
ABSTRACT
Introduction
Pancreas ductal adenocarcinoma (PDAC) is a frequently lethal malignancy that poses unique therapeutic challenges. The current mainstay of therapy for metastatic PDAC (mPDAC) is cytotoxic chemotherapy. NALIRIFOX (liposomal irinotecan, fluorouracil, leucovorin, oxaliplatin) is an emerging standard of care in the metastatic setting. An evolving understanding of PDAC pathogenesis is driving a shift toward targeted therapy. Olaparib, a poly-ADP-ribose polymerase (PARP) inhibitor, has regulatory approval for maintenance therapy in BRCA-mutated mPDAC along with other targeted agents receiving disease-agnostic approvals including for PDAC with rare fusions and mismatch repair deficiency. Ongoing research continues to identify and evaluate an expanding array of targeted therapies for PDAC.
Areas covered
This review provides a brief overview of standard therapies for PDAC and an emphasis on current and emerging targeted therapies.
Expert opinion
There is notable potential for targeted therapies for KRAS-mutated PDAC with opportunity for meaningful benefit for a sizable portion of patients with this disease. Further, emerging approaches are focused on novel immune, tumor microenvironment, and synthetic lethality strategies.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common malignancy arising from the pancreas. The American Cancer Society estimates that 64,050 new cases of pancreatic cancer will be diagnosed in the United States in 2023, and 50,550 people will die from the disease [Citation1]. The incidence of PDAC has increased by approximately 1% per year since the late 1990s in both men and women and it is expected to become the second-leading cause of cancer-related mortality by 2030 [Citation2]. Cigarette smoking accounts for 20–25% of cases and is the lifestyle factor most strongly associated with PDAC, particularly PDAC harboring a Kirsten rat sarcoma virus (KRAS) G12C mutation [Citation3–6]. Other lifestyle risk factors for PDAC are obesity, alcohol consumption (>30 g/day), chronic pancreatitis, and diets high in processed meats, high-fructose beverages, and saturated fat [Citation7–10]. The median age at diagnosis in the US is 71 years with a slight preponderance for males over females (5.5 vs 4.0 per 100,000 individuals) [Citation11]. Notably, the incidence in people younger than 30 years is increasing [Citation9]. This may in part be attributed to the increasing prevalence of obesity and diabetes in this population, both of which are associated with PDAC [Citation12].
The overall survival (OS) for PDAC has gradually improved with the five-year relative survival rate for all stages combined increasing to 12% between 2012 and 2018 [Citation13]. However, this remains significantly lower than other solid tumor malignancies. Furthermore, the five-year survival falls to 3% for patients presenting with metastatic PDAC (mPDAC), which accounts for approximately 50% of patients [Citation1]. Another 30–35% present with locally advanced disease and only 10–15% of patients have localized disease amenable to surgery at the time of presentation [Citation14]. Factors contributing to the poor prognosis of PDAC include advanced stage at presentation, early micro-metastatic spread, poor response to cytotoxic agents with de novo and acquired resistance, low tumor immunogenicity, immunosuppressive tumor microenvironment, and lack of effective screening.
2. Current standards of care
2.1. First-Line cytotoxic chemotherapy
The current American Society of Clinical Oncology (ASCO) and National Comprehensive Cancer network (NCCN) guidelines for treatment of mPDAC recommend cytotoxic chemotherapy [Citation15,Citation16]. In the first-line setting, standard of care (SOC) is FOLFIRINOX (leucovorin, fluorouracil, irinotecan, and oxaliplatin) often used in a modified version (mFOLFIRINOX; with reduced dose irinotecan and/or omission of bolus 5-fluorouracil or other variations) or gemcitabine combined with nanoparticle albumin-bound paclitaxel (nab-paclitaxel) for patients with good performance status (PS) of eastern cooperative oncology group (ECOG) score of 0 to 1, or KPS (Karnofsky Performance status) 70–100%. These regimens received approval based on the findings of two landmark trials [Citation17,Citation18].
In 2011, the Partenariat de Recherche en Oncologie Degestive (PRODIGE-4/ACCORD-11) Group trial, a multicenter, randomized, phase II/III trial compared FOLFIRINOX with single-agent gemcitabine [Citation17]. The primary endpoint of median overall survival (mOS) was 11.1 months (95% confidence interval [CI], 9.0 to 13.1) in the FOLFIRINOX group compared with 6.8 months (95% CI, 5.5 to 7.6) for gemcitabine alone (hazard ratio (HR), 0.57; 95% CI, 0.45 to 0.73; p < 0.001). Median progression-free survival (mPFS) was 6.4 months (95% CI, 5.5 to 7.2) in the FOLFIRINOX group and 3.3 months (95% CI, 2.2 to 3.6) in the gemcitabine group (HR for disease progression, 0.47; 95% CI, 0.37 to 0.59; p < 0.001). The objective response rate (ORR) was 31.6% in the FOLFIRINOX group versus 9.4% in the gemcitabine group (p < 0.001). This was followed in 2013 by the Metastatic Pancreatic Adenocarcinoma Clinical Trial (MPACT), an international, multicenter, open-label, randomized, phase III study comparing gemcitabine with gemcitabine and nab-paclitaxel (GnP) in patients with KPS 70–100% [Citation18]. The primary endpoint of mOS was 8.5 months (95% CI, 7.89 to 9.53) in the combination group versus 6.7 months (95% CI, 6.01 to 7.23) in the gemcitabine group (HR for death, 0.72; 95% CI, 0.62 to 0.83; p < 0.001).
The choice of first-line cytotoxic chemotherapy regimen in clinical practice depends on patient PS, preexisting comorbidities, patient, and physician preferences related to toxicity profiles, central venous access port necessity, and lifestyle considerations. For patients with an ECOG PS of 2, GnP is considered in selected patients, and gemcitabine monotherapy is a preferred option for patients with a less favorable performance status [Citation15,Citation16]. The addition of nab-paclitaxel to gemcitabine in patients with an ECOG of 2 was found to be well tolerated in the FRAGRANCE trial [Citation19]. Patients with an ECOG PS of 3 or with poorly controlled comorbid conditions despite ongoing active medical care should be considered for cancer-directed therapy only on a case-by-case basis with emphasis on optimal symptomatic and supportive management [Citation16].
A notable subpopulation is patients with a germline or somatic mutation affecting genes involved in homologous recombination repair (HRR) such as breast cancer gene 1/2 (BRCA1/2) and Partner and Localizer of BRCA2 (PALB2). The NCCN guidelines recommend these patients receive a platinum containing regimen such as FOLFIRINOX or gemcitabine plus cisplatin in the first-line setting (ECOG 0–1) [Citation16].
2.2. Emerging cytotoxic chemotherapy approaches in mPDAC
The recent global, open-label, phase III NAPOLI-3 trial compared NALIRIFOX (liposomal irinotecan, FU, leucovorin, and oxaliplatin) administered over 28-day cycle and SOC GnP in previously untreated patients with mPDAC [Citation20]. The intention-to-treat population of 770 patients was randomized to NALIRIFOX (n = 383) or GnP (n = 387). At median follow-up of 16.1 months, the primary endpoint of mOS was 11.1 months (95% CI, 10.0–12.1) for NALIRIFOX and 9.2 months (95% CI, 8.3–10.6) for GnP (HR 0.83 [95% CI 0.70–0.99]; p = 0·036). Additionally, mPFS was 7.4 months (95% CI, 6.0–7.7) in the NALIRIFOX group and 5.6 months (95% CI, 5.3–5.8) in the GnP group (HR 0.69 [0.58–0.83]; p < 0.0001) and ORR numerically favored NALRIFOX (ORR, 41.8% versus 36.2%). The findings of the NAPOLI-3 trial support the use of NALIRIFOX as a new SOC and reference regimen for the first-line treatment of mPDAC [Citation21].
In cross-trial comparison, rates of grade 3–4 peripheral sensory neuropathy and neutropenia with NALIRIFOX were lower than FOLFIRINOX (3.5% versus 9% and 23.8% versus 45.7%, respectively), due to the lower cumulative dose of oxaliplatin with the NALIRIFOX regimen and use of growth factors [Citation17,Citation20]. Cross-trial comparison of OS in the NALIRIFOX arm in NAPOLI-3 to the FOLFIRINOX arm of the PRODIGE-4/ACCORD-11 shows notable similarities in mOS at 11.1 months in both regimens [Citation17,Citation20]. Comparison between the GnP arms of NAPOLI-3 and MPACT trials showed slightly longer OS (9.2 versus 8.5 months) and higher ORR (41% versus 23%) in the NAPOLI-3 cohort [Citation18,Citation20]. In the absence of direct head-to-head trials, these cross-trial comparisons also appear to favor triplet FOLFIRINOX/NALIRIFOX over doublet GnP in terms of OS. However, the superiority of FOLFIRINOX/NALIRIFOX over GnP remains an open question, particularly in Japan given the antithetical data from a recent interim analysis of the phase II/III, JCOG1611-GENERATE trial presented at the 2023 European Society of Medical Oncology (ESMO) annual meeting [Citation22]. Preliminary OS results showed more favorable mOS of 17.0 months with GnP compared to 14.0 months with mFOLFIRINOX (HR 1.29; 95% CI 0.98–1.70) and 13.6 months with S-IROX (oxaliplatin, irinotecan) (HR 1.35, 95% CI 1.00–1.82), possibly related to toxicity and early cessation of the non-GnP treatment regimens [Citation22].
2.3. Other variations on standard of care
The sequential cycling of cytotoxic chemotherapy agents has been explored to address resistance in PDAC. The SEQUENCE trial, a phase I/II, randomized trial, investigated a regimen of GnP followed by modified FOLFOX versus GnP alone [Citation23]. Patients received either a 6-week cycle GnP and modified (m)FOLFOX-6 (oxaliplatin, leucovorin, 5-fluorouracil (5-FU)) or a 4-week cycle of GnP. A total of 157 patients were randomized to GnP/mFOLFOX (n = 78) and GnP (n = 79). For the primary endpoint of 12-month survival (55.3% v 35.4%), 24-month survival (22.4% v 7.6), and median OS (13.2 v 9.7 months) favored GnP/mFOLFOX over GnP alone. Safety was comparable except for a higher incidence of grade ≥ 3 neutropenia (46.1% versus 24.1%) and thrombocytopenia (23.7% versus 7.6%) in the GnP/FOLFOX group [Citation24].
The FOOTPATH trial, a randomized, open-label, phase II trial compared GnP against the NAPOLI (liposomal irinotecan, folinic acid, 5-FU) regimen, and NAPOLI plus mFOLFOX6 in previously untreated mPDAC [Citation25]. Two hundred and seventy-four patients were enrolled and randomized 1:1:1. For the primary endpoint of progression-free survival (PFS), the full set analysis did not show statistically significant improvement in PFS with NAPOLI (3.1 months, HR 1.224, p = 0.2123) or NAPOLI/FOLFOX6 (6.0 months, HR 0.864, p = 0.0720) compared with GnP alone (4.3 months). The NAPOLI/FOLFOX6 regimen had an OS of 11 months, greater than GnP (8.7 months) and NAPOLI regimen (7.9 months) but did not reach statistical significance. Correlative analyses from this study are underway.
2.4. Maintenance therapy
The NCCN guidelines recommend consideration of maintenance therapy for patients with mPDAC that is stable or responsive after 4–6 months of chemotherapy [Citation16]. Considerations regarding the choice of maintenance regimen include first-line regimen (FOLFIRINOX vs GnP), patient PS, patient and physician preference, and germline BRCA1/2 mutation status.
Patients treated with first-line GnP can continue on maintenance gemcitabine or a modified schedule of GnP (NCCN, Category 2B recommendation) [Citation16]. Patients treated with first-line FOLFIRINOX can be transitioned to a simplified version of this regimen, including 5FU + leucovorin, FOLFOX, FOLFIRI (5-FU, leucovorin, irinotecan) or monotherapy with capecitabine [Citation16].
