
ABSTRACT
Introduction
Forced vital capacity (FVC) decline is predictive of mortality in patients with idiopathic pulmonary fibrosis (IPF) and has been used as a clinical trial endpoint to define disease progression. How to interpret FVC findings in an individual patient with IPF in the real-world setting amid uncertainty about the measurement accuracy and variability has not been well established.
Areas covered
This review highlights the challenges and limitations of using FVC in the clinic to monitor disease progression in patients with IPF. Spirometry is noninvasive, relatively simple, and inexpensive. FVC measurements provide evidence for trends over time in patients with IPF. When using FVC in the clinic, several important challenges and limitations, including visit-to-visit variability, dependence on patient effort, inconsistent quality control, limitations on accuracy, and the influence of comorbidities and pretest factors, must be considered. Recent studies suggest the potential for home spirometry devices to facilitate more frequent collection of data and perhaps demonstrate more accurate trends.
Expert opinion
Measuring FVC decline in the clinic has an important role in monitoring disease progression in patients with IPF, but additional measures of disease progression should be considered along with FVC to facilitate decision-making about disease management.
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a debilitating, progressive, and fatal fibrotic lung disease [Citation1]. The rate of disease progression in IPF is variable and unpredictable [Citation2]. During the course of IPF progression, patients experience irreversible loss of lung function [Citation1,Citation2].
Forced vital capacity (FVC) is well established as a practical endpoint in clinical trials in patients with IPF to measure disease progression, but FVC measurements are highly variable from visit to visit in individual patients [Citation3–6]. Considerable variability has been observed in FVC measurements across visits in individual patients with IPF from the placebo groups in randomized, controlled clinical trials [Citation3–6]. At the population level in patients with IPF, robust trends for decline in FVC are clearly observed [Citation1,Citation2]. Furthermore, data from many studies in patients with IPF demonstrate that absolute or relative decline in FVC (mL) or % predicted FVC is associated with short-term mortality [Citation7–9]. However, in individual patients, decline in % predicted FVC is a poor predictor of future decline [Citation4,Citation10].
Pirfenidone and nintedanib are antifibrotic therapies that slow disease progression in patients with IPF [Citation11–15]. In Phase III trials, both pirfenidone and nintedanib reduced the rate of decline in FVC in patients with IPF [Citation13,Citation14,Citation15]. These medications were approved by the US Food and Drug Administration in 2014 to treat patients with IPF [Citation11,Citation12].
Despite evidence that patients with IPF will lose lung function over time, many physicians delay initiation of antifibrotic therapy until after disease progression is observed, most commonly as a decline in lung function [Citation16]. Some physicians may stop or switch therapies in an individual patient with detectable disease progression, despite evidence that supports the continued use of antifibrotic therapy in this context [Citation4].
Clear and robust population-level trends of FVC decline in patients with IPF make FVC an appropriate endpoint in clinical trials. However, a better understanding of how to interpret serial FVC findings in an individual patient with IPF is needed. This review, representing clinical experience from pulmonology practice and respiratory therapy, aims to highlight the value as well as the challenges and limitations of using FVC in the clinic to monitor disease progression in individual patients with IPF.
2. Advantages of using FVC in the clinic to monitor disease progression in patients with IPF
2.1. Spirometry
Spirometry is simple, noninvasive, and relatively inexpensive [Citation17]. Properly performed, it readily distinguishes normal from abnormal lung volumes and obstructive from restrictive ventilatory defects. Serial spirometry values provide evidence of disease trends over time. Spirometry also aids in the diagnosis of a range of respiratory diseases, and results are immediately available [Citation17,Citation18].
The long history and widespread use of spirometry to monitor general respiratory health have led to the development of evidence-based standards for best practices [Citation19]. Spirometry was first described by John Hutchinson in 1846, and the timed and forced expiratory maneuver was described by Robert Tiffeneau and Andre Pinelli in 1947 [Citation20,Citation21]. Standardization of spirometry has been an important activity of continued engagement by the American Thoracic Society (ATS) and European Respiratory Society (ERS) for decades [Citation19,Citation22,Citation23,Citation24,Citation25]. The 2019 update of the ATS/ERS standards provides guidance for instrument manufacturers, clinicians, operators, and researchers to ensure quality control and improve patients’ experiences based on available evidence and expert recommendations () [Citation19].
