
ABSTRACT
Introduction: Muscle impairments are prevalent in COPD and have adverse clinical implications in terms of physical performance capacity, disease burden, quality of life and even mortality. During acute exacerbations of COPD (AECOPDs) the respiratory symptoms worsen and this might also apply to the muscle impairments.
Areas covered: This report includes a review of both clinical and pre-clinical peer-reviewed literature of the past 20 years found in PubMed providing a comprehensive view on the role of AECOPD in muscle dysfunction in COPD, the putative underlying mechanisms and the treatment perspectives.
Expert opinion: The contribution of AECOPD and its recurrent nature to muscle impairment in COPD cannot be ignored and can be attributed to the acutely intensifying and converging disease-related drivers of muscle deterioration, in particular disuse, systemic inflammation and corticosteroid treatment. The search for novel treatment options should focus on the AECOPD-enhanced drivers of muscle dysfunction as well as on the underlying, mainly catabolic, mechanisms. Considering the impact of AECOPD on muscle function, and that of muscle impairment on the recurrence of exacerbations, counteracting muscle deterioration in AECOPD provides an unprecedented therapeutic opportunity.
1. Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by respiratory symptoms, such as dyspnea or cough, and airflow limitation as a consequence of airway and/or alveolar abnormalities [Citation1]. It is caused by exposure to noxious particles or gases, mainly tobacco smoking, but also biomass fuel exposure and air pollution [Citation1]. COPD is a common respiratory disease and a leading cause of morbidity and mortality worldwide. Global prevalence of COPD is estimated be around 12% [Citation2]. Moreover, according to the WHO COPD already ranked the 3rd leading cause of death worldwide in 2016 [Citation3]. COPD puts a heavy burden of health care systems, accounting for 3% of total direct costs of the total health care budget in the European Union [Citation1]. Historically, COPD was mainly considered as a pulmonary disease and treatment focused on the lung only. Inhaled bronchodilators are indeed still the cornerstone of COPD treatment [Citation1]. In the past decades, large cohort studies have shown that COPD is a complex, heterogeneous and multicomponent disease in which both pulmonary and extrapulmonary manifestations contribute to the disease burden [Citation4]. Alterations in body composition, such as muscle, fat and bone wasting, is one cluster of extrapulmonary manifestations in COPD [Citation4,Citation5]. Muscle wasting is frequently seen in COPD patients and its frequency varies depending on the setting. In population-base studies muscle weakness can be seen in 8% of patients, but this may be as high as 28% in patients referred for pulmonary rehabilitation and 63% in COPD patients residing in nursing homes [Citation4,Citation6]. The prevalence of muscle wasting also increases with airflow severity. One study showed that the prevalence of muscle weakness rose from 25% to 38% in patients with GOLD 1 to GOLD 4 COPD, respectively [Citation7]. Recent insights have also shown that this extrapulmonary tissue loss is not uniformly distributed between COPD patients, but might be centered around patients with emphysema. Patients with more severe emphysema had excessive loss of both pulmonary and extrapulmonary tissue and different metabolomics profile, suggesting an abnormal tissue maintenance is in this group of patients. The authors proposed that this subgroup be named the multi-organ loss of tissue (MOLT) COPD phenotype [Citation8].
In particular muscle weakness has a serious impact on the disease burden of COPD patients. Firstly, muscle weakness is an important determinant of exercise capacity; patients with a reduced quadriceps force have a shorter the 6-minute walking distance [Citation9]. Secondly, muscle weakness is associated with an increased risk of mortality. A study by Marquis and colleagues showed that mid-thigh muscle cross-sectional area had strong impact on mortality in patients with severe COPD and was a better predictor for mortality than the classically known low body weight [Citation10]. Finally, muscle weakness is related to an increased utilization of health care resources in patients with COPD [Citation11]. Moreover, Attaway et al recently reported that in-hospital mortality, LOS and healthcare costs are higher in patients with COPD exacerbations and low muscle mass [Citation12]. This review addresses the role of acute exacerbations in muscle dysfunction in COPD. For this PubMed was searched for relevant clinical as well as pre-clinical peer-reviewed papers from the past 20 years, with a focus on those of the last decade.
2. Muscle impairments in COPD
Skeletal muscle dysfunction is a major debilitating feature of COPD as it directly affects physical performance and ambulatory capacity having a large impact on quality of life [Citation13]. Muscle function comprises a strength (i.e. force generating capacity) and an endurance (i.e. capacity to sustain a submaximal force over time) component and both are compromised in COPD. Upper as well as lower limb and respiratory muscles are affected, albeit lower limbs to a greater extent [Citation14,Citation15]. The pathophysiology and underlying mechanisms will be discussed below (see for the processes and pathways involved).
Figure 1. Schematic overview of the main processes and pathways potentially involved in muscle dysfunction in COPD. Autophagy lysosomal pathway (ALP), ubiquitin–proteasome system (UPS), Forkhead box O (FOXO), muscle atrophy F-box (Atrogin-1), muscle-specific ring finger 1 (MuRF1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), insulin-like growth factor (IGF), phosphatidylinositide 3-kinases (PI-3 K) serine/threonine protein kinase Akt (Akt), mammalian target of rapamycin (mTOR), eIF4E-binding protein 1 (4E-BP1), ribosomal protein S6 kinase (P70S6K), peroxisome proliferator-activated receptor-delta (PPAR-δ), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), and mitochondrial transcription factor A (TFAM)
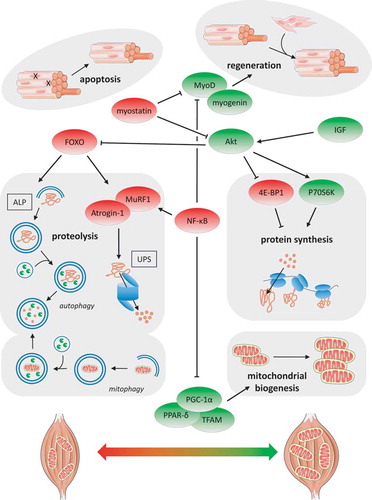
Muscle weakness can largely be attributed to muscle wasting. The golden standard for muscle mass assessment is the deuterium dilution method, but in practice it is mostly assessed as fat-free mass by means of dual-energy X-ray absorptiometry and bioelectrical impedance analysis, or muscle cross-sectional area/volume by means of MRI and ultrasonography [Citation13]. Loss of strength independent of the loss of muscle mass seems unlikely as most available research indicates preserved muscle contractility in COPD [Citation13]. This applies in particular to the relatively larger locomotor muscles; whether loss of contractility of the diaphragm occurs independently of muscle atrophy still remains unclear [Citation15,Citation16]. Muscle atrophy, in underweight patients but even in patients with normal-to-high BMI, is a frequent extrapulmonary manifestation of COPD and is also a strong predictor of mortality. Sarcopenia, i.e. the gradual loss of muscle mass during aging, is highly prevalent in COPD and therefore an important contributor to muscle atrophy [Citation17]. Another key factor in muscle wasting in COPD is cachexia, i.e. the active and more acute process of unvoluntary weight loss that often accompanies this disease [Citation18]. Both may be worsened or accelerated during an acute exacerbation of COPD (AECOPD) as will be discussed below. Detailed examination of the muscle architecture has revealed that muscle fiber atrophy, especially of the fast-twitch and mainly force-generating type IIx fibers, underlies low muscle mass in COPD, which generally applies to peripheral muscles as well as the diaphragm [Citation15,Citation19]. The underlying mechanisms of muscle wasting in COPD have only been partly elucidated. Taken together the available literature points toward increased expression levels of the muscle specific atrogenes Atrogin-1 and MuRF1 (which target proteins for degradation in the ubiquitin proteasome system (UPS)), as well as of their upstream transcription factors FOXO-1/3 and NF-κB, in COPD [Citation20]. Findings for altered muscle protein synthesis in COPD are less consistent albeit overall also point toward an enhancement, based on increased IGF-1 expression levels and IGF-1/Akt signaling (increased phosphorylation of the mRNA translation mediators 4E-BP1 and P70S6K) in patients characterized by muscle atrophy [Citation20]. With respect to myonuclear turnover as an essential process in muscle homeostasis, literature is not clear about whether apoptosis is increased in atrophied muscle of COPD patients [Citation20]. In contrast, more evidence exists indicative of impaired myonuclear accretion based on reports revealing altered muscle satellite (precursor) cell behavior, satellite cell senescence, decreased expression levels of myogenic factors (i.e. myogenin and MyoD) as well as increased myostatin signaling in patients with low muscle mass [Citation20]. Overall, whole body protein turnover is increased in COPD feasibly due to increased muscle proteolysis and a seemingly inadequate adaptive increase of protein synthesis [Citation20]. This increased muscle turnover as part of a seemingly futile adaptive response has been confirmed by an extensive analysis of, amongst others, the abovementioned molecular markers in COPD patients with low muscle mass [Citation21].
