
ABSTRACT
Introduction
New targets are needed to enable more accurate diagnosis, monitoring and effective therapy in uncontrolled asthma and chronic obstructive pulmonary disease (COPD), two disorders characterized by pathogenic alterations in the innate immune response. Interestingly, the IL-10-related cytokine IL-26 has been found to be abundantly expressed in human airways and alterations in its expression have been linked to reduced lung function and markers of neutrophilic inflammation in patients with uncontrolled asthma or COPD.
Areas covered
Literature search was conducted on PubMed to identify articles in the field of IL-26 immunology, as well as clinical studies on IL-26 in asthma and COPD, published between 2000 and 2021. We outline the main sources of IL-26 in human airways, as well as the effect of this cytokine on relevant immune and structural cells. Finally, we discuss the potential involvement of IL-26 in the pathophysiology of uncontrolled asthma and COPD.
Expert opinion
IL-26 constitutes a potential target for diagnostic purposes and therapeutic modulation of the innate immune response in the airways of patients with asthma and COPD. It seems reasonable to expect more conclusive evidence of its clinical utility for personalized medicine within the coming 5-year period.
KEYWORDS:
1. Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are heterogenous, chronic, and highly prevalent inflammatory lung diseases in need of new therapeutic targets. Corticosteroids have long been used to successfully treat mild-to-moderate cases of eosinophilic asthma, and the recently developed biological therapies targeting IgE and Th2 cytokines (i.e. IL-4, IL-5, and IL-13) have proven highly effective at reducing symptoms and exacerbations in severe Th2-high endotypes [Citation1,Citation2]. However, up to 60% of patients with severe asthma [Citation3] do not benefit from corticosteroid treatment or anti-Th2 approaches. Similarly, COPD patients do not benefit from corticosteroid treatment in the absence of exacerbations and, most likely, neither do they from anti-Th2 therapy [Citation4]. This is because patients with severe uncontrolled asthma or COPD present an endotype characterized by neutrophil accumulation [Citation5,Citation6] and increased expression of Th1 and Th17 cytokines (e.g. IFN-γ and IL-17) [Citation7–11].
Biological therapies targeting IL-17 have proven beneficial for the clinical treatment of certain chronic inflammatory diseases, namely psoriasis and two of its associated manifestations: psoriatic arthritis and ankylosing spondylitis [Citation12]. Nevertheless, according to the first phase 2a trials for each condition, anti-IL-17 therapy does not yield any effect on lung function in patients with moderate-to-severe asthma [Citation13] or COPD [Citation14]. However, it should be kept in mind that the recruitment criteria in these studies did not specify endotypes displaying airway neutrophilia or high IL-17 levels for the selection of patients. The same reservation can be made against an even more recent phase 2a trial, which forwarded evidence that biological therapy targeting IL-23 (the master Th17 regulator) yields no effect on lung function in patients with severe asthma, even among those with airway neutrophilia [Citation15]. Because of this, it seems necessary to continue to improve our understanding of the cellular and molecular mechanisms that underlie relevant endotypes of asthma and COPD.
Interestingly, the cytokine IL-26 is co-expressed with IFN-γ in Th1 cells [Citation16–18] and with IL-17 and IL-22 in Th17 cells [Citation16,Citation19,Citation20]. IL-26 has potentially important antimicrobial properties and is abundantly expressed in human airways by several cell types. Moreover, alterations in the expression of IL-26 have been associated with reduced lung function and neutrophil accumulation in patients with uncontrolled asthma [Citation21–24] or COPD [Citation25,Citation26]. In this article, we summarize the cellular immunology of IL-26 and review its potential involvement in the pathophysiology of uncontrolled asthma and COPD. We conclude our review by discussing the clinical relevance of these findings.
2. Cellular immunology of IL-26
IL-26 is a homodimeric protein (36 kDa) originally termed AK155 following its discovery in human T cells transformed with herpesvirus saimiri [Citation27]. Given its 25% homology and 47% similarity to IL-10, it is generally considered a member of the IL-10 family of cytokines [Citation27], specifically the IL-20 subfamily. The IL26 gene is flanked by the genes encoding IFNγ and IL22 on chromosome 12q15 [Citation27,Citation28], and to date, expression of IL-26 has been reported in several immune and structural cell types from both healthy and ill individuals. IL-26 has important antimicrobial properties that have been reviewed in detail elsewhere [Citation29,Citation30]. Namely, IL-26 can kill extracellular bacteria via membrane-pore formation [Citation31], reduce the viability of intracellular bacteria [Citation32], form complexes with extracellular DNA (bacterial and self) that can be recognized by Toll-like receptors (TLRs) [Citation31], and modulate viral infectivity [Citation33]. In addition, IL-26 can play more traditional ‘cytokine-like’ roles via the heterodimeric (IL-10R2/IL-20R1) IL-26 receptor complex, which results in activation of the transcription factors STAT1 and STAT3 [Citation34]. Although the expression of IL-10R2 is broad, IL-20R1 is mostly expressed by non-hematopoietic cells [Citation17]. This limited expression pattern should restrict the action of IL-26 but this cytokine can paradoxically act on cells that do not express the archetypical IL-26 receptor complex (discussed below).