Table 1. Summary of recommendations for quality assurance of spirometry [Citation19]
The pathophysiology of IPF involves the overproduction of extracellular matrix in the lung. This lays the foundation for progressive fibrosis with the loss of lung units and their associated contribution to the FVC. Extracellular matrix deposition results in stiffened and less compliant lung tissue, which cannot expand to the same extent during inspiration as a normal healthy lung; this further contributes to the characteristic decline in FVC in patients with IPF [Citation26].
Widespread experience with spirometry among clinicians has established decline in % predicted FVC as a practical method to monitor disease progression in patients with IPF. Other measures of disease progression, including diffusing capacity of the lung for carbon monoxide (DLco), 6-minute walk distance (6MWD), and high-resolution computed tomography (HRCT), are also used, albeit less frequently than FVC [Citation1,Citation27,Citation28].
2.2. Home-based spirometry
Recent ongoing work has explored the potential role for home spirometry to monitor disease progression in patients with IPF and in patients with unclassifiable interstitial lung disease (ILD) [Citation29–33]. In complement with in-clinic measurements, home-based spirometry would allow more frequent measurements that could promote early detection of changes in disease status in individual patients. Small single-center studies found adherence with home spirometry and high correlations with in-clinic measurements, suggesting that home spirometry may be useful in patients with IPF () [Citation29,Citation30,Citation31]. However, in large multicenter studies of IPF and unclassifiable ILD, poor adherence and technical issues demonstrated the need for further studies to determine how to best facilitate home spirometry with easy-to-use and reliable home devices. This may entail more robust software, improved patient training, and when necessary direct patient observation, albeit remotely () [Citation32,Citation33].
Table 2. Studies of home spirometry in patients with IPF and unclassifiable ILD
3. Challenges and limitations of using FVC in the clinic to monitor disease progression in patients with IPF
3.1. Visit-to-visit variability in FVC measurements over time
Visit-to-visit variability in FVC measurements is well known to clinicians and can make interpreting results challenging in an individual patient [Citation3–6]. Notably, a post hoc analysis of pooled data from 4 clinical trials in 954 patients with IPF found that 41% of 3-month changes in FVC (mL) were stable or improvements (absolute change ≥ 0 mL), including 7.3% of improvements ≥ 200 mL [Citation6]. However, over the same period, 59% of changes in FVC were declines, including 14.2% < −200 mL () [Citation5]. The most frequently recorded relative 3-month changes in FVC (mL) were ≥ −5% to < 0% (31.7% of intervals), ≥ 0% to ≤ 5% (27.7%), and ≥ −10% to < −5% (17.5%) () [Citation5]. However, a relative decline in FVC (mL) over 3 months was a poor predictor of further decline, as many patients had a relative increase in FVC (mL) at their next 3-month visit after an initial 3-month relative decline [Citation4,Citation6]. These findings demonstrate high intra patient visit-to-visit variability in FVC measurements over time.
Figure 1. Distribution of absolute (a) and relative (b) changes in FVC (mL) over 3-month intervals in patients with IPF enrolled in randomized, controlled trials [Citation5]. Histograms depict FVC changes over 3966 intervals of 3 months in 954 patients with IPF. The analysis population comprised 624 patients randomized to receive placebo in ASCEND (Study 016; NCT01366209) and CAPACITY (Studies 004 and 006; NCT00287716 and NCT00287729) and 330 patients randomized to receive interferon γ-1b or placebo in GIPF-001 (NCT00047645). FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis
![Figure 1. Distribution of absolute (a) and relative (b) changes in FVC (mL) over 3-month intervals in patients with IPF enrolled in randomized, controlled trials [Citation5]. Histograms depict FVC changes over 3966 intervals of 3 months in 954 patients with IPF. The analysis population comprised 624 patients randomized to receive placebo in ASCEND (Study 016; NCT01366209) and CAPACITY (Studies 004 and 006; NCT00287716 and NCT00287729) and 330 patients randomized to receive interferon γ-1b or placebo in GIPF-001 (NCT00047645). FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis](/cms/asset/7d6a479e-5966-4f53-adb8-d4c4d91e6297/ierx_a_1816831_f0001_oc.jpg)
3.2. Factors impacting spirometry results