Recent trials investigating maintenance regimens include the PANOPTIMOX/PRODIGE-35 trial, which demonstrated that in PDAC either responding or stable after 4 months of FOLFIRINOX, maintenance therapy with leucovorin plus fluorouracil had comparable OS of 11.2 months (95% CI, 9.0 to 13.1) compared with 10.1 months (95% CI, 8.5 to 12.2) for 6 months of FOLFIRINOX therapy [Citation26]. However, incidence of the Grade 3 or 4 neurotoxicity was higher at 19.8% in the FOLFIRINOX plus leucovorin/fluorouracil maintenance group than the 10.2% in the FOLFIRINOX only group, due to reintroduction of oxaliplatin at the time point of disease progression.
Patients with mPDAC harboring a germline BRCA1/2 mutation stable or responding after 16-weeks of platinum-based chemotherapy are candidates for maintenance therapy with the Poly ADP-ribose polymerase (PARP) inhibitor, olaparib, based on the findings of the practice changing POLO trial [Citation27]. The POLO (Pancreas Olaparib Ongoing) study was a phase III, randomized, double-blind, placebo-controlled trial investigating olaparib as maintenance therapy in platinum-sensitive mPDAC with germline BRCA1/2 mutations [Citation27]. After screening, 154 patients underwent randomization to olaparib (n = 92) or placebo (n = 62). The primary endpoint was PFS, which was significantly improved with olaparib compared with placebo (7.4 versus 3.8 months; HR for disease progression or death, 0.53; 95% CI, 0.35 to 0.82; p = 0.004) [Citation27]. No statistically significant OS benefit was observed between olaparib (19.0 months) and placebo (19.2 months) (HR, 0.83; 95% CI, 0.56 to 1.22; p = 0.3487) [Citation28]. The POLO trial led to the Food and Drug Administration (FDA) approval of olaparib as maintenance therapy for germline BRCA1/2 mutated PDAC in 2019. The phase Ib/II PARPVAX trial randomized patients with mPDAC (with or without a germline BRCA1, BRCA2, or PALB2 mutation) with stable or responding disease after 16 weeks of platinum-based chemotherapy to receive niraparib plus immune checkpoint blockade (ICB) with either nivolumab (anti-PDL1) or ipilimumab (anti-CTLA4) [Citation29]. The primary end point was PFS at 6 months and after median follow-up of 23 months (interquartile range (IQR) 15.0–31.5), the 6-month PFS was 20.6% (95% CI, 8.3–32.9; p = 0.0002 vs the null hypothesis of 44%) in the niraparib/nivolumab group and 59.6% (95% CI, 44.3–74.9; p = 0.045) in the niraparib/ipilimumab group. The ongoing Pembrolizumab and OLApaRib (POLAR) trial is evaluating the safety and efficacy of olaparib plus ICB pembrolizumab (anti-PD-1) as maintenance therapy in mPDAC with either a known mutation causing homologous recominbation repair deficiency (HRD) (Cohort A, B) or response to platinum-based chemotherapy (Cohort C) (NCT04666740) (discussed further in section 4.4). Selected completed clinical trials of targeted therapy in PDAC are outlined in .
Table 1. Selected completed trials of cytotoxic chemotherapy and targeted therapy in pancreatic cancer.
3. Molecular classification and subtyping of PDAC
The pathogenesis of PDAC is characterized by a multistep transformation from precancerous lesions such as pancreatic intraepithelial neoplasia, mucinous cystic neoplasms, and intra-ductal papillary mucinous neoplasm to PDAC. This process is characterized by a complex stepwise accumulation of somatic genetic mutations in key oncogene and tumor suppressor genes; most frequently KRAS, cyclin-dependent kinase inhibitor 2A (CDKN2A), tumor protein 53 (TP53), and SMAD Family Member 4 (SMAD4) [Citation30–32]. Overall, PDAC has been found to exhibit significant genetic heterogeneity with a mean of 50–60 somatic mutations [Citation33].
Several groups have performed genomic and molecular analysis for the classification and subtyping of PDAC. In 2011, Collision et al. performed combined analysis of transcriptional profiles of primary PDAC samples to define three PDAC subtypes: classical, quasi-mesenchymal, and exocrine-like [Citation34]. In 2015, Waddell et al. identified four PDAC subtypes based on chromosomal structural variations: stable (20%), locally rearranged (30%), scattered (36%), and unstable (14%) [Citation31]. In 2016, Waddell et al. performed further characterization via whole-genome, deep-exome sequencing, and RNA expression profiling, revealing four histopathological subtypes: squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine and exocrine (ADEX) [Citation35]. Moffitt et al. combined virtual microdissection with diverse pancreatic gene expression microarray data from normal pancreas, primary and metastatic cancer samples, identifying two tumor-specific subtypes: classical-like and basal-like [Citation36]. Given the rapid progress in this area, Rashid et al. performed a metanalysis of publicly available expression data in 2020 and published the PurIST (Purity Independent Subtyping of Tumors) tool, a single-sample signature tool supporting the classical or basal-like subtype system [Citation37].
The prognostic significance of the classical and basal-like subtypes and their role in treatment selection is an area of active research. The Comprehensive Molecular Characteri-zation of Advanced Pancreatic Ductal Adenocarcinomas for Better Treatment Selection (COMPASS) trial is a completed prospective cohort study evaluating response to first-line chemotherapy with FOLFIRINOX versus GnP [Citation38]. The primary endpoint was the feasibility of reporting whole-genome sequencing (WGS) results prior to first disease assessment scan at 8 weeks. The main secondary endpoint was the discovery of patient subsets with predictive mutational and transcriptional signatures. The classical subtype treated with mFOLFIRINOX (n = 55) achieved the best PFS (8.5 months; 95% CI, 6.5–not reached), while the least favorable PFS was observed in the basal-like subtype treated with mFOLFIRINOX (2.7 months; 95% CI, 2.1–not reached). Further analysis of the COMPASS data found that increased GATA6 expression was highly correlated with the classical subtype (p < 0.001) [Citation39]. GATA6 may represent a surrogate and prognostic marker for the classical subtype. The Pancreatic Adenocarcinoma Signature Stratification for Treatment (PASS-01) trial, a randomized phase II, multicenter clinical trial is assessing mFOLFIRINOX versus GnP in de novo mPDAC with a primary endpoint of mPFS and a blinded analysis of GATA6 as a surrogate biomarker of classical subtype. Extensive immunogenomic and other correlative analyses are incorporated in this study (NCT04469556) [Citation40].
4. DNA damage repair (DDR) deficiency
4.1. Homologous recombination repair deficiency (HRD)
DNA is continuously damaged by environmental exposures and endogenous cellular processes, resulting in genetic lesions such as single-stranded breaks (SSBs) and double-stranded breaks (DSBs) [Citation41,Citation42]. Loss of DDR pathways, particularly those responsible for DSB repair, can result in genomic instability and malignant transformation [Citation41,Citation43]. DSBs are repaired by HRR and non-homologous end joining (NHEJ) [Citation42]. HRR facilitates high fidelity DSB repair by employing the homologous sequence from the sister chromatid as a template [Citation42]. However, NHEJ involves little or no homology and is an error-prone process that can produce chromosomal rearrangements or translocations [Citation42,Citation44,Citation45].
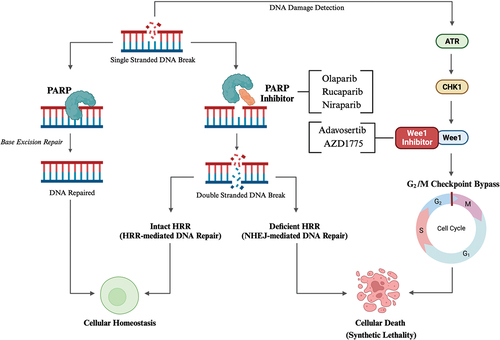
HRR is undertaken by a multi-protein complex including BRCA1, BRCA2, PALB2, Ataxia Telangiectasia Mutated (ATM), RAD51 and others [Citation46,Citation47]. Germline BRCA mutations have long been associated with a predisposition to breast, ovary, prostate, and pancreatic cancers [Citation41,Citation48,Citation49]. The prevalence of BRCA mutations in the general population is 0.1–0.3% [Citation50,Citation51]. In unselected cohorts of PDAC, there is a higher prevalence of BRCA1 (0.7–5.7%) and BRCA2 (0.3–2.3%) mutations. In familial PDAC, the prevalence of BRCA2 mutations is as high as 17% in some cohorts [Citation52]. Notably, other genes involved in the HRR such as PALB2 and ATM are also mutated in PDAC, highlighting the importance of HRR integrity in determining PDAC risk [Citation31,Citation53]. HRD renders cells more sensitive to therapies which induce DNA damage or inhibit other DNA repair pathways. The Poly (ADP-ribose) polymerase (PARP) enzyme detects and initiates repair of SSBs via base excision repair (BER) () [Citation54,Citation55]. Inhibition of PARP causes SSB accumulation, replication folk collapse and formation of DSBs [Citation56]. HRD cells rely on error-prone NHEJ for DSB repair, further disrupting the DNA sequence, compromising genomic stability and eventually inducing synthetic lethality [Citation57]. In addition to PARP, emerging therapeutic targets in DDR PDAC include the G2/M checkpoint cell cycle regulator Wee1, and DNA-dependent protein kinase (DNA – PK). Pre-clinical and early clinical data has demonstrated the potential of these proteins as targets for monotherapies, or in combination with preexisting therapies, such counteracting resistance to PARP inhibitors [Citation58–62].
Figure 1. Outlines (1) the impact of poly-ADP-ribose polymerase (PARP) inhibition in cells with intact homologous recombination repair (HRR) versus homologous recombination repair deficiency (HRD) cells and (2) the impact of Wee1 protein inhibition following DNA damage. First, in the absence of PARP inhibition, DNA single stranded breaks (SSBs) are detected and repaired via base excision repair (BER). PARP inhibition impairs SSB repair and culminates in the formation of DNA double stranded breaks (DSBs). Cells with intact HRR repair DNA DSBs via HRR. However, HRD cells rely on error-prone non-homologous end joining (NHEJ) which further destabilizes DNA and results in synthetic lethality. Second, DNA damage is detected by ataxia telangiectasia and Rad3-related protein (ATR), which signals through checkpoint kinase 1 (CHK1) to the Wee1 protein, which functions to halt the cell cycle at the G2/M checkpoint until DNA repair is performed. However, inhibition of Wee1 facilitates bypass of the G2/M checkpoint prior to DNA repair, inappropriately initiating mitosis and causing cellular death. Created with BioRender.com.
4.2. Platinum-based chemotherapy
The cytotoxic effect of platinum-based chemotherapy agents (oxaliplatin and cisplatin) is exerted primarily through DNA damage, including the induction of DNA crosslinks (intra- and interstrand) and DNA strand breaks [Citation63,Citation64]. BRCA1/2 mutated PDAC shows increased sensitivity to platinum-containing chemotherapy [Citation65,Citation66]. Patients with germline BRCA1/2 mutations and PDAC have improved OS compared to BRCA wild-type when treated with platinum-based chemotherapy [Citation65]. The current NCCN guidelines recommend that patients with mPDAC, germline mutation of the HRR genes (BRCA1/2, PALB2) and good performance status receive first-line platinum-containing chemotherapy with FOLFIRINOX, mFOLFIRINOX, or gemcitabine with cisplatin [Citation16].
4.3. PARP inhibitors
The POLO trial demonstrated clinical activity of olaparib as maintenance therapy in platinum-sensitive mPDAC with germline BRCA1/2 mutations [Citation27].
The RUCAPANC2 trial, a single-arm, phase II trial investigated maintenance rucaparib in platinum-sensitive mPDAC with germline or somatic pathogenic variants in BRCA or PALB2 [Citation67]. A total of 42 patients were enrolled to receive rucaparib 600 mg. The primary endpoint of PFS at 6 months was 59.5% (95% CI, 44.6 to 74.4). The mPFS was 13.1 months (95% CI, 4.4 to 21.8) and mOS was 23.5 months (95% CI, 20 to 27). The NCCN guidelines incorporate rucaparib maintenance therapy as an option for germline or somatic BRCA1/2 or PALB2 mutated and platinum sensitive PDAC [Citation16].