2.1. IL-26 expression in human airways
Constitutive expression of IL-26 has been reported in bronchial epithelial cells [Citation35], lung fibroblasts [Citation36], alveolar macrophages, and CD4+ and CD8+ T cells [Citation37] isolated from human airways, as well as neutrophils isolated from blood [Citation38], of healthy donors (). Expression of IL-26 has also been detected in NK and NKT cells found in tuberculous pleural effusion [Citation39], but whether they represent important sources of IL-26 in the airways in the absence of infection has not been investigated. Consistent with its postulated role in host defense, in vitro stimulation with endotoxin increased the expression of IL-26 in alveolar macrophages [Citation37], lung fibroblasts [Citation36], bronchial epithelial cells (preliminary data) [Citation40], and blood neutrophils [Citation38]. A similar response was observed when bronchial epithelial cells and lung fibroblasts were stimulated with several other TLR agonists (preliminary data) [Citation40], when CD4+ T cells isolated from tuberculous pleural effusion were incubated with ESAT-6/CFP-10 (an M. tuberculosis antigen) [Citation39], and when blood neutrophils were exposed to K. pneumoniae [Citation38]. Furthermore, co-stimulation with a TLR3 agonist (poly-IC) and IL-17A synergistically increased the release of IL-26 in bronchial epithelial cells, which was further enhanced in the presence of the Th17 cytokine IL-22 [Citation35]. Monocytes, which like neutrophils rapidly mobilize to the airway lumen upon infection, have been shown to express IL-26 only when stimulated with a combination of endotoxin, IFNγ, and an anti-IL-10 antibody [Citation17], or on-going tuberculosis infection [Citation41] (). However, expression of IL-26 can also be enhanced in response to noninfectious compounds, as in the case of Th17 cells exposed to IL-1β [Citation42], bronchial epithelial cells exposed to IL-17A [Citation35], and alveolar macrophages exposed to water-soluble tobacco smoke components [Citation25]. Finally, it should be kept in mind that TLRs can also be activated by damage-associated molecular patterns (DAMPs), as in the case of histones (TLR2 and 4) or mitochondrial RNA (TLR3) released by dying cells, and thus DAMPs can likely trigger and/or enhance IL-26 expression. Nonetheless, further research into the role of DAMPs in IL-26 expression is warranted.
Figure 1. Clinical immunology of IL-26 in the airways. IL-26 is abundantly expressed in human airways. To date, constitutive expression of IL-26 has been reported in both immune and structural cells relevant to human airways. Moreover, the stimuli shown in red can considerably enhance IL-26 expression in these cells. Although IL-26 stimulates the release of pro-inflammatory cytokines (incl. IL-8, TNFα, GM-CSF, and interferons) in alveolar macrophages, monocytes, T cells, and NK cells, it appears to have more complex effects on bronchial epithelial cells and neutrophils. In bronchial epithelial cells, IL-26 stimulation inhibits the release of pro-inflammatory cytokines, but upregulates the IL-26 receptor complex, and its associated transcription factors STAT1 and STAT3. Meanwhile, IL-26 stimulated neutrophils present enhanced chemotaxis, decreased chemokinesis, reduced secretion of myeloperoxidase (MPO) and elastase (NE), and downregulation of the IL-26 receptor complex, STAT1 and STAT3. These varied outcomes suggest that IL-26 induces cell type-specific effects.
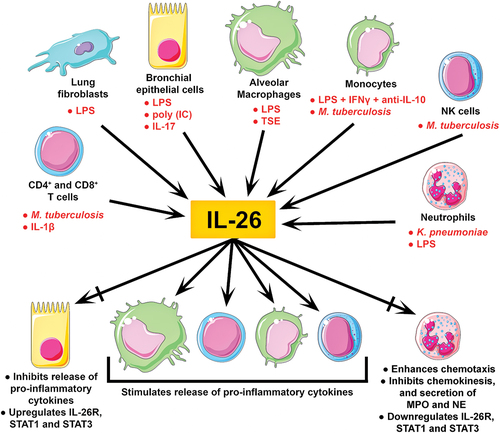
To date, little is known regarding endogenous inhibitors of IL-26 expression. However, a study on human lung fibroblasts found that the endotoxin-enhanced release of IL-26 could be inhibited in vitro by the glucocorticoid hydrocortisone and, to a lesser extent, by the muscarinic receptor antagonist tiotropium (but not aclidinium) and the β2-adrenoreceptor agonist salbutamol [Citation36]. These findings are of particular interest, given that these three medications are widely used for the treatment of COPD and asthma.
