at cancer onset is around 55 years in FCCTX vs. 45 years in LS [Citation67].
Table 3. Two main subgroups of hereditary non-polyposis colorectal cancer (HNPCC).
Table 4. Proposed novel high-penetrance susceptibility genes for FCCTX.
Apart from MMR status, the epigenomic pattern differs between FCCTX and LS (). Colorectal carcinomas from FCCTX families display a remarkably low LINE-1 methylation, which is a surrogate marker of global hypomethylation [Citation68]. In contrast, extensive LINE-1 hypomethylation is not characteristic of LS-colorectal cancers; a subset associated with a poor prognosis may constitute an exception [Citation69]. In FCCTX patients, low LINE-1 methylation is a feature of normal mucosae, too [Citation70] and may, therefore, represent a field defect or a constitutional alteration. The mechanistic basis of FCCTX-associated hypomethylation is unknown.
2.2. Candidate susceptibility genes for FCCTX
Genome-wide sequencing and other approaches have resulted in the discovery of novel candidate genes for FCCTX. Selected candidate genes with functional proof available are listed in . Each gene appears to be involved in only a small fraction of families and no major FCCTX susceptibility genes, analogous to MMR genes in LS, have been identified to date. The genes cluster into some preferential biological pathways to be described in detail below (, ).
Figure 1. Pathways affected and interactions between genes involved in FCCTX predisposition. Generalized pathways for the FCCTX-associated genes were constructed according to the WikiPathways database and are indicated with colored circles. Only pathways with at least two affected genes were considered. Gene interactions with at least medium confidence according to the STRING database are indicated with black lines.
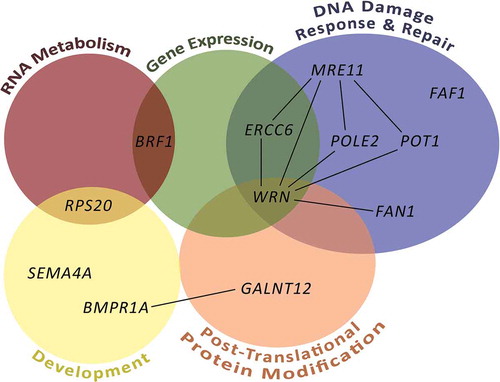
2.2.1. Protein glycosylation
2.2.1.1. GALNT12
Genetic linkage studies have provided evidence of the existence of a colorectal cancer susceptibility gene on chromosome 9q22 but the responsible genes remain elusive [Citation71–Citation73]. GALNT12 is one of the genes located in that chromosomal region and codes for the N-acetylgalactosaminyltransferase 12 enzyme involved in O-linked glycosylation of mucin-type glycans. GALNT12 was originally identified as a possible colon cancer susceptibility gene by a candidate gene approach, based on the knowledge of aberrant glycosylation as a hallmark of colon cancers and the high level of expression of GALNT12 in normal colon [Citation74]. Germline mutations of mainly missense type and leading to lost or reduced enzymatic activity of GALNT12 were identified in 2–3% of colorectal cancer patients with variable family histories, ranging from apparently sporadic cases to families referred to a cancer genetics clinic (; refs [Citation74–Citation76].). No GALNT12 germline mutations were detected in 103 families meeting the stringent Amsterdam criteria [Citation77]. The results argue against GALNT12 as a major high-penetrance susceptibility gene for FCCTX and provide evidence to support a role for GALNT12 as a moderate penetrance gene for colorectal cancer.
2.2.2. Ribosome biosynthesis
2.2.2.1. RPS20
Genome-wide linkage analysis in a multi-generation Finnish FCCTX family fulfilling the Amsterdam criteria identified an area of linkage around the centromeric region of chromosome 8, and the RPS20 gene from that region (8q12.1) was subsequently found to be mutant in four colon cancer-affected siblings by exome sequencing (; ref. [Citation78]). The mutation (c.147dupA) shared by the siblings was predicted to cause a frameshift and premature truncation. RPS20 encodes a component of the 40 S small ribosomal subunit. RNA analyses revealed a defect in the late steps of ribosome biogenesis affecting the formation of mature 18 S rRNA [Citation78]. Later investigations identified germline mutations (truncating or missense) in three additional probands, two of which represented familial colon cancer cases of European ancestry belonging to the UK National Study of Colorectal Cancer Genetics [Citation79] while the third one represented an Amsterdam criteria-positive MMR-proficient family from Utah [Citation80]. Studies on colorectal cancers from RPS20-associated families [Citation78,Citation80] revealed no loss of the wild-type allele, consistent with observations from zebrafish showing that ribosomal protein genes act as haploinsufficient suppressors of tumorigenesis [Citation81].
Ribosome dysgenesis is connected to cancer in many ways, often involving p53 activation [Citation82,Citation83]. Western blot analysis of lymphoblastoid cells from RPS20 mutation carriers showed increased expression of p53 protein [Citation78]. The same applies to germline mutation carriers of several other ribosomal proteins, such as RPL26 [Citation84]; unlike RPS20 mutations, heterozygous germline mutations of other ribosomal protein genes are not associated with FCCTX but Diamond-Blackfan anemia (DBA), a rare bone marrow failure syndrome with red cell aplasia and congenital anomalies [Citation83]. It is unknown why germline mutations of RPS20 underlie FCCTX while mutations of other ribosomal protein genes cause DBA. Some clinical overlap does exist: no significant anemia appears to accompany RPS20 mutation, but mutations of DBA-associated ribosomal proteins cause significantly increased risk of colon cancer [Citation85]. It is possible that different ribosomal proteins are rate-limiting in different cells, that different types of cells possess different sensitivity to p53 activation, or that p53 pathway activation has different downstream effects in different cells [Citation83,Citation86,Citation87], thereby explaining tissue-specific manifestations of mutant ribosomal protein genes.
2.2.2.2. BRF1
An exome sequencing study on three colon cancer affected relatives from an Amsterdam-positive FCCTX family from Spain identified a shared heterozygous splice site alteration (c.1459 + 2 T > C) that led to the skipping of exon 13 of BRF1 (; ref. [Citation88]). The protein product of BRF1 is part of the TFIIB complex that initiates transcription by the RNA Pol III. Among other RNAs, RNA Pol III synthesizes 5 S ribosomal RNA, a limiting molecule required for the formation of the large 60 S ribosomal subunit [Citation89]. A validation series of 502 individuals with familial or early-onset colorectal cancer sequenced for the coding region and exon-intron boundaries of BRF1 revealed 10 independent families with rare heterozygous germline mutations that affected protein expression and/or function of BRF1 [Citation88]. Thus, BRF1 was mutant in a total of 2% (11/503) of all cases investigated, most of which met at least the revised Bethesda criteria [Citation90] for hereditary non-polyposis colorectal cancer. In analogy to RPS20, colorectal tumors from BRF1-associated families showed no loss of the wild-type allele, supporting haploinsufficiency [Citation88]. Taken together, cosegregation and functional evidence derived from several families having mutant RPS20 or BRF1 with converging functions, link ribosomal biosynthesis to colorectal cancer predisposition, and RPS20 and BRF1 can be considered as high-penetrance susceptibility genes for FCCTX.
