
ABSTRACT
Introduction
Gastrointestinal (GI)-related symptoms, complications, and comorbidities in cystic fibrosis (CF) are common and research to reduce their burden is a priority for the CF community. To enable future research, this review aimed to summarize the range of GI symptoms, complications and comorbidities seen in CF, the underlying pathophysiology, and treatments.
Areas covered
This was a rapid systematic review undertaken using the recommendations from the Cochrane Rapid Reviews Methods Group. We searched databases including PubMed, Embase, Medline and the Cochrane database and identified those studies reporting GI-related symptoms, complications, or comorbidities in CF or their treatment. Our searches identified 2,930 studies and a total 119 studies met our inclusion criteria. Where a prevalence could be determined, GI symptoms were reported in 33.7% of study participants. The range of symptoms reported was broad and the highest median prevalence included flatulence (43.5%), bloating and abdominal distension (36%), and fatty stool (36%). Meconium ileus was reported in 12% and distal intestinal obstruction syndrome in 8.5%
Expert opinion
GI-related symptoms, complications, and comorbidities in CF are common. More consistent characterization and recording of these symptoms in clinical studies may help achieve the priority of reducing the burden of GI disease in CF.
1. Introduction
Cystic fibrosis (CF) is the most common life limiting, autosomal recessive inherited disease affecting people of North European decent [Citation1]. As of 2021, in the United Kingdom CF affects 10,908 people with just over 6,000 of the population aged 16 years or older [Citation2]. CF is a multisystem disease that is caused by mutations in the gene coding for the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) which is located on the long arm of chromosome 7. CFTR is an anion channel which is responsible for chloride and bicarbonate transport across the apical membrane of cells.
Early reports of cystic fibrosis describe: intestinal obstruction in the newborn period; pancreatic insufficiency causing steatorrhea; deficiency of fat soluble vitamins; and early onset of purulent bronchitis [Citation3]. In the early years of CF care, diagnosis was made through examination of duodenal fluid for evidence of pancreatic insufficiency [Citation4]. Seventy years ago, most children with CF did not reach their fifth birthday [Citation5].
The effects of CFTR mutation are best understood within the respiratory system, but less is known about the pathophysiology of CF in the gut. Through animal models, it is understood that abnormal chloride and bicarbonate secretion causes an acidic, dehydrated environment and increased mucus viscosity [Citation6,Citation7]. The thickened chyme resulting from the abnormal environment and thick mucus may then cause obstruction within the terminal ileum. Throughout this review, the theories explaining gastrointestinal (GI) dysfunction in CF will be explored.
Reducing the burden of GI symptoms and complications is important to the patient and clinical communities in CF. Work done through a James Lind Alliance Priority Setting Partnership asked people with CF (pwCF), families, and healthcare professionals to determine areas of research in CF which should be prioritized. This work was undertaken in 2017 and refreshed in 2022 and at both times ‘how can we relieve gastro-intestinal (GI) symptoms, such as stomach pain, bloating, and nausea in people with cystic fibrosis’ has placed within the top 3 priorities [Citation8,Citation9]. From the 2022 refresh, there is also a new priority asking for research to determine the extra-pulmonary effects of modulators including their effects on the GI system [Citation9].
The development of CFTR modulators has revolutionized the landscape of treatment in CF, working through both correcting defective trafficking of CFTR to the cell surface and potentiating the function of CFTR on the cell surface. The first CFTR modulator to be developed and used clinically, ivacaftor, was shown in trials to significantly improve respiratory function, as assessed by FEV1, reduce pulmonary exacerbations and improve body mass index (BMI) [Citation10]. With the development of the triple combination therapy elexacaftor/tezacaftor/ivacaftor (ETI) 90% of pwCF have genotypes eligible for treatment with modulators, although access to modulators varies across healthcare systems [Citation11].
The effects that modulators have on the GI tract is less clear. With ivacaftor, there is limited evidence from case reports and case series suggesting a restoration in exocrine pancreatic function after starting ivacaftor, particularly within the pediatric population [Citation12–17]. During the KIWI and KLIMB studies, both open-label phase 3 studies in children aged 2–5 years, fecal elastase was found to improve after 24 weeks and 84 weeks of ivacaftor, respectively, suggesting some recovery of pancreatic exocrine function [Citation18,Citation19]. For ETI, a large, multicenter trial used the CFAbd-score (a CF-specific patient reported outcome measure) to measure abdominal symptoms and associated quality of life before and after starting ETI. This study found significant improvements in all domains after 24 weeks of ETI treatment, compared to baseline [Citation20]. However, there are a limited number of trials and other well-designed studies, leaving many gaps in the evidence for treatments to reduce the GI symptoms and complications in CF [Citation21].
Closing these evidence gaps relating to GI disease in CF has become particularly important as the life expectancy of pwCF continues to increase and respiratory disease has a less damaging effect on quality of life. To enable this, an up-to-date, objective review of the range and causes of GI symptoms, complications, and comorbidities experienced by pwCF is needed.
Therefore, the aim of this review was to;
Provide an overview of the pathophysiology underlying GI dysfunction in CF
Present the range and prevalence of GI symptoms, complications, and comorbidities associated with CF
Describe the evidence for treating GI symptoms, complications, and comorbidities in CF.
2. Methods
This review was conducted using the guidance from the Cochrane Rapid Reviews Methods Group [Citation22]. This approach was adopted to allow for the expected large number of studies.
The full review protocol can be found on the Open Science Framework (DOI 10.17605/OSF.IO/2SVPX). We searched Medline, Embase, PubMed, and the Cochrane Library for keywords related to cystic fibrosis and GI symptoms and complications from January 2012 to December 2022 (see supplement S1 for full search strategy). Searches were also conducted on clinicaltrials.gov and clinical trials registries to identify the protocols for ongoing studies.
We aimed to capture data from a broad range of study designs. To do this, we included systematic reviews, randomized control trials, cohort studies, case-control studies, prospective observational studies, retrospective observational studies, case series of ≥10 participants and registry studies. We excluded case reports, case series with <10 patients, conference abstracts, and Phase I and II clinical trials.
The rapid review was undertaken by two reviewers (AY and DS). To aid with screening, both reviewers created and piloted screening forms used during the title and abstract and full-text review stages. At each stage, a portion of the studies would be screened using the screening forms (20% in title and abstract and 10% full text). Only after both reviewers were satisfied with the studies selected for inclusion using the screening forms were the remaining studies screened by the first reviewer. As a check, the second reviewer reviewed every study that was excluded by the first reviewer to ensure accuracy.
A data extraction collection form was created by the reviewers and the first reviewer extracted data using the agreed data extraction form. The second reviewer independently used the form to extract data from 10% of the included studies to ensure correctness and completeness. If the two reviewers also agreed that in this 10% the data collected was in keeping with what had been agreed during trialing, then the data extraction of the remaining 90% continued and completed by the first reviewer.
We aimed to create an estimate of the percentage prevalence for each symptom, complication, or comorbidity identified. To do this, for each individual study, the number of patients reported to have a symptom, complication, or comorbidity was determined. This number was then divided by the total number of participants reported in the same study. Then, in those studies where a prevalence could be determined, the calculated prevalence from each study would be grouped together by symptom, complication, or comorbidity, and a median and interquartile range calculated. If a prevalence for a symptom, complication, or comorbidity was reported in one of the systematic reviews, these data were not included in the median prevalence and analysis to avoid accidental duplication of data. Instead, they have been used to provide context and aid with narrative synthesis.
3. Results
The searches identified 2,930 articles, of which 119 articles met our criteria for inclusion (see supplementary table S1 for full list of the references included). We excluded 2,438 articles during title and abstract screening. During the full-text review, we excluded 380 articles and the reason for exclusion recorded, as outlined in :
Figure 1. PRISMA flow diagram of included and excluded studies.
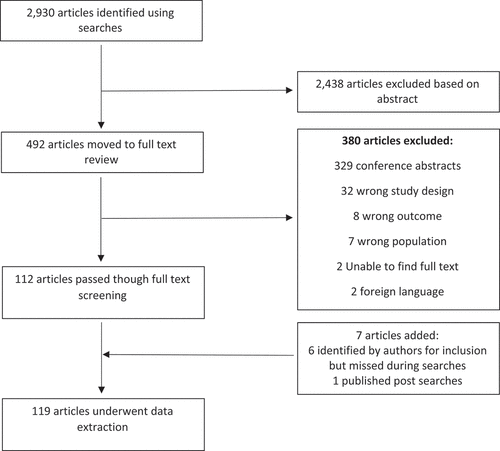
A total of 119 articles were included after 6 studies and 1 systematic review, not identified by the search, were added by the reviewers [Citation18,Citation19,Citation23–27].
Within the included articles (excluding systematic reviews) there were a total of 60,192 participants reported within the included articles. It is possible that the same individual may have taken part in more than one study. The focus and aims of the studies included were varied, ranging from determining the prevalence of symptoms and complications within a set population, using ultrasonography to measure pancreatic fluid flow to reviewing previous abdominal surgeries performed at a center [Citation28–30]. Studies included were published around the world including in North America, Europe, Iran, India, China, and Brazil.
In total, 12 systematic reviews met our inclusion criteria [Citation24,Citation31–41]. Within the included reviews, there were a total of 124 studies and 105,610 participants with CF (with one review contributing 99,925 [Citation39]). Of the systematic reviews, two were Cochrane Reviews. The included systematic reviews covered a wide variety of areas: four assessing the effects of probiotics, one assessing the risk of GI cancers in CF, two looking at CF-related reflux disease and treatment, one focusing on the incidence of celiac disease in CF, one focusing on the prevalence of constipation in CF, one on the treatment of distal intestinal obstruction syndrome (DIOS), one on CF-related gut dysbiosis, and one on neonatal fecal peritonitis.
Within the section below we will discuss our findings starting with pathophysiology followed by symptoms, complications, and comorbidities and finally treatments.
3.1. Pathophysiology
In CF, symptoms and comorbidities are experienced in both the upper and lower GI tract as the result of GI tract dysfunction. However, analysis of the included studies suggests that how this dysfunction occurs is different between the upper and lower GI tract.