Reduced endurance (or increased fatigability) of the upper and lower limb muscles in COPD can largely be explained by the loss of muscle quality rather than the loss of muscle quantity of which a shift in muscle fiber type proportion from oxidative fatigue-resistant slow-twitch type I toward fast-twitch type II fibers is most prominent [Citation13,Citation14,Citation22]. This is paralleled by a lower mitochondrial capacity (i.e. reduced mitochondrial content, OXPHOS complexes and enzyme activities of citrate synthase and 3-hydroxyacyl-CoA dehydrogenase) as well as mitochondrial dysfunction [Citation23–25]. This overall loss of muscle oxidative phenotype (OXPHEN) also renders the muscle less efficient from a metabolic perspective in that more oxygen is required for a certain amount of work. This subsequently may increase respiratory workload and worsens the dyspnea. Furthermore, reduced expression levels of important OXPHEN regulators like PGC-1α, PPAR-δ and TFAM have also been found in muscles of patients with COPD [Citation26], whereas regulators of mitophagy (the autophagosomal breakdown of mitochondria) are upregulated [Citation27]. In addition to loss of muscle OXPHEN an impaired capillarization and hence oxygenation of the muscle might further contribute to its fatigability although literature is not consistent on this aspect [Citation13]. Noteworthy, whereas the diaphragm seems to largely follow the pattern of limb muscle wasting, its metabolic changes are opposite: compared to healthy controls, the diaphragm of COPD patients is characterized by a higher proportion of type I fibers and a higher mitochondrial capacity which is likely the consequence of a quasi-endurance-training effect induced by the elevated breathing activity [Citation15,Citation16].
3. Acute exacerbations of COPD
Periodically, patients with COPD present with a deterioration of respiratory symptoms such as increased sputum or dyspnea. Such events are called acute exacerbations of COPD (AECOPDs). They are common in patients with COPD and have a significant impact on patients and health care services [Citation1]. AECOPDs are mostly caused by respiratory tract infections. Both viruses and bacteria can provoke an AECOPD. Around 20–30% of AECOPDs remain without an infectious cause; in such cases, AECOPDs can be triggered by air pollution, pulmonary embolism or heart failure [Citation28]. Bafadhel and colleagues have shown that AECOPDs can be grouped in four different biological clusters with distinct inflammatory signals: bacterial, viral, eosinophilic and pauci-inflammatory cluster [Citation29]. This clustering might have some predictive value for treatment and prognosis in AECOPD. First, follow-up studies have shown that bacterial and eosinophilic exacerbations show a high within person stability and a higher stability over time [Citation30,Citation31]. Second, eosinophilic exacerbations are associated with a favorable treatment outcome with steroid treatment but are also associated with an increased risk of exacerbation relapse [Citation32]. The presence of respiratory viruses at exacerbation are predictive for a higher symptom burden and a delayed recovery [Citation33].
Certain risk factors predispose COPD patients to an AECOPD. In general, AECOPDs become more frequent as the severity of the airflow limitation increases [Citation34,Citation35]. Moreover, certain features on chest CT scan, such as extent of emphysema and airway thickness, also predispose patients to recurrent exacerbations [Citation34]. However, a history of past exacerbations is the best predictor for future exacerbations [Citation35,Citation36]. This frequent exacerbator phenotype of COPD has poorer outcomes, including worse health status and higher mortality [Citation35,Citation37]. As mentioned previously, higher peripheral eosinophil counts are another important risk factor for recurrent exacerbations and finally, some comorbidities, such as anxiety and depression associate with exacerbation risk [Citation34].
There are distinct time courses in onset and recovery of AECOPDs. At onset, AECOPDs exhibit two different patterns; a sudden onset and a gradual onset AECOPD [Citation38]. Gradual onset AECOPDs are characterized by a slower onset of symptoms (median 4 days) and associate with less severe respiratory symptoms and longer recovery times. Symptoms of a common cold associate with longer recovery times, suggesting that viral infections may play a role [Citation39]. The longer prodromal time course, and the subsequent deleterious effect on respiratory fitness and muscle strength might be another explanation. Recovery of AECOPDs also shows different time courses. Multiple studies show a rapid improvement of respiratory symptoms over 2–4 weeks and then a slower improvement which can last up to 6 months [Citation35]. It has been shown that in the first weeks following an AECOPD there is a high risk of exacerbation recurrence. This risk of recurrence is estimated to be around 27%, which is 20% higher than would be expected if AECOPDs occurred randomly [Citation35,Citation36]. These AECOPD recurrences or relapses strongly impact patients with COPD. Suissa and colleagues have shown that after a second hospitalization the risk of death increases with each subsequent exacerbation [Citation40]. Furthermore, irrespective of hospitalization status, BMI, obstruction, dyspnea, and exercise capacity worsen when patients experience an exacerbation and this does not return to baseline after 1 year [Citation41]. Likewise, patients who experience recurrent exacerbations seem to have a poorer health status and prospective studies have shown that, after an initial event, COPD patients who relapse do not return to their baseline health status [Citation37]. The worsening of pulmonary and extrapulmonary manifestations of COPD are probably accelerated during (recurrent) exacerbations when disease-related factors such as systemic inflammation, hypoxia, inactivity, and corticosteroid treatment intensify and converge to exert their detrimental effects on skeletal muscle [Citation42].
Treatment of AECOPD is aimed at symptom relief and the prevention of disease deterioration. Inhaled bronchodilator therapy is the mainstay of treating AECOPD [Citation1]. Noninvasive ventilation in AECOPD with hypercapnic respiratory failure is a very important therapy as it prevents the need for invasive ventilation and reduces mortality in AECOPD [Citation43]. Antibiotics reduce treatment failure [Citation44], but probably have limited benefit in outpatient settings [Citation45]. Most AECOPD patients receive corticosteroid treatment, though recent studies have shown that corticosteroid treatment can be guided by peripheral eosinophil count [Citation46]. Short-courses, e.g. 5 days, of systemic steroids improve lung function, provide symptom relieve and prevent treatment failure [Citation47].
For longer courses of treatment with systemic corticosteroids little beneficial effects with regard to relapses or lung function have been documented and recent treatment guidelines even include a conditional recommendation against the use of maintenance oral corticosteroids in patients with COPD [Citation48]. Also, recent studies suggest higher risk of pneumonia and mortality with longer durations of steroid treatment [Citation49]. Moreover, there are substantial side effects when using steroids with the diaphragm and peripheral muscles being particularly affected [Citation47,Citation50].
4. Clinical evidence for a role of AECOPD in muscle impairments
Loss of muscle mass is a risk factor for (recurrent) exacerbations. Already 20 years ago it was reported that weight loss during an AECOPD hospitalization and low BMI on admission were related to the increased risk of unplanned readmission [Citation51]. Similarly, the likelihood of readmission within 1 year for an AECOPD is correlated to low muscle mass at discharge of an AECOPD-related hospitalization [Citation52]. In a larger longitudinal study including 78 patients, low muscle mass was also associated with the number of days hospitalized for an exacerbation as well as with readmission within the following 3 months [Citation53]. Likewise, in a retrospective cross-sectional study Guerri et al showed that reduced cross-sectional area of several limb, abdominal and intercostal (respiratory) muscles was lower in patients who suffered from four or more AECOPD admissions in the previous year than those that exacerbated less frequent [Citation54]. Moreover, a high prevalence (~50%) of sarcopenia and malnutrition was found in 54 patients during AECOPD hospitalization, which was still ~30% a half year later [Citation55] and also the ratio of circulating creatinine/cystatin C (a novel serum-based sarcopenia index) was recently found to be an independent predictor of hospitalizations in COPD [Citation56,Citation57]. In a recent post hoc analysis of a prospective observational cohort with 102 male COPD patients it was also shown that the average decline rate of the cross-sectional area of the erector spinae muscles was 5.21% over a 3-year interval and it was found that the fast decliners, i.e. those with a rate of decline > 10.42% (2 x average), had a significantly higher frequency of moderate-to-severe exacerbations [Citation58]. Finally, it is also very important to realize that subjects admitted with AECOPD may be overweight or even obese, yet frequently still have a below normal fat-free mass [Citation59].
In addition to loss of muscle mass, AECOPD is also associated with muscle dysfunction. In a prospective study with 52 patients with AECOPD, muscle strength was significantly impaired during hospitalization and further deteriorated after a 1-month follow-up [Citation60]. Handgrip strength in COPD was inversely related to exacerbation frequency in two retrospective studies [Citation61,Citation62]. Patients also demonstrate balance impairments and higher fall incidence (with increased risk for bone fractures [Citation63]) during an AECOPD which was associated with (and feasibly can be attributed to) the reduced quadriceps strength that was observed as well [Citation64]. In a prospective study (a one year follow-up) published in 2007 Hopkinson et al observed that COPD patients with frequent exacerbations (>1 per year) experienced a greater decline in fat-free mass over time compared to infrequent exacerbators [Citation65]. Cause and consequence in the interaction between AECOPD and muscle wasting are hard to disentangle. In fact, their reciprocal actions most likely result in a downward spiral as illustrated by a cross-sectional study by Vilaro et al who found that muscle dysfunction was associated with an increased risk hospital admission and that recurrent exacerbations further deteriorated muscle function of both respiratory and peripheral muscles [Citation66]. That an exacerbation may indeed lead to (further) functional decline is also illustrated by a prospective study showing that exercise capacity and muscle strength were deteriorated in COPD patients several days after presenting with a moderate exacerbation (out-patient managed) as compared to when they were stable [Citation67]. Likewise, it has been shown that muscle strength actually declines during an AECOPD hospitalization (between day 3 and 8 of the hospital stay), which was associated with by increased circulating levels of IL-8 and decreased levels of IGF-1 [Citation68]. Moreover, muscle force only partially recovered during the subsequent 3-month follow-up. The latter is actually an aspect that should not be neglected because regardless of their hospitalization status, patients with COPD who exacerbate have deterioration not only of pulmonary function but also of BMI and exercise capacity which does not return to baseline even after 1 year [Citation41].