2.2.3. DNA repair and damage response
2.2.3.1. FAN1
Sequencing of the exomes of three colon cancer affected members from an Amsterdam-positive FCCTX family from Spain led to the discovery of a nonsense mutation (c.141 C > A, p.C47*) in FAN1 which was shared by all affected family members and was absent in control individuals (; ref. [Citation91]). FAN1 encodes FANCD2/FANCI-associated nuclease 1 involved in interstrand cross-link repair. Lymphoblastoid cells from a heterozygous mutation carrier showed increased sensitivity to mitomycin C which is characteristic of defects in Fanconi anemia-related genes and indicates impaired interstrand cross-link repair [Citation91]. Homozygous FAN1 germline mutations underlie karyomegalic interstitial nephritis, where chronic kidney failure is a predominating feature and possible cancer-associations are unknown due to the rarity of the syndrome [Citation92]. A subsequent screen of 176 MMR-proficient Amsterdam-positive families identified 4 additional families with FAN1 germline mutations co-segregating with colorectal cancer in the families and having low mutant allele frequencies (<0.1%) in the control individuals [Citation91]. Thus, FAN1 germline mutations accounted for a total of 5/177 (3%) of Spanish FCCTX families. Colorectal tumors from FAN1 mutation carriers showed no evidence of somatic second hits, suggesting haploinsufficiency. The overall somatic mutational load in tumor tissue was in the non-hypermutant range [Citation91].
Interpretations of the role of FAN1 mutations in FCCTX susceptibility are complicated by the fact that some population-based control series, too, have been found to show disruptive FAN1 germline variants with frequencies less than 1% [Citation79,Citation91]. Analysis of the exomes of 863 familial colorectal cancer cases and 1604 controls from the UK National Study of Colorectal Cancer Genetics detected no significant increase of (likely) damaging FAN1 variants in cases (15/863, 1.7%) vs. controls (17/1604, 1.1%, p = 0.19). Therefore, additional investigations seem necessary to settle the role of defective FAN1 in FCCTX predisposition.
2.2.3.2. WRN and ERCC6
Arora et al. [Citation93] pursued the hypothesis that constitutional defects in double-strand break (DSB) repair may underlie familial colorectal carcinomas of an undefined genetic basis. In addition to colorectal cancer, 25 patients studied by Arora et al. [Citation93] had cancers of several other organs and some also had (mild) polyposis. Following the observation that peripheral blood cells from familial colorectal cancer patients vs. controls showed higher levels of DSB-associated γ(phospho)-H2AX foci after treatment with DNA-damaging agents, the patients were subjected to exome sequencing. Compared to controls, a significantly higher frequency of disruptive variants in genes relevant to DSB repair, nucleotide excision repair (NER), or other repair mechanisms were found. These included the WRN (RecQ-helicase-like) gene involved in DSB and the ERCC6 (excision repair cross-complementation group 6) gene involved in transcription-coupled NER. The variants occurred in a heterozygous state in the colon cancer patients and their consequences were validated by functional experiments. Homozygous null mutations in WRN are known to cause Werner’s syndrome, while homozygous ERCC6 mutations cause Cockayne’s syndrome; Werner syndrome is associated with increased risks of various tumors, including gastrointestinal cancers [Citation94], whereas in Cockayne’ syndrome, cells die prematurely which may offer protection against cancer development [Citation95].
2.2.3.3. POT1, POLE2, and MRE11
Chubb et al. [Citation96] hypothesized that rare high-impact alleles might contribute to the ‘missing heritability’ of colorectal cancer and conducted an exome sequencing-based epidemiological investigation on the occurrence of such alleles in 1006 early-onset familial colorectal cancer cases vs. 1609 healthy controls from the UK population. Highly penetrant rare mutations were identified in 16% of familial colorectal cancer cases. Excluding established colon cancer susceptibility genes (such as MMR genes and APC), POT1 (Protection of Telomeres 1), POLE2 (DNA Polymerase Epsilon 2, Accessory Subunit), and MRE11 (MRE11 Homolog, Double-Strand Break Repair Nuclease) were identified as new candidate colon cancer susceptibility genes, by virtue of truncating germline mutations that were significantly more frequent in cases than controls. The protein product of POT1 is part of the shelterin complex required for telomere maintenance. Heterozygous germline mutations have been associated with an autosomal dominant cancer family syndrome with diverse malignancies, such as glioma, angiosarcoma, and melanoma [Citation97]. POLE2 is part of the accessory (non-catalytic) subunit of the polymerase epsilon enzyme complex. In a previous study, targeted sequencing of polymerase genes resulted in the identification of rare POLE2 variants (one truncating and one missense) in two patients with colorectal polyposis [Citation98]. The MRE11 protein is involved in DSB repair, homologous recombination, and telomere length maintenance. Heterozygous germline mutations of MRE11 may cause increased breast cancer risk [Citation99], whereas homozygous mutations underlie ataxia-telangiectasia-like disorder [Citation100].
2.2.3.4. FAF1
Exome sequencing on 40 FCCTX families identified a missense variant (c.1111 G > A, p.Asp371Asn) in the FAF1 (Fas-associated factor 1) gene in an Amsterdam-positive family of Spanish origin [Citation101]. The variant was present in all four affected family members tested, including three with colorectal cancer and one with advanced adenoma. Targeted sequencing of FAF1 was subsequently conducted on 473 additional families mostly fulfilling the revised Bethesda criteria, and another presumably pathogenic missense variant (c.254 G > C, p.Arg85Pro) was discovered. No co-segregation studies were possible in the latter family. The two missense variants together explained a 0.4% fraction (2/513) of colorectal cancer families investigated [Citation101].