For the upper GI tract, there is a combination of factors that may have an impact on upper GI tract function. These include both respiratory factors and esophageal dysfunction. Using high-resolution manometry impedance, pwCF have been found to generate greater negative inspiratory intrathoracic pressures by comparison to healthy controls [Citation42]. This difference in pressure may contribute to an increased gastro-esophageal pressure gradient and result in reflux of stomach contents up into the esophagus [Citation42]. Compounding this is a combination of other areas of dysfunction within the esophagus and stomach. Studies have identified that the lower esophageal sphincter in pwCF generates less pressure than healthy controls, and is more likely to relax during inspiration [Citation42]. For the stomach, studies have suggested that gastric emptying may be delayed in pwCF [Citation43,Citation44], although not all data support this [Citation45].
This dysfunction means that pwCF are more likely to experience a greater degree of reflux up to the proximal esophagus, leading to prolonged acid exposure within the esophagus [Citation42,Citation43]. This results in an increased incidence of symptoms of gastroesophageal reflux disease (GERD) and may contribute to exacerbations of pulmonary symptoms [Citation35,Citation43,Citation46].
In the lower GI tract, there are also multiple important factors impacting upon GI dysfunction, chiefly amongst these a triad of GI dysmotility, dysbiosis, and increased inflammation.
Using both wireless pH motility capsules and Magnetic Resonance Imaging (MRI), the orocecal transit time (OCTT), which is the time taken for food to pass from the mouth to the cecum, can be measured. In CF, the OCTT has been found to be significantly prolonged, taking around 360 minutes [Citation45,Citation47,Citation48] vs. healthy controls (median time 210 minutes) [Citation45,Citation49]. The causes for the prolonged OCTT seen in pwCF remain to be determined. One contributor may be the impaired and sometimes incomplete neutralization of the acidic stomach content seen within the proximal duodenum in CF [Citation47]. Acidic stomach content should be neutralized by bicarbonate secreted by the pancreas, which enables a more optimal pH for pancreatic lipases to function (although gastric lipases function better in a more acidic environment). Both pancreatic lipase and bicarbonate secretion are deficient in CF. A high fat content within the lumen of the ileum delays transit in the jejunum through a reflex mechanism, termed the ”ileal brake” [Citation50], mediated in part by the release of peptide YY (PYY) [Citation51]. Another reason for the delayed OCTT may be dysfunction at the terminal ileum, a recognized site of pathology in CF. This is the result of a combination of factors including inflammation and partial physical or functional blockage [Citation45,Citation52]. In response to a meal, the terminal ileum contracts and expels contents into the cecum, this reflex is called the ‘gastro-ileal reflex’ [Citation53,Citation54]. Using MRI, it has been found that pwCF have a significant reduction in the volume of ileal content that is expelled from the terminal ileum into the cecum, in response to a meal [Citation45]. These delays in transit have been linked with bacterial dysbiosis within the GI tract in CF through correlating microbiota findings and MRI measurements of intestinal motility [Citation55].
CF-related gut dysbiosis is common and is characterized by reduced gut bacterial diversity, an increase in the number of pro-inflammatory bacteria and a reduction in anti-inflammatory bacteria [Citation24]. It is thought that this dysbiosis is linked to extensive antibiotic use. In CF, studies have found an increase in antibiotic resistant E. coli in children with CF compared to controls and an increase in the carriage of Clostridium difficile [Citation56–58]. The dysbiosis is also linked to increased inflammation (both locally and systemically), which, in turn, leads to further impairment of intestinal motility. The composition of bacteria in the gut in CF has been described as being similar to that seen in people with inflammatory bowel disease, particularly Crohn’s disease [Citation59]. In CF, it has also been found that there is a high prevalence of small intestinal bacterial overgrowth [Citation60]. However, in a recent systematic review, there are few studies linking gut dysbiosis to both systemic and lung inflammation. Further research is needed to determine whether CFTR modulator treatment results in changes to the microbiome [Citation24,Citation61].
Increases in inflammatory markers, particularly fecal calprotectin (FC), have been seen repeatedly in pwCF compared to healthy controls [Citation26,Citation62–65]. FC levels in CF, however, are not as high as levels expected in inflammatory bowel disease, which may indicate less severe inflammation [Citation66]. FC has also been shown to be increased in those with CF who are pancreatic insufficient, which could be linked to the absence of exocrine pancreatic function resulting in dysmotility, gut dysbiosis, and inflammation [Citation64,Citation67,Citation68]. In studies investigating the concurrent use of probiotics and ivacaftor, FC has been used as a measure of inflammation in studies [Citation31,Citation69]. These have suggested that FC may be reduced in those taking probiotics and ivacaftor [Citation31,Citation69].
3.2. Symptoms
Among the studies included, GI-related symptoms were commonly reported in 2,439 out of 7,245 participants (33.7%). A broad variety of symptoms were identified in these studies (32 symptoms in total). Of these symptoms, 13 were attributable to the upper GI tract, 9 to lower GI tract, 2 were symptoms related to the respiratory tract (wheezing and cough), 6 were related to weight and appetite, and 2 were more general including embarrassment and sleeping difficulties and their impact on a patient’s quality of life. Supplementary table S2 shows the number of studies in which each symptom was reported. The systematic reviews that reached our inclusion criteria have not been included in the results below to avoid duplication of studies.
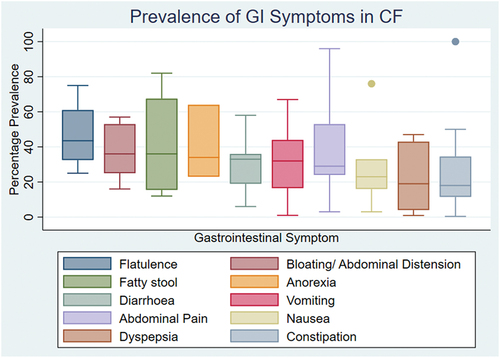
The most frequently reported symptoms were abdominal pain (550 participants) [Citation23,Citation30,Citation63,Citation67,Citation68,Citation70–82], constipation (311 participants) [Citation23,Citation30,Citation68,Citation70,Citation71,Citation73–76,Citation79–85] bloating and abdominal distension (272 participants) [Citation23,Citation71,Citation75,Citation76,Citation80] diarrhea (265 participants) [Citation23,Citation68,Citation70,Citation71,Citation74,Citation75,Citation80–82] and flatulence (225 participants) [Citation23,Citation75,Citation77,Citation80]. shows a box and whisker plot that summarizes the median percentage prevalence we have calculated for the 10 most common symptoms and further details can be found in supplementary table S3. It is likely that pwCF experience a combination of these symptoms. Although the percentage prevalence of weight loss was high (62%), it is likely that this may have been over-represented in our results due to the small number of studies reporting it (2 studies), one of which reported weight loss as a presenting feature in DIOS [Citation78].
Figure 2. Box and whisker chart demonstrating the percentage prevalence expressed as a median (horizontal line within the box) and interquartile range (whiskers). Dots represent outliers within an individual gastrointestinal symptom.
Within the studies reviewed, we identified an inconsistency with terms used to describe symptoms, particularly those related to symptoms in the upper GI tract. In two studies, the broad category of ‘gastrointestinal symptoms’ was used and in one study the category ‘digestive symptoms’ used [Citation86–88]. Between reports multiple synonyms would be used for example, symptoms of dysphagia were described as dysphagia, difficulty swallowing, gagging, and choking.
In addition to the inconsistent use of synonyms, there was inconsistency with the definitions used to classify functional GI disorders, such as constipation. In those studies that reported participants having constipation, diarrhea, and bloating (58 studies) we reviewed the studies again to identify whether they contained a definition for those symptoms using a diagnostic criteria such as the ROME IV criteria [Citation89]. Of the 58, 1 study made reference to the use of the ROME III criteria for characterizing functional constipation [Citation73]. In this study, the authors discussed that of the 15 participants who were reported to have constipation, when the ROME III criteria were applied, only 4 of the 15 would be defined as having constipation [Citation73,Citation90]. This would result in a possible over reporting of constipation in studies.
Further study is also needed to better understand the mechanisms behind GI dysfunction in CF, such as constipation, as the mechanisms underlying the dysfunction may differ in CF. One possibility is that, as well as dysmotility, abnormal stool viscosity may contribute to constipation in CF (as suggested by MRI data) [Citation91].
3.3. Complications and comorbidities
A total of 16 GI-related complications and comorbidities of CF were reported in the included studies. Out of the 16,096 participants include in studies where a prevalence of complications and comorbidities could be calculated (excluding systematic reviews), 3,347 participants (20.8%) were reported to be experiencing a GI-related complication or comorbidity.
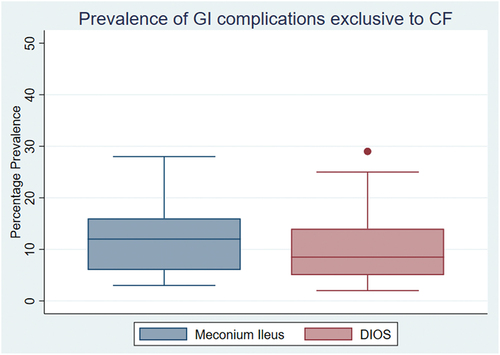
Meconium ileus was reported in 1,694 infants with CF in 25 studies [Citation30,Citation44,Citation47,Citation70,Citation73,Citation78,Citation79,Citation82,Citation83,Citation92–107] and DIOS was reported in 254 pwCF in 17 studies [Citation73,Citation76,Citation78–80,Citation83,Citation85,Citation94,Citation99,Citation101,Citation102,Citation105,Citation108–112]. The box and whisker chart in summarizes the calculated median prevalence of DIOS and meconium ileus.
Figure 3. Box and whisker chart showing the percentage prevalence of the complications associated specifically with CF: DIOS and meconium ileus. The percentage prevalence is expressed as a median (horizontal line within the box) and interquartile range (whiskers). Dots represent outliers.