Muscle biopsy data explaining the underlying molecular mechanisms of AECOPD-induced muscle impairments are very limited. Crul et al obtained quadriceps samples from 14 patients admitted to the hospital for an AECOPD and 11 stable COPD patients. They found reduced IGF-1 mRNA and MyoD protein expression levels in the AECOPD group, whereas myogenin could not be detected [Citation69]. Subsequently, using a microarray gene expression profiling approach in muscle biopsies, they also showed increased gene expression of the atrogenes Atrogin-1 and MuRF1 as well as a reduced expression of the OXPHEN marker COX6C (which encodes an important protein for mitochondrial respiration) in 4 AECOPD versus 5 stable COPD patients [Citation70]. Based on that same dataset using an in silico approach Duan et al identified several differentially expressed genes (e.g., NCL, GOT1, SOD2, TSPO and TMOD1) and miRNAs (e.g., miR-1, miR-9 and miR-23a) which could be involved in muscle impairments in AECOPD [Citation71]. Clearly, more studies with larger group sizes and longitudinal sampling are required for a better understanding the biological mechanisms associated with AECOPD-induced muscle impairments.
Indirect evidence for AECOPD affecting skeletal muscle also comes from disease-related factors associated with muscle impairment in COPD as outlined previously [Citation20]. As mentioned above, these triggers intensify and converge during an exacerbation. First of all COPD patients are less physically active during an exacerbation or even inactive when hospitalized [Citation72,Citation73]. Disuse of muscle inevitably and rapidly leads to loss of muscle mass and OXPHEN [Citation74]. Although not very consistent and mainly based on cross-sectional study designs in stable COPD, numerous associations between systemic inflammation and muscle impairments have been reported [Citation75]. An AECOPD is often accompanied by increased systemic inflammation (e.g. elevated circulating CRP, IL-6, IL-8, and TNFα levels) and indeed an inverse relationship between muscle strength and blood IL-8 levels was found during hospital admission [Citation68,Citation76]. In addition, COPD patients are likely in a negative energy balance during an exacerbation as it was found that resting energy expenditure was increased, while dietary caloric intake was reduced with 28%, feasibly mediated by increased serum levels of the appetite-suppressing hormone leptin and soluble TNFα receptor 55 [Citation77,Citation78]. Corticosteroid treatment is standard care for AECOPD despite the fact that in particular oral corticosteroids are known for their negative side effects including muscle wasting in COPD [Citation50,Citation65,Citation79]. Whether corticosteroids also affect muscle OXPHEN in COPD requires further investigation although one study suggests this might not be the case [Citation80]. Finally, as a consequence of worsened pulmonary failure hypoxemia and hypercapnia often develop which are known to affect muscle function, mass and muscle OXPHEN as well [Citation81–83].
5. Experimental evidence and mechanisms of AECOPD-induced muscle impairment
As outlined above, multiple triggers of muscle dysfunction are present during AECOPD, prompting a better understanding of their individual and converging actions and how these impact on skeletal muscle during these episodes of the disease. Experimental models, e.g. muscle cell culture and mainly animal models have provided evidence and insights in how inflammation, undernutrition, glucocorticoids, hypoxia and inactivity contribute to skeletal muscle dysfunction [Citation20], of which examples will be discussed below.
The actions of pro-inflammatory cytokines like TNFα, IL-1β and IFNγ on skeletal muscle have been well-documented in a variety of experimental settings. Raising circulatory levels of TNFα artificially using transgenic mice, or by implantation of TNFα- or IFNγ-secreting tumors, results in skeletal muscle dysfunction and atrophy of both limb and respiratory muscles [Citation84–88]. Cell studies using cultured myotubes (in vitro myofiber surrogates) have demonstrated that inflammatory cytokines directly impact on skeletal muscle. Administration of combinations of TNFα, IL-1, and IFNγ, or TNFα alone to myotube cultures is sufficient to induce atrophy [Citation89,Citation90]. Cytokine-driven muscle dysfunction involves increased proteolysis mediated by E3 Ub-ligases including MuRF1 and Atrogin-1 [Citation91,Citation92]. In addition, inflammatory cytokines impair satellite (i.e. muscle progenitor) cell-dependent myogenesis by affecting essential regulatory molecules like MyoD [Citation89,Citation93]. NF-κB activation has been shown as a required step in inflammation-induced muscle atrophy, both in vitro and in vivo, and muscle-specific activation of NF-κB using genetic modification is sufficient to induce a strong muscle wasting phenotype [Citation89,Citation94,Citation95]. Pro-inflammatory cytokines also cause a reduction in muscle OXPHEN, which primarily appears to rely on reduced mitochondrial biogenesis [Citation96,Citation97]. Moreover, TNFα or IL-1-elicited loss of OXPHEN requires NF-κB activation, implicating this intracellular inflammatory signaling pathway as a central mediator of inflammation-induced muscle dysfunction [Citation98].
The effect of undernutrition has been studied in fasting or starvation models, in which adipose and muscle tissue is affected. Despite increased lipolysis as an early adaptive response to starvation, simultaneous activation of muscle proteolysis has been reported [Citation99]. In homeostatic conditions, insulin suppresses the activity of the transcription factor FOXO-3, which has been implicated as a central transcriptional regulator of muscle protein degradation [Citation100]. FOXO-3 activation increases the expression of rate limiting constituents of the proteolytic UPS and autophagy lysosomal pathway (ALP). In addition, in conditions of sustained starvation, protein synthesis stimulatory signaling through the mTOR/4E-BP1/eIF4 and mTOR/p70S6K1/S6 pathways is reduced due to low insulin serum levels and limited amino acid availability [Citation101]. Combined, this shift in the balance between proteolysis and protein synthesis strongly favors muscle atrophy. Interestingly, skeletal muscle fibers with high oxidative capacity are relatively spared from starvation-induced increases in proteolysis, in line with the preservation of slow-twitch fibers in response to food deprivation [Citation102,Citation103]. Although increased mitophagy has been described following fasting, these observations were made in muscles of mixed fiber type and resulting changes in OXPHEN were not reported [Citation104]. Moreover, stimulation of mitochondrial biogenesis was observed in muscle of hibernating bears [Citation105], indicating that the net effect of starvation on skeletal muscle OXPHEN remains to be determined.
Glucocorticoids are a class of hormones with potent anti-inflammatory properties, which are released endogenously (e.g. cortisol) or applied as synthetic variants (e.g. dexamethasone or prednisolone) to suppress inflammatory responses [Citation106]. Importantly, in addition to inflammation, cortisol (or corticosterone as the predominant glucocorticoid in rodents) levels increase as well as the central part of a systemic stress response to triggers like fasting and hypoxemia [Citation107]. The acute and chronic administration of glucocorticoids is sufficient to induce muscle wasting and the multiple mechanisms might be involved: Reduced anabolic IGF-1- (and increased anti-anabolic myostatin-) receptor activation, resulting in a reduction in the mTOR signaling pathway, have been described for limb and respiratory muscle [Citation108,Citation109]. Moreover, induction of UPS and autophagy mediated muscle proteolysis, exemplified by the requirement for MuRF1 and Atrogin-1, occur in dexamethasone-induced myotube atrophy [Citation110,Citation111]. Moreover, suppression of myogenesis has also been reported, indicating that glucocorticoids disrupt multiple processes involved in muscle mass maintenance [Citation112]. Specific effects of glucocorticoids on muscle OXPHEN have only been reported in few studies with contradictory findings, including a decreased oxidative capacity observed in the limb muscles but an increase thereof in the diaphragm [Citation113,Citation114]. However, the consistently observed reduction in overall protein synthesis following glucocorticoids may impact on mitochondrial biogenesis as this relies on protein synthesis [Citation115]. As glucocorticoids were previously shown to induce mitophagy in cultured skeletal muscle cells, the combined actions of glucocorticoid signaling may culminate in loss of OXPHEN [Citation116].
Although hypoxemia and subsequent tissue hypoxia may occur as a consequence of respiratory insufficiency induced by various conditions, decreasing ambient oxygen levels has been used most frequently to model the impact of tissue hypoxia on skeletal muscle function. In line with findings in men and rats, hypoxia induces muscle atrophy in mice, which was only in part attributable to hypoxia-induced anorexia [Citation20,Citation117]. Muscle proteolytic signaling that was uniquely responsive to hypoxia included FOXO and activation of downstream UPS and ALP targets, Atrogin-1 and LC-3B respectively. In addition, muscle protein synthesis signaling is also impaired by hypoxia, which sensitizes skeletal muscle to coinciding atrophy stimuli [Citation118]. Interestingly, muscle differences in oxidative capacity correspond with their sensitivity to hypoxia-induced atrophy, which may be related to the relative preservation of muscle OXPHEN in response to hypoxia [Citation119,Citation120]. Little information is available regarding the effects of hypoxia on myonuclear turnover. No evidence for in vivo myofiber apoptosis was observed following hypoxia, while decreased MyoD expression and impaired satellite cell regenerative capacity were observed in response to hypoxia [Citation121–123].