The FAF1 gene product is a tumor suppressor with diverse functions; among others, it is a negative regulator of NF-κB and Wnt signaling [Citation101]. Fas-associated factor 1 also interacts with proteins involved, for example, in TFGβ signaling, Fas-mediated apoptosis, and ubiquitin-related functions [Citation102]. The two missense variants detected by Bonjoch et al. [Citation101] rendered the resulting proteins unstable. When FAF1 was knocked out in DLD1 colorectal cancer cells, cellular proliferation increased, and the cells became resistant to programmed cell death. Transient expression of the missense variants in DLD1 knockout cells recapitulated the features mentioned above and revealed NF-κB and Wnt signaling activation, as judged from increased nuclear translocation of NF-κB, and impaired degradation and cytoplasmic accumulation of β-catenin, respectively. As pointed out by Bonjoch et al. [Citation101], investigations of additional cohorts are necessary to confirm and further evaluate the significance of FAF1 as a FCCTX susceptibility gene.
2.2.4. Other pathways
2.2.4.1. SEMA4A
Combined linkage analysis and exome sequencing in an Amsterdam-positive FCCTX family from Austria identified a missense variant (c.232 G > A, p.Val78 Met) in SEMA4A as the only variant shared by all four colorectal cancer-affected members tested [Citation103]. The gene is located on chromosome 1q22 and encodes semaphorine 4A, a membrane protein with various functions (e.g., in neuronal axon extension, immunomodulation, and cell proliferation). The Val78 Met mutation was associated with increased MAPK/Erk and PI3 K/Akt signaling. Semaphorins have been found to have pro- and anti-carcinogenic functions; here, the evidence as a whole suggested loss of function, which was further supported by homozygosity of the variant in colorectal tumor tissues from two heterozygous mutation carriers [Citation103]. A targeted screen of 53 additional Amsterdam-positive FCCTX families from Austria, Germany, and the United States identified three further missense changes in SEMA4A; among these, the rare G484A and S326 F variants were predicted to be deleterious by in silico tools, whereas a case–control association study suggested a significant enrichment of the single nucleotide polymorphism p.Pro682 Ser in FCCTX cases (6%) vs. controls (1%, p = 0.0008). However, an attempt to validate the proposed cancer-predisposing role of the recurrent G484A and P682S variants in an independent case–control study involving ~7,000 colorectal cancer cases and 10,000 controls from six European populations failed to do so [Citation104]. Furthermore, the entire coding region of SEMA4A was sequenced in 158 FCCTX families, which resulted in the detection of protein changing variants in 16% of FCCTX families, compared with 14% of controls, suggesting no significantly increased frequency of SEMA4A variants in FCCTX families [Citation104]. In conclusion, additional investigations are needed to resolve the role of SEMA4A variants in FCCTX predisposition.
2.2.4.2. BMPR1A
Genome-wide linkage scan conducted on a large Amsterdam-positive FCCTX family from Finland resulted in the observation of tentative linkage on 10q23 [Citation105]. Sequencing of genes from the linked region revealed an in-frame germline mutation (c.264_266del, p.Glu88del) in exon 3 of BMPR1A which co-segregated with colon carcinoma and/or adenoma in the family [Citation105]. The protein product of the BMPR1A gene is a type I bone morphogenetic protein receptor that belongs to a family of serine/threonine kinases. BMPR1A was subsequently sequenced in 17 additional Finnish families fulfilling the Amsterdam or Bethesda criteria and having MMR-proficient tumors. The proband of an Amsterdam-positive family revealed a truncating splicing mutation; no other affected members from that family were available for co-segregation studies [Citation105]. Neither of the two predisposing mutations, each occurring in one family [Citation105], have been found in healthy reference populations.
Loss of function mutations of BMPR1A are known to underlie juvenile polyposis syndrome, characterized by hamartomatous polyps and increased risk of gastrointestinal cancer [Citation106]. Interplay between epithelial and stromal cells is believed to be important for the development of hamartomatous polyps, and loss of BMP signaling in the epithelial compartment alone may be insufficient [Citation107,Citation108]. For comparison, polyps from the two BMPR1A-linked FCCTX families described by Nieminen et al. [Citation105] were of adenomatous histology and the highest combined number of adenomatous and hyperplastic polyps per colonoscopy screen per person was 4; thus, the clinical picture was that of a non-polypotic colorectal cancer rather than polyposis. When bulk tumor tissues were investigated, the FCCTX-associated BMPR1A germline mutations were accompanied by loss of heterozygosity in colorectal tumors: the splicing mutation was associated with wild-type allele loss, whereas wild-type and mutant allele losses were equally common in tumors from the in-frame deletion carriers [Citation105].
Additional phenotypes that BMPR1A germline mutations have been linked to include early-onset MMR-proficient colorectal cancer [Citation109] and unexplained adenomatous polyposis [Citation110]. The basis of the reported genotype-phenotype associations is unknown. In the BMP signaling pathway, multiple ligand, receptor, and downstream effector combinations are possible and might allow for tissue or cell-specific responses. Furthermore, the seemingly different phenotypes may overlap. For example, Lieberman et al. [Citation111] evaluated seven unrelated families sharing a genomic deletion of the entire coding region of BMPR1A and found extremely variable phenotypes despite the same predisposing mutation and the same genetic background (Bukharin Jewish ancestry). Importantly, adenomatous polyps predominated over juvenile polyps in the families [Citation111]. Lastly, Evans et al. [Citation112] conducted a targeted investigation on 22 unrelated FCCTX families from Newfoundland to determine the frequency of BMPR1A germline mutations: no rare (mutant allele frequency < 1%) or novel variants were found. Therefore, studies on additional FCCTX families from different populations are warranted to define the relationship between BMPR1A mutations and FCCTX.
3. Prospects to identify novel cancer-predisposing genes – challenges and promises
A single biological pathway, MMR, with the major players already known () defines LS. The diagnosis of FCCTX on the other hand is based on the exclusion of LS and the demonstration of MMR-proficient tumors, until the responsible genes are identified. The available experience suggests that FCCTX is genetically heterogeneous and private genes and mutations may be the norm (). A burning question is how to best find the genes underlying colon cancer susceptibility in the majority of FCCTX families that currently lack molecular definition.