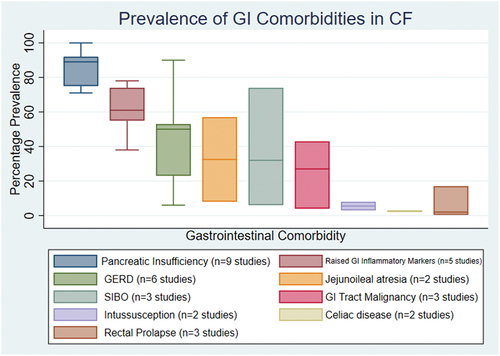
The GI comorbidities that were reported in the highest number of studies were raised intestinal inflammatory markers (16 studies) [Citation59,Citation61–65,Citation67,Citation69,Citation84,Citation101,Citation113–117], GERD (12 studies) [Citation42–44,Citation46,Citation71,Citation72,Citation80,Citation95,Citation118–121], pancreatic insufficiency (12 studies) [Citation34,Citation61,Citation69,Citation73,Citation80,Citation82,Citation97,Citation104,Citation105,Citation111,Citation121,Citation122], and GI tract malignancy (4 studies) [Citation29,Citation123–125]. The number of studies that each comorbidity was reported are shown in the supplementary table S4. The three comorbidities that were reported the most were pancreatic insufficiency (627 participants) [Citation73,Citation80,Citation82,Citation97,Citation104,Citation105,Citation111,Citation121,Citation122], raised intestinal inflammatory markers (266 participants) [Citation64,Citation65,Citation67,Citation68,Citation84], and GI tract malignancy (162 participants) [Citation29,Citation123,Citation125]. The calculated median percentage prevalence values for the comorbidities reported are shown in the box and whisker plot found in . Pancreatic insufficiency had the highest median prevalence (89%) followed by raised inflammatory markers at 61% and GERD at 50%. There are two complications where our methods for calculating prevalence have resulted in a higher prevalence than what is seen in practice. Jejunoileal atresia had a median prevalence of 32.5% but was only reported in two studies and the prevalence is likely raised due to the study design [Citation126,Citation127]. GI tract malignancy was calculated to have a median prevalence of 27% which is likely due to a selection bias (as a result of the study objectives) within the studies we have used to calculate this prevalence [Citation29,Citation123,Citation125]. A large systematic review including 99,925 pwCF (that was identified by our searches) reported the risk of GI tract malignancy to be significantly higher in pwCF compared to controls with a standardized incidence ratio of 8.13 [Citation39]. One comorbidity was reported in only one systematic review. This was meconium peritonitis which occurred in 11 neonates over a 10-year period [Citation40]. Two studies investigated whether pwCF had an increased risk of carrying Clostridium difficile [Citation57,Citation58]. Both studies determined that pwCF were more likely to carry Clostridium difficile and were more likely to have toxigenic strains [Citation57,Citation58]. A study from Brazil investigated whether the carriage of Helicobacter pylori was increased in CF, however the prevalence found was in keeping with the general population in Brazil [Citation75].
Figure 4. Box and whisker chart demonstrating the percentage prevalence expressed as a median (horizontal line within the box) and interquartile range (whiskers). Whiskers are missing where there are ≤ 3 studies contributing to the calculated prevalence due to the lower and upper quartile value being the same as minimum and maximum value.
Studies have researched whether there is an increased prevalence of celiac disease in pwCF with a median percentage prevalence of 2% [Citation81,Citation128]. In these studies, the prevalence of celiac disease in CF was reported as either being similar to or significantly higher compared to the general population [Citation81,Citation128]. However, the prevalence may be higher than that reported due to only the small number of participants undergoing endoscopy with a higher number having raised antigliadin antibodies and anti-transglutaminase IgA (tTGA) [Citation81].
There are three complications that are unique to either CF or its treatment: meconium ileus, DIOS, and fibrosing colonopathy. Meconium ileus and DIOS are caused by the dysfunction of the bowel in CF leading to complete or incomplete small bowel obstruction. Meconium ileus occurs within the neonatal period and is also seen antenatally. It is often the first presenting feature of CF. DIOS presents later in childhood or in adult life. Fibrosing colonopathy was in the early 1990s thought to be associated with high-dose preparations of pancreatic enzyme replacement therapy (PERT), resulting in the progressive accumulation of submucosal fibrosis and damage within the colon, usually beginning in the ascending colon [Citation129–135]. The mechanism behind the development of fibrosing colonopathy is controversial [Citation136], with several factors implicated, including the enteric coating of PERT [Citation132]. Since this mechanism was described [Citation131,Citation132], an upper limit of 10,000 lipase units/kg/day has been recommended for pwCF. Today, fibrosing colonopathy is now rarely seen [Citation137]. From our searches, we did not identify any participants with fibrosing colonopathy within the studies reviewed despite fibrosing colonopathy being contained within our search strategy. This is likely due to the rarity of the disease following the recommendation on maximum PERT dosage and our search timeframe being nearly 20 years after the first reports of fibrosing colonopathy in 1994 and 1995 [Citation131,Citation132]. In the 2021 annual patient registry data reports, in the US and the UK the incidence for fibrosing colonopathy was <0.1% and 0%, respectively [Citation2,Citation138].
Both the US and UK national CF registries report the annual incidence of GI complications and comorbidities. The complications and comorbidities common to both annual reports are DIOS, GERD, meconium ileus, peptic ulcer, fibrosing colonopathy and rectal prolapse. However, how the data on the numbers of people who suffer from these GI complications and comorbidities are reported vary between the registries. For example, in the CF Trust annual registry report the incidence of DIOS in the preceding 12 months is reported, whereas meconium ileus is reported as the number of pwCF who present with meconium ileus when those who were diagnosed with CF through newborn screening are excluded [Citation2]. This leads to challenges in directly comparing the data found in this review to both registry reports.
In we compare the prevalence of complications and comorbidities found in the studies we have reviewed against the prevalence or incidence reported in the UK CF Trust and U.S. CF Foundation registry annual data report 2021 [Citation2,Citation138]. The prevalence we have calculated is higher than the incidence in both registries for DIOS, GERD, rectal prolapse, pancreatitis, and GI malignancy. However, the prevalence of meconium ileus is harder to compare due to the differences in how the data are presented between the annual registry reports [Citation2,Citation138].
Table 1. Comparison of reported data on the incidence and prevalence of GI complications and comorbidities in CF. The first column reports the median percentage prevalence calculated from the studies included in the review. The second column shows the incidence reported in the CF Foundation registry report 2021. The third column shows the incidence reported in the CF Trust registry report 2021. *Likely much higher due to selection bias.
Another contributing factor that may result in the prevalence we have determined being higher is due to a possible selection bias among the studies included.
In the post-CFTR modulator era, the life expectancy for a person with CF is set to continue to increase. This will change the landscape of how pwCF will be treated. The types of comorbidities are likely to change as disease associated with adult and older age groups may become more prevalent in the CF population. This is particularly likely with GI-tract-related malignancies. A large systematic review with a population of a 99,925 pwCF compared the risk of developing GI tract malignancy between pwCF and the general population [Citation39]. Through meta-analysis they reported that pwCF were at significantly higher risk of developing a GI tract cancer by comparison to the general population. Small bowel cancer had the highest increase with a risk nearly 20 times higher than the general population and colon cancer had a risk 10 times higher [Citation39]. A pooled incidence rate was also described with small bowel cancer of 0.13 per 1000 person-years and colon cancer 0.39 per 1000 person-years [Citation39]. Another large retrospective registry study (41,188 pwCF) found that the risk of GI tract malignancy increased in those with a severe genotype, a previous history of DIOS and in those post-organ transplant [Citation124]. In the coming decades, it should become clear whether modulators reverse the abnormalities in GI function seen in pwCF and if this will decrease the risk of GI cancer in pwCF.
3.4. Treatments
Treatments for GI symptoms found within the articles reviewed included: CFTR modulators, pancreatic enzyme replacement therapy (PERT), laxatives, proton pump inhibitors (PPI), H2 receptor antagonists, and probiotic supplementation. Grading of the certainty of evidence has been performed where possible and a summary of the results can be found in supplementary table S5.
The use of gastric acid reducing medication was reported in 16 studies. One study reported that up to 76% of the pwCF in their study population were taking regular antacids or gastric acid suppressing medications [Citation139]. One systematic review recommends that in those participants with uncomplicated GERD a trial of an empiric medication such as a PPI for 8 weeks to assess for improvement of symptoms [Citation35]. The same review states that in those with severe, complicated GERD, medical therapies will often be insufficient and surgical intervention such as a Nissen fundoplication may be needed to improve pulmonary and nutritional status [Citation35]. In one small study investigating rates of Barrett’s esophagus, five pwCF who were started on a PPI for 2 months were found to have a significant improvement in total acid exposure index and DeMeester score [Citation120]. Some caution however should be considered when using PPIs to improve respiratory outcomes, as their use has been linked with increased pulmonary exacerbations and may increase airway inflammation [Citation25,Citation27]. Another reason for the use of gastric acid suppressing medications is to improve the effectiveness of PERT by increasing the pH of gastric contents before they pass into the duodenum [Citation140,Citation141]. The certainty of evidence for the use of gastric acid suppressing medication is very low due to study design and indirectness.
The use of laxatives was reported in five studies, but no assessment of their effectiveness was undertaken. The use of laxatives ranged from 20% to 52% [Citation44,Citation74].
In the studies included in this review, the median prevalence of pancreatic exocrine insufficiency was 89%. This would suggest that in 89% of pwCF, PERT would need to be used with meals and snacks. This medication replaces absent pancreatic enzymes, allowing for the breakdown of fat in the small bowel. PERT dose may vary between individuals and depending on the fat content of a meal. One study we reviewed suggested that men were more likely to take higher doses of PERT with meals than women and that higher doses were associated with participants reporting more normal stool consistency [Citation74]. A randomized control trial looked at whether altering the timing of when PERT is taken at mealtime (before or after eating) altered symptoms or stool frequency [Citation77]. Results found that overall, there was no difference in abdominal pain, bowel habit, and quality of life when compared to taking PERT before or after meals [Citation77]. However, some consideration may be taken on an individual or case by case basis. A Cochrane review tried to bring together multiple studies to provide guidance on when PERT should be given, but no studies met the Cochrane review’s inclusion criteria of being a randomized control trial (RCT) or randomized cross over trial [Citation142]. Overall, the grading of the certainty of evidence around timing of PERT was low due to the risk of bias and imprecision (see supplementary table S5).