2.2. Effect of IL-26 on structural and immune cells of the airways
So far, the expression of the IL-26 receptor complex in human airways has been reported in bronchial epithelial cells [Citation35] and alveolar macrophages only [Citation25], though it has also been identified in blood neutrophils from healthy donors [Citation37] and in CD4+ T cells isolated from tuberculous pleural effusion [Citation39]. Previous studies found that IL-26 induced the expression of TNFα, IL-8, and IL-10 in different human epithelial cell lines and primary keratinocytes via the IL-26 receptor complex [Citation43,Citation44]. Intriguingly, in vitro stimulation with IL-26 increased the transcription of pro-inflammatory mediators (e.g. TNFα, IL-1β, and IL-8) in alveolar macrophages [Citation25] but inhibited the production of these same mediators in bronchial epithelial cells [Citation37], all cells being of human origin. Moreover, bronchial epithelial cells responded to IL-26 stimulation by upregulating the expression of the IL-26 receptor complex and its associated transcription factors STAT1 and STAT3 [Citation37]. In CD4+ T cells from tuberculous pleural effusion, exposure to IL-26 resulted in increased IL-22, TNFα, and IL-6 release [Citation39]. Blood neutrophils, on the other hand, showed enhanced IL-8- and fMLP-dependent chemotaxis, reduced chemokinesis, decreased myeloperoxidase and elastase secretion, as well as downregulation of the IL-26 receptor complex, STAT1, and STAT3 in vitro in response to IL-26 [Citation37,Citation38]. These varied outcomes highlight the cell type- and tissue-specific effects of IL-26 ().
Nevertheless, the results from several studies suggest that IL-26 has important effects on immune cells that do not express the IL-26 receptor complex. Unlike alveolar macrophages, human monocytes do not seem to express the IL-26 receptor complex under homeostatic conditions, but they still upregulate pro-inflammatory mediators in response to IL-26 [Citation45]. Furthermore, IL-26-primed monocytes induced differentiation of naïve CD4+ T cells into Th17 cells in vitro [Citation45]. IL-26 was also shown to inhibit the production of IgA and IgG in anti-CD40-treated B cells [Citation46] and induce the expression of inflammatory cytokines and interferons in NK cells [Citation47] independently of the IL-26 receptor. Although these studies suggest the existence of a hitherto unidentified IL-26 receptor, it is also possible that IL-26 acts on these cells via an altogether different mechanism. For instance, IL-26 was shown to bind and shuttle extracellular DNA (bacterial and self) into the cytoplasm of human myeloid cells, and IL-26/DNA complexes were shown to activate plasmacytoid dendritic cells (pDCs) and monocytes via the TLR9, stimulator of interferon genes (STING), and inflammasome pathways [Citation31,Citation48]. Although different mechanisms have been put forward to explain how IL-26 might enter the cytoplasm [Citation29], it remains to be determined whether IL-26 could shuttle other DAMPs besides extracellular DNA and whether internalized IL-26 could have effects by itself even in the absence of DAMPs.
2.3. In vivo studies
IL-26 is constitutively expressed in human airways, and intra-bronchial exposure to endotoxin increased the concentration of IL-26 in bronchoalveolar lavage (BAL) fluid from healthy human subjects [Citation37]. Given that mice lack the IL26 gene [Citation28], mechanistic studies regarding the function of this cytokine in health and disease have been hampered substantially. Nonetheless, the archetypical IL-26 receptor complex is expressed in mouse airways [Citation49,Citation50] and the effect of human IL-26 on mice has been investigated. Intranasal instillation of recombinant human IL-26 (rhIL-26) enhanced the endotoxin-mediated accumulation of leukocytes (mainly neutrophils and macrophages) and expression of pro-inflammatory mediators in the airway lumen of wildtype mice [Citation49]. This confirms in vivo the enhanced chemotaxis of neutrophils observed in vitro [Citation37]. However, a single instillation of rhIL-26 alone exerted no clear effect on wildtype mice in the first 72 hours post-administration. In a model of transplant-related obliterative bronchiolitis, mice engineered to express human IL-26 showed increased IL-26 expression and collagen deposition in airway tissue 4 weeks after transplantation, which was inhibited by neutralizing anti-IL-26 antibodies [Citation50]. These studies suggest that while IL-26 might not have any substantial short-term effects in vivo by itself, the long-term upregulation of IL-26 can lead to airway remodeling. Despite these findings, most animal models of disease have not been used to investigate whether IL-26 may play a role in asthma or COPD.
2.3.1. IL-26 in asthma
Among patients with asthma, distinct inflammatory profiles (i.e. endotypes) are associated with specific levels of disease control and response to treatment (i.e. phenotype). The endotype with high eosinophil counts (> 3%) in induced sputum can present mild-to-severe disease but responds well to corticosteroids and more specific anti-Th2 therapies [Citation51]. However, up to 50% of all patients with asthma have a non-eosinophilic endotype [Citation52–54], displaying neutrophilic or paucigranulocytic inflammation. The neutrophilic endotype of asthma (defined as > 61 or 76% neutrophils [depending on the study] in induced sputum) accounts for 18–25% of all cases [Citation53,Citation54], does not respond to available treatments [Citation55], and is associated with more severe and poorly controlled disease (asthma control test [ACT] < 20) [Citation56,Citation57]. The paucigranulocytic endotype of asthma, on the other hand, shows no sputum eosinophilia or neutrophilia and manifests milder symptoms and better lung function than the eosinophilic and neutrophilic endotypes [Citation58]. Given that IL-26 can be involved in neutrophil mobilization and Th17-mediated inflammation, key features of certain endotypes, this cytokine harbors therapeutic potential for the phenotype of uncontrolled asthma.