Inactivity, muscle disuse and unloading are well-documented triggers of muscle atrophy. These have been modeled in animals and humans by limb immobilization or suspension, or by enforcing sustained bed rest, which is followed by significant muscle atrophy and reduced muscular strength. Both decreased protein synthesis and increased proteolysis have been reported in skeletal muscle during disuse atrophy, although these changes are only apparent during progressive loss of muscle mass [Citation124,Citation125]. Activation of the ALP as well as UPS have been implicated in inactivity-induced proteolysis [Citation125,Citation126]. Besides MuRF1 and Atrogin-1, which are also transcriptionally activated in response to the other triggers of atrophy discussed here, Nedd4 is an E3 Ub-ligase specifically activated in inactivity-induced muscle atrophy [Citation127]. FOXO and NF-κB, including non-canonical NF-κB signaling, have been implicated in the transcriptional regulation of proteolysis during disuse atrophy [Citation20]. Inactivity also has a profound impact on myonuclear turnover, as it induces apoptosis and affects satellite cell proliferation and muscle regeneration [Citation128,Citation129]. Muscle disuse is a potent trigger of OXPHEN loss [Citation130]. Extensive alterations in mitochondrial biogenesis and mitophagy occur rapidly in response to inactivity, underpinning these OXPHEN decreases [Citation126]. Interestingly, overexpression of PGC-1α prevents disuse-induced muscle OXPHEN [Citation131,Citation132], implying this exercise-responsive master regulator of mitochondrial health as an integrative switch between muscle OXPHEN and mass during muscle adaptive responses to altered activity levels.
Various experimental animal disease models exist to investigate COPD lung and systemic pathology. Chronic cigarette smoke (CS)-induced emphysema is the most frequently used COPD model. Although skeletal muscle impairment has been reported in these chronic models mimicking stable COPD, the extent to which muscle dysfunction, muscle OXPHEN and muscle mass are affected varies between no to modest impairment [Citation133,Citation134]. Moreover, whether muscle dysfunction develops progressively in those chronic models is not clear, and in contrast to clinical COPD it appears to be reversible upon cessation of smoke exposure [Citation135]. Similarly, in elastase-induced emphysema models the alterations in muscle mass and function were absent or minor [Citation136–138]. In the CS- and elastase-induced COPD models, intra-tracheal instillation of live bacteria or other bacterial or viral components has been used to mimic AECOPDs [Citation139–141]. While the impact of such an AECOPD on skeletal muscle has not yet been investigated in the CS-based models, Ceelen et al demonstrated rapid loss of muscle function and mass in an AECOPD-model that consists of a single bolus of intra-tracheal LPS in elastase-induced emphysematous mice [Citation136]. The acute muscle impairments were accompanied by inhibition of protein synthesis signaling and activation of proteolytic pathways, including the ALP and UPS. A functional UPS was shown to be required for acute pulmonary inflammation-induced muscle alterations, as transgenic overexpression of mutant, inhibitory ubiquitin molecules attenuated muscle atrophy [Citation142]. Increased expression of the E3-Ub ligase MuRF1 appears essential for skeletal muscle wasting in AECOPD, as its genetic blockade or suppression by upstream inhibition of muscle NF-κB activity prevents skeletal muscle atrophy in response to lung inflammation [Citation94,Citation143]. Moreover, besides limb muscle impairment, diaphragm atrophy has also been described in response to LPS-induced pulmonary inflammation [Citation144]. Besides inflammatory signaling, induction of glucocorticoid-responsive gene expression is also evident in AECOPD mice, and the contribution of malnutrition [Citation94] in pulmonary inflammation-driven muscle wasting has been well documented. Combined, in this experimental model which consists of multiple converging AE-related triggers, the acute impact of AECOPD on skeletal muscle mass appears mainly the result of increased proteolysis.
Following resolution of pulmonary inflammation in healthy mice after one or more boluses of intra-tracheal LPS, the imbalance in protein turnover is restored and muscle mass loss is reversed [Citation142]. This reflects the normal recovery of transient decreases in muscle function following disease in men. In contrast, skeletal muscle mass did not completely recover in emphysematous mice following repetitive LPS boluses (i.e. reflecting recurrent exacerbations), despite normalization of proteolysis signaling [Citation145]. In these AECOPD-mice, aberrant myogenic responses are observed in skeletal muscle, and serum transfer experiments suggest that systemic, emphysema-related factors may impede appropriate recovery of atrophied muscle. This notion is supported by other studies revealing impaired muscle regenerative responses in skeletal muscle of chronic pulmonary inflammation-induced emphysema or CS induced-emphysema in mice [Citation84,Citation146]. In other models of chronic or repetitive pulmonary inflammation in presence of emphysema, skeletal muscle dysfunction has also been reported [Citation147,Citation148]. Combined, these studies indicate that chronic, progressive muscle atrophy in COPD may reflect the cumulative loss of muscle mass and impaired muscle regenerative capacity caused by acute recurrent exacerbations. However, whether this also applies to the loss of muscle OXPHEN as the other hallmark of skeletal muscle dysfunction in COPD remains to be determined.
6. Treatment of muscle impairment in AECOPD
Despite the varying training modalities applied, pulmonary rehabilitation (PR) and exercise training directly after AECOPD hospital admission, i.e. to enhance recovery, certainly may have added value on physical performance and muscle mass/function as reviewed elsewhere [Citation149–152]. It is already fairly common practice and the current recommendation in the ‘Management of COPD exacerbations: a European Respiratory Society/American Thoracic Society guideline’ is to initiate PR within 3 weeks after hospitalization [Citation79]. However, since prevention is better than cure, attempting to avoid muscle deterioration by PR during AECOPD hospitalization in the first place would make sense (as a side note, the term ‘rehabilitation’ during the admission phase does not seem appropriate and maybe should be reconsidered in this context). Recently, Dobler et al conducted a systematic review including a meta-analysis on non-pharmacologic therapies and showed that exercise interventions and pulmonary rehabilitation programs may indeed ameliorate functional decline in patients hospitalized for an AECOPD [Citation153]. In-hospital rehabilitation (with focus on exercise training) during admission attenuates muscle mass decline during AECOPD [Citation154,Citation155] and aerobic exercise training during admission, despite being experienced as intense, was found to be safe [Citation156]. However, in a recent RCT no beneficial effect of rehabilitation including neuromuscular electrostimulation (NMES) during an AECOPD admission was found on recovery of physical function (e.g. quadriceps force), and mortality was even higher in the intervention group after one year [Citation157]. As such, the recent guideline on management of COPD exacerbations conditionally recommended against PR during hospitalization [Citation79]. This undoubtedly will remain under debate and more high quality RCTs are required [Citation152,Citation158]. Moreover, prevention of muscle deterioration should not only be considered during hospital admission as a recent pilot study suggests that adding community-based PR to the outpatient management during a mild-to-moderate AECOPD might also be beneficial in terms of quadriceps muscle strength [Citation159].
Muscle biopsy data on the effect of the abovementioned treatments is very limited and we found only two studies. Troosters et al found that myostatin expression was lower and the myogenin/MyoD ratio tended to be higher after resistance training in AECOPD, indicative of a pro-anabolic response [Citation154]. In another small study it was found that NMES at 35 Hz (versus sham treatment) following an AECOPD had positive results in terms of muscle strength and 6-min walking distance accompanied by a proportional increase of type I fibers [Citation160].
Data on the effect of nutritional support during AECOPD are still very scarce, but the few trials that have been conducted failed at least from the muscle perspective. In one RCT supplementation with extra calories (10 kcal/kg/day) for 2 weeks had no effect on muscle strength in patients with an AECOPD, which feasibly could be attributed to increased corticosteroid intake because the latter was negatively correlated to the nitrogen balance in these patients [Citation161]. In another RCT the effect of an energy- and also protein-rich nutritional supplement was therefore studied during hospitalization, but here too no improvements in muscle function were found [Citation162]. Similarly, supplementation with 1 g/day of eicosapentaenoic acid (EPA), known for its anti-inflammatory and muscle OXPHEN promoting properties, failed to preserve lean body and muscle mass during hospital admission for AECOPD [Citation163].
To our knowledge there are no RCTs aimed at investigating the effect of pharmaca to counteract muscle impairment during AECOPDs. The myostatin inhibitor Bimagrumab was recently tested in stable COPD and it lead to a 7% increase in thigh muscle mass albeit without any functional improvements [Citation164]. Trials in stable COPD with androgens (e.g. nandrolone and oxandrolone), testosterone, growth hormone, and grehlin had mixed and rather disappointing results as reviewed elsewhere [Citation149,Citation165]. Interestingly though, re-analysis of data of an earlier clinical trial investigating the efficacy of anabolic steroid supplementation (nandrolone) on muscle recovery in muscle-wasted patients with stable COPD revealed that the anabolic response in terms of muscle mass and respiratory muscle strength was enhanced in the subgroup on maintenance glucocorticoid treatment as compared to patients that received no glucocorticoids [Citation112]. Furthermore, in an in vitro experiment it was previously shown that exposure of cultured muscle cells to glucocorticoids together with IGF-1 (an anabolic) had an even higher anabolic response than that of IGF-1 alone (and with glucocorticoids alone having the anticipated catabolic effects). These findings might thus be of interest from an AECOPD treatment perspective.