A number of factors can impact spirometry results from one visit to the next, including the equipment, the technician, patient effort, test performance, result selection, number of measurements, change in disease severity, and comorbid conditions [Citation19,Citation24]. The dependence of test results on patient effort underscores the need for appropriate training of the patient by the test operator. Coughing during FVC maneuvers occurs frequently in patients with IPF and renders the test results unusable. Fatigue during repeated forced expiratory maneuvers is also common, especially in patients with significant cough or those who require multiple attempts to achieve acceptable and repeatable data. The use of different spirometers can also cause variability, and it is not unusual for different well-trained technicians to obtain differing results. Comparing FVC results from different pulmonary function laboratories is therefore potentially risky, particularly when trying to interpret changes or trends in FVC over time. Adherence to published standards is important to minimize variability between laboratories or clinics due to differences in personnel or equipment [Citation19]. Comorbidities, such as cardiovascular disease, in patients with IPF also pose a diagnostic challenge for clinicians [Citation35]. Patients with IPF and comorbid heart failure can be misdiagnosed, as these conditions share several symptoms. Diagnostic findings shared by both conditions include impaired lung function on spirometry (reduced FVC and other lung volumes) and reduced exercise tolerance [Citation35,Citation36]. In addition, some evidence suggests that up to 1 in 10 patients with IPF have co existing reversible airflow limitation. A significant mean intra test increase in FVC was observed with bronchodilator use; however, there was no significant inter-test difference in change in FVC over time between pre- and post-bronchodilator results [Citation37].
3.3. Accuracy and repeatability of spirometry measurements
Quality control issues are critically important in the accurate and repeatable measurement of FVC, and all pulmonary function laboratories should adopt and follow the most recent professional guidelines [Citation19]. A 2016 study of spirometers in use in primary care offices in the Salt Lake City metropolitan area found that only 1 of 17 spirometers met the 2005 ATS/ERS accuracy criteria [Citation38]. The implications of inaccurate measurements of FVC for patient care may be profound. When antifibrotic therapies were in Phase II and III clinical development, the 2005 ATS/ERS standards were in effect and accuracy of spirometry was considered to be ± 3.5% [Citation24]. Current ATS/ERS spirometry standards recommend that equipment should adhere to the 2016 International Organization for Standardization 26782 standards, with a permissible error of 2.5% [Citation19,Citation39]. Thus, with the estimated annual FVC decline in patients with IPF of approximately 150–200 mL, an inaccuracy of up to 2.5% may be sizable [Citation2]. Furthermore, the minimal clinically important difference in % predicted FVC in patients with IPF is 2–6%, placing the lower range very near the accuracy limit of spirometers [Citation40]. In addition to inaccuracy of single measurements, repeatability of results is also affected by the variability in FVC measurements over time. Findings from an analysis of data from three Phase III trials showed significant intra and inter patient variability in magnitude and direction of change [Citation4].
Concurrent comorbidities and changes in a patient’s health can influence spirometry results. For example, concomitant emphysema and IPF can result in preservation of lung volumes and an FVC that might not reflect the severity of disease or demonstrate progression over time [Citation41,Citation42,Citation43]. Weight loss may improve FVC, as demonstrated in a study of men with obesity who experienced a substantial reduction in body weight [Citation44]. However, the impact of weight loss on FVC in patients with IPF has not been carefully investigated. Age-related FVC decline (≈ 30–35 mL/year in healthy adults aged 60–80 years), while notably less than FVC decline due to IPF progression, should also be considered in interpreting serial FVC results [Citation45].
In clinical experience, some patients with IPF cannot perform the recommended number of acceptable FVC maneuvers per the ATS/ERS criteria [Citation19] and cannot provide repeatable FVC values. In these patients, disease progression cannot be monitored with FVC measurements alone. Furthermore, although FVC maneuvers may not meet acceptability criteria, they may still be usable, with appropriate notes added to the spirometry report [Citation19]. The slow vital capacity [SVC] maneuver, which requires less patient effort, might be useful as a secondary measure of lung function in patients with IPF who have difficulty with the FVC maneuver [Citation19], including cough. However, there is little experience with SVC measurement in patients with IPF [Citation46]. DLco measurements can also be useful, as progressive, sustained declines in the DLco are associated with disease progression [Citation1]. Changes in 6MWD from one visit to the next might corroborate changes in FVC as evidence of meaningful disease progression [Citation5,Citation6,Citation47,Citation48]. Other measures of disease progression, including increased dyspnea, cough, oxygen desaturation at rest and with walking, or resting oxygen requirements, may also be useful.