To date, the role of IL-26 in asthma has been the subject of four observational studies. A large case-control study showed that the concentration of IL-26 is higher in serum from adults with asthma than in healthy subjects regardless of disease severity, degree of control, or atopic status [Citation24]. Another study focused exclusively on women with severe uncontrolled asthma and found that the concentration of IL-26 is increased not only in blood, but also in induced sputum (i.e. proximal airways) [Citation23]. In contrast, our group has shown that the concentration of IL-26 is downregulated in BAL fluid (i.e. distal airways) of adult patients with mild-to-moderate asthma [Citation22]. This discrepancy might be explained by the fact that each study probed different airway compartments and focused on populations with distinct disease severity. It is known that proximal and distal airways have unique immune and structural cell microenvironments [Citation59,Citation60], which might present different alterations in IL-26 expression during a complex inflammatory disease such as asthma. In addition, the downregulation of IL-26 observed in BAL fluid from patients with mild-to-moderate asthma might be associated with a better response to glucocorticoid therapy, which has been shown to inhibit IL-26 production in vitro [Citation36]. Severe and uncontrolled cases of asthma are resistant to glucocorticoid therapy and might favor IL-26 production. Indeed, when comparing the protein concentration of IL-26 in patients with controlled (ACT ≥ 20) versus uncontrolled (ACT < 20) asthma, IL-26 was found to be upregulated in sputum from children with severe uncontrolled non-eosinophilic asthma [Citation21] and in BAL fluid from adults with mild-to-moderate uncontrolled asthma [Citation22]. In summary, while changes in the expression of IL-26 appear to be compartment-specific and dependent on disease severity in asthma in general, evidence suggests that IL-26 is always higher in the airways of patients with uncontrolled asthma, particularly in non-eosinophilic endotypes ().
Figure 2. Principal involvement of IL-26 in asthma. One study showed that the concentration of IL-26 in is higher in patients with asthma regardless of disease-severity, degree of control, or atopic status. Furthermore, another study found that patients with severe asthma have increased levels of IL-26 in induced sputum (i.e. proximal airways). In contrast, another study showed that patients with mild-to-moderate asthma have decreased levels of IL-26 in BAL fluid (i.e. distal airways). Thus, alterations in IL-26 expression appear to be compartment-specific and disease severity-dependent in patients with asthma. However, the concentration of IL-26 in induced sputum and BAL fluid is higher in children and adults with uncontrolled asthma, which hints at a potential link between IL-26 and glucocorticoid-resistant phenotypes of asthma. Although it is unclear whether alterations in IL-26 expression could be a causative factor, a consequence, or a defense mechanism of asthma, these changes have been associated with reduced lung function and increased markers of neutrophilic inflammation.
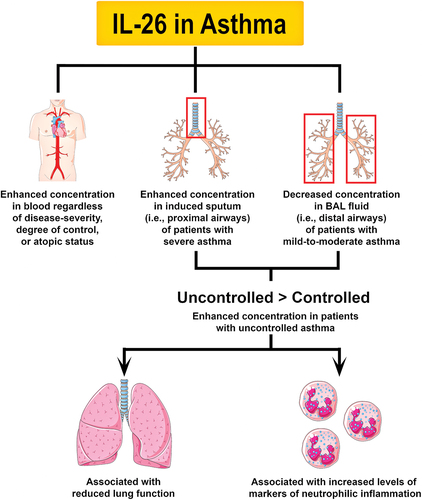
Although the role of IL-26 in asthma has not been verified in interventional studies, some important correlations have been identified (). Thus, IL-26 in induced sputum [Citation23] and BAL fluid [Citation22] correlated with reduced lung function (i.e. FEV1% or FEV1/FVC ratio) in adult patients with asthma. IL-26 also correlated with increased IL-8 in BAL fluid from adults with asthma [Citation22], and with increased blood neutrophils in children with asthma [Citation21]. Among adults with uncontrolled asthma, IL-26 correlated with increased IL-17A and neutrophil counts in sputum [Citation23] and reduced eosinophils and lymphocytes in BAL samples [Citation22]. Of note, adult patients with uncontrolled asthma who had low expression of IL-26 (< median) also had worse asthma control (i.e. lower ACT score) [Citation22]. Finally, the inclusion of IL-26 in a predictive disease model resulted in higher accuracy when discriminating severe asthma from other inflammatory airway diseases [Citation61]. Taken together, the evidence put forward by these studies suggests that IL-26 is linked to uncontrolled asthma, but it is unclear whether IL-26 is a causative factor, a consequence, or even a natural defense mechanism against disease progression. Notably, a recently identified IL26 gene polymorphism (rs7134599 A) was shown to reduce the risk of developing severe asthma, while another (rs2870946 CC) was shown to increase the risk of asthma in patients who smoke [Citation24]. Although this study forwards IL-26 as a potential causative factor in asthma development, the underlying mechanisms are yet to be defined.