3.1. Methodological considerations
Over the years, the discovery of new cancer predisposing genes has been based on three main strategies: candidate gene analysis, genetic linkage analysis, and most recently, genome-wide sequencing [Citation113]. Large families with multiple cases of preferably early-onset colorectal cancer affecting several generations would provide convincing evidence of a hereditary nature of cancer predisposition and would obviously be ideal for searches for novel susceptibility genes. This setting was utilized in previous linkage-based studies that led to the identification of many currently known colorectal cancer predisposition genes [Citation114]. Genetic linkage analysis was also part of the strategies used to identify many of the FCCTX candidate genes listed in ; however, genome-wide sequencing, usually of the whole exomes, is nowadays the main approach. While the available methods are constantly improving, some challenges remain and may guide searches for predisposing mutations that have possibly escaped detection. First, not all relevant regions of the genome may be covered. High-penetrance mutations are assumed to mainly reside in the coding portions of the genome, which is why exome sequencing is the preferred method. Exome sequencing interrogates the exons and their immediate flanking regions (~ 2% of the human genome). However, some high-penetrance mutations may involve promoter regions, introns, or non-coding RNA. For example, mutations residing deep in the introns can activate sequences (pseudoexons) flanked by splice sites, which can result in the insertion of intronic sequences in the mature mRNA [Citation115]. In such cases, whole-genome sequencing and RNA-sequencing would be required to catch the alteration [Citation115]. Likewise, for the detection of constitutional epimutations, a combination of techniques (genetic and epigenetic) would be needed. Second, gaps may remain in data analysis. While single nucleotide variants (SNVs) are effectively detected by the available variant calling pipelines, the ability to accurately call insertion-deletion type changes (indels) remains imperfect [Citation116]. The same applies to copy number variant (CNV) analysis, although CNV detection algorithms have been developed for whole genome, whole exome, and targeted deep sequencing data [Citation117].
3.2. Data interpretation
Protein truncating variants, which predominate among known disease-causing germline mutations of cancer genes [Citation118], are considered deleterious by nature. However, panel sequencing studies suggest that up to 40% of patients suspected to have a hereditary cancer syndrome may carry a variant of uncertain significance, VUS [Citation119]. VUSes typically consist of single amino acid substitution (missense) changes which can be classified neither pathogenic nor harmless a priori. Even synonymous nucleotide changes (i.e., nucleotide substitutions which do not result in a coding change) can be pathogenic if they, for example, disrupt motifs for proper splicing [Citation120]. Thus, if a novel candidate susceptibility gene is affected by non-truncating alterations alone, the cancer predisposing role of the gene may remain unknown in cases where confirmatory studies (e.g., co-segregation or functional investigations) are not possible. Occasionally, two or more variants (of the same gene or different genes) may co-exist and even co-segregate with disease phenotype in a single family, which could be compatible with several alternative interpretations, including a true predisposing change coexisting with spurious variants, or a possibly multigenic basis of colon cancer susceptibility (see below).
3.3. Genotypic and phenotypic heterogeneity
A plausible reason why searches for new predisposition genes for FCCTX often fail is that FCCTX families do not have any pathognomonic features, either clinical (such as multiplicity and/or distinct histology of polyps that may aid the dissection of polyposis syndromes) or molecular (such as deficient MMR characteristic of LS), which could guide gene discovery efforts and distinguish true gene carriers from phenocopies (non-carriers). The possibility to investigate multiple (affected and unaffected) family members or utilize other hallmarks of hereditary cancer besides family history (such as multiple tumors in a single individual or the combination of specific tumor types) can be important advantages. Genome-wide sequencing studies have demonstrated that the inclusion of multiple affected family members who can reasonably be assumed to share the same high-penetrance susceptibility variant can significantly reduce the number of candidate variants worth considering [Citation78]. Tumor genotype and phenotype can likewise be informative, including the type of genomic instability and epigenomic pattern () or mutational signature [Citation121]. Since each mutational process leaves a specific imprint in the genome of a cell, somatic mutational signatures can offer clues to the predisposing genes involved. Diaz-Kay et al. [Citation122] performed an integrated whole-exome analysis of germline and tumor DNA to detect novel tumor suppressor genes underlying familial colorectal cancer. They required that genes of interest should be altered in both normal and tumor DNA according to Knudson’s two-hit hypothesis. The strategy pinpointed DNA repair genes, including BRCA2, BLM, ERCC2, RECQL, REV3 L, and RIF1, and mutational signatures additionally provided evidence of the existence of novel predisposition genes to be defined by further investigations [Citation122].
3.4. Polygenic basis of colon cancer susceptibility
Rare high-penetrance variants in known susceptibility genes account for only ~5% of the total colorectal cancer burden [Citation123]. Genome-wide association studies of common colorectal cancers have identified over 40 independent loci of low-penetrance risk variants frequent in the average population [Citation124]. Each low-penetrance locus is estimated to increase colorectal cancer risk no more than up to 1.5-fold [Citation123]. Although mostly residing outside coding regions, low-penetrance variants can influence the expression of genes from pathways associated with high-penetrance susceptibility genes (including, for example, the TGFβ/BMP pathway [Citation125]). Thus, epistatic (synergistic non-additive) interactions between two or more variants from different loci may account for some of the currently unexplained colorectal cancer susceptibility. Rare variants with moderate effects are another worthwhile possibility in searches for novel colon cancer predisposition genes. This is because such variants are easily overlooked in family-based studies due to incomplete co-segregation, and the same could happen with genome-wide association studies because large sample sizes would be required to reach statistical significance [Citation123].
4. Expert opinion
Hereditary non-polyposis colorectal cancer offers an illustrative example of the remarkable progress that, along with technical developments, has taken place in understanding the genetic basis of human disorders in recent years. LS is well-defined in terms of predisposing genes, and genetic challenges that remain mainly concern optimal detection methods of predisposing variants and interpretation of clinical significance of the variants. It is currently recommended that all new patients with colorectal or endometrial cancer should undergo systematic screening for LS, consisting of MSI and MMR protein tests on tumor tissue followed by further analyses (for MLH1 methylation and additionally, BRAF-V600E mutation for colorectal cancers), and ultimately, germline mutation testing when appropriate [Citation126]. This ‘universal’ screening, combined with high-throughput sequencing that is replacing gene-based methods in germline mutation detection, is likely to multiply the numbers of MMR gene variants that need to be annotated for pathogenic significance. While various in silico tools [Citation127] may be helpful for a preliminary assessment, there is an urgent need for functional tests robust enough that could unequivocally distinguish between normal and abnormal MMR gene function. LS carrier tests based on the effect of MMR protein dosage on MMR capability [Citation128,Citation129] are under development and/or already available for clinical diagnostics (DiagMMR®).
Increased knowledge of genotype-phenotype correlations in LS is expected to improve the possibilities of tailoring cancer prevention and treatment according to the predisposing gene and variant [Citation7,Citation9] as well as the genetic, epigenetic, or immunological type of tumors [Citation130,Citation131]. Clinical trials conducted to date have already established the responsiveness of LS and other hypermutant/MSI tumors to immune checkpoint inhibitors, motivating the use of such drugs to treat metastatic/advanced disease [Citation132]. At the same time, immunovaccination [Citation133] and chemoprevention [Citation134] represent promising new approaches for cancer prevention in LS.