As well as the timing of PERT, there have been questions about the correct dosage. This question was particularly important following the emergence of fibrosing colonopathy in the early 1990s around the time of high strength PERT preparation roll out [Citation129–135]. Current ECFS and Cystic Fibrosis Foundation recommendations suggest dosing per gram of fat ingested, starting at 500 units lipase/kg/meal and titrating up to a maximum of 10,000 units lipase/kg/day in children aged 4 years and above and in adults [Citation143,Citation144]. One study compared the difference in the coefficient of fat absorption when PERT doses had high variability to when there was low variability and found that with low variability there was a greater coefficient of fat absorption [Citation122]. Previous work has been conducted to develop an app (MyCyFapp) to aid patients with the correct dosing and timings of PERT which showed encouraging data on improving abdominal symptom scores, however this app is unfortunately no longer being developed at the time of writing [Citation145].
The use of probiotics was reported in three studies and their use was assessed in three systematic reviews. Multiple systematic reviews have been undertaken in order to determine the effect of probiotics on both respiratory exacerbations and intestinal inflammation [Citation32,Citation33]. One review identified three studies where FC reduced with the use of probiotics for 6 months and one study demonstrated an increase in microbiota diversity following probiotic use (although this was not significant) [Citation31]. The second found that in 4 out of 5 of the included studies, there was an improvement in intestinal inflammation and a reduction in pulmonary exacerbation frequency [Citation33]. Another systematic review, however, summarized the evidence for the use of probiotics and determined that due to a lack of high quality randomized control trials, it was not possible to support a general recommendation for the use of probiotics to prevent pulmonary exacerbations and reduce intestinal inflammation [Citation32].
Our search revealed little information of prebiotics, defined as a dietary food source (such as fiber) favoring beneficial gut bacteria. In one systematic review included, prebiotics were a part of search strategies however no further information was included [Citation33].
The effects that modulators have on the GI tract are under investigation but are yet to be determined. We identified 11 studies that investigated the effects modulators have on the GI tract, three measuring the effect of ETI, four lumacaftor/ivacaftor, three ivacaftor, and one tezacaftor/ivacaftor [Citation20,Citation61,Citation69,Citation113,Citation117,Citation118,Citation146–150]. We also identified two phase 3 open-label studies that observed an improvement in exocrine pancreatic function in 2- to 5-year-olds after 24 and 84 weeks of ivacaftor [Citation18,Citation19]. One prospective study found that symptoms of GERD, measured using two reflux symptom scores, improved after 3 and 6 months of ETI [Citation118]. Another prospective study used the GI specific CFAbd-score in a large, international study to determine whether GI symptom scores improved 24 weeks after starting ETI [Citation20]. From the cohort from Germany, CFAbd-scores were found to fall significantly after 24 weeks of ETI, both overall and in specific domains including pain, GERD, bowel movement, disorders of appetite, quality of life impairment [Citation20]. The tolerability of ETI in those who have received a lung transplant was assessed in a retrospective study [Citation146]. Four participants found an improvement in GI symptoms and three participants reported self-decreased or self-stopping of PERT. Three of the 13 participants had their ETI temporarily paused due to abdominal complaints, but were successfully restarted and four had to stop altogether due to reduced pulmonary function or mood disturbance [Citation146]. The certainty of evidence for ETI improving GI symptoms was graded as low.
As well as a small number of studies reporting an improvement in symptoms, one retrospective study identified a significant reduction in FC after 3 months of lumacaftor/ivacaftor [Citation117]. There is also a growing number of case series, particularly in children, that showed an improvement in exocrine pancreatic function after starting ivacaftor [Citation12–15]. However, most of these studies were excluded from our analysis due to small numbers. The most robust evidence for improvement in exocrine function was an observation made in children aged 2–5 years taking ivacaftor as part of an open-label phase 3 trial (KIWI and KLIMB studies). In these studies, fecal elastase was found to increase after 24 weeks of ivacaftor and this improvement was sustained after 84 weeks [Citation18,Citation19]. An ongoing registry study, the CFTR-MAGIC study (NCT05253859), aims to determine the effect of ivacaftor of GI complications and PERT use in the UK and the US over a 10-year period [Citation151].
Research has been undertaken to determine treatment options for both meconium ileus and DIOS. For meconium ileus, a non-surgical treatment used was a hyperosmolar contrast enema, such as diatrizoate (gastrografin®), but in the majority of cases surgical intervention was needed [Citation99,Citation152]. In a retrospective study, one tertiary center reported that in the majority of cases DIOS could be managed with conservative management, such as simple laxatives, enema, or bowel preparations [Citation76]. In those where DIOS did not resolve, surgical intervention, particularly laparotomy would be required [Citation76]. A working group review looked at the treatment of pancreatitis in children, including those with CF, but this concluded that there is limited evidence on nutrition strategies in children with pancreatitis [Citation153].
Overall, all the interventions above had either a low or very low quality of evidence. This is due to several factors including only a small number of randomized control trials (which were not blinded), imprecision due to small numbers of participants and heterogeneity of reported outcomes.
4. Conclusion
GI symptoms, complications, and comorbidities are commonly experienced by pwCF and reducing their burden is a priority to the CF community [Citation8,Citation9]. GI symptoms are caused by dysfunction throughout the GI tract with pwCF experiencing symptoms from both the upper and lower GI tract. The symptoms with the highest median prevalence included flatulence, bloating and abdominal distension, fatty stool, anorexia, vomiting, diarrhea, abdominal pain, nausea, and constipation. CF specific complications including meconium ileus and DIOS were also commonly experienced, with a median prevalence of 12% and 8.5%, respectively.
Current literature suggests that the underlying mechanisms for the symptoms, complications, and comorbidities differ for the upper and lower GI tract. Upper GI tract dysfunction may be due to a combination of a large gastro-esophageal pressure gradient, lower esophageal sphincter dysfunction and esophageal dysmotility, whereas lower GI dysfunction is due to a triad of dysmotility, gut microbiota dysbiosis, and inflammation.
There is a growing body of research on the use of treatments to improve GI symptoms in pwCF and medications used include PPIs, H2 receptor antagonists, PERT, probiotics, and CFTR modulators. However, further high-quality studies are needed to provide recommendations for treatment of symptoms and to inform guidelines.
A difficulty within this review was combining the results of the different studies due to the inconsistencies in the reporting of outcomes and the tools used to measure these differed between studies. To aid and drive future research, there is a need to standardize which outcomes are reported when researching GI symptoms in CF and how symptoms are classified to allow for easier comparison between studies. This is due to the heterogeneity of data reported within the studies included in this review, both in terms of what is reported and how symptoms are classified. This can be addressed through the ongoing development and increasing use of standardized patient reported outcome measures (PROMs) for GI symptoms in CF. Current validated patient reported outcome measures include the CFAbd-score, PedsQL-GI, and CFQ-R, with the CFAbd-score being designed specifically for measuring abdominal symptoms in pwCF [Citation154]. A currently ongoing study called CARDS-CF (NCT05251467) is developing a new patient reported outcome measure for monitoring abdominal symptoms on a daily basis and uses an app-based questionnaire [Citation155,Citation156]. However, these patients reported outcome measures can only be used in prospective studies due to a limited recall period and therefore cannot be used in all study designs. In addition to PROMs to promote consistency and standardization in reporting of outcomes and to allow for the results of multiple studies to be combined, there is a need for the development of a core outcome sets in CF to ensure consistency and standardization in reporting of outcomes and to allow for the results of multiple studies to be combined. To improve consistency and comparability of data between studies, greater adoption of criteria for diagnosing functional GI disorders, such as the ROME criteria could help to ensure that symptoms are not over reported in studies.
5. Expert opinion
This review adds to the knowledge that GI disease in CF is common and adds to both the symptom burden and burden of treatment in pwCF. There is a wide range of symptoms, complications, and comorbidities experienced by pwCF. There is heterogeneity in reports of these symptoms, complications, and comorbidities in the literature due to differences in study design. Methodological improvements in the design of clinical studies of GI disease in CF would facilitate the interpretation of study findings and allow comparisons between studies.
There are a number of strategies that can be employed to achieve this, but this will require further development and the building of a consensus. One option is the creation of a core outcome set, specific to GI disease in CF. This core outcome set should identify outcome measures that are both repeatable and correlate to meaningful changes to patients in terms of the burden from the GI dysfunction they experience. Another option would be to have an increase in the use of CF specific patient reported outcome measures, particularly in studies using a prospective study design. Finally, to ensure that all studies have used a consistent definition for symptoms, such as constipation, and therefore reduce the likelihood of bias from over reporting, classification systems for functional bowel disorders, such as the ROME IV criteria could be used [Citation89]. This would allow for the prevalence of specific symptoms in CF to be compared with other conditions where the same definition has been used.
Reducing the burden of GI disease in CF and understanding the extrapulmonary effects of CFTR modulators are research priorities which have been identified by the CF community [Citation8,Citation9]. Although there is an expanding literature on treatments for GI dysfunction in CF, more research needs to be undertaken to understand the pathophysiology underlying this GI dysfunction and what treatments can be used to reduce the associated symptom burden. CFTR modulators have transformed the landscape for the treatment of CF by improving respiratory function and BMI. These changes are expected to result in a rise in life expectancy and have improved quality of life. However, we do not yet fully understand the effects that CFTR modulators have on the gut and whether their use will lead to improvements in GI symptoms, complications, and comorbidities.
The expected increase in the age of the CF population due to CFTR modulators and improved care will likely result in an increase in age-related complications and comorbidities. From the perspective of the GI tract, an important comorbidity is the increased risk that pwCF have of developing GI malignancy. Reducing this will require greater understanding of the underlying GI dysfunction associated with CF, including inflammation, mucosal barrier dysfunction, dysbiosis, and oncogene activation (related to CFTR gene mutations).
Reducing the burden of GI disease in CF is an important area for future research within CF and will require further high-quality studies to solve. Areas of research may include the development of a core outcome set, patient reported outcome measures and the creation of new or repurposed medications or dietary interventions that reduce GI symptoms. Therefore, as stated by the CF community, reducing the burden of GI disease is one of the next and greatest challenges to overcome within CF.