The ongoing APOLLO trial is a randomized, phase II study investigating adjuvant olaparib for BRCA1/2 or PALB2 mutated PDAC (NCT04858334) [Citation68]. The primary endpoint is relapse-free survival (RFS). Selected ongoing trials of PARP inhibitors in the PDAC maintenance setting are outlined in .
Table 2. Selected ongoing trials of PARP inhibitors and PARP inhibitors plus Immune Checkpoint Blockade in pancreatic cancer.
4.4. PARP inhibitors in combination with immune checkpoint blockade
In preclinical models, PARP inhibition combined with ICB has activity in DDR deficient tumors [Citation69,Citation70]. The PARPVAX trial demonstrated clinical activity of niraparib plus ipilimumab in platinum-sensitive mPDAC with or without an HRR mutation [Citation29]. The ongoing phase II POLAR trial is evaluating the safety and efficacy of olaparib and pembrolizumab in patients with mPDAC and either known core HRD mutation (BRCA1/2 or PALB2), non-core HRD mutations or response to platinum-based chemotherapy (NCT04666740). Patients are stratified into three cohorts in the order of canonical HRR gene order; Cohort A consists of patients with pathogenic germline or somatic alterations of 3 core HRR-genes (BRCA1/2, or PALB2), Cohort B of patients with pathogenic somatic or germline alterations in 15 non-core HRR-genes and Cohort C of patients without any of the HRR-gene alterations included in Cohort A and B who have platinum-sensitivity. The primary endpoint is PFS. Preliminary data for cohorts B and C were presented at the ASCO 2023 annual meeting [Citation71]. Cohorts B and C enrolled 15 patients each with 25 patients evaluable by RECIST. Median follow-up was 9.9 (95% CI, 1.3–22.8) and 11.3 (95% CI, 5.8–23) months for cohort B and C, respectively. Disease control rate (DCR) was 60% and 46.5%, and ORR 0% and 13% for cohorts B and C, respectively. mPFS was 4 months (95% CI, 2.1–5.4) with the mOS of 14 months (95% CI, 10 - not reached) for cohorts B and C combined. Cohort A is actively accruing, and the estimated study completion is 2024. Selected trials of PARP inhibitors in combination with ICBs are outlined in .
5. Molecular targets
5.1. KRAS
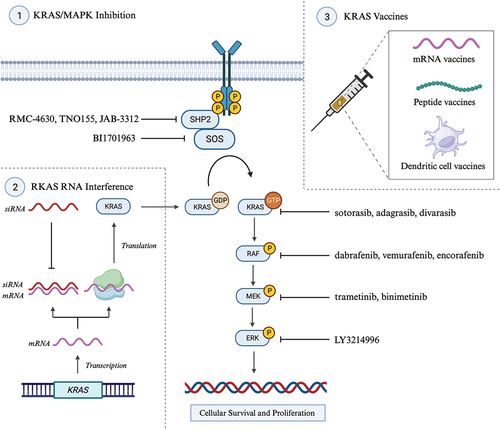
The Kirsten rat sarcoma virus (KRAS) protein is a GTPase enzyme and central mediator of intracellular signaling pathways and transcription factors promoting cell proliferation, migration, transformation, and survival. KRAS induces downstream activation the RAF-MEK-ERK (v raf murine sarcoma viral oncogene homologue B1, mitogen activated protein kinase, extracellular signal-regulated kinase) pathway and PIK3-AKT-mTOR (phosphatidylinositol-3-kinase, protein kinase B, mammalian target of rapamycin) pathways [Citation72,Citation73]. KRAS mutations typically occur early in PDAC pathogenesis and 90–95% of PDAC cases harbor a KRAS mutation [Citation31,Citation32]. Codon 12 is most frequently mutated with subtype allele frequencies of G12D (~40%), G12V (~ 34%), G12R (~ 16%) and G12C (~ 1%) [Citation74,Citation75]. A KRAS mutation is associated with poorer OS compared with KRAS wild type (WT) [Citation76,Citation77]. The ubiquity of KRAS mutations in PDAC makes it an attractive therapeutic target. However, for decades the KRAS protein was considered ‘undruggable,’ until in 2013, Shokat and colleagues published a landmark paper identifying the novel KRAS G12C cysteine-containing switch II pocket and demonstrating the first selective inhibition of KRAS G12C [Citation78]. The translation of KRAS inhibitors to the clinical setting has yielded FDA approval for sotorasib and adagrasib in KRAS G12C-mutated non-small cell lung cancer (NSCLC) [Citation79]. Sotorasib and adagrasib are included in the NCCN guidelines for KRAS G12C-mutated locally advanced and mPDAC [Citation16]. Renewed interested in KRAS-targeted therapies has seen an expanding array of small-molecule inhibitors, interfering RNA (SiRNA), KRAS vaccines, and downstream pathway inhibitors ().
5.1.1. Targeting KRAS G12C
The initial clinical study on KRAS G12C inhibitors was the CodeBreaK 100, a multicenter, phase I/II trial assessing the safety and response rates of sotorasib (AMG510) in a variety of previously treated, KRAS G12C-mutated advanced solid cancers [Citation80]. In total, 38 patients with KRAS G12C-mutated mPDAC received sotorasib [Citation81]. The confirmed ORR was 21% (95% CI, 10 to 37), mPFS 4.0 months (95% CI, 2.8 to 5.6), and mOS was 6.9 months (95% CI, 5.0 to 9.1) [Citation81]. The follow-up CodeBreaK 101 trial investigating sotorasib in combination with other therapies is currently underway (NCT04185883). Preliminary data from the ongoing KRYSTAL-1 study investigating adagrasib (MRTX849) in advanced solid tumors harboring a KRAS G12C mutation showed that among the 21 patients with PDAC, ORR was 33.3% (7/21); DCR was 81% (17/21), mPFS was 5.4 months (95% CI 3.9–8.2), and mOS was 8 months (95% CI 5.2–11.8) [Citation82]. Selected ongoing trials of KRAS G12C-mutated PDAC are listed in .
Table 3. Selected ongoing trials of KRAS-directed therapy in pancreatic cancer.
5.1.2. Allele specific targeting and Pan/All RAS inhibitors
KRAS G12C mutations account for 1–2% of KRAS mutation in PDAC and additional trials are investigating inhibitors targeting more frequently occurring KRAS mutations (G12D, G12V, and G12R) and pan/all-RAS inhibitors [Citation76]. The switch II pocket of KRAS G12D protein lacks a reactive cysteine, rendering it a more challenging target for covalent binding and inhibition [Citation83]. A non-covalent KRAS G12D inhibitor, MRTX1133, was developed by Wang et al. in 2021 [Citation84]. MRTX1133 prevents downstream signaling through inhibition of KRAS nucleotide exchange and binding of downstream effector RAF1 [Citation84]. Preclinical data show that MRTX1133 reverses early PDAC growth and favorably influences tumor microenvironment (TME) by increasing intratumoral CD8+ effector T cells, decreasing myeloid infiltration, and reprograming cancer-associated fibroblasts [Citation85,Citation86]. An ongoing phase I/II trial evaluating several oral formulations of MRTX1133 in KRAS G12D-mutated advanced solid tumors is recruiting (NCT05737706). Another novel KRAS G12D inhibitor, RMC-9805 reacts covalently with Asp12, thereby attenuating KRAS G12D downstream signaling [Citation87]. An ongoing phase I trial is investigating the safety and tolerability of RMC-9805 in adults with KRAS G12D-mutated solid tumors (NCT06040541). Other KRAS G12D inhibitors include TH-Z827 and TH-Z835, which interact via the Asp12 within the KRAS G12D switch II pocket and are yet to undergo investigation in clinical trials [Citation88].
Beyond targeting specific KRAS mutant alleles, the development of pan-RAS inhibitors is under investigation. The primary challenge with pan-RAS inhibition is likely toxicity resulting from off-target inhibition of RAS WT in normal tissues [Citation89]. A key point in this context is the distinction between the likely toxicities associated with isoform-specific pan-KRAS inhibition compared with isoform-agnostic pan-RAS drugs that inhibit all three RAS isoforms (KRAS, HRAS, and NRAS). It is likely that isoform-specific pan-RAS inhibitors represent the best path forward. Preliminary data were presented at ESMO 2023 from a phase I trial investigating pan/multi-KRAS inhibitor RMC-6236 in patients with previously treated mPDAC (n = 22) and non-small cell lung cancer (NSCLC) (n = 11) harboring different KRAS mutations, excluding G12C (NCT05379985) [Citation90]. Among 14 patients (10 PDAC, 4 NSCLC), the ORR was 36% (2 confirmed and 3 unconfirmed partial response (PR); 2/10 (20%) PDAC and 3/4 NSCLC) and DCR was 86% (12/14; 8/10 PDAC and 4/4 NSCLC) [Citation90].
5.1.3. Targeting KRAS upstream pathways
Indirect pan-KRAS inhibition may be achieved through targeting the upstream signaling proteins SHP2 (Src homology-2 domain-containing protein tyrosine phosphatase-2) and SOS1 (Son of Sevenless 1) involved in KRAS activation. The rationale is based on the observation that KRAS oncoproteins continue to cycle between an inactive and active state, thereby relying on upstream activation and nucleotide exchange [Citation91–93]. SHP2 inhibitors stabilize the inactive conformation of KRAS and thereby disrupt SOS1-mediated nucleotide exchange [Citation92]. Several SHP2 inhibitors are in development and three (RMC-4630, TNO155, and JAB-3068) have proceeded to phase I/II clinical trials (NCT03634982, NCT04330664, and NCT05288205). The GEF (Guanine nucleotide Exchange Factor) SOS1 protein facilitates nucleotide exchange, GTP loading, and activation KRAS [Citation93]. BI1701963 is a SOS1 inhibitor being investigated in a phase I trial to determine dosing as monotherapy and in combination with trametinib for KRAS-mutated solid tumors (NCT04111458). Selected ongoing clinical trials targeting proteins upstream of KRAS are outlined in .
5.1.4. KRAS vaccines
Peptide-based vaccines aim to stimulate a tumor-specific host immune response against neoantigens unique to the tumor [Citation94]. Ongoing peptide-based vaccine trials targeting KRAS-mutated PDAC include the AMPLIFY-7P, a phase II, multicenter trial investigating a lipophilic modified RAS peptide vaccine (ELI-002) (NCT05726864). The ELI-002 vaccine contains seven lipid-conjugated peptide-based KRAS antigens (G12D/R/V/A/C/S/D) (Amph-Peptides) plus a lipid-conjugated immune-stimulatory oligonucleotide (Amph-CpG-7909). ELI-002 is also being investigated in the adjuvant setting for the treatment of minimal residual disease (MRD) in subjects with KRAS/NRAS mutated PDAC or other solid tumors (NCT04853017). Preliminary data for this trial were presented at the ASCO 2023 annual meeting showed a biomarker reduction in 15/19 (79%), including 5/5 (100%) at highest doses of the immune adjuvant, and MRD biomarker or circulating DNA (ctDNA) clearance in 4/19 (21%, n = 2 pancreatic, n = 2 colorectal). Additionally, polyfunctional KRAS-specific T cell responses were observed in 80% of patients (n = 12/15). Vaccines utilizing mRNA technology for KRAS-mutated PDAC include mRNA-5671, which encodes KRAS G12D, G12V, G13D, and G12C-specific peptides are currently under investigation in combination with pembrolizumab in a phase I trial in KRAS-mutant advanced cancer (NCT03948763). Selected ongoing clinical trials of KRAS vaccines are outlined in .