3. IL-26 in COPD
Although available therapies can reduce exacerbations and certain symptoms, COPD remains a deadly disease, in fact the third deadliest according to the World Health Organization (WHO) [Citation62]. Long-term smoking is the predominant risk factor associated with COPD development, which often coincides with the development of its two major comorbidities: chronic bronchitis and/or emphysema. Besides tobacco smoking, COPD may also develop due to long-term inhalation of biomass smoke from indoors cooking [Citation63] or occupational exposure to noxious particles (e.g. silica, asbestos, and asphalt) [Citation64–66]. The clearest genetic predisposition for COPD is found in patients deficient in α1-antitrypsin, a potent inhibitor of neutrophil elastase, which affects approximately 1% of all COPD patients [Citation67]. It seems feasible that the causative factor may relate to immunological endotypes and clinical phenotypes of COPD. For instance, exposure to biomass smoke has been linked to more chronic bronchitis [Citation68], less emphysema [Citation69], and more serum IgE than exposure to cigarette smoke [Citation70]. To date, the role of IL-26 in COPD has only been addressed in relation to COPD among long-term smokers. Just like in uncontrolled asthma, IL-26 represents a potential therapeutical target for COPD among patients with the endotypes displaying neutrophilic airway inflammation and/or enhanced Th17 signaling.
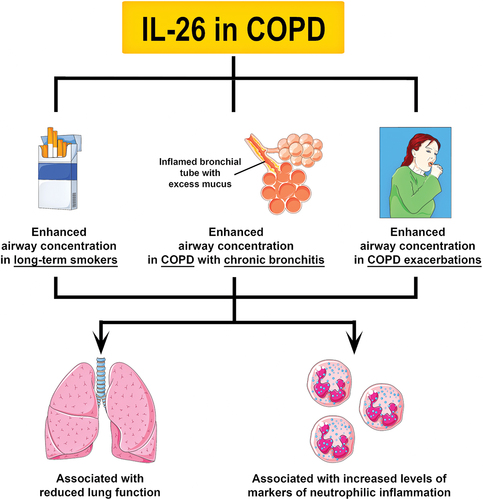
Two independent studies recently showed that the concentration of IL-26 is increased in the airways of COPD patients and that this increase correlated with reduced lung function (i.e. FEV1% and FEV1/FVC ratio), increased neutrophil mobilization, enhanced expression of pro-inflammatory mediators, and higher body mass index (BMI) [Citation25,Citation26] (). Nevertheless, from a statistical point-of-view, the increase in IL-26 seemed to be a consequence of long-term smoking rather than the disease itself. When all smokers with and without COPD were grouped together, it became apparent that increased IL-26 levels were associated with chronic bronchitis and colonization by pathogenic bacteria [Citation25]. However, it is important to consider that these analyses were carried out in samples obtained during stable clinical conditions. During exacerbations, long-term smokers with COPD displayed even higher IL-26 levels in induced sputum. In fact, a detectable increase in IL-26 preceded the exacerbations by an average of 17 days [Citation25]. This finding suggests that IL-26 is involved in the actual course of COPD and, potentially, that it may even bear potential as a biomarker of exacerbations. Intriguingly, inflammatory cells isolated from the airways of smokers with and without COPD expressed less IL-26 receptor complex and its associated transcription factors STAT1 and STAT3 [Citation25]. It is still unclear whether this finding represents a protective mechanism against the pro-inflammatory activity of IL-26 or a defect that drives an increase in IL-26 production.
Figure 3. Principal involvement of IL-26 in COPD. To date, two studies have shown that the concentration of IL-26 is increased in induced sputum and BAL fluid from long-term smokers, and COPD patients with chronic bronchitis or exacerbations. Like in asthma, an increase in IL-26 expression has been associated with reduced lung function and increased markers of neutrophilic inflammation in long-term smokers and COPD patients. Notably, an increase in IL-26 in the airways of COPD patients was shown to precede exacerbations. Further research is needed to determine the specific role of IL-26 in COPD.
4. Conclusion
IL-26 has both direct and indirect antimicrobial functions and is constitutively expressed by many different immune and structural cell types in human airways. The local expression of IL-26 is upregulated in response to pathogen-associated molecular patterns, and IL-26 may enhance neutrophil mobilization to sites of infection. Interestingly, this IL-10-related cytokine exerts different inhibitory and pro-inflammatory effects on epithelial and immune cells from human airways, which highlights the importance of improving our understanding of its complete receptor biology. Based on a limited number of clinical studies on asthma and COPD, it can be concluded that local IL-26 is enhanced in the airways of patients with uncontrolled asthma, long-term smokers without COPD, and in long-term smokers with COPD who are undergoing exacerbations or have chronic bronchitis. Notably, these clinical studies have linked the upregulation of IL-26 with reduced lung function and markers of neutrophilic inflammation in patients with uncontrolled asthma and in patients with COPD and chronic bronchitis or exacerbations.
