
ABSTRACT
Introduction
The use and generation of gene signatures have been established as a method to define molecular endotypes in complex diseases such as severe asthma. Bioinformatic approaches have now been applied to large omics datasets to define the various co-existing inflammatory and cellular functional pathways driving or characterizing a particular molecular endotype.
Areas covered
Molecular phenotypes and endotypes of Type 2 inflammatory pathways and also of non-Type 2 inflammatory pathways, such as IL-6 trans-signaling, IL-17 activation, and IL-22 activation, have been defined in the Unbiased Biomarkers for the Prediction of Respiratory Disease Outcomes dataset. There has also been the identification of the role of mast cell activation and of macrophage dysfunction in various phenotypes of severe asthma.
Expert opinion
Phenotyping on the basis of clinical treatable traits is not sufficient for understanding of mechanisms driving the disease in severe asthma. It is time to consider whether certain patients with severe asthma, such as those non-responsive to current therapies, including Type 2 biologics, would be better served using an approach of molecular endotyping using gene signatures for management purposes rather than the current sole reliance on blood eosinophil counts or exhaled nitric oxide measurements.
1. Toward molecular phenotypes and endotypes
Analysis of clinical, physiologic, and inflammatory features of asthma is not sufficient to derive mechanism-based clusters or to identify the molecular interactions that underlie these features of the disease in an individual person. Recent emphasis on the treatment of readily identifiable treatable traits in respiratory disease has led to a path toward personalized medicine but does not address the issue of the underlying mechanisms of disease. In order to understand such mechanisms, one needs to approach the disease as a complex dynamic system with many interacting pathways that represent even more complexity than analyzing only each of its component parts separately. Thus, it is becoming clear that different clinical traits associated with severe asthma, such as airflow obstruction or blood eosinophilia, can be linked to different molecular pathways. Systems biology models biologic systems and processes such as those occurring in asthma through multi-scale mathematical and computing methods that integrate the biologic networks and pathways involved [Citation1].
This approach is key to achieving the practice of precision medicine, which is ‘an approach to treat and prevent disease by taking into consideration the individual variability in genes, environment and lifestyle for each individual.’ This leads to the concept of treating ‘the right patient with the right drug at the right time,’ using treatments that are targeted for each particular individual [Citation2]. Thus, the composite analysis of the genes, proteins, lipids, and metabolites in samples obtained from an individual is more likely to indicate potential pathogenic and causative pathways that would be necessary for the definition of molecular phenotypes or endotypes based on the identification of the driving mechanisms [Citation3]. Targeting the mechanisms and pathways driving these phenotypes represents a more successful strategy to achieve effective treatments than an analytical approach to clinical phenotypes, as exemplified by the success of personalized therapies in oncology.
Molecular phenotypes or endotypes are distinct from clinical phenotypes and represent the immune-inflammatory links between clinical features and disease-driving mechanisms. An endotype was defined in 2008 as ‘a subtype of disease defined functionally and pathologically by a molecular mechanism or by treatment response… ’ [Citation4]. Due to the heterogeneity of asthma and the genetic and environmental factors involved in its pathogenesis, asthma is not composed of a single endotype, and many distinct endotypes may (co)-exist, each associated with specific clinical features but reflecting differing molecular causes and clinical responses to defined therapies. This may explain why not all severe asthma patients stratified to a Type 2 (T2) biomarker-high asthma phenotype (high levels of blood eosinophils and fractional exhaled nitric oxide, FeNO) respond similarly to anti-T2 biologics [Citation5].
To approach the definition of asthma endotypes, the focus has been on the integration of multi-omics data, which included genetics, epigenetics, transcriptomics (gene array and bulk and single-cell RNA-sequencing), proteomics (unbiased and focused multiplexing), lipidomics, metabolomics, and metagenomics, of various samples not only from the airways (bronchial biopsy, bronchial brushings, nasal brushings, bronchoalveolar lavage cells, and sputum samples) but also from other compartments such as blood, urine, and breath from patients with asthma of differing asthma severities or clinical characteristics. This approach has been successful in identifying subgroups of Chronic Obstructive Pulmonary Disaese (COPD) patients within a Swedish cohort using an -omics fusion method known as Similarity Network Fusion [Citation6]. A similar approach is being taken in the European asthma consortium of the Unbiased Biomarkers for the Prediction of Respiratory Disease Outcomes (U-BIOPRED) [Citation7].
2. Signatures and gene expression profiling
The advent of high-throughput sequencing has enabled simultaneous assessment of the expression of thousands of genes in a given sample and has led to an increased interest in the identification and generation of gene signatures. The identification of disease mechanisms has been made possible through the statistical analysis of different levels of -omics data, such as transcriptomics or proteomics. This can be followed by annotation with up-to-date ontologies to generate biomarker signatures derived from data collected from a single -omics platform or those biomarker signatures derived from data collected from more than one -omics platform [Citation8].
Gene signatures are specific group of genes that exhibit a coordinated gene expression pattern or alteration associated with, or in response to, a particular drug treatment, disease state, or biological process [Citation9,Citation10]. These gene signatures can therefore be used to classify groups of samples in any independent dataset. In the context of lung diseases, gene signatures can be derived from one’s own cell or tissue data or be knowledge-based and obtained from accessible gene ontology or pathway databases including newly described single-cell lung atlases [Citation11,Citation12]. These latter datasets define individual lung cell types and activation types. Thus, these signatures have been used to interpret the results of analyses of gene expression data such as differential expression analysis and clustering approaches.