5.1.5. KRAS RNA interference
RNA interference (RNAi) utilizes short non-coding strands of RNA, known as short interfering RNA (SiRNA) or microRNA (miRNA), to selectively block the translation of messenger RNA (mRNA) from specific oncogenes [Citation95]. Despite encouraging findings from pre-clinical studies, clinical success has been limited to date [Citation96]. Several challenges to the successful delivery to siRNA to target tumor exist including low in vivo siRNA stability, and limited delivery through lipid vascular and cellular membranes [Citation97]. Delivery systems using exosomes, liposomes, and biodegradable polymers such as the low drug-eluter (LODER) have been investigated, as has the direct delivery of siRNA to the target tumor. In an early-phase study by Golan et al., siRNAs against KRAS G12D mRNA were implanted directly into PDAC tumors via endoscopic intervention in patients receiving concurrent systemic chemotherapy [Citation98]. The results showed promising PFS without adverse outcomes [Citation98]. The phase II (PROTRACT) trial investigated siG12D (Loder™) in combination with standard of care chemotherapy for patients with KRAS G12D/V mutated, locally advanced PDAC. There was a non-statistically significant numerical improvement in mOS of 21.1 months for Loder™ plus mFOLFIRINOX or GnP (n = 18) versus 13.8 months for GnP alone (n = 5). A novel formulation of siG12D is under development. A phase I trial investigating mesenchymal stromal cell-derived exosomes (iExosomes) for delivery of KRAS G12D siRNA in mPDAC is underway (NCT01676259, NCT03608631).
The ongoing investigation of multiple KRAS-targeting therapeutic modalities () represents a new era in PDAC research that is likely to have practice changing implications. Given the early and ubiquitous role of KRAS mutations in PDAC, a sizable proportion of patients may benefit from KRAS-targeted therapies in the neoadjuvant, adjuvant, and metastatic setting.
Figure 2. Selected KRAS (Kirsten rat sarcoma virus)-targeted therapeutic modalities under investigation or with disease agnostic-approval pancreatic ductal adenocarcinoma (PDAC). Section 1 outlines therapies targeting KRAS and upstream (SHP, src homology-2 domain-containing protein tyrosine phosphatase-2; SOS1, son of sevenless 1) and downstream (RAF, v raf murine sarcoma viral oncogene homologue B1; MEK, mitogen activated protein kinase kinase; ERK, extracellular signal-regulated kinase) signaling pathways. Section 2 outlines the mechanism of small interfering RNA in selectively silencing KRAS expression by inhibiting translation of KRAS mRNA into KRAS protein. Section 3 outlines the three primary vaccine modalities under investigation in KRAS-mutated PDAC: mRNA vaccines, peptides vaccines and dendritic cell vaccines (DCVs). Created with BioRender.com.
5.2. KRAS wild-type pancreatic cancer
Four–10.7% of PDAC tumors do not contain KRAS mutations and are KRAS WT [Citation99–101]. KRAS WT PDAC is associated with a better prognosis and is enriched for potentially targetable oncogenic mutations including epidermal growth factor receptor (EGRF), BRAF, neuregulin 1 (NRG1) rearrangements, neurotrophic tropomyosin kinase receptors (NTRK) and other rare fusions and anaplastic lymphoma kinase (ALK) rearrangements [Citation102–105].
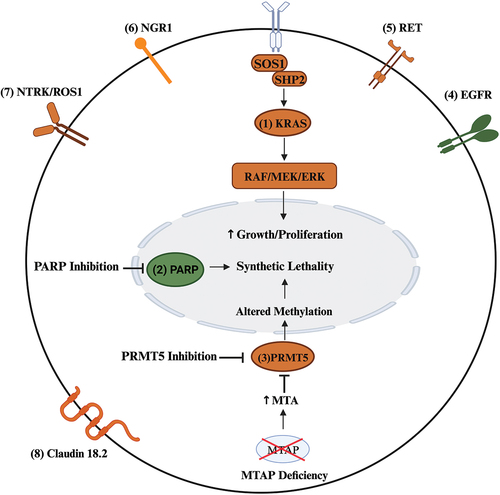
Figure 3. Summary of selected current and future therapeutic targets located on the tumor cell in PDAC. Therapeutic targets under investigation or with disease-agnostic approval are colored orange while those with FDA approval for PDAC are colored green (PARP inhibitor – olaparib, EGRF inhibitor – erlotinib). Multiple target therapy modalities are under investigation KRAS (Kirsten rat sarcoma virus) including allele-specific and pan/all KRAS enzyme inhibitors, vaccines, and small interfering RNA, as are therapies targeting the upstream (SOS1, SHP2) and downstream (RAF/MEK/ERK) signaling proteins of KRAS. In PDAC cells with methylthioadenosine phosphorylase (MTAP) deficiency, the accumulation of methylthioadenosine (MTA) causes endogenous inhibition of protein arginine methyltransferases 5 (PRMT5), a protein central to histone and non-histone methylation. Targeted inhibition of PRMT5 in MTAP-deficient PDAC may induce synthetic lethality. Poly-ADP-ribose polymerase (PARP) inhibition also induces synthetic lethality through impaired detection and repair of single-stranded DNA. BRAF (v-raf murine sarcoma viral oncogene homolog B1); EGFR (epidermal growth factor receptor); ERK (extracellular signal-regulated kinase); MEK (mitogen-activated protein kinase kinase); Neuregulin 1 (NRG1); NTRK (neurotrophic tropomyosin kinase receptors); PRMT5 (protein arginine methyltransferases 5); RET (rearranged during transfection); SHP2 (src homology-2 domain-containing protein tyrosine phosphatase-2); SOS1 (son of sevenless 1). Created with BioRender.com.
5.3. EGFR
The EGFR is overexpressed in most cases of PDAC. The EFGR inhibitor erlotinib received FDA approval after a double-blind, placebo-controlled, phase 3 trial of erlotinib and gemcitabine versus gemcitabine monotherapy demonstrated an improved mOS of 6.24 months v 5.91 months, respectively (estimated HR, 0.82, 95% CI, 0.69 to 0.99; p = 0.038); however, the clinical signal was considered very modest and the trial was conducted in a genomically unselected population [Citation106]. The recent NOTABLE III trial, a phase III, randomized trial investigated the anti-EGFR monoclonal antibody, nimotuzumab, combined with gemcitabine in locally advanced and metastatic KRAS WT PDAC [Citation107]. The primary endpoint was OS, and 480 patients were randomized. The mOS was 10.9 months (95% CI, 5.6 to 16.3) in the nimotuzumab-gemcitabine group and 8.5 months (95% CI, 5.7 to 10.0) in the placebo-gemcitabine group (Cox proportional HR, 0.66; 95% CI, 0.42 to 1.05; log-rank p = 0.08). PFS was longer in the nimotuzumab-gemcitabine group than in the placebo-gemcitabine group, with median survival time of 4.2 months (95% CI, 2.7 to 7.3) versus 3.6 months (95% CI, 2.0 to 5.0; HR, 0.60; 95% CI, 0.37 to 0.99; log-rank p = 0.04), respectively. Overall, these data suggest utility to targeting EGFR in KRAS WT PDAC.
5.4. BRAF/MEK/ERK
One of the primary downstream signaling pathways of RAS is composed of BRAF, MEK, and ERK [Citation108]. Together RAS-RAF-MEK-ERK comprises the MAPK (mitogen activated protein kinase) pathway. BRAF mutations occur in 2–3% of PDAC with BRAF V600E as the predominant allele [Citation109]. Multiple case reports have demonstrated responses to MAPK pathway inhibition exceeding 6-month duration in PDAC harboring BRAF mutations [Citation110–112]. Additionally, a retrospective case series of BRAF-mutated PDAC showed clinical benefit from BRAF/MEK/ERK inhibitors in three of three patients within the V600E subgroup (two PRs), four of six with fusions (two PRs), two of ix with Exon 11 mutations (one PR), and 0 of 3 with confounding drivers [Citation109,Citation113]. Clinical trials of MAPK inhibition in PDAC include an ongoing, phase II multicenter, single-arm clinical trial investigating the combination of BRAF inhibitor encorafenib and the MEK inhibitor binimetinib in advanced BRAF V600E-mutated PDAC with ORR as the primary endpoint (NCT04390243). The results of two large basket trials of BRAF V600E mutated solid tumors, the Rare Oncology Agnostic Research (ROAR) and National Cancer Institute-Molecular Analysis for Therapy Choice (NCI-MATCH) supported the tissue-agnostic FDA approval of combination dabrafenib and trametinib for BRAF V600E-positive solid tumors [Citation114,Citation115]. Selected ongoing trials of MAPK inhibition in PDAC are outlined in .
5.5. RET
RET fusions or alterations occur in 0.6–1.3% of PDAC and 1.35% of KRAS WT disease [Citation101,Citation102,Citation116]. The LIBRETTO-001 trial was an open-label, phase I/II, basket trial assessing the ORR of RET fusion – containing malignancies treated with the RET inhibitor selpercatinib [Citation117]. Twelve patients with mPDAC where enrolled for which an ORR of 54.5% (95% CI, 23.4 to 83.3) was observed. Selpercatinib was granted accelerated tissue-agnostic approval for patients with solid tumors harboring a RET fusion by the FDA [Citation118]. The ARROW study is a multi-cohort, open-label, phase I/II study investigating pralsetinib in RET-altered solid tumors, and included four patients with KRAS WT PDAC [Citation119]. Responses were seen in all four patients with PDAC (including an ongoing CR with three treatment duration of 33.1 months) [Citation119]. There are several ongoing trials of selpercatinib, pralsetinib and next-generation RET inhibitors (EP0031 and KL590586) in patients with solid tumors (NCT05443126, NCT05265091, and NCT05265091).
5.6. NRG1
Neuregulin-1 (NRG1) fusions are rare in PDAC, occurring in 0.6–1.3% of unselected patients although in up to 17% of patients with KRAS WT PDAC [Citation101]. These fusions cause near-constitutive activation of human epidermal growth factor receptor (HER) 3 and promote tumor proliferation through downstream activation of the MAPK and PI3K pathways [Citation120]. An ongoing phase II, multicenter trial is investigating zenocutuzumab in solid tumors harboring NRG1 fusions including PDAC (NCT02912949). Preliminary data presented at the ESMO annual meeting in 2023 outlined that 38 patients with PDAC harboring NRG1 fusions had received zenocutuzumab, with efficacy assessed in 27 patients [Citation121]. The ORR was 44% (95% CI, 26–65), with 1 CR and 11 PRs. Most (22 of 27; 81%) patients had target lesion reduction and 16/19 (84%) patients had CA 19–9 decline ≥ 50% from baseline. Median duration of response (DOR) was 9.1 months with responses ongoing in 4/12 (33%) patients. The FDA granted breakthrough therapy designation for zenocutuzumab in PDAC with NRG1 fusions in June 2023. Preliminary data for the ongoing CRESTONE phase II trial investigating seribantumab presented at the ASCO 2022 annual meeting showed ORR of 30% (3/10) and a DCR of 90% (1 complete response (CR), 2 PRs, 6 (SD), 1 progressive disease (POD)) in mPDAC [Citation122]. Additional phase I/II trials investigating the anti-HER3 monoclonal antibody HMBD-001 in solid tumors with NRG1 fusions including PDAC are ongoing (NCT05919537).
5.7. NTRK/ROS1
The neurotrophic tropomyosin kinase receptors (NTRKs) are a family of transmembrane tyrosine kinases responsible for neuronal development [Citation123]. Activating NTRK fusions occurs in < 0.5% of all PDAC [Citation124–126]. The RTK inhibitor larotrectinib received accelerated FDA approval for the treatment of advanced solid tumors harboring a NTRK fusion in November 2018 following the results of a basket study [Citation127]. The STARTRK-1 (NCT02097810), ALKA-372-001 (NCT02097810) and STARTRK-2 (NCT02568267) resulted in the August 2019 FDA approval of entrectinib, an oral pan-TRK, ROS1, and ALK inhibitor, in patients with advanced solid malignancies harboring molecular aberrations in NTRK1/2/3, ROS1, or ALK. The STARTRK-2 is ongoing and preliminary data released at the ASCO 2022 annual meeting showed that four patients with PDAC had enrolled [Citation128].