4. Conclusions
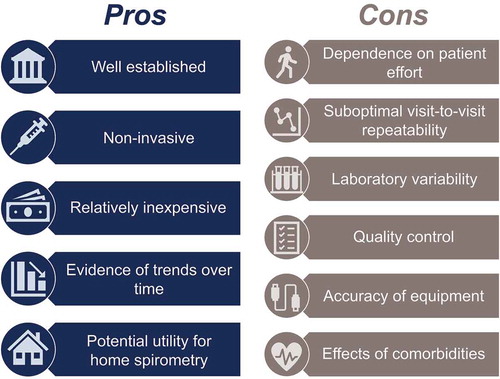
In large IPF clinical trial populations, decline in FVC over ≥ 12 months has clear value in predicting mortality [Citation7,Citation8]. Despite these consistent trends in the IPF population, how an observed change in FVC in an individual patient informs clinical practice decisions has not been well established or fully explored in the literature. This review highlights key strengths and limitations of using FVC in the clinic (). Changes in FVC from one clinic visit to the next may not account for the entirety of changes in a patient’s disease activity, and concurrent changes in other measures of disease activity, such as symptoms or 6MWD, might be useful in validating small changes in FVC. Caution is advised in making decisions to stop or switch antifibrotic therapy based solely on visit-to-visit changes in FVC.
Figure 2. Using FVC in the clinic to monitor patients with IPF: pros and cons. FVC, forced vital capacity; IPF, idiopathic pulmonary fibrosis
5. Expert opinion
Although spirometry remains a valuable lung-function testing tool in patients with IPF, its limitations related to the interpretation of intra patient variability in FVC results over time pose a challenge to its clinical usefulness in the assessment of IPF progression. A more reliable method for assessment of disease progression would enable physicians to make clinical decisions based on clear parameters to guide patient management. This could be achieved through improvements and technological advances in existing spirometry equipment, strict quality control in pulmonary function testing (PFT) laboratories, and ensuring that training of spirometry operators is compliant with the ATS/ERS guidelines. Currently, trying to interpret changes in FVC over time in individual patients by comparing results obtained from different PFT laboratories can be problematic. In addition, the use of different types of spirometers in epidemiological and clinical studies and in clinical practice also poses a challenge, as they may present different findings, which ultimately affects the interpretation of results. Development of a new, comprehensive, more sensitive, and more effective assessment tool and achievement of consensus on how to better integrate FVC with other measurements (such as DLco, 6MWD, and oxygen requirements) into the new composite tool would assist physicians in the accurate assessment of IPF progression. Furthermore, investigation of other possible metrics, such as quantitative HRCT parameters, would be advantageous. Additional research into home spirometry should be encouraged and supported to provide more robust data and eliminate variability caused by unpredictable events that may occur on the day a patient presents to the PFT laboratory. Current evidence shows promising results and good correlations with in-clinic tests; however, research has also identified several factors that can adversely affect clinical utility. Future studies of home spirometry need to focus on addressing technical issues, improving patient adherence, and determining its role in the assessment of IPF progression. In the intermediate to long term, further research into the development of novel technologies (such as portable, user-friendly, home monitoring devices connected to patients’ smartphones and physicians’ devices), with optimized capabilities to account for factors known to influence the variability in FVC results, could provide better insight into a patient’s condition and a more accurate assessment of IPF progression. Several comorbidities and changes in a patient’s health can influence FVC measurements in patients with IPF, so future research could focus on specific patient populations, such as those with heart failure. We know that in untreated patients with IPF, the annual decline in FVC averages 150–250 mL; however, in patients with IPF and comorbidities, this may be different. The findings from such studies could improve the interpretation of serial FVC results in the context of patient comorbidities or other influencing factors and enable a more accurate assessment of disease progression. Furthermore, they would provide guidance for the development of more reliable parameters for measuring disease progression in patients with IPF, which could be integrated into a composite assessment tool to support physicians in their decision-making for and counseling of patients.