Table of abbreviations
| BMI | = | body mass index |
| CF | = | cystic fibrosis |
| CFTR | = | cystic fibrosis transmembrane conductance regulator |
| DIOS | = | distal intestinal obstruction syndrome |
| ETI | = | elexacaftor/tezacaftor/ivacaftor |
| FC | = | fecal calprotectin |
| FEV1 | = | forced expiratory volume in one second |
| GI | = | gastrointestinal |
| GERD | = | gastro-esophageal reflux disease |
| MI | = | meconium ileus |
| MRI | = | magnetic resonance imaging |
| PERT | = | pancreatic enzyme replacement therapy |
| PROM’s | = | patient reported outcome measures |
| PPI | = | proton pump inhibitor |
| PYY | = | peptide YY |
| PROM’s | = | people with cystic fibrosis |
| RCT | = | randomized control trial |
| tTGA | = | anti-transglutaminase IgA |
Article highlights
GI symptoms, complications, and comorbidities are commonly experienced by pwCF.
Improving the burden of GI disease in CF is a priority for research according to the CF community.
The symptoms that are most commonly experienced by pwCF are flatulence, bloating and abdominal distension, fatty stool, anorexia, vomiting, diarrhea, abdominal pain, nausea, and constipation.
The reporting of symptoms in the literature and their classification is inconsistent. To help address this, the creation of a core outcome set would be beneficial alongside the increased use of CF specific patient reported outcome measures.
Gut dysfunction in CF differs in the upper and lower GI tract.
In CF, wide intrathoracic pressure excursions (due to lung disease), impaired lower esophageal sphincter function, and dysmotility all contribute to upper GI tract dysfunction.
Gut dysmotility, gut dysbiosis, and inflammation all contribute to lower GI tract dysfunction. Further research is required to determine the mechanism behind symptoms and hence to make a rational choice of new and repurposed treatment interventions to evaluate in clinical trials.
Declaration of interest
The clinical research fellow post of A Yule is funded through a grant provided by Vertex Pharmaceuticals on the GIFT-CF3 study (NCT04618185). R Spiller receives grants/research support from Sanofi-Aventis Deutschland GmbH and speaker fees from Ardelyx, F.Trenka, and Bi Pharma Consultants. AR Smyth has research grants (paid to the University of Nottingham) from Vertex Pharmaceuticals and payment for an advisory board (paid to the University of Nottingham) from Viatris Pharmaceuticals, all outside the current work. AR Smyth has patents issued (Camara M, Williams P, Barrett D, Halliday N, Knox A, Smyth A, Fogarty A, Barr H, Forrester D.) Alkyl quinolones as biomarkers of Pseudomonas aeruginosa infection and uses thereof. US2016131648-A1; https://pubchem.ncbi.nlm.nih.gov/patent/US-2016131648-A1 Outside the current work, AR Smyth reports participation on a Data Safety Monitoring Board for the North American Cystic Fibrosis Foundation Therapeutic Development Network. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Supplemental Material
Download Zip (106 KB)Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/17476348.2023.2228194.
Additional information
Funding
References
- Farrell P, Férec C, Macek M, et al. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis. Eur J Hum Genet. 2018;26(12):1832–1839. doi: 10.1038/s41431-018-0234-z
- Cystic Fibrosis Trust. UK Cystic Fibrosis Registry 2021 Annual Data Report: Cystic Fibrosis Trust; 2022. [cited Sep 2022]. Available from: https://www.cysticfibrosis.org.uk/sites/default/files/2022-10/CFT_2021-Annual-Data-Report-WEB.pdf.
- Andersen DH. Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am J Dis Child. 1938;56(2):344–399. doi: 10.1001/archpedi.1938.01980140114013
- Wilmers MJ, Mackay HM, Anderson IM. Five cases of cystic fibrosis of the pancreas. Proc R Soc Med. 1950;43(11):829–832.
- Andersen DH. Therapy and prognosis of fibrocystic disease of the pancreas. Pediatrics. 1949;3(4):406–417. doi: 10.1542/peds.3.4.406
- Sathe M, Houwen R. Meconium ileus in cystic fibrosis. J Cyst Fibros. 2017;16:S32–S9. doi: 10.1016/j.jcf.2017.06.007
- Carlyle BE, Borowitz DS, Glick PL. A review of pathophysiology and management of fetuses and neonates with meconium ileus for the pediatric surgeon. J Pediatr Surg. 2012;47(4):772–781. doi: 10.1016/j.jpedsurg.2012.02.019
- Rowbotham NJ, Smith S, Leighton PA, et al. The top 10 research priorities in cystic fibrosis developed by a partnership between people with CF and healthcare providers. Thorax. 2018;73(4):388–390. doi: 10.1136/thoraxjnl-2017-210473
- Rowbotham NJ, Smith S, Elliott ZC, et al. A refresh of the top 10 research priorities in cystic fibrosis. Thorax. 2023:thorax-2023–220100. doi: 10.1136/thorax-2023-220100
- Ramsey BW, Davies J, McElvaney NG, et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. NEJM. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185
- Middleton PG, Mall MA, Dřevínek P, et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. NEJM. 2019;381(19):1809–1819. doi: 10.1056/NEJMoa1908639
- Nichols AL, Davies JC, Jones D, Carr SB, et al. Restoration of exocrine pancreatic function in older children with cystic fibrosis on ivacaftor. Paediatr Respir Rev. 2020;35:99–102. doi: 10.1016/j.prrv.2020.04.003
- Munce D, Lim M, Akong K. Persistent recovery of pancreatic function in patients with cystic fibrosis after ivacaftor. Pediatr Pulmonol. 2020;55(12):3381–3383. doi: 10.1002/ppul.25065
- Megalaa R, Gopalareddy V, Champion E, et al. Time for a gut check: pancreatic sufficiency resulting from CFTR modulator use. Pediatr Pulmonol. 2019;54(8):E16–E8. doi: 10.1002/ppul.24353
- Hamilton JL, Zobell JT, Robson J. Pancreatic insufficiency converted to pancreatic sufficiency with ivacaftor. Pediatr Pulmonol. 2019;54(11):1654–. doi: 10.1002/ppul.24454
- Smith H, Rayment JH. Sustained recovery of exocrine pancreatic function in a teenager with cystic fibrosis treated with ivacaftor. Pediatr Pulmonol. 2020;55(10):2493–2494. doi: 10.1002/ppul.24952
- Hutchinson I, Mcnally P. Appearance of pancreatic sufficiency and discontinuation of pancreatic enzyme replacement therapy in children with cystic fibrosis on ivacaftor. Ann Am Thorac Soc. 2021;18(1):182–183. doi: 10.1513/AnnalsATS.202006-614RL
- Davies JC, Cunningham S, Harris W, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2-5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4(2):107–115. doi: 10.1016/S2213-2600(15)00545-7
- Rosenfeld M, Cunningham S, T HW, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2-5 years (KLIMB). J Cyst Fibros. 2019;18(6):838–843. doi: 10.1016/j.jcf.2019.03.009
- Mainz JG, Zagoya C, Polte L, et al. Elexacaftor-Tezacaftor-Ivacaftor Treatment Reduces Abdominal Symptoms in Cystic Fibrosis-Early results Obtained with the CF-Specific CFAbd-Score. Front Pharmacol. 2022;13. doi: 10.3389/fphar.2022.877118
- Rowbotham NJ, Smith S, Prayle AP, et al. Gaps in the evidence for treatment decisions in cystic fibrosis: a systematic review. Thorax. 2019;74(3):229–236. doi: 10.1136/thoraxjnl-2017-210858
- Garritty C, Gartlehner G, Nussbaumer-Streit B, et al. Cochrane rapid reviews methods group offers evidence-informed guidance to conduct rapid reviews. J Clin Epidemiol. 2021;130:13–22. doi: 10.1016/j.jclinepi.2020.10.007
- Smith S, Rowbotham N, Davies G, et al. How can we relieve gastrointestinal symptoms in people with cystic fibrosis? An international qualitative survey. BMJ Open Respir Res. 2020;7(1):e000614. doi: 10.1136/bmjresp-2020-000614
- Caley LR, White H, de Goffau MC, et al. Cystic fibrosis-related gut dysbiosis: a systematic review. Dig Dis Sci. 2023;68(5):1797–1814. doi: 10.1007/s10620-022-07812-1
- Dimango E, Walker P, Keating C, et al. Effect of esomeprazole versus placebo on pulmonary exacerbations in cystic fibrosis. BMC Pulm Med. 2014;14(1):21. doi: 10.1186/1471-2466-14-21
- Garg M, Leach ST, Coffey MJ, et al. Age-dependent variation of fecal calprotectin in cystic fibrosis and healthy children. J Cyst Fibros. 2017;16(5):631–636. doi: 10.1016/j.jcf.2017.03.010
- Pauwels A, Verleden S, Farre R, et al. The effect of gastric juice on interleukin-8 production by cystic fibrosis primary bronchial epithelial cells. J Cyst Fibros. 2013;12(6):700–705. doi: 10.1016/j.jcf.2013.03.006
- Engjom T, Erchinger F, Tjora E, et al. Diagnostic accuracy of secretin-stimulated ultrasonography of the pancreas assessing exocrine pancreatic failure in cystic fibrosis and chronic pancreatitis. Scand J Gastroenterol. 2015;50(5):601–610. doi: 10.3109/00365521.2015.1004363
- Hite MA, Gaertner WB, Garcia B, et al. Abdominal Surgical Procedures in Adult Patients with Cystic Fibrosis: what are the Risks? Dis Colon Rectum. 2022;65(8):e805–e15. doi: 10.1097/DCR.0000000000002162
- Sharma A, Morton A, Peckham D, et al. Gastrointestinal surgery in adult patients with cystic fibrosis. Frontline Gastroenterol. 2012;3(4):242–247. doi: 10.1136/flgastro-2012-100184
- Van Biervliet S, Declercq D, Somerset S. Clinical effects of probiotics in cystic fibrosis patients: a systematic review. Clin Nutr ESPEN. 2017;18:37–43. doi: 10.1016/j.clnesp.2017.01.007
- Nikniaz Z, Nikniaz L, Bilan N, et al. Does probiotic supplementation affect pulmonary exacerbation and intestinal inflammation in cystic fibrosis: a systematic review of randomized clinical trials. World J Pediatr. 2017;13(4):307–313. doi: 10.1007/s12519-017-0033-6
- Neri LDCL, Taminato M, Silva Filho LVRFD. Systematic review of probiotics for cystic fibrosis patients: moving forward. J Pediatr Gastroenterol Nutr. 2019;68(3):394–399. doi: 10.1097/MPG.0000000000002185
- Anderson JL, Miles C, Tierney AC. Effect of probiotics on respiratory, gastrointestinal and nutritional outcomes in patients with cystic fibrosis: a systematic review. J Cyst Fibros. 2017;16(2):186–197. doi: 10.1016/j.jcf.2016.09.004
- Bongiovanni A, Manti S, Parisi GF, et al. Focus on gastroesophageal reflux disease in patients with cystic fibrosis. World J Gastroenterol. 2020;26(41):6322–6334. doi: 10.3748/wjg.v26.i41.6322
- Imrei M, Németh D, Szakács Z, et al. Increased prevalence of celiac disease in patients with cystic fibrosis: a systematic review and meta-analysis. J Pers Med. 2021;11(9):859. doi: 10.3390/jpm11090859
- Gilchrist FJ, Green J, Carroll W. Interventions for treating distal intestinal obstruction syndrome (DIOS) in cystic fibrosis. Cochrane Database Syst Rev. 2018;8(8):Cd012798. doi: 10.1002/14651858.CD012798.pub2
- Stefano MA, Poderoso RE, Mainz JG, et al. Prevalence of constipation in cystic fibrosis patients: a systematic review of observational studies. J Pediatr (Rio J). 2020;96(6):686–692. doi: 10.1016/j.jped.2020.03.004
- Yamada A, Komaki Y, Komaki F, et al. Risk of gastrointestinal cancers in patients with cystic fibrosis: a systematic review and meta-analysis. Lancet Oncol. 2018;19(6):758–767. doi: 10.1016/S1470-2045(18)30188-8
- Shinar S, Agrawal S, Ryu M, et al. Fetal meconium peritonitis - prenatal findings and postnatal outcome: a case series systematic review, and meta-analysis. Ultraschall Med. 2022;43(2):194–203. doi: 10.1055/a-1194-4363
- Ng SM, Franchini AJ. Drug therapies for reducing gastric acidity in people with cystic fibrosis. Cochrane Database Syst Rev. 2021;2014(7):Cd003424.