5.8. PRMT5/CDKN2A/MAT2A axis
A novel synthetic lethality strategy is emerging in methylthioadenosine phosphorylase (MTAP) deficient PDAC. The MTAP gene is located at chromosome 9p21 and deletion has been observed in 15–20% of PDAC [Citation129]. The MTAP protein functions to salvage adenine and methionine from methylthioadenosine (MTA), a byproduct of the polyamine pathway [Citation129]. MTA accumulates in MTAP-deficient cells and acts as an endogenous inhibitor of protein arginine methyltransferases 5 (PRMT5), which has critical roles in diverse biological processes through regulation of histone methylation [Citation130]. Notably, MTAP-deficient tumor cells exhibit increased sensitivity to inhibition of PRMT5 and related protein cyclin-dependent kinase inhibitor 2A (CDKN2A) and methionine adenosyltransferase 2A (MAT2A) [Citation131]. Organoid models have demonstrated activity of the PRMT5 inhibitor EZP015556 in both MTAP-deficient and a subset of MTAP-proficient cells with elevated MTA levels [Citation132]. Initial results from a first-in-human study of AMG-193, an MTA-cooperative PRMT5 inhibitor, were presented at the 2023 AACR-NCI-EORTC (American Association for Cancer Research, National Cancer Institute, European Organization for Research and Treatment of Cancer) conference (NCT05094336) [Citation133]. This phase I trial enrolled 48 patients with advanced solid tumors with MTAP/CDKN2A loss including 10 patients with PDAC. Five patients had a radiology confirmed PR by RECIST v1.1, including one patient with PDAC.
5.9. Claudin 18.2
Claudin 18.2 (CLDN18.2) is a tight junction protein that is overexpressed in gastric cancer and PDAC [Citation134,Citation135]. An ongoing phase II study is investigating the efficacy and safety of zolbetuximab, a monoclonal antibody against CLDN18.2, in combination with GnP as first-line therapy for CLDN18.2 positive mPDAC (NCT03816163). Additional CLDN18.2 targeted therapies for PDAC in phase I/II clinical trials include the anti-CLDN18.2/CD47 bispecific antibody PT886 (NCT05482893) and chimeric antigen receptor therapy (CAR-T) (NCT04404595, NCT05620732, NCT05472857, and NCT05539430).
6. Immunotherapy
6.1. Immune checkpoint blockade
DNA mismatch repair (MMR) corrects inappropriate nucleotide insertions, deletions, and single nucleotide mismatched incorporations [Citation136–138]. The primary genes involved in MMR are MutL protein homolog 1 (MLH-1), PMS1 homolog 2 (PMS-2), MutS homolog 2 (MSH-2) and MutS homolog 6 (MSH-6) [Citation139]. Germline or somatic mutation of these genes causing MMR deficiency (dMMR) are associated with both sporadic malignancies and hereditary cancer syndromes. Loss of MMR leads to expansion DNA microsatellites, producing a state of microsatellite instability (MSI) [Citation139]. MSI disrupts protein synthesis by inducing transcriptional frameshifts, thereby increasing neoantigen formation, which likely contributes to the efficacy of immunotherapy in dMMR/MSI-H tumors [Citation140].
Immune checkpoints protect from autoimmunity but are also exploited by malignant cells to evade immune detection. ICB includes therapeutic monoclonal antibodies against immune checkpoints such as programmed cell death 1 (PD1) and cytotoxic T-lymphocyte – associated antigen 4 (CTLA4). Despite encouraging results from other solid tumor malignancies, the role ICB currently remains limited in PDAC following a series of initial trials of both ICB monotherapy and combination therapy with anti-PD1 (durvalumab) plus anti-CTLA4 (tremelimumab) [Citation141,Citation142]. The NCCN guidelines currently recommend pembrolizumab for mPDAC that is dMMR, MSI-H or with a tumor mutational burden (TMB) >10 mut/Mb [Citation16]. Dostarlimab received tissue-agonistic FDA approval for dMMR solid tumors in 2021 [Citation143]. The focus of clinical trials has since shifted to investigating ICB in combination with other therapies such as PARP inhibitors, personalized neoantigen and public neoantigen vaccines, and adoptive cell transfer and modulators of the TME.
6.2. Personalized neoantigen vaccination approaches in PDAC
Personalized neoantigen vaccines (PNV) are being investigated in combination with ICB with a rationale that concurrent ICB administration may enhance vaccine response. In a PDAC adjuvant setting, a phase I trial of autogene cevumeran, an individualized mRNA neoantigen vaccine, was conducted in patients with resectable PDAC [Citation144]. Atezolizumab (anti-PDL1) was administered 6 weeks post-operatively, followed by nine doses of autogene cevumeran and standard adjuvant mFOLFIRINOX followed by a solitary ‘booster’ vaccine on completion of SOC therapy. high-magnitude neoantigen-specific T cells in occurred in 50% of patients (8/16) with half targeting more than one vaccine neoantigen. At median follow-up of 18 months, evaluable vaccine responders (n = 8) had longer recurrence-free survival versus non-responders (n = 8) (median not reached versus 13.7 months; HR 0.08, 95% CI, 0.01–0.4; p = 0.003). A randomized, phase II trial evaluating mFOLFIRINOX with or without autogene cevumeran (IMCODE003) in resected PDAC is actively recruiting (NCT05968326). In the metastatic setting, there is an ongoing Phase Ia/Ib dose-escalation study evaluating autogene cevumeran as a single agent and in combination with atezolizumab in locally advanced or mPDAC (NCT03289962).
The GVAX (granulocyte-macrophage colony-stimulating factor vaccine) is long-studied in PDAC and is currently under investigation in combination with urelumab (an anti-CD137 agonist) and nivolumab in patients with resectable PDAC (NCT02451982) [Citation145,Citation146]. Additional selected trials of GVAX are outlined in .
Table 4. Selected ongoing Trials in pancreatic cancer investigating vaccines, adoptive cell Transfer and tumor microenvironment.
Dendritic cell vaccines (DCVs) represent another vaccine modality in clinical trials for PDAC following initial reports of clinical benefit in NSCLC (NCT03329248) [Citation147]. A Phase Ib trial evaluating an autologous DCV loaded with Personalized Peptides (PEP) in combination with standard of care adjuvant chemotherapy and nivolumab is underway in patients with PDAC (NCT04627246). Another single arm open-label trial assessing the safety and tolerability of adjuvant mature DC (mDC3/8) vaccine (primer and booster) in the adjuvant setting for resected PDAC is underway (NCT03592888).
In summary, multiple-vaccine strategies are being investigated in a minimal residual disease state in PDAC where utility is likely to be greatest given highest potential for functional immune system, minimal tumor burden, preservation of human leucocyte antigen (HLA) expression and other factors. The combination of vaccines with other immune modulating therapies may enhance vaccine responses and efficacy.
6.3. Adoptive cell transfer
There continues to be significant interest in adoptive cellular therapy with chimeric antigen receptor T cells (CAT-T) and T cell receptor therapy (TCR-T) in PDAC. Both approaches aim to generate cellular and humoral immunity against selected tumor-associated proteins such as KRAS mutant alleles (G12D, G12V), CD133, CD137, EGFR, or mesothelin [Citation148]. A phase I trial of anti-CD133, a cancer stem cell marker highly expressed in PDAC, CAR-T included seven patients with mPDAC and showed that three individuals achieved SD, 2 PR, and 2 patients with POD [Citation149]. There are multiple ongoing CAR-T trials in PDAC with target antigens including KRAS, CD70 (NCT02830724), CEA (NCT04037241) and others (NCT03323944) as selected and outlined in .
Encouraging data is emerging for TCR therapy in highly select patients with PDAC. A case report of autologous T-cell therapy expressing HLA-C * 08:02–restricted TCRs targeting KRAS G12D indicated efficacy in two patients with chemotherapy-refractory PDAC [Citation150]. Pre-conditioning with tocilizumab and cyclophosphamide followed by TCR therapy and high-dose interleukin-2 post-infusion, resulted in an initial response in both patients and one maintained a durable PR lasting more than 6 months [Citation150]. There are several ongoing phase I/II clinical trials evaluating TCR therapy directed against KRAS G12V (NCT04146298, NCT03190941), KRAS G12D (NCT03745326), TP53 (NCT05877599) and mesothelin (NCT04809766) in PDAC as selected and outlined in .
7. Modulating the tumor microenvironment
The PDAC TME supports tumor growth while facilitating primary and adaptive resistance to cytotoxic chemotherapies, targeted therapies and immune-modulating treatments [Citation151]. The PDAC TME is a complex, desmoplastic, and heterogenous network composed of extra-cellular and cellular components [Citation152,Citation153]. The extracellular matrix consists of hyaluronic acid, collagens, fibronectin, laminin, and proteoglycans, among others. Cellular components include abundant cancer associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), tumor-associated neutrophils (TANs) and regulatory T cells [Citation154]. These cells produce an immunosuppressive milieu and support tumor growth through the secretion of chemokines and growth factors [Citation155]. Infiltration of antigen presenting cells (APCs) or CD8+ T cells is limited [Citation156,Citation157].
CCR2 interacts primarily with the multifunctional ligand CCL2 and functions to recruit monocytes to the TME [Citation158]. In mouse models of PDAC, CCR2 inhibition reduced TME infiltration of TAMs, reduced tumor progression, and enhanced the effectiveness of anti-PD-1 therapy [Citation158,Citation159]. In early clinical trials, combinations of CCR2 inhibitors, PF-04136309 and CCX872, with standard chemotherapy showed mixed outcomes in PDAC patients [Citation160–162]. A phase Ib study combining PF-04136309 and GnP highlighted the potential of pulmonary toxicity and did not exhibit a clear efficacy signal over GnP alone [Citation162]. However, more promising safety data emerged from combination of PF-04136409 and CCX872 with FOLFIRINOX [Citation163]. A phase Ib/II trial evaluating BMS-813160, a small-molecule CCR2 and CCR5 antagonist, as monotherapy or in combination with chemotherapy or nivolumab in advanced PDAC or colorectal cancer is ongoing (NCT03184870).
CD40 is a transmembrane receptor of the tumor necrosis factor (TNF) super-family expressed on APCs including dendritic cells, macrophages, and B cells. CD40 facilitates CD8+ T cell activation and effector functions [Citation164]. In one phase Ib study, sotigalimab (APX005M, CD40 agonist), was combined with chemotherapy and nivolumab for mPDAC [Citation165]. Among 24 patients, the ORR was 58% and mPFS was 11.7 months [Citation165]. The subsequent PRINCE trial was a randomized, multicenter, phase II trial of 105 patients with mPDAC treated with GnP with or without sotigalimab and with or without nivolumab [Citation165]. The primary endpoint of 1-year OS was met for nivolumab/GnP (57.7%, p = 0.006 compared to historical 1-year OS of 35%, n = 34) but was not met for sotigalimab/GnP (48.1%, p = 0.062, n = 36) or sotigalimab/nivolumab/GnP (41.3%, p = 0.223, n = 35). Further investigation of early immune and correlative signatures is underway in the GnP and immunotherapy REVOLUTION platform trial (NCT04787991).
The CSF-1 R is a member of the receptor protein tyrosine kinase family and is expressed by TAMs and MDSCs. Inhibition of CSF-1 R in mouse models reduced TAMs and increased CD8+:CD4+ T cell ratio in the TME [Citation166]. In PDAC models, CSF-1 R inhibition increased PD-L1 and CTLA-4 expression and improved response to PD-L1 and CTLA-1 directed therapies [Citation167].
8. Conclusion
The incidence of PDAC continues to increase and outcomes are wanting, highlighting the urgent need for more effective therapies across the neoadjuvant, adjuvant, and metastatic setting. Although cytotoxic chemotherapy remains the mainstay systemic therapy for PDAC with choice of regimen based primarily on performance status, biomarker, and targeted therapy era is firmly underway. This review outlines a forward-looking perspective on promising avenues of targeted therapy research and development in PDAC with a focus of KRAS-targeted therapies, DNA damage repair therapies and immunotherapies. The domain of KRAS-targeted therapies has seen the convergence of work on several treatment modalities and holds notable promise of yielding practice changing findings for a notable proportion of patients with PDAC. The role of ICB as part of combination therapies may enhance response to vaccines and TME modulating agents.
9. Expert opinion
The treatment landscape for PDAC is undergoing a paradigm shift toward an era of biomarker-based treatment selection and targeted therapies. Whereas past treatment selection was guided primarily by clinical factors, an increasing array of biomarker-based targeted therapeutic options are emerging and endorsed by NCCN guidelines including olaparib (BRCA1/2 mutated), pembrolizumab (MSI-H/dMMR/TMB-H), and therapies for rare fusions (NTRK, RET) and gene re-arrangements.