Despite recent progress, the underlying genes remain to be identified for most cases of FCCTX. Even for those few families with plausible candidate genes discovered, the true predisposing nature of the genes and variants is typically awaiting further proof. The decreasing costs for genome-wide sequencing make investigations of larger patient cohorts affordable, thereby facilitating new gene discoveries on the one hand and providing accurate information of the molecular epidemiology of existing candidate genes and variants on the other hand. Experience from pan-cancer analysis of whole genomes shows that common and rare germline variants influence the patterns of somatic mutations [Citation135]; hence, mutational signatures of tumor tissues are becoming an integral part of future searches for FCCTX susceptibility genes. Moreover, a closer scrutiny of the non-coding genome (comprising 98% of the whole genome) is anticipated to pinpoint new genes and mechanisms that may underlie colon cancer susceptibility or modify the disease phenotype.
Based on available knowledge (), a modified cancer prevention protocol for FCCTX is currently recommended [Citation136], consisting of colonoscopy once in every 5 years (vs. once in every 1–2 years in LS [Citation126]) and starting from the age of 45 years (vs. 20–35 years in LS [Citation126]). Cancer surveillance in FCCTX focuses on colon cancer alone since extracolonic cancers are considered rare. A reappraisal may be warranted since FCCTX families from a national (Danish) HNPCC register revealed an increased risk of five types of extracolonic cancers (cancers of the urinary tract, breast, stomach, and pancreas, as well as ocular melanoma) compared to the average population [Citation137]. In the study mentioned above [Citation137], information of possible predisposing genes was not available, and FCCTX families were ascertained through the Amsterdam criteria and MMR-proficient tumors A better understanding of the individual nature of the susceptibility genes, their biological function and associated clinical features (including tumor spectra), is likely to refine protocols for cancer surveillance and enable targeted therapy for subsets of FCCTX in the future.
Article highlights
Hereditary non-polyposis colorectal cancer refers to cancer predisposition caused by single genes affected with high-penetrance mutations. Transmission is usually autosomal dominant. While colorectal tumorigenesis may involve polyp precursors, no significant polyposis is present.
DNA mismatch repair deficiency vs. proficiency divides hereditary non-polyposis colorectal cancer into two genetically and clinically distinct categories, Lynch syndrome and familial colorectal cancer type X, respectively.
Germline mutations in MLH1, MSH2, MSH6, PMS2, and EPCAM cause predisposition to Lynch syndrome. Over 3,000 different germline mutations are known.
Following somatic inactivation of the wild-type allele, tumor tissues from Lynch syndrome patients show absent mismatch repair protein(s) and high-degree microsatellite instability. Defective mismatch repair results in increased numbers of somatic mutations in other genes (hypermutability).
Clinical phenotype of Lynch syndrome depends on the predisposing gene and typically involves colorectal and extracolonic cancers. Constitutional homozygosity for mismatch repair gene mutations gives rise to distinct clinical features and frequent polyposis.
No established major predisposition genes are known for familial colorectal cancer type X, but several candidate genes have been proposed, each affected in a few families or a single family. The genes may participate in protein glycosylation (GALNT12), ribosome biosynthesis (RPS20 and BRF1), DNA repair (FAN1), or various other biological pathways.
Tumors from patients with familial colorectal cancer type X are mismatch repair-proficient and microsatellite-stable. Somatic mutational signatures may provide clues to the predisposing genes involved.
While the low numbers of mutation carriers make assessments of tumor spectra and other clinical features unreliable, site-specific colorectal cancer diagnosed at somewhat later ages compared to Lynch syndrome appears true for most families with colorectal cancer type X.
Genome-wide profiling of constitutional and tumor tissues, combined with functional investigations, are likely to define genotype-phenotype correlations in the existing syndromes and identify novel high-penetrance susceptibility genes for familial colorectal cancer type X.
Declaration of interest
P Peltomäki receives research grant support from Jane and Aatos Erkko Foundation, the Academy of Finland (grant number 294,643 to P.P.); the Finnish Cancer Foundation; the Sigrid Juselius Foundation; the HiLIFE Fellows 2017-2020; T Nieminen is the recipient of research grants from Jane and Aatos Erkko Foundation and the Päivikki and Sakari Sohlberg Foundation. A Olkinuora is supported by the Doctoral Programme of Biomedicine of the University of Helsinki. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Thompson BA, Spurdle AB, Plazzer JP, et al. Application of a 5-tiered scheme for standardized classification of 2.360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46(2): 107–115.
- Haraldsdottir S, Rafnar T, Frankel WL, et al. Comprehensive population-wide analysis of lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat. Commun. 2017;8(1):14755.
- Hampel H, de la Chapelle A. How do we approach the goal of identifying everybody with lynch syndrome? Fam. Cancer. 2013;12(2):313–317.
- Manhart CM, Alani E. Roles for mismatch repair family proteins in promoting meiotic crossing over. DNA Repair (Amst). 2016;38:84–93.
- Jiricny J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 2013;5(4):a012633.
- Peltomäki P. Update on lynch syndrome genomics. Fam. Cancer. 2016;15(3):385–393.
- Dominguez-Valentin M, Sampson JR, Seppälä TT, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective lynch syndrome database. Genet Med. 2020;22(1): 15–25.
- Ten Broeke SW, van Bavel TC, Jansen AML, et al. Molecular background of colorectal tumors from patients with lynch syndrome associated with germline variants in PMS2. Gastroenterology. 2018;155(3): 844–851.
- Engel C, Ahadova A, Seppälä TT, et al. Associations of pathogenic variants in MLH1, MSH2, and MSH6 with risk of colorectal adenomas and tumors and with somatic mutations in patients with lynch syndrome. Gastroenterology. 2020;158(5): 1326–1333.
- Mangold E, Pagenstecher C, Friedl W, et al. Spectrum and frequencies of mutations inMSH2 andMLH1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int J Cancer. 2005;116(5):692–702.
- Lagerstedt Robinson K, Liu T, Vandrovcova J, et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J. Natl. Cancer Inst. 2007;99(4):291–299.
- Wijnen J, Khan PM, Vasen H, et al. Hereditary nonpolyposis colorectal cancer families not complying with the Amsterdam criteria show extremely low frequency of mismatch-repair-gene mutations. Am J Hum Genet. 1997;61(2):329–335.
- Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3(4):464.