7. Expert opinion
Muscle dysfunction due to muscle wasting and metabolic impairments is a hallmark of COPD which affects patients’ physical performance, daily activities, quality of life and is associated with increased mortality. Muscle wasting is very common in COPD with reported prevalence numbers ranging from 4 to 35% (depending on disease severity and the criteria applied) up to even 50% in female patients with GOLD 4 COPD [Citation13]. Moreover, Vanfleteren et al reported a muscle wasting prevalence of 28% making it one of the more prominent comorbidities of COPD together with hyperglycemia, atherosclerosis, hypertension, dyslipidemia, and osteoporosis [Citation166]. Disease-related factors such as systemic inflammation, undernutrition, glucocorticoids, hypoxia and physical inactivity are putative drivers of muscle deterioration in COPD. These factors intensify and converge during acute exacerbations of COPD and therefore exacerbations play an important role in the muscle impairments in COPD as substantiated by the clinical and experimental evidence discussed above. Although muscle dysfunction of the upper limbs seems to be relatively preserved as compared to that of the lower limbs, AECOPDs seem to affect both because scarce data suggest the respiratory muscles are not spared either. Whether an AECOPD affects all muscle compartments to the same extent however remains to be elucidated.
Although the development of muscle impairment in COPD may appear a gradual process in the slipstream of disease progression, this decline may actually often follow a step-wise course which is caused by accelerated wasting followed by incomplete recovery of muscle as a consequence of exacerbations [Citation20,Citation42], as depicted in . Firstly, muscle recovery after an AE appears compromised in COPD because of the disease-related factors still being present (to a lower extent) in combination with, or leading to impaired muscle homeostasis. Currently, this notion, as well as further insights in the potential underlying intracellular mechanisms and contribution of AECOPD-associated triggers of skeletal muscle, is mainly based on research using experimental models. Secondly, muscle recovery may simply be incomplete because of a recurrent exacerbation. Whereas disuse-induced muscle loss has been estimated around 0.5–0.6% of total muscle mass per day and thus several percentages of muscle mass can be lost in a matter of days of being bedridden for example, recovery thereof may take several months even in healthy persons: In elderly it is estimated that just two short periods of illness per year are sufficient to prevent complete recovery of disuse-induced muscle loss, which is thought to play an important role in the development of age-related sarcopenia [Citation167].
Figure 2. Conceptual visualization of the step-wise decline of muscle mass, OXPHEN (reflected by the mitochondria) and function in COPD due to (recurrent) exacerbations. Muscle wasting and loss of function are accelerated during an exacerbation in response to acutely increased levels of COPD-related triggers of muscle dysfunction. During the recovery phase muscle regeneration is intrinsically impaired in COPD or incomplete due to a recurrent exacerbation
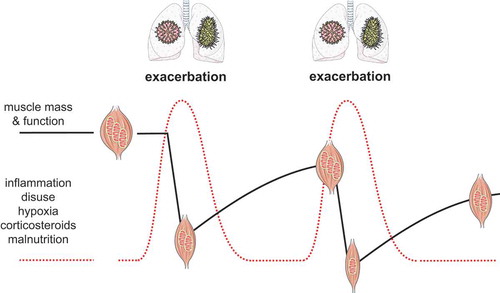
Muscle impairment and AECOPD appear to have amplificatory interactions since low muscle mass is a determinant of AE-frequency and hospitalization status, whereas muscle deterioration is accelerated by exacerbations. More pre-clinical (i.e. using AECOPD animal models) and prospective clinical studies are required to confirm that muscle dysfunction is indeed a consequence of an AECOPD. Nevertheless, the AE-associated muscle impairment provides a promising window of opportunity from a treatment perspective to break the vicious circle. However, due to a lack of muscle biopsy data of patients suffering from an AECOPD, it remains largely unclear what the precise underlying mechanisms of loss of muscle mass and OXPHEN are in terms of anabolic and catabolic processes, and mitochondrial turnover respectively. Moreover, longitudinal studies with consecutive muscle biopsies in the course of the AE and recovery phases would reveal when these processes are affected most and indicate the optimal time to intervene, e.g. pharmacologically. Oral corticosteroids, which are applied to suppress excess inflammation, should be administered with caution because of their negative impact on muscle. Interestingly, although the use of anabolic steroids during an AECOPD has not been studied yet, it could be speculated that administration of anabolic steroids as an adjunct to conventional glucocorticoid treatment could prevent or slow down the decline in muscle mass and strength during an AECOPD, based on the observed pro-anabolic effect of nandrolone in combination with the corticosteroids mentioned above. This however must be further confirmed by preclinical research and proper clinical trials. Finally, future research should also focus on timely recognition of the distinct AECOPD-associated triggers of muscle dysfunction in an attempt to minimize their impact. The development of interventions (e.g. pulmonary ‘rehabilitation’, NMES, nutritional support, medication) targeting AECOPD-associated muscle impairments is however still in its infancy. Pre-clinical studies and clinical trials aimed at such intervention strategies are of utmost importance because although AECOPDs clearly have a large impact on the muscle, they at the same time also provide a unique window of opportunity for treatment of muscle impairment in COPD.
Article highlights
Muscle dysfunction is a hallmark of COPD with many adverse clinical implications
Clinical evidence indicates an important role for acute exacerbations of COPD (AECOPDs) in the muscle impairment in COPD, which probably often progresses in a step-wise decline due to the recurrent nature of AECOPDs.
Muscle impairment in turn is associated with a higher exacerbation frequency and longer hospital stays and therefore counteracting muscle deterioration during an AECOPD provides an unprecedented therapeutic opportunity.
Experimental data from animal models provide crucial insights into how an AECOPD or its accompanying individual disease-related triggers (e.g. disuse, systemic inflammation and corticosteroid treatment) may lead to muscle dysfunction, offering novel therapeutic perspectives.
Future research should include 1) prospective studies to reveal the cause and/or consequence relationship between muscle dysfunction and AECOPD, and 2) timed muscle biopsy data during the course of the AECOPD to precisely map the underlying molecular processes thereby providing information on how and when to intervene.
Declaration of interest
S Simons has received speaker fees from AstraZeneca and research grants from GlaxoSmithKline, all unrelated to the submitted work. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease 2020 report. 2020; Cited 2020 Aug 25 https://goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf).
- Adeloye D, Chua S, Lee C, et al. Global and regional estimates of COPD prevalence: systematic review and meta-analysis. J Glob Health. 2015;5(2):020415
- WHO. The top 10 causes of death 2018 Cited 2020 Oct 21 2018. https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- Vanfleteren L, Spruit MA, Wouters EFM, et al. Management of chronic obstructive pulmonary disease beyond the lungs. Lancet Respir Med. 2016;4(11):911–924.
- Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1(1):73–83.
- Benz E, Trajanoska K, Lahousse L, et al. Sarcopenia in COPD: a systematic review and meta-analysis. Eur Respir Rev. 2019;28(154):154.
- Seymour JM, Spruit MA, Hopkinson NS, et al. The prevalence of quadriceps weakness in COPD and the relationship with disease severity. Eur Respir J. 2010;36(1):81–88.
- Celli BR, Locantore N, Tal-Singer R, et al., Emphysema and extrapulmonary tissue loss in COPD: a multi-organ loss of tissue phenotype. Eur Respir J. 51(2): 2. 2018. .
- Gosselink R, Troosters T, Decramer M. Peripheral muscle weakness contributes to exercise limitation in COPD. Am J Respir Crit Care Med. 1996;153(3):976–980.
- Marquis K, Debigare R, Lacasse Y, et al. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(6):809–813.
- Decramer M, Gosselink R, Troosters T, et al. Muscle weakness is related to utilization of health care resources in COPD patients. Eur Respir J. 1997;10(2):417–423.
- Attaway AH, Welch N, Hatipoglu U, et al. Muscle loss contributes to higher morbidity and mortality in COPD : an analysis of national trends. Respirology. 2020. DOI:10.1111/resp.13877
- Maltais F, Decramer M, Casaburi R, et al. An official American thoracic society/European respiratory society statement: update on limb muscle dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;189(9):e15–62.
- Gosker HR, Lencer NH, Franssen FM, et al. Striking similarities in systemic factors contributing to decreased exercise capacity in patients with severe chronic heart failure or COPD. Chest. 2003;123(5):1416–1424.
- Caron MA, Debigare R, Dekhuijzen PN, et al. Comparative assessment of the quadriceps and the diaphragm in patients with COPD. J Appl Physiol. 2009;107(3):952–961.
- Jaitovich A, Barreiro E. Skeletal muscle dysfunction in chronic obstructive pulmonary disease. what we know and can do for our patients. Am J Respir Crit Care Med. 2018;198(2):175–186.
- Sepulveda-Loyola W, Osadnik C, Phu S, et al. Diagnosis, prevalence, and clinical impact of sarcopenia in COPD: a systematic review and meta-analysis. J Cachexia Sarcopenia Muscle. 2020;11(5):1164-1176..
- Sanders KJ, Kneppers AE, van de Bool C, et al. Cachexia in chronic obstructive pulmonary disease: new insights and therapeutic perspective. J Cachexia Sarcopenia Muscle. 2016;7(1):5–22.
- Gosker HR, Engelen MPKJ, van Mameren H, et al. Muscle fiber type IIX atrophy is involved in the loss of fat-free mass in chronic obstructive pulmonary disease. Am J Clin Nutr. 2002;76(1):113–119.
- Langen RC, Gosker HR, Remels AH, et al. Triggers and mechanisms of skeletal muscle wasting in chronic obstructive pulmonary disease. Int J Biochem Cell Biol. 2013;45(10):2245–2256.