5. Expert opinion
From an immunological point of view, IL-26 emerges as a somewhat unique cytokine, given its capacity to kill bacteria directly, through a bactericidal effect, and indirectly, by promoting the mobilization of neutrophils. Clearly, due to its strong mechanistic link to the innate immune response in the airways, this ‘antimicrobial cytokine’ is interesting for asthma and COPD, two airway disorders in which the clinical course is impaired by bacterial colonization and infection. Moreover, the proven involvement of IL-26 in uncontrolled asthma, in COPD with chronic bronchitis or exacerbations, as well as its association with impaired lung function in these disorders, forwards it as a target of interest. Because IL-26 is abundantly expressed in human airways, this protein is also an accessible target for diagnostic purposes. However, it remains unclear whether the alterations in local IL-26 found in asthma and COPD represent a reactive and protective response or a truly pathogenic one, a player that contributes to the progress of the referred airway diseases.
6. Five-year view
It seems both important and feasible that the intriguing receptor biology behind the dualistic effects of IL-26 can be dissected and better understood within the coming five-year period. We think that this will be necessary to determine how IL-26 should be targeted for clinical utility, whether in asthma or COPD. Likewise, it will be important to improve the understanding of which specific phenotypes of these two disorders may benefit from targeting IL-26. Notably, this key area of ‘target research’ will require extensive and costly clinical studies, combining a careful in-depth characterization of immunology, bacteriology, virology, and critical clinical features such as impaired lung function, symptoms, and exacerbation frequency in the coming years. In addition, it will be important to improve the understanding of how existing pharmacotherapy, including corticosteroids, affect immune signaling via IL-26 in the airways. Hopefully, this type of clinical research, with a stringent biological focus, can teach us how IL-26 can be utilized as a biomarker for diagnosis and monitoring or even as therapeutic target in two of the most prevalent and costly airway diseases at the global level.
Article highlights
IL-26 is constitutively expressed by both structural and immune cells in human airways.
Expression of IL-26 is upregulated in response to pathogen-associated molecular patterns, tobacco smoke, and Th17 cytokines.
IL-26 may enhance neutrophil mobilization to the airways in response to infections or chronic inflammation.
IL-26 exerts pro-inflammatory and inhibitory effects on structural and immune cells that may be cell type-specific and not always occur via the canonical IL-26 receptor complex.
IL-26 is upregulated in patients with uncontrolled asthma, long-term smokers without COPD, and long-term smokers with COPD who are undergoing exacerbations or have chronic bronchitis.
This IL-26 upregulation has been linked to reduced lung function and markers of neutrophilic inflammation.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The free-of-charge and non-copyright support of “smart.servier.com” in the processing of figures is gratefully acknowledged.
Additional information
Funding
References
- Farne HA, Wilson A, and Powell C, et al. Anti-IL5 therapies for asthma. Cochrane Database Syst Rev. 2017;9:(9):CD010834 .
- Bagnasco D, Ferrando M, Varricchi G, et al. A critical evaluation of Anti-IL-13 and Anti-IL-4 strategies in severe asthma. Int Arch Allergy Immunol. 2016;170(2):122–131.
- Wang E, Wechsler ME, Tran TN, et al. Characterization of severe asthma worldwide: data from the international severe asthma registry. Chest. 2020;157(4):790–804.
- Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131(3):636–645.
- Snelgrove RJ, Patel DF, Patel T, et al. The enigmatic role of the neutrophil in asthma: friend, foe or indifferent? Clinical and Experimental Allergy: Journal of the British Society for Allergy and Clinical Immunology. 2018;48:1275–1285.
- Butler A, Walton GM, Sapey E. Neutrophilic inflammation in the pathogenesis of chronic obstructive pulmonary disease. COPD. 2018;15(4):392–404.
- Newcomb DC, Peebles RS. Th17-mediated inflammation in asthma. Curr Opin Immunol. 2013;25(6):755–760.
- le Rouzic O, Pichavant M, and Frealle E, et al. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50 4 1602434 .
- Xu W, Li R, Sun Y. Increased IFN-γ-producing Th17/Th1 cells and their association with lung function and current smoking status in patients with chronic obstructive pulmonary disease. BMC Pulm Med. 2019;19(1):137.
- Yu Y, Zhao L, Xie Y, et al. Th1/Th17 cytokine profiles are associated with disease severity and exacerbation frequency in COPD patients. Int J Chron Obstruct Pulmon Dis. 2020;15:1287–1299.
- Corrigan CJ, Kay AB. CD4 T-lymphocyte activation in acute severe asthma. Relationship to disease severity and atopic status. Am Rev Respir Dis. 1990;141(4_pt_1):970–977.
- Reich K, Warren RB, Coates LC, et al. Long-term efficacy and safety of secukinumab in the treatment of the multiple manifestations of psoriatic disease. J Eur Acad Dermatol Venereol JEADV. 2020;34(6):1161–1173.
- Busse WW, Holgate S, and Kerwin E, et al. Randomized, double-blind, placebo-controlled study of brodalumab, a human Anti–IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188(11):1294–1302.