- Espenschied CR, LaDuca H, Li S, et al. Multigene panel testing provides a new perspective on lynch syndrome. J. Clin. Oncol. 2017;35(22):2568–2575.
- Ligtenberg MJL, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009;41(1):112–117.
- Kovacs ME, Papp J, Szentirmay Z, et al. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009;30(2):197–203.
- Cini G, Carnevali I, Quaia M, et al. Concomitant mutation and epimutation of the MLH1 gene in a lynch syndrome family. Carcinogenesis. 2015;36(4):452–458.
- Morak M, Koehler U, Schackert HK, et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 2011;48(8):513–519.
- Morak M, Ibisler A, Keller G, et al. Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J Med Genet. 2018;55(4):240–248.
- Gylling A, Ridanpää M, Vierimaa O, et al. Large genomic rearrangements and germline epimutations in Lynch syndrome. Int. J. Cancer. 2009;124(10):2333–2340.
- Morak M, Schackert HK, Rahner N, et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur. J. Hum. Genet. 2008;16(7):804–811.
- Ward RL, Dobbins T, Lindor NM, et al. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet. Med. 2013;15(1):25–35.
- Niessen RC, Hofstra RMW, Westers H, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes. Chromosomes Cancer. 2009;48(8):737–744.
- Leclerc J, Flament C, Lovecchio T, et al. Diversity of genetic events associated with MLH1 promoter methylation in Lynch syndrome families with heritable constitutional epimutation. Genet Med. 2018;20(12): 1589–1599.
- Castillejo A, Hernández-Illán E, Rodriguez-Soler M, et al. Prevalence of MLH1 constitutional epimutations as a cause of Lynch syndrome in unselected versus selected consecutive series of patients with colorectal cancer. J Med Genet. 2015;52(7):498–502.
- Dámaso E, Canet-Hermida J, Vargas-Parra G, et al. Highly sensitive MLH1 methylation analysis in blood identifies a cancer patient with low-level mosaic MLH1 epimutation. Clin Epigenetics. 2019;11(1):171.
- Liu Y, Chew MH, Goh XW, et al. Systematic study on genetic and epimutational profile of a cohort of amsterdam criteria-defined lynch syndrome in Singapore. Suzuki H, editor. PLoS ONE. 2014;9(4):e94170.
- Kidambi TD, Blanco A, Van Ziffle J, et al. Constitutional MLH1 methylation presenting with colonic polyposis syndrome and not Lynch syndrome. Fam Cancer. 2016;15(2):275–280.
- Kempers MJ, Kuiper RP, Ockeloen CW, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 2011;12(1):49–55.
- Hitchins M, Williams R, Cheong K, et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129(5):1392–1399.
- Huth C, Kloor M, Voigt AY, et al. The molecular basis of EPCAM expression loss in Lynch syndrome-associated tumors. Mod. Pathol. 2012;25(6):911–916.
- Nagasaka T, Rhees J, Kloor M, et al. Somatic hypermethylation of MSH2 Is a frequent event in lynch syndrome colorectal cancers. Cancer Res. 2010;70(8):3098–3108.
- Hitchins MP, Wong JJL, Suthers G, et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N. Engl. J. Med. 2007;356(7):697–705.
- Sloane MA, Nunez AC, Packham D, et al. Mosaic epigenetic inheritance as a cause of early-onset colorectal cancer. JAMA Oncol. 2015;1(7):953–957.
- Dámaso E, Castillejo A, Arias M, et al. Primary constitutional MLH1 epimutations: a focal epigenetic event. Br J Cancer. 2018;119(8):978–987.
- Hitchins MP, Rapkins RW, Kwok C-T, et al. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5′UTR. Cancer Cell. 2011;20(2):200–213.
- Porkka N, Lahtinen L, Ahtiainen M, et al. Epidemiological, clinical and molecular characterization of Lynch‐like syndrome: A population‐based study. Int. J. Cancer. 2019;145(1):87–98.
- Pasanen A, Loukovaara M, Bützow R. Clinicopathological significance of deficient DNA mismatch repair and MLH1 promoter methylation in endometrioid endometrial carcinoma. Mod Pathol. 2020; In press. doi:10.1038/s41379-020-0501-8.
- Rodríguez–Soler M, Pérez–Carbonell L, Guarinos C, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013;144(5):926–932.
- Geurts-Giele WR, Leenen CH, Dubbink HJ, et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers: somatic MMR aberrations cause Lynch syndrome-like tumours. J. Pathol. 2014;234(4):548–559.
- Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308–1316.
- Mensenkamp AR, Vogelaar IP, van Zelst–stams WAG, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in lynch syndrome-like tumors. Gastroenterology. 2014;146(3):643–646.
- Pearlman R, Haraldsdottir S, de la Chapelle A, et al. Clinical Characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. J. Med. Genet. 2019;56(7):462–470.
- Mas-Moya J, Dudley B, Brand RE, et al. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum. Pathol. 2015;46(11):1616–1625.
- Picó MD, Castillejo A, Murcia Ó, et al. Clinical and pathological characterization of lynch-like syndrome. Clin. Gastroenterol. Hepatol. 2020;18(2):368–374.
- Vargas-Parra GM, González-Acosta M, Thompson BA, et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis: molecular basis of MSH2-deficient tumors. Int. J. Cancer. 2017;141(7):1365–1380.
- Xicola RM, Clark JR, Carroll T, et al. Implication of DNA repair genes in Lynch-like syndrome. Fam. Cancer. 2019;18(3):331–342.
- Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum. Genet. 2008;124(2):105–122.
- Bakry D, Aronson M, Durno C, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014;50(5):987–996.
- Bodo S, Colas C, Buhard O, et al. Diagnosis of constitutional mismatch repair-deficiency syndrome based on microsatellite instability and lymphocyte tolerance to methylating agents. Gastroenterology. 2015;149(4):1017–1029.
- Wang Q, Montmain G, Ruano E, et al. Neurofibromatosis type 1 gene as a mutational target in a mismatch repair-deficient cell type. Hum. Genet. 2003;112(2):117–123.
- Gallon R, Mühlegger B, Wenzel -S-S, et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum. Mutat. 2019;40(5):649–655.
- González-Acosta M, Marín F, Puliafito B, et al. High-sensitivity microsatellite instability assessment for the detection of mismatch repair defects in normal tissue of biallelic germline mismatch repair mutation carriers. J. Med. Genet. 2020;57(4):269–273.
- Goodenberger ML, Thomas BC, Riegert-Johnson D, et al. PMS2 monoallelic mutation carriers: the known unknown. Genet. Med. 2016;18(1):13–19.