- Pauwels A, Blondeau K, Dupont JL, et al. Mechanisms of increased gastroesophageal reflux in patients with cystic fibrosis. Am J Gastroenterol. 2012;107(9):1346–1353. doi: 10.1038/ajg.2012.213
- Mendez BM, Davis CS, Weber C, et al. Gastroesophageal reflux disease in lung transplant patients with cystic fibrosis. Am J Surg. 2012;204(5):e21–6. doi: 10.1016/j.amjsurg.2012.07.019
- Hauser B, De Schepper J, Malfroot A, et al. Gastric emptying and gastro-oesophageal reflux in children with cystic fibrosis. J Cyst Fibros. 2016;15(4):540–547. doi: 10.1016/j.jcf.2015.12.015
- Ng C, Dellschaft NS, Hoad CL, et al. Postprandial changes in gastrointestinal function and transit in cystic fibrosis assessed by Magnetic Resonance Imaging. J Cyst Fibros. 2021;20(4):591–597. doi: 10.1016/j.jcf.2020.06.004
- Safe M, Cho J, Krishnan U. Combined multichannel intraluminal impedance and pH measurement in detecting gastroesophageal reflux disease in children. J Pediatr Gastroenterol Nutr. 2016;63(5):e98–e106. doi: 10.1097/MPG.0000000000001396
- Gelfond D, Ma C, Semler J, et al. Intestinal pH and gastrointestinal transit profiles in cystic fibrosis patients measured by wireless motility capsule. Dig Dis Sci. 2013;58(8):2275–2281. doi: 10.1007/s10620-012-2209-1
- Hedsund C, Gregersen T, Joensson IM, et al. Gastrointestinal transit times and motility in patients with cystic fibrosis. Scand J Gastroenterol. 2012;47(8–9):920–926. doi: 10.3109/00365521.2012.699548
- Kokubo T, Matsui S, Ishiguro M. Meta-analysis of oro-cecal transit time in fasting subjects. Pharm Res. 2013;30(2):402–411. doi: 10.1007/s11095-012-0882-6
- Spiller RC, Trotman IF, Adrian TE, et al. Further characterisation of the ‘ileal brake’ reflex in man–effect of ileal infusion of partial digests of fat, protein, and starch on jejunal motility and release of neurotensin, enteroglucagon, and peptide YY. Gut. 1988;29(8):1042–1051. doi: 10.1136/gut.29.8.1042
- Wen J, Phillips SF, Sarr MG, et al. PYY and GLP-1 contribute to feedback inhibition from the canine ileum and colon. Am J Physiol. 1995;269(6 Pt 1):G945–52. doi: 10.1152/ajpgi.1995.269.6.G945
- Abraham JM, Taylor CJ. Cystic fibrosis & disorders of the large intestine: DIOS, constipation, and colorectal cancer. J Cyst Fibros. 2017;16(Suppl 2):S40–s9. doi: 10.1016/j.jcf.2017.06.013
- Deiteren A, Camilleri M, Burton D, et al. Effect of meal ingestion on ileocolonic and colonic transit in health and irritable bowel syndrome. Dig Dis Sci. 2010;55(2):384–391. doi: 10.1007/s10620-009-1041-8
- Coffin B, Lémann M, Flourié B, et al. Ileal tone in humans: effects of locoregional distensions and eating. Am J Physiol. 1994;267(4 Pt 1):G569–74. doi: 10.1152/ajpgi.1994.267.4.G569
- Marsh R, Gavillet H, Hanson L, et al. Intestinal function and transit associate with gut microbiota dysbiosis in cystic fibrosis. J Cyst Fibros. 2022;21(3):506–513. doi: 10.1016/j.jcf.2021.11.014
- Knudsen PK, Brandtzaeg P, Høiby EA, et al. Impact of extensive antibiotic treatment on faecal carriage of antibiotic-resistant enterobacteria in children in a low resistance prevalence setting. PLoS One. 2017;12(11):e0187618. doi: 10.1371/journal.pone.0187618
- Burke DG, Harrison MJ, Fleming C, et al. Clostridium difficile carriage in adult cystic fibrosis (CF); implications for patients with CF and the potential for transmission of nosocomial infection. J Cyst Fibros. 2017;16(2):291–298. doi: 10.1016/j.jcf.2016.09.008
- Deane J, Fouhy F, Ronan NJ, et al. A multicentre analysis of Clostridium difficile in persons with Cystic Fibrosis demonstrates that carriage may be transient and highly variable with respect to strain and level. J Infect. 2021;82(3):363–370. doi: 10.1016/j.jinf.2020.12.027
- Enaud R, Hooks KB, Barre A, et al. Intestinal Inflammation in Children with Cystic Fibrosis is Associated with Crohn’s-Like Microbiota Disturbances. J Clin Med. 2019;8(5):645. doi: 10.3390/jcm8050645
- Furnari M, De Alessandri A, Cresta F, et al. The role of small intestinal bacterial overgrowth in cystic fibrosis: a randomized case-controlled clinical trial with rifaximin. J Gastroenterol. 2019;54(3):261–270. doi: 10.1007/s00535-018-1509-4
- Ronan NJ, Einarsson GG, Deane J, et al. Modulation, microbiota and inflammation in the adult CF gut: a prospective study. J Cyst Fibros. 2022;21(5):837–843. doi: 10.1016/j.jcf.2022.06.002
- Adriaanse MP, Van Der Sande LJTM, Van Den Neucker AM, et al. Evidence for a Cystic Fibrosis Enteropathy. PLoS One. 2015;10(10):e0138062. doi: 10.1371/journal.pone.0138062
- Jaudszus A, Pfeifer E, Lorenz M, et al. Abdominal symptoms assessed with the CFAbd-score are associated with intestinal inflammation in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2022;74(3):355–360. doi: 10.1097/MPG.0000000000003357
- Dhaliwal J, Leach S, Katz T, et al. Intestinal inflammation and impact on growth in children with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2015;60(4):521–526. doi: 10.1097/MPG.0000000000000683
- Bruzzese E, Callegari ML, Raia V, et al. Disrupted intestinal microbiota and intestinal inflammation in children with cystic fibrosis and its restoration with Lactobacillus GG: a randomised clinical trial. PLoS One. 2014;9(2):e87796. doi: 10.1371/journal.pone.0087796
- Bruzzese E, Raia V, Gaudiello G, et al. Intestinal inflammation is a frequent feature of cystic fibrosis and is reduced by probiotic administration. Aliment Pharmacol Ther. 2004;20(7):813–819. doi: 10.1111/j.1365-2036.2004.02174.x
- Ellemunter H, Engelhardt A, Schüller K, et al. Fecal calprotectin in cystic fibrosis and its relation to disease parameters: a longitudinal analysis for 12 years. J Pediatr Gastroenterol Nutr. 2017;65(4):438–442. doi: 10.1097/MPG.0000000000001544
- Roda J, Maia C, Almeida S, et al. Faecal calprotectin and rectal histological inflammatory markers in cystic fibrosis: a single-centre study. BMJ Paediatr Open. 2022;6(1):e001422. doi: 10.1136/bmjpo-2022-001422
- Stallings VA, Sainath N, Oberle M, et al. Energy Balance and Mechanisms of Weight Gain with Ivacaftor Treatment of Cystic Fibrosis Gating Mutations. J Pediatr. 2018;201:229–37.e4. doi: 10.1016/j.jpeds.2018.05.018
- Freeman AJ, Huang R, Heltshe SL, et al. Association between stool consistency and clinical variables among infants with cystic fibrosis: findings from the BONUS study. J Cyst Fibros. 2022;21(5):830–836. doi: 10.1016/j.jcf.2022.05.003
- Fraquelli M, Baccarin A, Corti F, et al. Bowel ultrasound imaging in patients with cystic fibrosis: relationship with clinical symptoms and CFTR genotype. Dig Liver Dis. 2016;48(3):271–276. doi: 10.1016/j.dld.2015.09.010
- Woodley FW, Machado RS, Hayes D, et al. Children with cystic fibrosis have prolonged chemical clearance of acid reflux compared to symptomatic children without cystic fibrosis. Dig Dis Sci. 2014;59(3):623–630. doi: 10.1007/s10620-013-2950-0
- De Sillos MD, Chiba SM, Soares AC, et al. Colonic Transit Time and Fecal Impaction in Children and Adolescents with Cystic Fibrosis-associated Constipation. J Pediatr Gastroenterol Nutr. 2021;73(3):319–324. doi: 10.1097/MPG.0000000000003202
- Olsen MF, Kjøller-Svarre MS, Møller G, et al. Correlates of Pancreatic Enzyme Replacement Therapy Intake in Adults with Cystic Fibrosis: results of a Cross-Sectional Study. Nutrients. 2022;14(7):1330. doi: 10.3390/nu14071330
- Ramos AF, de Fuccio MB, Moretzsohn LD, et al. Cystic fibrosis, gastroduodenal inflammation, duodenal ulcer, and H. pylori infection: the “cystic fibrosis paradox” revisited. J Cyst Fibros. 2013;12(4):377–383. doi: 10.1016/j.jcf.2012.11.001
- Subhi R, Ooi R, Finlayson F, et al. Distal intestinal obstruction syndrome in cystic fibrosis: presentation, outcome and management in a tertiary hospital (2007-2012). ANZ J Surg. 2014;84(10):740–744. doi: 10.1111/ans.12397
- Raun AM, Brekke G, Mølgaard C, et al. Impact of timing of PERT on gastrointestinal symptoms in Danish children and adolescents with CF. Acta Paediatr. 2022;111(2):432–439. doi: 10.1111/apa.16143
- Munck A, Alberti C, Colombo C, et al. International prospective study of distal intestinal obstruction syndrome in cystic fibrosis: associated factors and outcome. J Cyst Fibros. 2016;15(4):531–539. doi: 10.1016/j.jcf.2016.02.002
- Malagelada C, Bendezú RA, Seguí S, et al. Motor dysfunction of the gut in cystic fibrosis. Neurogastroenterol Motil. 2020;32(9):e13883. doi: 10.1111/nmo.13883
- Anderson JL, Tierney AC, Miles C, et al. Probiotic use in adults with cystic fibrosis is common and influenced by gastrointestinal health needs: a cross-sectional survey study. J Hum Nutr Diet. 2022;35(3):444–454. doi: 10.1111/jhn.12991
- Banjar H, Bawazir A, Ghomraoui F, et al. The first report on the association of celiac disease and cystic fibrosis in a tertiary care center in Saudi Arabia. Int J Pediatr Adolesc Med. 2022;9(1):56–61. doi: 10.1016/j.ijpam.2021.05.001
- Shah N, Tan HL, Sebire N, et al. The role of endoscopy and biopsy in the management of severe gastrointestinal disease in cystic fibrosis patients. Pediatr Pulmonol. 2013;48(12):1181–1189. doi: 10.1002/ppul.22697
- Farjadian S, Moghtaderi M, Kashef S, et al. Clinical and genetic features in patients with cystic fibrosis in southwestern iran. Iran J Pediatr. 2013;23(2):212–215.