- Eich A, Urban V, and Jutel M, et al. A randomized, placebo-controlled phase 2 trial of CNTO 6785 in chronic obstructive pulmonary disease. COPD. 2017;14(5):476–483.
- Brightling CE, Nair P, and Cousins DJ, et al. Risankizumab in severe asthma - a phase 2a, placebo-controlled trial. N Engl J Med. 2021;385(18):1669–1679.
- Collins PL, Henderson MA, Aune TM. Lineage-specific adjacent IFNG and IL26 genes share a common distal enhancer element. Genes Immun. 2012;13(6):481–488.
- Nagalakshmi ML, Murphy E, McClanahan T, et al. Expression patterns of IL-10 ligand and receptor gene families provide leads for biological characterization. Int Immunopharmacol. 2004;4(5):577–592.
- Wolk K, Kunz S, Asadullah K, et al. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Iimmunol. 2002;168(11):5397–5402.
- Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8(9):950–957.
- Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9(6):641–649.
- Konradsen JR, Nordlund B, Levänen B, et al. The cytokine interleukin-26 as a biomarker in pediatric asthma. Respir Res. 2016;17(1):32.
- Tufvesson E, Jogdand P, Che KF, et al. Enhanced local production of IL-26 in uncontrolled compared with controlled adult asthma. J Allergy Clin Immunol. 2019;144(4):1134–1136.e10.
- Louhaichi S, Mlika M, Hamdi B, et al. Sputum IL-26 is overexpressed in severe asthma and induces proinflammatory cytokine production and Th17 cell generation: a case-control study of women. J Asthma Allergy. 2020;13:95–107.
- Salhi M, Tizaoui K, and Louhaichi S, et al. IL-26 gene variants and protein expression in Tunisian asthmatic patients. Cytokine. 2020;134:155206.
- Che KF, Tufvesson E, Tengvall S, et al. The neutrophil-mobilizing cytokine interleukin-26 in the airways of long-term tobacco smokers. Clin Sci. 2018;132(9):959–983.
- Savchenko L, Mykytiuk M, Cinato M, et al. IL-26 in the induced sputum is associated with the level of systemic inflammation, lung functions and body weight in COPD patients. Int J Chron Obstruct Pulmon Dis. 2018;13:2569–2575.
- Knappe A, Hör S, Wittmann S, et al. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J Virol. 2000;74(8):3881–3887.
- Dumoutier L, van Roost E, Ameye G, et al. IL-TIF/IL-22: genomic organization and mapping of the human and mouse genes. Genes Immun. 2000;1(8):488–494.
- Larochette V, Miot C, Poli C, et al. IL-26, a cytokine with roles in extracellular DNA-induced inflammation and microbial defense. Front Immunol. 2019;10:204.
- Che KF, Tengvall S, Lindén A. Interleukin-26 in host defense and inflammatory disorders of the airways. Cytokine Growth Factor Rev. 2021;57:1–10.
- Meller S, Di Domizio J, Voo KS, et al. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. 2015;16(9):970–979.
- Dang AT, Teles RM, Weiss DI, et al. IL-26 contributes to host defense against intracellular bacteria. J Clin Invest. 2019;129(5):1926–1939.
- Braum O, Klages M, Fickenscher H. The cationic cytokine IL-26 differentially modulates virus infection in culture. PloS one. 2013;8(7):e70281.
- Sheikh F, Baurin VV, Lewis-Antes A, et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J Iimmunol. 2004;172(4):2006–2010.
- Che KF, Kaarteenaho R, Lappi-Blanco E, et al. Interleukin-26 production in human primary bronchial epithelial cells in response to viral stimulation: modulation by Th17 cytokines. Mol Med. 2017;23(1):247–257.
- Che KF, Sun J, Linden A. Pharmacological modulation of endotoxin-induced release of IL-26 in human primary lung fibroblasts. Front Pharmacol. 2019;10:956.
- Che KF, Tengvall S, and Levänen B, et al. Interleukin-26 in antibacterial host defense of human lungs. Effects on neutrophil mobilization. Am J Respir Crit Care Med. 2014;190(9):1022–1031.
- Che KF, Paulsson M, Piersiala K, et al. Complex involvement of interleukin-26 in bacterial lung infection. Front Immunol. 2021;12:761317.
- Zhang M, Niu Y-R, Liu J-Y, et al. Interleukin-26 upregulates interleukin-22 production by human CD4+ T cells in tuberculous pleurisy. J Mol Med. 2019;97(5):619–631.
- Che KF, Ji J, and Palmberg L, et al. Signaling mechanisms and release of IL-26 in human structural lung cells in response to pathogen associated molecular patterns. In: A32 MICROBIAL INTERACTIONS AND HOST DEFENSE. New York: American Thoracic Society;2015:A1309–A1309.
- Guerra-Laso JM, Raposo-García S, García-García S, et al. Microarray analysis of Mycobacterium tuberculosis -infected monocytes reveals IL26 as a new candidate gene for tuberculosis susceptibility. Immunology. 2015;144(2):291–301.