- Levi Z, Kariv R, Barnes-Kedar I, et al. The gastrointestinal manifestation of constitutional mismatch repair deficiency syndrome: from a single adenoma to polyposis-like phenotype and early onset cancer: the gastrointestinal phenotype of CMMRD. Clin. Genet. 2015;88(5):474–478.
- Durno C, Boland CR, Cohen S, et al. Recommendations on surveillance and management of biallelic mismatch repair deficiency (BMMRD) syndrome: a consensus statement by the US multi-society task force on colorectal cancer. Gastroenterology. 2017;152(6):1605–1614.
- Adam R, Spier I, Zhao B, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet. 2016;99(2):337–351.
- Olkinuora A, Nieminen TT, Mårtensson E, et al. Biallelic germline nonsense variant of MLH3 underlies polyposis predisposition. Genet. Med. 2019;21(8):1868–1873.
- Wu Y, Berends MJ, Sijmons RH, et al. A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat. Genet. 2001;29(2):137–138.
- Liu H-X, Zhou X-L, Liu T, et al. The role of hMLH3 in familial colorectal cancer. Cancer Res. 2003;63(8):1894–1899.
- Shlien A, Campbell BB, de Borja R, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 2015;47(3):257–262.
- Siraj AK, Masoodi T, Bu R, et al. The study of Lynch syndrome in a special population reveals a strong founder effect and an unusual mutational mechanism in familial adenomatous polyposis. Gut. 2020;18(2):368–374.e1.
- Vasen HF, Mecklin JP, Khan PM, et al. The international collaborative group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis. Colon Rectum. 1991;34(5):424–425.
- Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116(6):1453–1456.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293(16):1979–1985.
- Vasen HFA, Boland CR. Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA. 2005;293(16):2028–2030.
- Dominguez-Valentin M, Therkildsen C, Da Silva S, et al. Familial colorectal cancer type X: genetic profiles and phenotypic features. Mod. Pathol. 2015;28(1):30–36.
- Goel A, Xicola RM, Nguyen T, et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2010;138(5):1854–1862.
- Sahnane N, Magnoli F, Bernasconi B, et al. Aberrant DNA methylation profiles of inherited and sporadic colorectal cancer. Clin. Epigenetics. 2015;21(1):131.
- Pavicic W, Joensuu EI, Nieminen T, et al. LINE-1 hypomethylation in familial and sporadic cancer. J Mol Med. 2012;90(7):827–835.
- Skoglund J, Djureinovic T, Zhou X-L, et al. Linkage analysis in a large Swedish family supports the presence of a susceptibility locus for adenoma and colorectal cancer on chromosome 9q22.32-31.1. J Med Genet. 2006;43(8):685–690.
- Gray-McGuire C, Guda K, Adrianto I, et al. Confirmation of Linkage to and Localization of Familial Colon Cancer Risk Haplotype on Chromosome 9q22. Cancer Res. 2010;70(13):5409–5418.
- Thutkawkorapin J, Mahdessian H, Barber T, et al. Two novel colorectal cancer risk loci in the region on chromosome 9q22.32. Oncotarget. 2018;9(13):11170–11179.
- Guda K, Moinova H, He J, et al. Inactivating germ-line and somatic mutations in polypeptide N-acetylgalactosaminyltransferase 12 in human colon cancers. Proc. Natl. Acad. Sci. 2009;106(31):12921–12925.
- Clarke E, Green RC, Green JS, et al. Inherited deleterious variants in GALNT12 are associated with CRC susceptibility. Hum Mutat. 2012;33(7):1056–1058.
- Evans DR, Venkitachalam S, Revoredo L, et al. Evidence for GALNT12 as a moderate penetrance gene for colorectal cancer. Hum Mutat. 2018;39(8):1092–1101.
- Seguí N, Pineda M, Navarro M, et al. GALNT12 is not a major contributor of familial colorectal cancer type x. Hum Mutat. 2014;35(1):50–52.
- Nieminen TT, O’Donohue M-F, Wu Y, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology. 2014;147(3): 595–598. .
- Broderick P, Dobbins SE, Chubb D, et al. Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients-a systematic review. Gastroenterology. 2017;152(1):75–77.
- Thompson BA, Snow AK, Koptiuch C, et al. A novel ribosomal protein S20 variant in a family with unexplained colorectal cancer and polyposis. Clin Genet. 2020;97(6):943-944.
- Amsterdam A, Sadler KC, Lai K, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2(5):E139.
- Ajore R, Raiser D, McConkey M, et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP 53 mutations. EMBO Mol Med. 2017;9(4):498–507.
- Aspesi A, Ellis SR. Rare ribosomopathies: insights into mechanisms of cancer. Nat. Rev. Cancer. 2019;19(4):228–238.
- Gazda HT, Preti M, Sheen MR, et al. Frameshift mutation in p53 regulator RPL26 is associated with multiple physical abnormalities and a specific pre-ribosomal RNA processing defect in diamond-blackfan anemia. Hum Mutat. 2012;33(7):1037–1044.
- Vlachos A, Rosenberg PS, Atsidaftos E, et al. Increased risk of colon cancer and osteogenic sarcoma in Diamond-Blackfan anemia. Blood. 2018;132(20):2205–2208.
- McGowan KA, Li JZ, Park CY, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 2008;40(8):963–970.
- Khajuria RK, Munschauer M, Ulirsch JC, et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell. 2018;173(1):90–103.
- Bellido F, Sowada N, Mur P, et al. Association between germline mutations in BRF1, a subunit of the RNA polymerase iii transcription complex, and hereditary colorectal cancer. Gastroenterology. 2018;154(1): 181–194.
- Wang W, Nag S, Zhang X, et al. Ribosomal proteins and human diseases: pathogenesis, molecular mechanisms, and therapeutic implications. Med Res Rev. 2015;35:225–285.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004;96(4):261–268.
- Seguí N, Mina LB, Lázaro C, et al. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology. 2015;149(3):563–566.
- Zhou W, Otto EA, Cluckey A, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012;44(8):910–915.
- Arora S, Yan H, Cho I, et al. Genetic variants that predispose to DNA double-strand breaks in lymphocytes from a subset of patients with familial colorectal carcinomas. Gastroenterology. 2015;149(7):1872–1883.
- Lauper JM, Krause A, Vaughan TL, et al. Spectrum and risk of neoplasia in Werner syndrome: a systematic review. Burk RD, editor. PLoS ONE. 2013;8(4):e59709.