- Sathe M, Huang R, Heltshe S, et al. Gastrointestinal factors associated with hospitalization in infants with cystic fibrosis: results from the baby observational and nutrition study. J Pediatr Gastroenterol Nutr. 2021;73(3):395–402. doi: 10.1097/MPG.0000000000003173
- Ooi CY, Jeyaruban C, Lau J, et al. High ambient temperature and risk of intestinal obstruction in cystic fibrosis. J Paediatr Child Health. 2016;52(4):430–435. doi: 10.1111/jpc.13096
- Coutinho CA, Marson FA, Ribeiro AF, et al. Cystic fibrosis transmembrane conductance regulator mutations at a referral center for cystic fibrosis. J Bras Pneumol. 2013;39(5):555–561. doi: 10.1590/S1806-37132013000500005
- Farahmand F, Tajdini P, Falahi G, et al. Evaluation of serum adenosine deaminase in cystic fibrosis patients in an Iranian Referral Hospital. Innov J Pediatr. 2016;26(3):e2246. doi: 10.5812/ijp.2246v3
- Toth T, Mak E, Galio N, et al. Research on the quality of life of adult patients with cystic fibrosis in Hungary. New Med. 2016;20:53–58. doi: 10.5604/14270994.1206757
- Lacy BE, Mearin F, Chang L, et al. Bowel Disorders. Gastroenterology. 2016;150(6):1393–407.e5. doi: 10.1053/j.gastro.2016.02.031
- Rasquin A, Di Lorenzo C, Forbes D, et al. Childhood functional gastrointestinal disorders: child/adolescent. Gastroenterology. 2006;130(5):1527–1537. doi: 10.1053/j.gastro.2005.08.063
- Dellschaft NS, Ng C, Hoad C, et al. Magnetic resonance imaging of the gastrointestinal tract shows reduced small bowel motility and altered chyme in cystic fibrosis compared to controls. J Cyst Fibros. 2022;21(3):502–505. doi: 10.1016/j.jcf.2021.12.007
- Al-Baba R, Zetoune AB. A retrospective study of cases diagnosed with cystic fibrosis at a single care center in syria. Egypt J Med Hum Genet. 2021;22(1):59. doi: 10.1186/s43042-021-00178-5
- Sağlam D, Demirbaş F, Bilgici MC, et al. Can point shear wave elastography be used as an early indicator of involvement? J Ultrasound Med. 2020;39(9):1769–1776. doi: 10.1002/jum.15281
- Aziz DA, Billoo AG, Qureshi A, et al. Clinical and laboratory profile of children with Cystic Fibrosis: experience of a tertiary care center in Pakistan. Pak J Med Sci. 2017;33(3):554–559. doi: 10.12669/pjms.333.12188
- Caldaro T, Alghisi F, De Angelis P, et al. Cystic fibrosis: a surgical matter? J Pediatr Surg. 2014;49(5):753–758. doi: 10.1016/j.jpedsurg.2014.02.089
- Huang L, Lai HJ, Antos N, et al. Defining and identifying early-onset lung disease in cystic fibrosis with cumulative clinical characteristics. Pediatr Pulmonol. 2022;57(10):2363–2373. doi: 10.1002/ppul.26040
- Kristensen M, Prevaes S, Kalkman G, et al. Development of the gut microbiota in early life: the impact of cystic fibrosis and antibiotic treatment. J Cyst Fibros. 2020;19(4):553–561. doi: 10.1016/j.jcf.2020.04.007
- Baad M, Delgado J, Dayneka JS, et al. Diagnostic performance and role of the contrast enema for low intestinal obstruction in neonates. Pediatr Surg Int. 2020;36(9):1093–1101. doi: 10.1007/s00383-020-04701-4
- Farrelly PJ, Charlesworth C, Lee S, et al. Gastrointestinal surgery in cystic fibrosis: a 20-year review. J Pediatr Surg. 2014;49(2):280–283. doi: 10.1016/j.jpedsurg.2013.11.038
- Rafeey M, Jabarpoor-Bonyadi M, Vahedi L. Genotype-phenotype correlation for cystic fibrosis according to registry center of cystic fibrosis. Crescent J Med Biol Sci. 2020;7(1):124–129.
- Beaufils F, Mas E, Mittaine M, et al. Increased fecal calprotectin is associated with worse gastrointestinal symptoms and quality of life scores in children with cystic fibrosis. J Clin Med. 2020;9(12):4080. doi: 10.3390/jcm9124080
- Mentessidou A, Loukou I, Kampouroglou G, et al. Long-term intestinal obstruction sequelae and growth in children with cystic fibrosis operated for meconium ileus: expectancies and surprises. J Pediatr Surg. 2018;53(8):1504–1508. doi: 10.1016/j.jpedsurg.2017.11.040
- Guo X, Pace RG, Stonebraker JR, et al. Meconium ileus in cystic fibrosis is not linked to central repetitive region length variation in MUC1, MUC2, and MUC5AC. J Cyst Fibros. 2014;13(6):613–616. doi: 10.1016/j.jcf.2014.05.005
- Smith DJ, Klein K, Hartel G, et al. Mutations in the HFE gene can be associated with increased lung disease severity in cystic fibrosis. Gene. 2019;683:12–17. doi: 10.1016/j.gene.2018.10.002
- Thomas L, Kumar M, Lionel BA, et al. Pancreatic, hepatobiliary, and gastrointestinal manifestations of children with cystic fibrosis: a 10-year experience from a tertiary care center in southern India. Indian J Gastroenterol. 2022;41(3):266–272. doi: 10.1007/s12664-021-01225-0
- Aksit MA, Ling H, Pace RG, et al. Pleiotropic modifiers of age-related diabetes and neonatal intestinal obstruction in cystic fibrosis. Am J Hum Genet. 2022;109(10):1894–1908. doi: 10.1016/j.ajhg.2022.09.004
- Banjar H, Qeretli R, Ramadan A, et al. The first report on CFTR mutations of meconium ileus in cystic fibrosis population in Saudi Arabia: a single center review. Int J Pediatr Adolesc Med. 2022;9(1):32–35. doi: 10.1016/j.ijpam.2021.03.008
- Terlizzi V, Carnovale V, Castaldo G, et al. Clinical expression of patients with the D1152H CFTR mutation. J Cyst Fibros. 2015;14(4):447–452. doi: 10.1016/j.jcf.2014.12.012
- Kumar A, Aggarwal B, Bamal P, et al. Clinical profile of children with cystic fibrosis surviving through adolescence and beyond. Indian Pediatr. 2022;59(1):43–45. doi: 10.1007/s13312-022-2419-3
- Rovner AJ, Schall JI, Mondick JT, et al. Delayed small bowel transit in children with cystic fibrosis and pancreatic insufficiency. J Pediatr Gastroenterol Nutr. 2013;57(1):81–84. doi: 10.1097/MPG.0b013e318290d112
- Aghamohammadi A, Keivanfar M, Navaei S, et al. First cystic fibrosis patient registry annual data report - cystic fibrosis foundation of Iran. Acta Med Iran. 2019;57(1):33–41. doi: 10.18502/acta.v57i1.1751
- Declercq D, Van Biervliet S, Robberecht E. Nutrition and pancreatic enzyme intake in patients with cystic fibrosis with distal intestinal obstruction syndrome. Nutr Clin Pract. 2015;30(1):134–137. doi: 10.1177/0884533614551838
- Gabel ME, Wang H, Gelfond D, et al. Changes in glucose breath test in cystic fibrosis patients treated with 1 month of lumacaftor/ivacaftor. J Pediatr Gastroenterol Nutr. 2022;75(1):42–47. doi: 10.1097/MPG.0000000000003459
- Ooi CY, Pang T, Leach ST, et al. Fecal human β-defensin 2 in children with cystic fibrosis: is there a diminished intestinal innate immune response? Dig Dis Sci. 2015;60(10):2946–2952. doi: 10.1007/s10620-015-3842-2
- Del Campo R, Garriga M, Pérez-Aragón A, et al. Improvement of digestive health and reduction in proteobacterial populations in the gut microbiota of cystic fibrosis patients using a lactobacillus reuteri probiotic preparation: a double blind prospective study. J Cyst Fibros. 2014;13(6):716–722. doi: 10.1016/j.jcf.2014.02.007
- Bozic M, Goss CH, Tirouvanziam RM et al. Oral Glutathione and Growth in Cystic Fibrosis: a Multicenter, Randomized, Placebo-controlled, Double-blind Trial. J Pediatr Gastroenterol Nutr. 2020;71(6):771–777. doi: 10.1097/MPG.0000000000002948
- Tétard C, Mittaine M, Bui S, et al. Reduced intestinal inflammation with lumacaftor/ivacaftor in adolescents with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2020;71(6):778–781. doi: 10.1097/MPG.0000000000002864
- Shakir S, Echevarria C, Doe S, et al. Elexacaftor-tezacaftor-ivacaftor improve gastro-oesophageal reflux and sinonasal symptoms in advanced cystic fibrosis. J Cyst Fibros. 2022;21(5):807–810. doi: 10.1016/j.jcf.2022.06.003