- Weiss DI, Ma F, Merleev AA, et al. IL-1β induces the rapid secretion of the antimicrobial protein IL-26 from Th17 cells. The Journal of Immunology. 2019;203(4):911–921.
- Hör S, Pirzer H, Dumoutier L, et al. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem. 2004;279(32):33343–33351.
- Dambacher J, Beigel F, Zitzmann K, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58(9):1207–1217.
- Corvaisier M, Delneste Y, Jeanvoine H, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. 2012;10(9):e1001395.
- Hummelshoj L, Ryder LP, Poulsen LK. The role of the interleukin-10 subfamily members in immunoglobulin production by human B cells. Scand J Immunol. 2006;64(1):40–47.
- Miot C, Beaumont E, Duluc D, et al. IL-26 is overexpressed in chronically HCV-infected patients and enhances TRAIL-mediated cytotoxicity and interferon production by human NK cells. Gut. 2015;64(9):1466–1475.
- Poli C, Augusto JF, Dauvé J, et al. IL-26 confers proinflammatory properties to extracellular DNA. J Iimmunol. 2017;198(9):3650–3661.
- Bao A, Che KF, Bozinovski S, et al. Recombinant human IL-26 facilitates the innate immune response to endotoxin in the bronchoalveolar space of mice in vivo. PloS one. 2017;12(12):e0188909.
- Ohnuma K, Hatano R, Aune TM, et al. Regulation of pulmonary graft-versus-host disease by IL-26 + CD26 + CD4 T lymphocytes. J Iimmunol. 2015;194(8):3697–3712.
- Busse WW. Biological treatments for severe asthma: a major advance in asthma care. Allergol Int. 2019;68(2):158–166.
- McGrath KW, Icitovic N, Boushey HA, et al. A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med. 2012;185(6):612–619.
- Schleich FN, Manise M, Sele J, et al. Distribution of sputum cellular phenotype in a large asthma cohort: predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulm Med. 2013;13(1):11.
- Brooks CR, van Dalen CJ, Zacharasiewicz A, et al. Absence of airway inflammation in a large proportion of adolescents with asthma. Respirology. 2016;21(3):460–466.
- Crisford H, Sapey E, Rogers GB, et al. Neutrophils in asthma: the good, the bad and the bacteria. Thorax. 2021;76(8):835–844.
- Moore WC, Hastie AT, Li X, et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol. 2014;133(6):1557–63.e5.
- Ray A, Kolls JK. Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. 2017;38(12):942–954.
- Ntontsi P, Loukides S, Bakakos P, et al. Clinical, functional and inflammatory characteristics in patients with paucigranulocytic stable asthma: comparison with different sputum phenotypes. Allergy. 2017;72(11):1761–1767.
- Bieńkowska-Haba M, Cembrzyńska-Nowak M, Liebhart J, et al. Comparison of leukocyte populations from bronchoalveolar lavage and induced sputum in the evaluation of cellular composition and nitric oxide production in patients with bronchial asthma. Arch Immunol Ther Exp (Warsz). 2002;50(1):75–82.
- Hewitt RJ, Lloyd CM. Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol. 2021;21(6):347–362.
- Adel-Patient K, Grauso M, Abou-Taam R, et al. A comprehensive analysis of immune constituents in blood and bronchoalveolar lavage allows identification of an immune signature of severe asthma in children. Front Immunol. 2021;12:700521.
- World Health Organization. The top 10 causes of death. 2020 Accessed 01 October 2021. Available from: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
- Capistrano SJ, van Reyk D, Chen H, et al. Evidence of biomass smoke exposure as a causative factor for the development of COPD. Toxics. 2017;5(4):36.
- Hnizdo E, Vallyathan V. Chronic obstructive pulmonary disease due to occupational exposure to silica dust: a review of epidemiological and pathological evidence. Occup Environ Med. 2003;60(4):237–243.
- Huuskonen O, Kivisaari L, Zitting A, et al. Emphysema findings associated with heavy asbestos-exposure in high resolution computed tomography of finnish construction workers. J Occup Health. 2004;46(4):266–271.
- Grahn K, Gustavsson P, Andersson T, et al. Occupational exposure to particles and increased risk of developing chronic obstructive pulmonary disease (COPD): a population-based cohort study in Stockholm, Sweden. Environ Res. 2021;200:111739.
- Silverman EK, Sandhaus RA. Alpha 1 -antitrypsin deficiency. N Engl J Med. 2009;360(26):2749–2757.
- Pérez-Padilla R, Regalado J, Vedal S, et al. Exposure to biomass smoke and chronic airway disease in Mexican women. A case-control study. Am J Respir Crit Care Med. 1996;154(3):701–706.
- Camp PG, Ramirez-Venegas A, Sansores RH, et al. COPD phenotypes in biomass smoke- versus tobacco smoke-exposed Mexican women. Eur Respir J. 2014;43(3):725–734.
- Olloquequi J, Jaime S, Parra V, et al. Comparative analysis of COPD associated with tobacco smoking, biomass smoke exposure or both. Respir Res. 2018;19(1):13.