A notable focus has been on targeting mutant KRAS. Multiple developing therapeutic modalities including small-molecule enzyme inhibitors, siRNA, and vaccines have converged on this target. The high prevalence of KRAS mutations in PDAC, coupled with their central pathogenic role, underscores the potential for effective novel therapies to provide meaningful benefit for patients with PDAC across the neoadjuvant, adjuvant, and metastatic setting. Current challenges with KRAS-targeted therapeutics encompass the emergence of both de novo and acquired resistance. The latter may be overcome by combining KRAS-targeted modalities or by simultaneously targeting KRAS and upstream (SHP2 and SOS1) or downstream (RAF-MEK-ERK) pathways.
The identification of novel therapeutic targets such as claudin 18.2, and PRMT5 in MAT2A or CDKN2A deficient PDAC has yielded responses in early clinical trials, providing the impetus for further clinical evaluation and the potential for regulatory approval. As 15–20% of PDAC cases are MTAP-deficient, effective novel therapies targeting the PRMT5-CDKN2A-MAT2A axis could provide important additional therapeutic options for a considerable number of patients with PDAC.
Although initial clinical trials of ICB monotherapy in PDAC yielded limited benefit compared to other solid tumor malignancies – pembrolizumab has received regulatory approval for mPDAC that is dMMR, MSI-H or TMB > 10 mut/Mb -there are emerging data to support the role of ICB in combination strategies for PDAC, particularly when paired with PARP inhibitors, vaccines, or TME-directed therapies. Furthermore, combining ICB with therapies that mitigate the immunosuppressive nature of the PDAC TME may enhance responsiveness to these agents. The TME plays a central role in the recalcitrance of PDAC to systemic therapies including chemotherapy and immunotherapy. Effective TME-targeted therapies may serve as an important adjuvant to preexisting and future cytotoxic chemotherapy regimens or targeted therapies.
PDAC tumor subtype-profiling into classical and basal-like is anticipated to have a developing role in treatment selection and clinical trial design. Selection for first-line cytotoxic chemotherapy in mPDAC may be guided by the findings that classical subtype response is more favorable to FOLFIRINOX over GnP. The identification of subtype surrogate markers such as GATA6 expression and development of signature tools such as PurIST may streamline and make the subtyping process more widely available.
Although extensive discussion of this topic is beyond the scope of this review, it is worth briefly mentioning the importance of developing effective screening strategies for PDAC. This is crucial as most cases of PDAC are diagnosed at advanced stages when the disease is locally advanced, inoperable and/or metastatic. Early diagnosis enhances patient potential for surgical resection and significantly improves prognosis. The Pancreatic Cancer Early Detection (PRECEDE) Consortium is an observational prospective cohort study investigating novel early detection strategies by correlating clinical outcomes with 6–12 monthly biospecimen collection in defined high-risk groups (NCT04970056). The aims of the PRECEDE study include identifying individuals at high-risk of PDAC along with genetic and other biomarkers that increase risk. This is with a view to developing novel screening techniques and protocols. Targeted screening of high-risk individuals may prove a time and resource-efficient initial approach to screening and earlier detection of PDAC in the general population.
Finally, it is imperative to maximize enrollment of patients with PDAC in clinical trials given the low incidence of this disease and the medical exigency. Clinical trial national networks and clinical trials cooperative groups facilitate patient recruitment on a scale beyond what is typically achievable for a single institution. A continuing emphasis on quality control and standardization spanning the pre-analytical, analytical, and post-analytical aspects of clinical trial design and execution is essential across collaborating clinical trial sites. The harmonization of standards improves multi-center clinical trials enhance efficiency and reliability. The future success of clinical and translational research concerning novel therapies for PDAC will also depend on an ongoing national and international collaboration between academia, industry, and governmental and regulatory entities.
Article highlights
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy that poses unique therapeutic challenges.
Cytotoxic chemotherapy remains the mainstay of treatment for most patients with this disease. An evolving knowledge of PDAC pathogenesis, the tumor microenvironment and immunotherapy are driving a shift toward targeted therapies.
A notable focus has been on targeting mutant KRAS, where multiple therapeutic modalities including small and large-molecule enzyme inhibitors, siRNA, and vaccines have converged on this target, and which hold the potential to advance therapeutic paradigms.
There are emerging data to support the role of immune checkpoint blockade (ICB) in combination with other targeted strategies such as poly-ADP-ribose polymerase (PARP) inhibitors, vaccines, or tumor microenvironment (TME)-directed therapies in PDAC.
Many novel therapeutic agents are under evaluation, including strategies capitalizing on synthetic lethality through targeting protein arginine methyltransferases 5 (PRMT5) in methylthioadenosine phosphorylase (MTAP)-deficient PDAC, and targeting claudin 18.2.
Refined tumor molecular subtyping systems (classical and basal-like), subtype surrogate markers such as GATA6 expression and development of signature tools such as PurIST, are anticipated to inform future treatment selection and clinical trial design.
Declaration of interests
E O’Reilly declares the following: research funding to institution: Genentech/Roche, BioNTech, AstraZeneca, Arcus, Elicio, Parker Institute, NIH/NCI, Digestive Care. Consulting/DSMB: Arcus, Alligator, Agenus, BioNTech, Ipsen, Merck, Novartis, Syros, Leap Therapeutics, Astellas, BMS, Fibrogen, Revolution Medicine, Merus, Agios (spouse), Genentech-Roche (spouse), Eisai (spouse) Servier (Spouse).The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Society AC. Cancer facts and figures 2023. American Cancer Society. 2023.
- Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. doi: 10.1158/0008-5472.CAN-14-0155
- Owens DK, Davidson KW, Krist AH, et al. Screening for pancreatic cancer. JAMA. 2019;322(5):438. doi: 10.1001/jama.2019.10232
- Bosetti C, Lucenteforte E, Silverman DT, et al. Cigarette smoking and pancreatic cancer: an analysis from the International pancreatic cancer Case-Control Consortium (Panc4). Ann Oncol. 2012;23(7):1880–1888. doi: 10.1093/annonc/mdr541
- Salem ME, El-Refai SM, Sha W, et al. Landscape of KRAS G12C , associated genomic alterations, and interrelation with immuno-oncology biomarkers in KRAS -mutated cancers. JCO Precision Oncol. 2022;(6):6. doi: 10.1200/PO.21.00245
- Blackford A, Parmigiani G, Kensler TW, et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 2009 Apr 15;69(8):3681–3688. doi: 10.1158/0008-5472.CAN-09-0015
- Wang Y-T, Gou Y-W, Jin W-W, et al. Association between alcohol intake and the risk of pancreatic cancer: a dose–response meta-analysis of cohort studies. BMC Cancer. 2016;16(1). doi: 10.1186/s12885-016-2241-1
- Genkinger JM, Spiegelman D, Anderson KE, et al. Alcohol intake and pancreatic cancer risk: a pooled analysis of fourteen cohort studies. Cancer Epidemiol Biomarkers Prev. 2009 Mar;18(3):765–776.
- Sung H, Siegel RL, Rosenberg PS, et al. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health. 2019;4(3):e137–e147. doi: 10.1016/S2468-2667(18)30267-6
- Johansen D, Stocks T, Jonsson H, et al. Metabolic factors and the risk of pancreatic cancer: a prospective analysis of almost 580,000 men and women in the metabolic syndrome and cancer project. Cancer Epidemiol Biomarkers Prev. 2010;19(9):2307–2317. doi: 10.1158/1055-9965.EPI-10-0234
- Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10(1):10–27. doi: 10.14740/wjon1166
- Ben-Aharon I, van Laarhoven HWM, Fontana E, et al. Early-onset cancer in the gastrointestinal tract is on the rise—evidence and implications. Cancer Discovery. 2023;13(3):538–551. doi: 10.1158/2159-8290.CD-22-1038
- Cancer Facts & Figures 2023. American Cancer Society. 2023;23.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2021. Ca A Cancer J Clinicians. 2021;71(1):7–33. doi: 10.3322/caac.21654
- Sohal DPS, Kennedy EB, Cinar P, et al. Metastatic pancreatic cancer: ASCO guideline update. J Clin Oncol. 2020;38(27):3217–3230. doi: 10.1200/JCO.20.01364
- NCCN Clinical Practice Guidelines in Oncology, Pancreatic Adenocarcinoma (Version 2. 2023). 2023 June 19, [cited 2023 Oct 2]. Available from: https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf
- Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med. 2011;364(19):1817–1825. doi: 10.1056/NEJMoa1011923
- Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-Paclitaxel plus Gemcitabine. N Engl J Med. 2013;369(18):1691–1703. doi: 10.1056/NEJMoa1304369
- Macarulla T, Pazo-Cid R, Guillén-Ponce C, et al. Phase I/II trial to evaluate the efficacy and safety of nanoparticle albumin-bound paclitaxel in combination with gemcitabine in patients with pancreatic cancer and an ECOG performance status of 2. J Clin Oncol. 2019;37(3):230–238. doi: 10.1200/JCO.18.00089
- Wainberg ZA, Melisi D, Macarulla T, et al. NALIRIFOX versus nab-paclitaxel and gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (NAPOLI 3): a randomised, open-label, phase 3 trial. Lancet. 2023;402(10409):1272–1281. doi: 10.1016/S0140-6736(23)01366-1
- Hu ZI, O’Reilly EM. Therapeutic developments in pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2023 Oct 5;21: 7–24.
- Ohba A, Ozaka M, Ogawa G, et al. 1616O Nab-paclitaxel plus gemcitabine versus modified FOLFIRINOX or S-IROX in metastatic or recurrent pancreatic cancer (JCOG1611, GENERATE): A multicentred, randomized, open-label, three-arm, phase II/III trial. Ann Oncol. 2023;34:S894. doi: 10.1016/j.annonc.2023.09.2565
- Carrato A, Pazo-Cid R, Macarulla T, et al. Sequential nab-paclitaxel/gemcitabine followed by modified FOLFOX for first-line metastatic pancreatic cancer: the SEQUENCE trial. J Clin Oncol. 2022;40(16_suppl):4022. doi: 10.1200/JCO.2022.40.16_suppl.4022
- Carrato A, Vieitez JM, Benavides M, et al. Phase I/II trial of sequential treatment of nab-paclitaxel in combination with gemcitabine followed by modified FOLFOX chemotherapy in patients with untreated metastatic exocrine pancreatic cancer: phase I results. Eur J Cancer. 2020 Nov 1;139:51–58.
- Westphalen B, Gaska T, Reichert M, et al. FOOTPATH: a randomized, open-label phase-2 study of liposomal irinotecan + 5-FU and folinic acid (NAPOLI) versus sequential NAPOLI and mFOLFOX6 versus gemcitabine/nab-paclitaxel in treatment-naïve metastatic pancreatic cancer (mPDAC). J Clin Oncol. 2023;41(16_suppl):4021. doi: 10.1200/JCO.2023.41.16_suppl.4021
- Dahan L, Williet N, Le Malicot K, et al. Randomized phase II trial evaluating two sequential treatments in first line of metastatic pancreatic cancer: results of the PANOPTIMOX-PRODIGE 35 trial. J Clin Oncol. 2021;39(29):3242–3250. doi: 10.1200/JCO.20.03329
- Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019;381(4):317–327. doi: 10.1056/NEJMoa1903387
- Kindler HL, Hammel P, Reni M, et al. Overall survival results from the POLO trial: a phase III study of active maintenance olaparib versus placebo for germline BRCA-Mutated metastatic pancreatic cancer. J Clin Oncol. 2022 Dec 1;40(34):3929–3939. doi: 10.1200/JCO.21.01604
- Reiss KA, Mick R, Teitelbaum U, et al. Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum-sensitive advanced pancreatic cancer: a randomised, phase 1b/2 trial. Lancet Oncol. 2022 Aug;23(8):1009–1020. doi: 10.1016/S1470-2045(22)00369-2
- Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142(4):730–733.e9. doi: 10.1053/j.gastro.2011.12.042
- Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169
- SD AC, Forrester K, Martin J, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988 May 20;53(4):549–554. doi: 10.1016/0092-8674(88)90571-5
- Jones SN, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368
- Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011 Apr;17(4):500–503.
- Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016 Mar 01;531(7592):47–52. doi: 10.1038/nature16965
- Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nature Genet. 2015;47(10):1168–1178. doi: 10.1038/ng.3398
- Rashid NU, Peng XL, Jin C, et al. Purity Independent subtyping of tumors (PurIST), a clinically robust, single-sample Classifier for tumor subtyping in pancreatic cancer. Clin Cancer Res. 2020 Jan 1;26(1):82–92. doi: 10.1158/1078-0432.CCR-19-1467
- Aung KL, Fischer SE, Denroche RE, et al. Genomics-driven precision medicine for advanced pancreatic cancer: early results from the COMPASS trial. Clin Cancer Res. 2018;24(6):1344–1354. doi: 10.1158/1078-0432.CCR-17-2994
- O’Kane GM, Grünwald BT, Jang GH, et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in advanced pancreatic cancer. Clin Cancer Res. 2020 Sep 15;26(18):4901–4910. doi: 10.1158/1078-0432.CCR-19-3724
- Knox JJ, Jaffee EM, O’Kane GM, et al. PASS-01: Pancreatic adenocarcinoma signature stratification for treatment–01. J Clin Oncol. 2022;40(4_suppl):TPS635. doi: 10.1200/JCO.2022.40.4_suppl.TPS635
- Tutt A, Ashworth A. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med. 2002 Dec;8(12):571–576. doi: 10.1016/S1471-4914(02)02434-6
- DC VG, Hoeijmakers JHJ, Kanaar R. Chromosomal stability and the DNA double-stranded break connection.Nat Rev Genet. 2001 Mar 1;2(3):196–206. doi: 10.1038/35056049
- Walsh CS. Two decades beyond BRCA1/2: homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol Oncol. 2015 May;137(2):343–350. doi: 10.1016/j.ygyno.2015.02.017
- Shin DS, Chahwan C, Huffman JL, et al. Structure and function of the double-strand break repair machinery. DNA Repair. 2004 Aug;3(8–9):863–873.
- Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002 Jan 25;108(2):171–182. doi: 10.1016/S0092-8674(02)00615-3
- Chen CC, Feng W, Lim PX, et al. Homology-Directed Repair and the Role of BRCA1, BRCA2, and related proteins in genome integrity and cancer. Annu Rev Cancer Biol. 2018 Mar;2(1):313–336.
- Prakash R, Zhang Y, Feng W, et al. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015 Apr 1;7(4):a016600. doi: 10.1101/cshperspect.a016600
- Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994 Oct 7;266(5182):66–71. doi: 10.1126/science.7545954
- Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995 Dec 21-28;378(6559):789–792. doi: 10.1038/378789a0
- Maxwell KN, Domchek SM, Nathanson KL, et al. Population frequency of germline BRCA1/2 mutations. J Clin Oncol. 2016;34(34):4183–4185. doi: 10.1200/JCO.2016.67.0554
- Roa BB, Boyd AA, Volcik K, et al. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet. 1996 Oct;14(2):185–187.
- Hahn SA, Greenhalf B, Ellis I, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003 Feb 5;95(3):214–221. doi: 10.1093/jnci/95.3.214
- Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018 Jun 19;319(23):2401–2409. doi: 10.1001/jama.2018.6228
- Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005 Apr 1;434(7035):917–921. doi: 10.1038/nature03445
- Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Nat Acad Sci. 2011;108(8):3406–3411. doi: 10.1073/pnas.1013715108
- Haber JE. DNA recombination: the replication connection. Trends Biochem Sci. 1999 Jul;24(7):271–275. doi: 10.1016/S0968-0004(99)01413-9
- Ashworth A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break repair. J Clin Oncol. 2008;26(22):3785–3790. doi: 10.1200/JCO.2008.16.0812
- Burki TK. AZD1775 plus chemoradiotherapy for pancreatic cancer. Lancet Oncol. 2019 Sep;20(9):e472. doi: 10.1016/S1470-2045(19)30537-6
- Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011 May 1;17(9):2799–2806. doi: 10.1158/1078-0432.CCR-10-2580
- Cuneo KC, Morgan MA, Sahai V, et al. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J Clin Oncol. 2019 Oct 10;37(29):2643–2650. doi: 10.1200/JCO.19.00730
- Gupta N, Huang T-T, Horibata S, et al. Cell cycle checkpoints and beyond: exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor–resistant cancer. Pharmacol Res. 2022 Apr 1;178:106162. doi: 10.1016/j.phrs.2022.106162
- Fok JHL, Ramos-Montoya A, Vazquez-Chantada M, et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat Commun. 2019 Nov7;10(1):5065. doi: 10.1038/s41467-019-12836-9
- Faivre S, Chan D, Salinas R, et al. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem Pharmacol. 2003 Jul 15;66(2):225–237. doi: 10.1016/S0006-2952(03)00260-0
- Alcindor T, Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr Oncol. 2011 Jan;18(1):18–25. doi: 10.3747/co.v18i1.708
- Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111(6):1132–1138. doi: 10.1038/bjc.2014.418
- Reiss KA, Yu S, Judy R, et al. Retrospective survival analysis of patients with advanced pancreatic ductal adenocarcinoma and germline BRCA or PALB2 mutations. JCO Precision Oncol. 2018;2(2):1–9. doi: 10.1200/PO.17.00152
- Reiss KA, Mick R, O’Hara MH, et al. Phase II study of maintenance rucaparib in patients with Platinum-Sensitive Advanced pancreatic cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J Clin Oncol. 2021;39(22):2497–2505. doi: 10.1200/JCO.21.00003
- Reiss K, Hong S, Kasi A, et al. Apollo: a randomized phase II double-blind study of olaparib versus placebo following curative intent therapy in patients with resected pancreatic cancer and a pathogenic BRCA1, BRCA2, or PALB2 mutation—ECOG-ACRIN EA2192. JCO. 2023 Jun 1;41(16_suppl):TPS4202. doi: 10.1200/JCO.2023.41.16_suppl.TPS4202
- Higuchi T, Flies DB, Marjon NA, et al. CTLA-4 blockade synergizes therapeutically with PARP inhibition in BRCA1-deficient ovarian cancer. Cancer Immunol Res. 2015;3(11):1257–1268. doi: 10.1158/2326-6066.CIR-15-0044
- Balli D, Rech AJ, Stanger BZ, et al. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res. 2017 Jun 15;23(12):3129–3138. doi: 10.1158/1078-0432.CCR-16-2128
- Park W, O’Connor C, Chou JF, et al. Phase 2 trial of pembrolizumab and olaparib (POLAR) maintenance for patients (pts) with metastatic pancreatic cancer (mPDAC): two cohorts B non-core homologous recombination deficiency (HRD) and C exceptional response to platinum-therapy. J Clin Oncol. 2023;41(16_suppl):4140–4140. doi: 10.1200/JCO.2023.41.16_suppl.4140
- Wittinghofer A, Pai EF. The structure of Ras protein: a model for a universal molecular switch. Trends Biochem Sci. 1991 Oct;16(10):382–387. doi: 10.1016/0968-0004(91)90156-P
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003 Jan;3(1):11–22. doi: 10.1038/nrc969
- Bryant KL, Mancias JD, Kimmelman AC, et al. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39(2):91–100. doi: 10.1016/j.tibs.2013.12.004
- Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi: 10.1038/nature11547
- Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020;17(3):153–168. doi: 10.1038/s41575-019-0245-4
- Kim MK, Woo SM, Park B, et al. Prognostic implications of multiplex detection of KRAS mutations in Cell-Free DNA from patients with pancreatic ductal adenocarcinoma. Clin Chem. 2018 Apr;64(4):726–734.
- Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013 Nov 28;503(7477):548–551. doi: 10.1038/nature12796
- McCormick F. Targeting KRAS directly. Annual Rev Cancer Biol. 2018;2(1):81–90. doi: 10.1146/annurev-cancerbio-050216-122010
- Hong DS, Fakih MG, Strickler JH, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020 Sep 24;383(13):1207–1217. doi: 10.1056/NEJMoa1917239
- Strickler JH, Satake H, George TJ, et al. Sotorasib in KRAS p.G12C–mutated advanced pancreatic cancer. N Engl J Med. 2022;388(1):33–43. doi: 10.1056/NEJMoa2208470
- Bekaii-Saab TS, Yaeger R, Spira AI, et al. Adagrasib in advanced solid tumors harboring a KRASG12C mutation. J Clin Oncol. 2023;41(25):4097–4106. doi: 10.1200/JCO.23.00434
- Bannoura SF, Khan HY, Azmi AS. KRAS G12D targeted therapies for pancreatic cancer: has the fortress been conquered? Front Oncol. 2022;12:1013902. doi: 10.3389/fonc.2022.1013902
- Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. 2022 Feb 24;65(4):3123–3133. doi: 10.1021/acs.jmedchem.1c01688
- Mahadevan KK, McAndrews KM, LeBleu VS, et al. KRAS(G12D) inhibition reprograms the microenvironment of early and advanced pancreatic cancer to promote FAS-mediated killing by CD8(+) T cells. Cancer Cell. 2023 Sep 11;41(9):1606–1620.e8. doi: 10.1016/j.ccell.2023.07.002
- Hallin J, Bowcut V, Calinisan A, et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat Med. 2022 Oct;28(10):2171–2182.
- Knox JE, Jiang J, Burnett GL, et al. Abstract 3596: RM-036, a first-in-class, orally-bioavailable, Tri-Complex covalent KRASG12D(ON) inhibitor, drives profound anti-tumor activity in KRASG12D mutant tumor models. Cancer Res. 2022;82(12_Supplement):3596–3596. doi: 10.1158/1538-7445.AM2022-3596
- Mao Z, Xiao H, Shen P, et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022 Jan 25;8(1):5. doi: 10.1038/s41421-021-00368-w
- Punekar SR, Velcheti V, Neel BG, et al. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. 2022 Oct 1;19(10):637–655. doi: 10.1038/s41571-022-00671-9
- Arbour KC, Punekar S, Garrido-Laguna I, et al. 652O preliminary clinical activity of RMC-6236, a first-in-class, RAS-selective, tri-complex RAS-MULTI(ON) inhibitor in patients with KRAS mutant pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC). Ann Oncol. 2023;34:S458. doi: 10.1016/j.annonc.2023.09.1838
- Lito P, Solomon M, Li LS, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016 Feb 5;351(6273):604–608. doi: 10.1126/science.aad6204
- Nichols RJ, Haderk F, Stahlhut C, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol. 2018 Sep;20(9):1064–1073.
- Hofmann MH, Gmachl M, Ramharter J, et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-Driven cancers through combined MEK inhibition. Cancer Discov. 2021 Jan;11(1):142–157.
- Srivastava N, Srivastava PK, Rich B. Modeling the repertoire of true tumor-specific MHC I epitopes in a human tumor. PLoS One. 2009 Jul 10;4(7):e6094. doi: 10.1371/journal.pone.0006094
- Yuan TL, Fellmann C, Lee CS, et al. Development of siRNA payloads to target KRAS-mutant cancer. Cancer Discov. 2014 Oct;4(10):1182–1197.
- Ross SJ, Revenko AS, Hanson LL. et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci Transl Med. 2017 Jun 14;9(394). doi: 10.1126/scitranslmed.aal5253
- Tian Z, Liang G, Cui K, et al. Insight into the prospects for RNAi therapy of cancer. Front Pharmacol. 2021;12:644718. doi: 10.3389/fphar.2021.644718
- Golan T, Khvalevsky EZ, Hubert A, et al. Rnai therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015 Sep 15;6(27):24560–24570. doi: 10.18632/oncotarget.4183
- Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6(1):6744. doi: 10.1038/ncomms7744
- Raphael BJ, Aguirre A, Hruban R, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017 Aug 14;32(2):185–203.e13.