- Woodley FW, Hayes Jr D, Kopp BT, et al. Gastroesophageal reflux in cystic fibrosis across the age spectrum. Transl Gastroenterol Hepatol. 2019;4:69. doi: 10.21037/tgh.2019.08.11
- Brecelj J, Zidar N, Jeruc J, et al. Morphological and functional assessment of oesophageal mucosa integrity in children with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2016;62(5):757–764. doi: 10.1097/MPG.0000000000001131
- Doumit M, Krishnan U, Jaffé A, et al. Acid and non-acid reflux during physiotherapy in young children with cystic fibrosis. Pediatr Pulmonol. 2012;47(2):119–124. doi: 10.1002/ppul.21524
- Calvo-Lerma J, Martínez-Barona S, Masip E, et al. Pancreatic enzyme replacement therapy in cystic fibrosis: dose, variability and coefficient of fat absorption. Rev Esp Enferm Dig. 2017;109(10):684–689. doi: 10.17235/reed.2017.4951/2017
- Matson AG, Bunting JP, Kaul A, et al. A non-randomised single centre cohort study, comparing standard and modified bowel preparations, in adults with cystic fibrosis requiring colonoscopy. BMC Gastroenterol. 2019;19(1):89. doi: 10.1186/s12876-019-0979-z
- Maisonneuve P, Marshall BC, Knapp EA, et al. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J Natl Cancer Inst. 2013;105(2):122–129. doi: 10.1093/jnci/djs481
- Billings JL, Dunitz JM, Mcallister S, et al. Early colon screening of adult patients with cystic fibrosis reveals high incidence of adenomatous colon polyps. J Clin Gastroenterol. 2014;48(9):e85–8. doi: 10.1097/MCG.0000000000000034
- Schmedding A, Hutter M, Gfroerer S, et al. Jejunoileal atresia: a national cohort study. Front Pediatr. 2021;9:665022. doi: 10.3389/fped.2021.665022
- Siersma CL, Rottier BL, Hulscher JB, et al. Jejunoileal atresia and cystic fibrosis: don’t miss it. BMC Res Notes. 2012;5:677. doi: 10.1186/1756-0500-5-677
- Emiralioglu N, Ademhan Tural D, Hizarcioglu Gulsen H, et al. Does cystic fibrosis make susceptible to celiac disease? Eur J Pediatr. 2021;180(9):2807–2813. doi: 10.1007/s00431-021-04011-4
- Saxby NPC, Kench A, King S, et al. Nutrition Guidelines For Cystic Fibrosis In Australia And New Zealand Thoracic Society Of Australia And New Zealand, Sydney; 2017
- CF Trust Nutrition Working Group. Nutritional management of cystic fibrosis: cystic fibrosis trust; 2016. Available from: https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/Nutritional%20Management%20of%20cystic%20fibrosis%20Sep%2016.pdf.
- Smyth RL, Smyth AR, Lloyd DA et al. Strictures of ascending colon in cystic fibrosis and high-strength pancreatic enzymes. Lancet. 1994;343(8889):85–86. doi: 10.1016/S0140-6736(94)90817-6
- Smyth RL, O’hea U, Burrows E, et al. Fibrosing colonopathy in cystic fibrosis: results of a case-control study. Lancet. 1995;346(8985):1247–1251. doi: 10.1016/S0140-6736(95)91860-4
- Dodge JA. Pancreatic enzymes and fibrosing colonopathy. J Cyst Fibros. 2015;14(1):153. doi: 10.1016/j.jcf.2014.09.002
- Gelfond D, Heltshe SL, Skalland M, et al. Pancreatic enzyme replacement therapy use in infants with cystic fibrosis diagnosed by newborn screening. J Pediatr Gastroenterol Nutr. 2018;66(4):657–663. doi: 10.1097/MPG.0000000000001829
- Kashirskaya NY, Kapranov NI, Sander-Struckmeier S, et al. Safety and efficacy of Creon Micro in children with exocrine pancreatic insufficiency due to cystic fibrosis. J Cyst Fibros. 2015;14(2):275–281. doi: 10.1016/j.jcf.2014.07.006
- Dodge JA. Fibrosing colonopathy. Gut. 2000;46(2):152–153. doi: 10.1136/gut.46.2.152
- Committee on Safety of Medicines. Report of the pancreatic enzymes working party. Stationary Office: London; 1995.
- Cystic Fibrosis Foundation. Cystic fibrosis foundation patient registry 2021 annual data report. Bethesda (MD): Cystic Fibrosis Foundation; 2022.
- Knotts RM, Solfisburg QS, Keating C, et al. Cystic fibrosis is associated with an increased risk of Barrett’s esophagus. J Cyst Fibros. 2019;18(3):425–429. doi: 10.1016/j.jcf.2018.11.005
- Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335(7632):1255–1259. doi: 10.1136/bmj.39391.713229.AD
- National Institute for Health and Care Excellence. Exocrine pancreatic insufficiency; 2023. Available from: https://bnf.nice.org.uk/treatment-summaries/exocrine-pancreatic-insufficiency/#related-drugs.
- Ng C, Major G, Smyth AR. Timing of pancreatic enzyme replacement therapy (PERT) in cystic fibrosis. Cochrane Database Syst Rev. 2021;2021(8). doi: 10.1002/14651858.CD013488.pub2
- Turck D, Braegger CP, Colombo C, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr. 2016;35(3):557–577. doi: 10.1016/j.clnu.2016.03.004
- Borowitz D, Robinson KA, Rosenfeld M, et al. Cystic fibrosis foundation evidence-based guidelines for management of infants with cystic fibrosis. J Pediatr. 2009;155(6, Supplement):S73–S93. doi: 10.1016/j.jpeds.2009.09.001
- Boon M, Calvo-Lerma J, Claes I, et al. Use of a mobile application for self-management of pancreatic enzyme replacement therapy is associated with improved gastro-intestinal related quality of life in children with cystic fibrosis. J Cyst Fibros. 2020;19(4):562–568. doi: 10.1016/j.jcf.2020.04.001
- Doligalski CT, McKinzie CJ, Yang A, et al. Poor tolerability of cystic fibrosis transmembrane conductance regulator modulator therapy in lung transplant recipients. Pharmacotherapy. 2022;42(7):580–584. doi: 10.1002/phar.2710
- Gelfond D, Heltshe S, Ma C, et al. Impact of CFTR modulation on intestinal pH, motility, and clinical outcomes in patients with cystic fibrosis and the G551D mutation. Clin Transl Gastroenterol. 2017;8(3):e81. doi: 10.1038/ctg.2017.10
- Pope CE, Vo AT, Hayden HS, et al. Changes in fecal microbiota with CFTR modulator therapy: a pilot study. J Cyst Fibros. 2021;20(5):742–746. doi: 10.1016/j.jcf.2020.12.002
- Graeber SY, Dopfer C, Naehrlich L, et al. Effects of lumacaftor-ivacaftor therapy on cystic fibrosis transmembrane conductance regulator function in phe508del homozygous patients with cystic fibrosis. Am J Respir Crit Care Med. 2018;197(11):1433–1442. doi: 10.1164/rccm.201710-1983OC
- Smyth AR, Giles M Gut imaging for function and transit in cystic fibrosis study 2 (GIFT-CF2) clinicaltrials.Gov: clinicaltrials.Gov; 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT04006873.
- Smyth AR CFTR Modulators and Gastrointestinal Complications (CFTR-MAGIC) clinicaltrials.Gov: clinicaltrials.Gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT05253859.
- Padoan R, Cirilli N, Falchetti D, et al. Risk factors for adverse outcome in infancy in meconium ileus cystic fibrosis infants: a multicentre Italian study. J Cyst Fibros. 2019;18(6):863–868. doi: 10.1016/j.jcf.2019.07.003
- Abu-El-Haija M, Uc A, Werlin SL, et al. Nutritional considerations in pediatric pancreatitis: a position paper from the NASPGHAN pancreas committee and ESPGHAN Cystic Fibrosis/Pancreas Working Group. J Pediatr Gastroenterol Nutr. 2018;67(1):131–143. doi: 10.1097/MPG.0000000000002023
- Jaudszus A, Zeman E, Jans T, et al. Validity and reliability of a novel multimodal questionnaire for the assessment of abdominal symptoms in people with cystic fibrosis (CFAbd-score). Patient. 2019;12(4):419–428. doi: 10.1007/s40271-019-00361-2
- Smyth AR A comprehensive approach to relief of digestive symptoms in cystic fibrosis: CARDS-CF (CARDS-CF) clinicaltrials.Gov: clinicaltrials.Gov; 2022. Available from: https://clinicaltrials.gov/ct2/show/NCT05251467.
- Smyth AR The core outcome set taskforce for CF (COST-CF) comet initiative: comet initiative; 2022. Available from: https://www.comet-initiative.org/studies/details/882.