Article highlights
Although FVC is well established as a practical endpoint in IPF clinical trials as a measure of disease progression, FVC measurements are highly variable from visit to visit in individual patients in a real-world setting.
Advantages of using FVC include its noninvasiveness, simplicity, relatively small expense, and ability to monitor long-term trends in lung function. However, the measurement is limited by its visit-to-visit variability, dependence on patient effort, inconsistent quality control, limited accuracy, and impact of patient comorbidities.
Additional measures of disease progression, such as diffusing capacity for carbon monoxide (DLco) and 6-minute walk distance (6MWD), should be considered along with FVC to facilitate decision-making about disease management in patients with IPF.
Declaration of interest
SD Nathan was a member of the ASCEND study steering committee; has served on a scientific advisory board and received research funding from InterMune (a wholly owned subsidiary of Roche); has received research funding and served as a consultant for Boehringer Ingelheim, Gilead and Roche/Genentech and is on the speakers bureau for Roche/Genentech; and his institution has received research funding from Boehringer Ingelheim and Roche/Genentech. JD Wanger has served as a consultant for Genentech and Fibrogen. J Zibrak has served as a consultant for Boehringer Ingelheim and Roche/Genentech. ML Wencel has served as a consultant and is on the speakers bureau for Genentech, Inc. C Burg and JL Stauffer are employees of Genentech, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Support for third-party writing assistance, furnished by Health Interactions, Inc., was provided by F. Hoffmann-La Roche Ltd.
Additional information
Funding
References
- Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
- Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431–440.
- Martinez FJ, Safrin S, Weycker D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 Pt 1):963–967.
- Nathan SD, Albera C, Bradford WZ, et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429–435.
- Nathan SD, Yang M, Morgenthien EA, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: visit-to-visit variability and the role of 6-minute walk distance to validate changes [Abstract]. Am J Respir Crit Care Med. 2019;199:A7134.
- Nathan SD, Yang M, Morgenthien EA, et al. FVC variability in patients with idiopathic pulmonary fibrosis and role of 6-min walk test to predict further change. Eur Respir J. 2020;55(5):1902151.
- Durheim MT, Collard HR, Roberts RS, et al. Association of hospital admission and forced vital capacity endpoints with survival in patients with idiopathic pulmonary fibrosis: analysis of a pooled cohort from three clinical trials. Lancet Respir Med. 2015;3(5):388–396.
- Paterniti MO, Bi Y, Rekic D, et al. Acute exacerbation and decline in forced vital capacity are associated with increased mortality in idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2017;14(9):1395–1402.
- Raghu G, Collard HR, Anstrom KJ, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med. 2012;185(10):1044–1048.
- Schmidt SL, Tayob N, Han MK, et al. Predicting pulmonary fibrosis disease course from past trends in pulmonary function. Chest. 2014;145(3):579–585.
- Esbriet (Pirfenidone) [ package insert]. South San Francisco (CA): Genentech USA, Inc; 2019 [cited 2020 Aug 12]. Available from: https://www.gene.com/download/pdf/esbriet_prescribing.pdf
- Ofev (Nintedanib) [ package insert]. Ridgefield (CT): Boehringer Ingelheim Pharmaceuticals Inc; 2020 [cited 2020 Aug 12]. Available from: https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Ofev/ofev.pdf
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–1769.
- King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–2092.
- Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082.
- Maher TM, Swigris JJ, Kreuter M, et al. Identifying barriers to idiopathic pulmonary fibrosis treatment: a survey of patient and physician views. Respiration. 2018;96(6):514–524.
- Moore VC. Spirometry: step by step. Breathe. 2012;8(3):232–240.
- Dempsey TM, Scanlon PD. Pulmonary function tests for the generalist: a brief review. Mayo Clin Proc. 2018;93(6):763–771.
- Graham BL, Steenbruggen I, Miller MR, et al. Standardization of spirometry 2019 update. An official American Thoracic Society and European Respiratory Society technical statement. Am J Respir Crit Care Med. 2019;200(8):e70–e88.
- Yernault JC. The birth and development of the forced expiratory manoeuvre: a tribute to Robert Tiffeneau (1910–1961). Eur Respir J. 1997;10(12):2704–2710.
- Petty TL. John Hutchinson’s mysterious machine revisited. Chest. 2002;121(5Suppl):219s–223s.