- Kneppers AEM, Langen RCJ, Gosker HR, et al. Increased myogenic and protein turnover signaling in skeletal muscle of chronic obstructive pulmonary disease patients with sarcopenia. J Am Med Dir Assoc. 2017;18(7):637 e631–637 e611.
- Gosker HR, Zeegers MP, Wouters EF, et al. Muscle fibre type shifting in the vastus lateralis of patients with COPD is associated with disease severity: a systematic review and meta-analysis. Thorax. 2007;62(11):944–949.
- Gosker HR, van Mameren H, van Dijk PJ, et al. Skeletal muscle fibre-type shifting and metabolic profile in patients with chronic obstructive pulmonary disease. Eur Respir J. 2002;19(4):617–625.
- Rabinovich RA, Bastos R, Ardite E, et al. Mitochondrial dysfunction in COPD patients with low body mass index. Eur Respir J. 2007;29(4):643–650.
- van den Borst B, Slot IG, Hellwig VA, et al. Loss of quadriceps muscle oxidative phenotype and decreased endurance in patients with mild-to-moderate COPD. J Appl Physiol. 2013;114(9):1319–1328.
- Remels AH, Schrauwen P, Broekhuizen R, et al. Peroxisome proliferator-activated receptor expression is reduced in skeletal muscle in COPD. Eur Respir J. 2007;30(2):245–252.
- Leermakers PA, Schols A, Kneppers AEM, et al. Molecular signalling towards mitochondrial breakdown is enhanced in skeletal muscle of patients with chronic obstructive pulmonary disease (COPD). Sci Rep. 2018;8(1):15007.
- Celli BR, Barnes PJ. Exacerbations of chronic obstructive pulmonary disease. Eur Respir J. 2007;29(6):1224–1238.
- Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671
- Wang Z, Singh R, Miller BE, et al. Sputum microbiome temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax. 2018;73(4):331–338.
- Mayhew D, Devos N, Lambert C, et al. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax. 2018;73(5):422–430.
- Couillard S, Larivee P, Courteau J, et al. Eosinophils in COPD exacerbations are associated with increased readmissions. Chest. 2017;151(2):366–373.
- Mathioudakis AG, Janssens W, Sivapalan P, et al. Acute exacerbations of chronic obstructive pulmonary disease: in search of diagnostic biomarkers and treatable traits. Thorax. 2020;75(6):520–527.
- Viniol C, Vogelmeier CF. Exacerbations of COPD. Eur Respir Rev. 2018;27(147):147.
- Simons SO, Hurst JR. Acute exacerbations of pulmonary diseases (ERS monograph. In: Burgel P-R, Contoli M, López-Campos J, editors. COPD: definition, severity and impact of pulmonary exacerbations. Sheffield: European Respiratory Society; 2017. p. 13–24.
- Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med. 2010;363(12):1128–1138.
- Spencer S, Jones PW, Group GS. Time course of recovery of health status following an infective exacerbation of chronic bronchitis. Thorax. 2003;58(7):589–593.
- Aaron SD, Donaldson GC, Whitmore GA, et al. Time course and pattern of COPD exacerbation onset. Thorax. 2012;67(3):238–243.
- Seemungal TA, Donaldson GC, Bhowmik A, et al. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(5):1608–1613.
- Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax. 2012;67(11):957–963.
- Cote CG, Dordelly LJ, Celli BR. Impact of COPD exacerbations on patient-centered outcomes. Chest. 2007;131(3):696–704.
- Lainscak M, Gosker HR, Schols AM. Chronic obstructive pulmonary disease patient journey: hospitalizations as window of opportunity for extra-pulmonary intervention. Curr Opin Clin Nutr Metab Care. 2013;16(3):278–283.
- Osadnik CR, Tee VS, Carson-Chahhoud KV, et al. Non-invasive ventilation for the management of acute hypercapnic respiratory failure due to exacerbation of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2017;7:CD004104.
- Dobler CC, Morrow AS, Beuschel B, et al. Pharmacologic therapies in patients with exacerbation of chronic obstructive pulmonary disease: a systematic review with meta-analysis. Ann Intern Med. 2020;172(6):413–422.
- van Velzen P, Ter Riet G, Bresser P, et al. Doxycycline for outpatient-treated acute exacerbations of COPD: a randomised double-blind placebo-controlled trial. Lancet Respir Med. 2017;5(6):492–499.
- Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med. 2012;186(1):48–55.
- Walters JA, Tan DJ, White CJ, et al. Different durations of corticosteroid therapy for exacerbations of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2018;3:CD006897.
- Nici L, Mammen MJ, Charbek E, et al. Management of chronic obstructive pulmonary disease. An official American thoracic society clinical practice guideline. Am J Respir Crit Care Med. 2020;201(9):e56–e69.
- Sivapalan P, Ingebrigtsen TS, Rasmussen DB, et al. COPD exacerbations: the impact of long versus short courses of oral corticosteroids on mortality and pneumonia: nationwide data on 67 000 patients with COPD followed for 12 months. BMJ Open Respir Res. 2019;6(1):e000407.
- Decramer M, de Bock V, Dom R. Functional and histologic picture of steroid-induced myopathy in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1996;153(6 Pt 1):1958–1964.
- Pouw EM, Ten Velde GP, Croonen BH, et al. Early non-elective readmission for chronic obstructive pulmonary disease is associated with weight loss. Clin Nutr. 2000;19(2):95–99.
- Greening NJ, Harvey-Dunstan TC, Chaplin EJ, et al. Bedside assessment of quadriceps muscle by ultrasound after admission for acute exacerbations of chronic respiratory disease. Am J Respir Crit Care Med. 2015;192(7):810–816
- Giron R, Matesanz C, Garcia-Rio F, et al. Nutritional state during COPD exacerbation: clinical and prognostic implications. Ann Nutr Metab. 2009;54(1):52–58
- Guerri R, Gayete A, Balcells E, et al. Mass of intercostal muscles associates with risk of multiple exacerbations in COPD. Respir Med. 2010;104(3):378–388.
- Perrot L, Greil A, Boirie Y, et al. Prevalence of sarcopenia and malnutrition during acute exacerbation of COPD and after 6 months recovery. Eur J Clin Nutr. 2020. DOI:10.1038/s41430-020-0623-6.
- Amado CA, Garcia-Unzueta MT, Lavin BA, et al. The ratio serum creatinine/serum cystatin c (a surrogate marker of muscle mass) as a predictor of hospitalization in chronic obstructive pulmonary disease outpatients. Respiration. 2019;97(4):302–309.
- Hirai K, Tanaka A, Homma T, et al. Serum creatinine/cystatin C ratio as a surrogate marker for sarcopenia in patients with chronic obstructive pulmonary disease. Clin Nutr. 2020. DOI:10.1016/j.clnu.2020.08.010.
- Tanimura K, Sato S, Sato A, et al. Accelerated loss of antigravity muscles is associated with mortality in patients with COPD. Respiration. 2020;99(4):298–306.
- Horadagoda C, Dinihan T, Roberts M, et al. Body composition and micronutrient deficiencies in patients with an acute exacerbation of chronic obstructive pulmonary disease. Intern Med J. 2017;47(9):1057–1063.
- Torres-Sanchez I, Cabrera-Martos I, Diaz-Pelegrina A, et al. Physical and functional impairment during and after hospitalization in subjects with severe COPD exacerbation. Respir Care. 2017;62(2):209–214.
- Martinez CH, Diaz AA, Meldrum CA, et al. Handgrip strength in chronic obstructive pulmonary disease. associations with acute exacerbations and body composition. Ann Am Thorac Soc. 2017;14(11):1638–1645
- Ansari K, Keaney N, Taylor I, et al. Muscle weakness, health status and frequency of exacerbations in chronic obstructive pulmonary disease. Postgrad Med J. 2012;88(1041):372–376.
- Alajlouni D, Bliuc D, Tran T, et al. Decline in muscle strength and performance predicts fracture risk in elderly women and men. J Clin Endocrinol Metab. 2020;105: e3363–e3373.
- Oliveira CC, Lee AL, McGinley J, et al. Balance and falls in acute exacerbation of chronic obstructive pulmonary disease: a prospective study. COPD. 2017;14(5):518–525.
- Hopkinson NS, Tennant RC, Dayer MJ, et al. A prospective study of decline in fat free mass and skeletal muscle strength in chronic obstructive pulmonary disease. Respir Res. 2007;8:25.
- Vilaro J, Ramirez-Sarmiento A, Martinez-Llorens JM, et al. Global muscle dysfunction as a risk factor of readmission to hospital due to COPD exacerbations. Respir Med. 2010;104(12):1896–1902.
- Alahmari AD, Kowlessar BS, Patel AR, et al. Physical activity and exercise capacity in patients with moderate COPD exacerbations. Eur Respir J. 2016;48(2):340–349.
- Spruit MA, Gosselink R, Troosters T, et al. Muscle force during an acute exacerbation in hospitalised patients with COPD and its relationship with CXCL8 and IGF-I. Thorax. 2003;58(9):752–756.
- Crul T, Spruit MA, Gayan-Ramirez G, et al. Markers of inflammation and disuse in vastus lateralis of chronic obstructive pulmonary disease patients. Eur J Clin Invest. 2007;37(11):897–904.
- Crul T, Testelmans D, Spruit MA, et al., Gene expression profiling in vastus lateralis muscle during an acute exacerbation of COPD. Cell Physiol Biochem. 25(4–5): 491–500. 2010. .