- Hoeijmakers JHJ, Damage DNA. Aging, and Cancer. N. Engl. J. Med. 2009;361(15):1475–1485.
- Chubb D, Broderick P, Dobbins SE, et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun. 2016;7(1): 11883.
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1 -associated hereditary cancer syndrome families. J Med Genet. 2020;jmedgenet-2019-106657. In press. doi:10.1136/jmedgenet-2019-106657.
- Spier I, Holzapfel S, Altmüller J, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int J Cancer. 2015;137(2):320–331.
- Bartkova J, Tommiska J, Oplustilova L, et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2(4):296–316.
- Waltes R, Kalb R, Gatei M, et al. Human RAD50 deficiency in a nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009;84(5):605–616.
- Bonjoch L, Franch-Expósito S, Garre P, et al. Germline mutations in FAF1 are associated with hereditary colorectal cancer. Gastroenterology. 2020; In press. doi:10.1053/j.gastro.2020.03.015.
- Wang C-H, Hung P-W, Chiang C-W, et al. Identification of two independent SUMO-interacting motifs in Fas-associated factor 1 (FAF1): implications for mineralocorticoid receptor (MR)-mediated transcriptional regulation. Biochim Biophys Acta Mol Cell Res. 2019;1866(8):1282–1297.
- Schulz E, Klampfl P, Holzapfel S, et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat. Commun. 2014;5(1):5191.
- Kinnersley B, Chubb D, Dobbins SE, et al. Correspondence: SEMA4A variation and risk of colorectal cancer. Nat. Commun. 2016;7(1):10611.
- Nieminen TT, Abdel-Rahman WM, Ristimäki A, et al. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2011;141(1):23–26.
- Latchford AR, Neale K, Phillips RKS, et al. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Dis Colon Rectum. 2012;55(10):1038–1043.
- Jacoby RF, Schlack S, Cole CE, et al. A juvenile polyposis tumor suppressor locus at 10q22 is deleted from nonepithelial cells in the lamina propria. Gastroenterology. 1997;112(4):1398–1403.
- Auclair BA, Benoit YD, Rivard N, et al. Bone morphogenetic protein signaling is essential for terminal differentiation of the intestinal secretory cell lineage. Gastroenterology. 2007;133(3):887–896.
- Fernandez-Rozadilla C, Brea-Fernández A, Bessa X, et al. BMPR1A mutations in early-onset colorectal cancer with mismatch repair proficiency. Clin Genet. 2013;84(1):94–96.
- Rohlin A, Rambech E, Kvist A, et al. Expanding the genotype–phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam Cancer. 2017;16(2):195–203.
- Lieberman S, Beeri R, Walsh T, et al. Variable features of juvenile polyposis syndrome with gastric involvement among patients with a large genomic deletion of BMPR1A. Clin. Transl. Gastroenterol. 2019;10(7):e00054.
- Evans DR, Green JS, Woods MO. Screening of BMPR1a for pathogenic mutations in familial colorectal cancer type X families from Newfoundland. Fam. Cancer. 2018;17(2):205–208.
- Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505(7483):302–308.
- Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat. Rev. Cancer. 2010;10(5):353–361.
- Nieminen TT, Pavicic W, Porkka N, et al. Pseudoexons provide a mechanism for allele-specific expression of APC in familial adenomatous polyposis. Oncotarget. 2016;7(43):70685–70698.
- Kim B-Y, Park JH, Jo H-Y, et al. Optimized detection of insertions/deletions (INDELs) in whole-exome sequencing data. PloS One. 2017;12(8):e0182272.
- Serratì S, De Summa S, Pilato B, et al. Next-generation sequencing: advances and applications in cancer diagnosis. Oncol Targets Ther. 2016;9:7355–7365.
- Sokolenko AP, Suspitsin EN, Kuligina ES, et al. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015;369(2):274–288.
- Yurgelun MB, Allen B, Kaldate RR, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology. 2015;149(3):604–613.
- Aretz S, Uhlhaas S, Sun Y, et al. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat. 2004;24:370–380.
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421.
- Díaz-Gay M, Franch-Expósito S, Arnau-Collell C, et al. Integrated analysis of germline and tumor DNA identifies new candidate genes involved in familial colorectal cancer. Cancers (Basel). 2019;11(3):362.
- Peters U, Bien S, Zubair N. Genetic architecture of colorectal cancer. Gut. 2015;64(10):1623–1636.
- Zeng C, Matsuda K, Jia WH, et al. Identification of Susceptibility Loci and Genes for Colorectal Cancer Risk. Gastroenterology. 2016;150(7):1633–1645.
- Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat. Rev. Genet. 2009;10(6):353–358.
- Stjepanovic N, Moreira L, Carneiro F, et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019;30(10):1558–1571.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015;17(5):405–424.
- Kansikas M, Kasela M, Kantelinen J, et al. Assessing how reduced expression levels of the mismatch repair genesMLH1,MSH2, and MSH6 affect repair efficiency. Hum Mutat. 2014;35(9):1123–1127.
- Kasela M, Nyström M, Kansikas M. PMS2 expression decrease causes severe problems in mismatch repair.. Hum Mutat. 2019;40(7):904–907.
- Binder H, Hopp L, Schweiger MR, et al. Genomic and transcriptomic heterogeneity of colorectal tumours arising in Lynch syndrome: genomic and transcriptomic heterogeneity of Lynch syndrome in colon. J. Pathol. 2017;243(2):242–254.
- Ahtiainen M, Wirta E-V, Kuopio T, et al. Combined prognostic value of CD274 (PD-L1)/PDCDI (PD-1) expression and immune cell infiltration in colorectal cancer as per mismatch repair status. Mod. Pathol. 2019;32(6):866–883.
- Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair–deficient/microsatellite instability–high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779.
- von Knebel Doeberitz M, Kloor M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam. Cancer. 2013;12(2):307–312.
- Burn J, Sheth H, Elliott F, et al. Cancer prevention with aspirin in hereditary colorectal cancer (lynch syndrome), 10 year follow-up and registry based 20 year data in the CAPP2 study: a double-blind, randomised, placebo-controlled trial. Lancet. 2020;395(10240):1855–1863.
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82–93.
- Vasen HFA, Tomlinson I, Castells A. Clinical management of hereditary colorectal cancer syndromes. Nat. Rev. Gastroenterol. Hepatol. 2015;12(2):88–97.
- Therkildsen C, Rasmussen M, Smith-Hansen L, et al. Broadening risk profile in familial colorectal cancer type X; increased risk for five cancer types in the national Danish cohort. BMC Cancer. 2020;20(1):345.