- American Thoracic Society. Standardization of spirometry – 1987 update. Statement of the American Thoracic Society. Am Rev Respir Dis. 1987;136(5):1285–1298.
- American Thoracic Society. Lung function testing: selection of reference values and interpretative strategies. Am Rev Respir Dis. 1991;144(5):1202–1218.
- Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338.
- American Thoracic Society. Standardization of spirometry, 1994 update. Am J Respir Crit Care Med. 1995;152(3):1107–1136.
- Upagupta C, Shimbori C, Alsilmi R, et al. Matrix abnormalities in pulmonary fibrosis. Eur Respir Rev. 2018;27(148):180033.
- Jacob J, Bartholmai BJ, Rajagopalan S, et al. Predicting outcomes in idiopathic pulmonary fibrosis using automated computed tomographic analysis. Am J Respir Crit Care Med. 2018;198(6):767–776.
- Putman RK, Gudmundsson G, Axelsson GT, et al. Imaging patterns are associated with interstitial lung abnormality progression and mortality. Am J Respir Crit Care Med. 2019;200(2):175–183.
- Johannson KA, Vittinghoff E, Morisset J, et al. Home monitoring improves endpoint efficiency in idiopathic pulmonary fibrosis. Eur Respir J. 50(1):1602406. 2017.
- Russell AM, Adamali H, Molyneaux PL, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194(8):989–997.
- Moor CC, Wapenaar M, Miedema JR, et al. A home monitoring program including real-time wireless home spirometry in idiopathic pulmonary fibrosis: a pilot study on experiences and barriers. Respir Res. 2018;19(1):105.
- Swigris J, Nathan S, Tighe R, et al. STARMAP: an observational study to assess disease-relevant outcomes using home-monitoring devices in patients with idiopathic pulmonary fibrosis (IPF). Eur Respir J. 2019;54(Suppl 63):PA1333.
- Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8(2):147–157.
- Jenkins RG, Simpson JK, Saini G, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med. 2015;3(6):462–472.
- van Cleemput J, Sonaglioni A, Wuyts WA, et al. Idiopathic pulmonary fibrosis for cardiologists: differential diagnosis, cardiovascular comorbidities, and patient management. Adv Ther. 2019;36(2):298–317.
- Magnussen H, Canepa M, Zambito PE, et al. What can we learn from pulmonary function testing in heart failure? Eur J Heart Fail. 2017;19(10):1222–1229.
- Assayag D, Vittinghoff E, Ryerson CJ, et al. The effect of bronchodilators on forced vital capacity measurement in patients with idiopathic pulmonary fibrosis. Respir Med. 2015;109(8): 1058–1062.
- Hegewald MJ, Gallo HM, Wilson EL. Accuracy and quality of spirometry in primary care offices. Ann Am Thorac Soc. 2016;13(12):2119–2124.
- International Organization for Standardization. ISO 26782. Anaesthetic and respiratory equipment – spirometers intended for the measurement of time forced expired volumes in humans. Geneva (Switzerland): International Organization for Standardization; 2016.
- Du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184(12):1382–1389.
- Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26(4):586–593.
- Akagi T, Matsumoto T, Harada T, et al. Coexistent emphysema delays the decrease of vital capacity in idiopathic pulmonary fibrosis. Respir Med. 2009;103(8):1209–1215.
- Lin H, Jiang S. Combined pulmonary fibrosis and emphysema (CPFE): an entity different from emphysema or pulmonary fibrosis alone. J Thorac Dis. 2015;7(4):767–779.
- Womack CJ, Harris DL, Katzel LI, et al. Weight loss, not aerobic exercise, improves pulmonary function in older obese men. J Gerontol A Biol Sci Med Sci. 2000;55(8):M453–M457.
- Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. 1999;159(1):179–187.
- Rodriguez Moncalvo JJ, Ruiz JM, Mastroianni M, et al. Spirometric evaluation of idiopathic pulmonary fibrosis (IPF): forced or slow vital capacity? Eur Respir J. 2019;54(Suppl 63):PA4703.
- Du Bois RM, Weycker D, Albera C, et al. Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med. 2011;183(9):1231–1237.
- Du Bois RM, Albera C, Bradford WZ, et al. 6-Minute walk distance is an independent predictor of mortality in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2014;43(5):1421–1429.