- Duan Y, Zhou M, Xiao J, et al. Prediction of key genes and miRNAs responsible for loss of muscle force in patients during an acute exacerbation of chronic obstructive pulmonary disease. Int J Mol Med. 2016;38(5):1450–1462.
- Pitta F, Troosters T, Probst VS, et al. Physical activity and hospitalization for exacerbation of COPD. Chest. 2006;129(3):536–544.
- Donaldson GC, Wilkinson TM, Hurst JR, et al. Exacerbations and time spent outdoors in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171(5):446–452.
- Hyatt H, Deminice R, Yoshihara T, et al. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: A review of the causes and effects. Arch Biochem Biophys. 2019;662:49–60.
- Dalle S, Koppo K. Is inflammatory signaling involved in disease-related muscle wasting? Evidence from osteoarthritis, chronic obstructive pulmonary disease and type II diabetes. Exp Gerontol. 2020;137:110964.
- Wouters EF, Groenewegen KH, Dentener MA, et al. Systemic inflammation in chronic obstructive pulmonary disease: the role of exacerbations. Proc Am Thorac Soc. 2007;4(8):626–634.
- Vermeeren MAP, Schols A, Wouters EFM. Effects of an acute exacerbation on nutritional and metabolic profile of patients with copd. Eur Respir J. 1997;10:2264–2269.
- Creutzberg EC, Wouters EF, Vanderhoven-Augustin IM, et al. Disturbances in leptin metabolism are related to energy imbalance during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1239–1245.
- Wedzicha JAE-C-C, Miravitlles M, Hurst JR, et al., Management of COPD exacerbations: a European respiratory society/American thoracic society guideline. Eur Respir J. 49(3): 3. 2017. .
- Pouw EM, Koerts-de Lang E, Gosker HR, et al. Muscle metabolic status in patients with severe COPD with and without long-term prednisolone. Eur Respir J. 2000;16(2):247–252.
- Balnis J, Korponay TC, Jaitovich A. AMP-activated protein kinase (AMPK) at the crossroads between co2 retention and skeletal muscle dysfunction in chronic obstructive pulmonary disease (COPD). Int J Mol Sci. 2020;21(3):955.
- Gayan-Ramirez G, Decramer M. Mechanisms of striated muscle dysfunction during acute exacerbations of COPD. J Appl Physiol. 2013;114(9):1291–1299.
- de Theije C, Costes F, Langen RC, et al. Hypoxia and muscle maintenance regulation: implications for chronic respiratory disease. Curr Opin Clin Nutr Metab Care. 2011;14(6):548–553.
- Langen RC, Schols AM, Kelders MC, et al. Muscle wasting and impaired muscle regeneration in a murine model of chronic pulmonary inflammation. Am J Respir Cell Mol Biol. 2006;35(6):689–696.
- Zuo L, Nogueira L, Hogan MC. Effect of pulmonary TNF-alpha overexpression on mouse isolated skeletal muscle function. Am J Physiol Regul Integr Comp Physiol. 2011;301(4):R1025–1031.
- Li X, Moody MR, Engel D, et al. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation. 2000;102(14):1690–1696.
- Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. Embo J. 1996;15(8):1753–1765.
- Matthys P, Dijkmans R, Proost P, et al. Severe cachexia in mice inoculated with interferon-gamma-producing tumor cells. Int J Cancer. 1991;49(1):77–82.
- Guttridge DC, Mayo MW, Madrid LV, et al. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289(5488):2363–2366.
- Li YP, Schwartz RJ, Waddell ID, et al. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. Faseb J. 1998;12(10):871–880.
- Adams V, Mangner N, Gasch A, et al. Induction of MuRF1 is essential for TNF-alpha-induced loss of muscle function in mice. J Mol Biol. 2008;384(1):48–59.
- De Larichaudy J, Zufferli A, Serra F, et al. TNF-alpha- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet Muscle. 2012;2(1):2.
- Langen RCJ, Van Der Velden JLJ, Schols AMWJ, et al. Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. Faseb J. 2004;in press. DOI:10.1096/fj.03-0251com.
- Langen RC, Haegens A, Vernooy JH, et al. NF-kappaB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am J Respir Cell Mol Biol. 2012;47(3):288–297.
- Cai D, Frantz JD, Tawa NE Jr., et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119(2):285–298.
- Remels AH, Gosker HR, Schrauwen P, et al. TNF-alpha impairs regulation of muscle oxidative phenotype: implications for cachexia? Faseb J. 2010;24(12):5052–5062.
- Tang K, Wagner PD, Breen EC. TNF-alpha-mediated reduction in PGC-1alpha may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol. 2009;222(2):320–327.
- Remels AH, Gosker HR, Bakker J, et al. Regulation of skeletal muscle oxidative phenotype by classical NF-kappaB signalling. Biochim Biophys Acta. 2013;1832(8):1313–1325.
- Hakvoort TB, Moerland PD, Frijters R, et al. Interorgan coordination of the murine adaptive response to fasting. J Biol Chem. 2011;286(18):16332–16343.
- Zhao J, Brault JJ, Schild A, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6(6):472–483.
- Shah OJ, Anthony JC, Kimball SR, et al. 4E-BP1 and S6K1: translational integration sites for nutritional and hormonal information in muscle. Am J Physiol Endocrinol Metab. 2000;279(4):E715–729.
- Mofarrahi M, Guo Y, Haspel JA, et al. Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy. 2013;9(10):10.
- Henriksson J. The possible role of skeletal muscle in the adaptation to periods of energy deficiency. Eur J Clin Nutr. 1990;44(Suppl 1(4)):55–64.
- Oost LJ, Kustermann M, Armani A, et al. Fibroblast growth factor 21 controls mitophagy and muscle mass. J Cachexia Sarcopenia Muscle. 2019;10(3):630–642.
- Miyazaki M, Shimozuru M, Tsubota T. Skeletal muscles of hibernating black bears show minimal atrophy and phenotype shifting despite prolonged physical inactivity and starvation. PLoS One. 2019;14(4):e0215489.
- Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998;94(6):557–572.
- de Theije CC, Schols A, Lamers WH, et al. Glucocorticoid receptor signaling impairs protein turnover regulation in hypoxia-induced muscle atrophy in male mice. Endocrinology. 2018;159(1):519–534.
- Gayan-Ramirez G, Vanderhoydonc F, Verhoeven G, et al. Acute treatment with corticosteroids decreases IGF-1 and IGF-2 expression in the rat diaphragm and gastrocnemius. Am J Respir Crit Care Med. 1999;159(1):283–289.
- Gilson H, Schakman O, Combaret L, et al. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology. 2007;148(1):452–460.
- Verhees KJ, Schols AM, Kelders MC, et al. Glycogen synthase kinase-3beta is required for the induction of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2011;301(5):C995–C1007.
- Castillero E, Alamdari N, Lecker SH, et al. Suppression of atrogin-1 and MuRF1 prevents dexamethasone-induced atrophy of cultured myotubes. Metabolism. 2013;62(10):1495–1502.
- Pansters NA, Langen RC, Wouters EF, et al. Synergistic stimulation of myogenesis by glucocorticoid and IGF-I signaling. J Appl Physiol. 2013;114(9):1329–1339.
- van RH B, Dekhuijzen PN, Folgering HT, et al. Effects of long-term low-dose methylprednisolone on rat diaphragm function and structure. Muscle Nerve. 1997;20(8):983–990.
- Huang Y, Chen K, Ren Q, et al. Dihydromyricetin attenuates dexamethasone-induced muscle atrophy by improving mitochondrial function via the PGC-1alpha pathway. Cell Physiol Biochem. 2018;49(2):758–779.
- You YN, Short KR, Jourdan M, et al. The effect of high glucocorticoid administration and food restriction on rodent skeletal muscle mitochondrial function and protein metabolism. PLoS One. 2009;4(4):e5283.
- Troncoso R, Paredes F, Parra V, et al. Dexamethasone-induced autophagy mediates muscle atrophy through mitochondrial clearance. Cell Cycle. 2014;13(14):2281–2295.
- de Theije CC, Langen RC, Lamers WH, et al. Distinct responses of protein turnover regulatory pathways in hypoxia- and semistarvation-induced muscle atrophy. Am J Physiol Lung Cell Mol Physiol. 2013;305(1):L82–91.
- de Theije CC, Schols A, Lamers WH, et al. Hypoxia impairs adaptation of skeletal muscle protein turnover- and AMPK signaling during fasting-induced muscle atrophy. PLoS One. 2018;13(9):e0203630.
- de Theije CC, Langen RC, Lamers WH, et al. Differential sensitivity of oxidative and glycolytic muscles to hypoxia-induced muscle atrophy. J Appl Physiol. 2015;118(2):200–211.
- Slot IG, Schols AM, de Theije CC, et al. Alterations in skeletal muscle oxidative phenotype in mice exposed to 3 weeks of normobaric hypoxia. J Cell Physiol. 2016;231(2):377–392.
- Riva C, Chevrier C, Pasqual N, et al. Bcl- 2/Bax protein expression in heart, slow-twitch and fast-twitch muscles in young rats growing under chronic hypoxia conditions. Mol Cell Biochem. 2001;226(1–2):9–16.
- Di Carlo A, De Mori R, Martelli F, et al. Hypoxia inhibits myogenic differentiation through accelerated MyoD degradation. J Biol Chem. 2004;279(16):16332–16338.