- Philip PA, Azar I, Xiu J, et al. Molecular characterization of KRAS wild-type tumors in patients with pancreatic adenocarcinoma. Clin Cancer Res. 2022 Jun 13;28(12):2704–2714. doi: 10.1158/1078-0432.CCR-21-3581
- Singhi AD, George B, Greenbowe JR, et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas identifies genetic alterations that might Be Targeted with existing drugs or used as biomarkers. Gastroenterology. 2019;156(8):2242–2253.e4. doi: 10.1053/j.gastro.2019.02.037
- Guan M, Bender R, Pishvaian M, et al. Molecular and clinical characterization of BRAF mutations in pancreatic ductal adenocarcinomas (PDACs). JCO. 2018 Feb1;36(4_suppl):214. doi: 10.1200/JCO.2018.36.4_suppl.214
- Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main Driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4(3):e173420. doi: 10.1001/jamaoncol.2017.3420
- McIntyre CA, Lawrence SA, Richards AL, et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer. 2020;126(17):3939–3949. doi: 10.1002/cncr.33038
- Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer Institute of Canada Clinical Trials group. J Clin Oncol. 2007;25(15):1960–1966. doi: 10.1200/JCO.2006.07.9525
- Qin S, Li J, Bai Y, et al. Nimotuzumab plus gemcitabine for K-Ras wild-type locally advanced or metastatic pancreatic cancer. J Clin Oncol. 2023;(0):JCO.22.02630.
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007 May 1;26(22):3291–3310. doi: 10.1038/sj.onc.1210422
- Ciner AT, Jiang Y, Hausner P. BRAF-Driven Pancreatic Cancer: Prevalence, Molecular Features, and Therapeutic Opportunities. Mol Cancer Res. 2023 Apr 1;21(4):293–300. doi: 10.1158/1541-7786.MCR-22-0626
- Busch E, Kreutzfeldt S, Agaimy A, et al. Successful BRAF/MEK inhibition in a patient with BRAF V600E -mutated extrapancreatic acinar cell carcinoma. Cold Spring Harb Mol Case Stud. 2020 Aug;6(4):a005553.
- Li HS, Yang K, Wang Y. Remarkable response of BRAF (V600E)-mutated metastatic pancreatic cancer to BRAF/MEK inhibition: a case report. Gastroenterol Rep. 2022;10:goab031. doi: 10.1093/gastro/goab031
- Sasankan S, Rebuck L, Darrah G, et al. Metastatic pancreatic cancer with BRAF and P53 mutations: case report of therapeutic response to doublet targeted therapy. Case Rep Oncol. 2020 Sep;13(3):1239–1243.
- Hendifar A, Blais EM, Wolpin B, et al. Retrospective case series analysis of RAF family alterations in pancreatic cancer: Real-World Outcomes from targeted and standard therapies. JCO Precision Oncol. 2021;5(5):1325–1338. doi: 10.1200/PO.20.00494
- Subbiah V, Kreitman RJ, Wainberg ZA, et al. Dabrafenib plus trametinib in BRAFV600E-mutated rare cancers: the phase 2 ROAR trial. Nature Med. 2023 May 1;29(5):1103–1112. doi: 10.1038/s41591-023-02321-8
- Gouda MA, Subbiah V. Expanding the Benefit: Dabrafenib/Trametinib as Tissue-Agnostic Therapy for BRAF V600E–Positive Adult and Pediatric Solid Tumors. Am Soc Clin Oncol Educ Book. 2023;43(43):e404770. doi: 10.1200/EDBK_404770
- Kato S, Subbiah V, Marchlik E, et al. RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin Cancer Res. 2017 Apr 15;23(8):1988–1997. doi: 10.1158/1078-0432.CCR-16-1679
- Subbiah V, Wolf J, Konda B, et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): a phase 1/2, open-label, basket trial. Lancet Oncol. 2022 Oct;23(10):1261–1273.
- Duke ES, Bradford D, Marcovitz M, et al. FDA approval summary: selpercatinib for the treatment of advanced RET fusion-positive solid tumors. Clin Cancer Res. 2023 Jun 2;Of1–of6. doi: 10.1158/1078-0432.CCR-23-2268
- Subbiah V, Cassier PA, Siena S, et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat Med. 2022 Aug;28(8):1640–1645.
- Heining C, Horak P, Uhrig S, et al. NRG1 Fusions in KRAS wild-type pancreatic cancer. Cancer Discov. 2018 Sep;8(9):1087–1095.
- Schram A, Macarulla T, Cleary J, et al. 1618P Durable efficacy of zenocutuzumab, a HER2 x HER3 bispecific antibody, in advanced NRG1 fusion positive (NRG1+) pancreatic ductal adenocarcinoma (PDAC). Ann Oncol. 2023;34:S895–S896. doi: 10.1016/j.annonc.2023.09.2567
- Carrizosa DR, Burkard ME, Elamin YY, et al. CRESTONE: initial efficacy and safety of seribantumab in solid tumors harboring NRG1 fusions. J Clin Oncol. 2022;40(16_suppl):3006. doi: 10.1200/JCO.2022.40.16_suppl.3006
- Kaplan DR, Hempstead BL, Martin-Zanca D, et al. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991 Apr 26;252(5005):554–558. doi: 10.1126/science.1850549
- Pishvaian MJ, Garrido-Laguna I, Liu SV, et al. Entrectinib in TRK and ROS1 fusion-positive metastatic pancreatic cancer. JCO Precis Oncol. 2018 Nov;2(2):1–7.
- Okamura R, Boichard A, Kato S, et al. Analysis of NTRK Alterations in Pan-cancer Adult and pediatric malignancies: implications for NTRK-Targeted therapeutics. JCO Precision Oncol. 2018;2(2):1–20. doi: 10.1200/PO.18.00183
- Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33(1):38–46. doi: 10.1038/s41379-019-0324-7
- Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med. 2018 Feb 22;378(8):731–739. doi: 10.1056/NEJMoa1714448
- Krzakowski MJ, Lu S, Cousin S, et al. Updated analysis of the efficacy and safety of entrectinib in patients (pts) with locally advanced/metastatic NTRK fusion-positive (NTRK-fp) solid tumors. J Clin Oncol. 2022;40(16_suppl):3099. doi: 10.1200/JCO.2022.40.16_suppl.3099
- Fan N, Zhang Y, Zou S. Methylthioadenosine phosphorylase deficiency in tumors: a compelling therapeutic target. Front Cell Dev Biol. 2023;11:1173356. doi: 10.3389/fcell.2023.1173356
- Zhu F, Rui L. PRMT5 in gene regulation and hematologic malignancies. Genes Dis. 2019 Sep;6(3):247–257. doi: 10.1016/j.gendis.2019.06.002
- Marjon K, Cameron MJ, Quang P, et al. MTAP Deletions in cancer create vulnerability to targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016 Apr 19;15(3):574–587. doi: 10.1016/j.celrep.2016.03.043
- Driehuis E, van Hoeck A, Moore K, et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc Natl Acad Sci U S A. 2019 Dec 26;116(52):26580–26590. doi: 10.1073/pnas.1911273116
- Rodon J, Doi T, Noboru Y, et al. Editor poster (LB_B14): initial results from first-in-human study of AMG 193, an MTA-Cooperative PRMT5 inhibitor, in Biomarker-Selected Solid Tumors. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics; 2023 2023 Oct 13; Boston, MA, USA.
- Tanaka M, Shibahara J, Fukushima N, et al. Claudin-18 is an early-stage marker of pancreatic carcinogenesis. J Histochem Cytochem. 2011 Oct;59(10):942–952.
- Sahin U, Koslowski M, Dhaene K, et al. Claudin-18 splice variant 2 is a pan-cancer target suitable for therapeutic antibody development. Clin Cancer Res. 2008 Dec 1;14(23):7624–7634. doi: 10.1158/1078-0432.CCR-08-1547
- Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999 Feb;9(1):89–96. doi: 10.1016/S0959-437X(99)80013-6
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74(1):681–710. doi: 10.1146/annurev.biochem.74.082803.133243
- Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65(1):101–133. doi: 10.1146/annurev.bi.65.070196.000533
- Sahin IH, Akce M, Alese O, et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer. 2019 Nov 1;121(10):809–818. doi: 10.1038/s41416-019-0599-y
- Buchler T. Microsatellite instability and metastatic colorectal cancer - a clinical perspective. Front Oncol. 2022;12:888181. doi: 10.3389/fonc.2022.888181
- O’Reilly EM, Oh DY, Dhani N, et al. Durvalumab with or without tremelimumab for patients with metastatic pancreatic ductal adenocarcinoma: a phase 2 randomized clinical trial. JAMA Oncol. 2019 Oct 1;5(10):1431–1438. doi: 10.1001/jamaoncol.2019.1588
- Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010 Oct;33(8):828–833.
- FDA grants accelerated approval to dostarlimab-gxly for dMMR advanced solid tumors. [cited 2023 Oct 2]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors
- Rojas LA, Sethna Z, Soares KC, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144–150. doi: 10.1038/s41586-023-06063-y
- Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccin. 2019 Feb 8;4(1):7. doi: 10.1038/s41541-019-0103-y
- DD MR, Hartley ML, Noel MS. The role of immunotherapy in pancreatic cancer. Curr Oncol. 2022 Sep 23;29(10):6864–6892. doi: 10.3390/curroncol29100541
- Ding Z, Li Q, Zhang R, et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct Ther. 2021 Jan 20;6(1):26. doi: 10.1038/s41392-020-00448-5
- Beatty GL, O’Hara MH, Lacey SF, et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a phase 1 trial. Gastroenterology. 2018 Jul;155(1):29–32.
- Wang Y, Chen M, Wu Z, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7(7):e1440169. doi: 10.1080/2162402X.2018.1440169
- Leidner R, Sanjuan Silva N, Huang H, et al. Neoantigen T-Cell receptor gene therapy in pancreatic cancer. N Engl J Med. 2022;386(22):2112–2119. doi: 10.1056/NEJMoa2119662
- Sherman MH, Beatty GL. Tumor microenvironment in pancreatic cancer pathogenesis and therapeutic resistance. Annu Rev Pathol. 2023;18(1):123–148. doi: 10.1146/annurev-pathmechdis-031621-024600
- Grünwald BT, Devisme A, Andrieux G, et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell. 2021 Oct 28;184(22):5577–5592.e18. doi: 10.1016/j.cell.2021.09.022
- Park W, Chawla A, O’Reilly EM. Pancreatic Cancer: A Review. JAMA. 2021 Sep 7;326(9):851–862. doi: 10.1001/jama.2021.13027
- Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007 Oct 1;67(19):9518–9527. doi: 10.1158/0008-5472.CAN-07-0175
- Zhang Y, Velez-Delgado A, Mathew E, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2017 Jan;66(1):124–136.
- Hegde S, Krisnawan VE, Herzog BH, et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell. 2020 Mar 16;37(3):289–307.e9. doi: 10.1016/j.ccell.2020.02.008
- Lin JH, Huffman AP, Wattenberg MM. et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J Exp Med. 2020 Aug 3;217(8). doi: 10.1084/jem.20190673
- Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013 Feb 1;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731
- Janson C, Jung H, Ertl L, et al. Abstract 5655: Inhibition of CCR2 potentiates checkpoint inhibitor immunotherapy in murine model of pancreatic cancer. Cancer Res. 2017;77(13_Supplement):5655. doi: 10.1158/1538-7445.AM2017-5655
- Linehan D, Noel MS, Hezel AF, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: correlation with blood monocyte counts. Am Soc Clin Oncol. 2018;36(5_suppl):92. doi: 10.1200/JCO.2018.36.5_suppl.92
- Wang-Gillam A, Nywening TM, Sanford DE, et al. Phase IB study of FOLFIRINOX plus PF-04136309 in patients with borderline resectable and locally advanced pancreatic adenocarcinoma (PC). American Society of Clinical Oncology. 2015.
- Noel M, O’Reilly EM, Wolpin BM, et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2020 Jun;38(3):800–811.
- Nywening TM, Wang-Gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016 May;17(5):651–662.
- Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol. 2009 Oct;21(5):265–272. doi: 10.1016/j.smim.2009.05.010
- O’Hara MH, O’Reilly EM, Varadhachary G, et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol. 2021 Jan;22(1):118–131.
- Ries CH, Cannarile MA, Hoves S, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014 Jun 16;25(6):846–859. doi: 10.1016/j.ccr.2014.05.016
- Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014 Sep 15;74(18):5057–5069. doi: 10.1158/0008-5472.CAN-13-3723