- Mancinelli R, Pietrangelo T, La Rovere R, et al. Cellular and molecular responses of human skeletal muscle exposed to hypoxic environment. J Biol Regul Homeost Agents. 2011;25(4):635–645.
- Atherton PJ, Greenhaff PL, Phillips SM, et al. Control of skeletal muscle atrophy in response to disuse: clinical/preclinical contentions and fallacies of evidence. Am J Physiol Endocrinol Metab. 2016;311(3):E594–604.
- Theeuwes WF, Pansters NAM, Gosker HR, et al. Recovery of muscle mass and muscle oxidative phenotype following disuse does not require GSK-3 inactivation. Biochim Biophys Acta Mol Basis Dis. 2020;1866(6):165740.
- Leermakers PA, Kneppers AEM, Schols A, et al. Skeletal muscle unloading results in increased mitophagy and decreased mitochondrial biogenesis regulation. Muscle Nerve. 2019. DOI:10.1002/mus.26702.
- Koncarevic A, Jackman RW, Kandarian SC. The ubiquitin-protein ligase Nedd4 targets Notch1 in skeletal muscle and distinguishes the subset of atrophies caused by reduced muscle tension. Faseb J. 2007;21(2):427–437.
- Guo Y, Gosker HR, Schols AM, et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(11):1313–1320.
- Pansters NA, Schols AM, Verhees KJ, et al. Muscle-specific GSK-3beta ablation accelerates regeneration of disuse-atrophied skeletal muscle. Biochim Biophys Acta. 2015;1852(3):490–506.
- Gram M, Dahl R, Dela F. Physical inactivity and muscle oxidative capacity in humans. Eur J Sport Sci. 2014;14(4):376–383.
- Kang C, Ji LLPGC. 1alpha overexpression via local transfection attenuates mitophagy pathway in muscle disuse atrophy. Free Radic Biol Med. 2016;93:32–40.
- Cannavino J, Brocca L, Sandri M, et al. PGC1-alpha over-expression prevents metabolic alterations and soleus muscle atrophy in hindlimb unloaded mice. J Physiol. 2014;592(20):4575–4589.
- Gosker HR, Langen RC, Bracke KR, et al. Extrapulmonary manifestations of chronic obstructive pulmonary disease in a mouse model of chronic cigarette smoke exposure. Am J Respir Cell Mol Biol. 2009;40(6):710–716.
- Rinaldi M, Maes K, De Vleeschauwer S, et al. Long-term nose-only cigarette smoke exposure induces emphysema and mild skeletal muscle dysfunction in mice. Dis Model Mech. 2012;5(3):333–341.
- Caron MA, Morissette MC, Theriault ME, et al. Alterations in skeletal muscle cell homeostasis in a mouse model of cigarette smoke exposure. PLoS One. 2013;8(6):e66433.
- Ceelen JJM, Schols A, van Hoof SJ, et al. Differential regulation of muscle protein turnover in response to emphysema and acute pulmonary inflammation. Respir Res. 2017;18(1):75.
- Mattson JP, Poole DC, Hahn SA, et al. Maximal force is unaffected by emphysema-induced atrophy in extensor digitorium longus. Respir Physiol Neurobiol. 2008;161(2):119–124.
- Zhang XL, Pang BS, Hou XL, et al. Oxidative stress and peripheral skeletal muscle dysfunction in rats with emphysema. Chin Med J (Engl). 2010;123(1):40–44.
- Kobayashi S, Fujinawa R, Ota F, et al., A single dose of lipopolysaccharide into mice with emphysema mimics human chronic obstructive pulmonary disease exacerbation as assessed by micro-computed tomography. Am J Respir Cell Mol Biol. 49(6): 971–977. 2013. .
- Gou X, Zhang Q, More S, et al., Repeated exposure to streptococcus pneumoniae exacerbates chronic obstructive pulmonary disease. Am J Pathol. 189(9): 1711–1720. 2019. .
- Vlahos R, Bozinovski S. Preclinical murine models of chronic obstructive pulmonary disease. Eur J Pharmacol. 2015;759:265–271.
- Ceelen JJM, Schols A, Thielen NGM, et al. Pulmonary inflammation-induced loss and subsequent recovery of skeletal muscle mass require functional poly-ubiquitin conjugation. Respir Res. 2018;19(1):80.
- Files DC, D’Alessio FR, Johnston LF, et al. A critical role for muscle ring finger-1 in acute lung injury-associated skeletal muscle wasting. Am J Respir Crit Care Med. 2012;185(8):825–834.
- Haegens A, Schols AM, Gorissen SH, et al. NF-kappaB activation and polyubiquitin conjugation are required for pulmonary inflammation-induced diaphragm atrophy. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L103–110.
- Ceelen JJM, Schols A, Kneppers AEM, et al., Altered protein turnover signaling and myogenesis during impaired recovery of inflammation-induced muscle atrophy in emphysematous mice. Sci Rep. 8(1): 10761. 2018. .
- Chan SMH, Cerni C, Passey S, et al. Cigarette smoking exacerbates skeletal muscle injury without compromising its regenerative capacity. Am J Respir Cell Mol Biol. 2020;62(2):217–230.
- Balnis J, Korponay TC, Vincent CE, et al. IL-13-driven pulmonary emphysema leads to skeletal muscle dysfunction attenuated by endurance exercise. J Appl Physiol. 2020;128(1):134–148.
- Graber TG, Rawls BL, Tian B, et al. Repetitive TLR3 activation in the lung induces skeletal muscle adaptations and cachexia. Exp Gerontol. 2018;106:88–100.
- Abdulai RM, Jensen TJ, Patel NR, et al. Deterioration of limb muscle function during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(4):433–449.
- Gayan-Ramirez G. Relevance of nutritional support and early rehabilitation in hospitalized patients with COPD. J Thorac Dis. 2018;10(Suppl 12):S1400–S1414.
- Jones SE, Barker RE, Nolan CM, et al. Pulmonary rehabilitation in patients with an acute exacerbation of chronic obstructive pulmonary disease. J Thorac Dis. 2018;10(Suppl 12):S1390–S1399.
- Spencer L. Pulmonary rehabilitation for patients with acute chronic obstructive pulmonary disease exacerbations: is the evidence strengthening? Curr Opin Pulm Med. 2018;24(2):147–151.
- Dobler CC, Morrow AS, Farah MH, et al. Nonpharmacologic therapies in patients with exacerbation of chronic obstructive pulmonary disease: a systematic review with meta-analysis. Mayo Clin Proc. 2020;95(6):1169–1183.
- Troosters T, Probst VS, Crul T, et al. Resistance training prevents deterioration in quadriceps muscle function during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(10):1072–1077.
- Martin-Salvador A, Colodro-Amores G, Torres-Sanchez I, et al. Physical therapy intervention during hospitalization in patients with acute exacerbation of chronic obstructive pulmonary disease and pneumonia: a randomized clinical trial. Med Clin (Barc). 2016;146(7):301–304.
- Knaut C, Mesquita CB, Caram LM, et al. Assessment of aerobic exercise adverse effects during COPD exacerbation hospitalization. Can Respir J. 2017;5937908:2017.
- Greening NJ, Williams JEA, Hussain SF, et al. An early rehabilitation intervention to enhance recovery during hospital admission for an exacerbation of chronic respiratory disease: randomised controlled trial.BMJ. 2014; 349:g4315.
- Spruit MA, Singh SJ, Rochester CL, et al., Pulmonary rehabilitation for patients with COPD during and after an exacerbation-related hospitalisation: back to the future? Eur Respir J. 51(1): 1. 2018. .
- Machado A, Oliveira A, Valente C, et al. Effects of a community-based pulmonary rehabilitation programme during acute exacerbations of chronic obstructive pulmonary disease - A quasi-experimental pilot study. Pulmonology. 2020;26(1):27–38.
- Abdellaoui A, Prefaut C, Gouzi F, et al. Skeletal muscle effects of electrostimulation after COPD exacerbation: a pilot study. Eur Respir J. 2011;38(4):781–788.
- Saudny Unterberger H, Martin JG, Gray Donald K. Impact of nutritional support on functional status during an acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1997;156(3 Pt 1):794–799.
- Vermeeren MA, Wouters EF, Geraerts-Keeris AJ, et al. Nutritional support in patients with chronic obstructive pulmonary disease during hospitalization for an acute exacerbation; a randomized controlled feasibility trial. Clin Nutr. 2004;23(5):1184–1192.
- Ogasawara T, Marui S, Miura E, et al. Effect of eicosapentaenoic acid on prevention of lean body mass depletion in patients with exacerbation of chronic obstructive pulmonary disease: A prospective randomized controlled trial. Clin Nutr ESPEN. 2018;28:67–73.
- Polkey MI, Praestgaard J, Berwick A, et al. Activin type II receptor blockade for treatment of muscle depletion in chronic obstructive pulmonary disease. A randomized trial. Am J Respir Crit Care Med. 2019;199(3):313–320
- Passey SL, Hansen MJ, Bozinovski S, et al. Emerging therapies for the treatment of skeletal muscle wasting in chronic obstructive pulmonary disease. Pharmacol Ther. 2016;166:56–70.
- Vanfleteren LE, Spruit MA, Groenen M, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(7):728–735.
- Wall BT, Dirks ML, van Loon LJ. Skeletal muscle atrophy during short-term disuse: implications for age-related sarcopenia. Ageing Res Rev. 2013;12(4):898–906.