Differentially expressed gene (DEG) analysis is one such method and is often employed to compare gene expression levels between different groups or conditions to identify genes that are differentially expressed. The approaches used to identify these signatures typically start by defining a null hypothesis, generating a p-value, and then applying a significance threshold that considers multiple comparisons to minimize the risk of Type 1 error [Citation13]. Several common techniques apart from the Wilcoxon rank-sum test are available to calculate different methods of dispersion to moderate gene-specific variance to give more accurate DEGs, especially for genes with low counts [Citation14–16], and these DEGs can then be used as a candidate gene signature. A similar approach, albeit more computationally sophisticated, is used to define single-cell signatures and activation states and may possibly be used in the future to help define or predict super-responders to therapies.
Pathway analysis methods for utilizing gene signatures and assessing their enrichment in other samples include Gene Set Enrichment Analysis (GSEA) [Citation17] and Gene Set Variation Analysis (GSVA) [Citation18]. GSEA tests the hypothesis that none of the gene signatures is associated with the phenotype group. The genes are ranked by the mean expression level for the phenotype group and the control group. Each gene in the gene signature is then matched up to the rank of the gene from the previous step. If a gene is in the gene signature, the aggregate gene expression value for that group is added to the running sum of expression score, and if it is not in the gene signature, then a set value is subtracted from the running sum. The peak value of the running sum is set as the enrichment score of that phenotype.
GSVA is used to calculate the sample-wise enrichment (gene set score) of a set of genes (gene signatures) in a normalized gene expression matrix. First, the genes are ordered by the rank of the expression levels for each sample [Citation18]. Then, for each gene signature, the cumulative distribution function of the ranked expression values of genes within the gene signature and genes not in the gene signature is calculated for each sample. A p-value gives the probability they come from the same distribution. GSVA can also be applied using an unsupervised approach where sample-wise enrichment scores are calculated irrespective of group labels that express the variation in activity of a set of genes that represents a specific cell activation state or pathway over the whole sample population.
3. Identification of molecular phenotypes in U-BIOPRED
All the analyses of gene signatures in severe asthma that will be presented in this review will come from the U-BIOPRED database, which is a unique database of patients with severe asthma with comparative groups of mild-moderate asthma and non-asthmatic controls [Citation19]. In addition, readily available biomarkers, samples of blood, sputum, urine, nasal brushings, bronchoscopic specimens, bronchial brushings, and bronchial biopsies were also obtained for various omics platform analyses. Bioinformatic ‘large data’ analytical approaches were undertaken in order to link the clinical, physiologic, and inflammatory characteristics with the omics data so that mechanistic pathways could be derived [Citation8]. Hierarchical clustering of the DEGs in an unbiased manner and supervised machine learning algorithms to refine any signatures that are used to identify molecular clsuters have been used in the analysis of U-BIOPRED omics data. The molecular clusters so-derived are then characterized according to their clinico-physiologic features. One reason for focusing on the U-BIOPRED database apart from its extensive rich -omics data is that this single database has been analyzed in many different ways. This review will also bring in published analyses from other severe asthma cohorts if only to compare the molecular phenotype findings.
3.1. Hierarchical clustering of DEGs in sputum to yield gene signatures of endotypes
A semi-biased transcriptomic analysis together with unsupervised machine learning analysis of asthma sputum eosinophilic samples compared with non-eosinophilic samples identified three transcriptome-associated clusters (TAC) [Citation20] (). The first cluster, TAC1, was characterized by frequent exacerbations, severe airflow obstruction, oral corticosteroid-dependent asthma, high sputum eosinophilia, and high FeNO, together with the expression of IL33R, CCR3, and TSLPR genes. Using GSVA, this TAC1 cluster had the highest expression enrichment for the gene signatures of the IL-13-induced epithelial cell transcripts and of ILC2 activation transcripts, both representative of T2-inflammation gene signature. This molecular phenotype forms the basis for the severe eosinophilic asthma phenotype.
Figure 1. List of gene signatures associated with each of the transcriptome-associated clusters (TACs) and GSVA heatmap of signatures associated with specific pathways. Heatmap showing the GSVA enrichment score of the pathway signatures for IL13-Th2, ILC2, inflammasome, neutrophil, aging, and OXPHOS grouped by the TACs. (Data taken from Kuo et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. 2017;49(2): 1602135.).
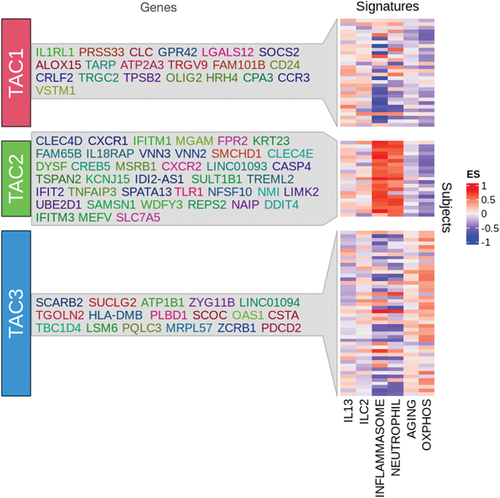
In contrast, TAC2 was characterized by sputum neutrophilia, inflammasome activation, interferon (IFN)-α and tumor necrosis factor-α–associated genes, and high serum C-reactive protein levels, and clinically, these subjects had a higher prevalence of eczema and a moderate degree of airflow obstruction. Signatures of inflammasome and neutrophil activation were by far mostly expressed in this molecular phenotype, but there was also high expression of the IL-13 T2 signature, indicating that this phenotype is driven by both T1 and T2 mechanisms.
TAC3 was associated with pauci-granulocytic asthma and the highest expression of metabolic, mitochondrial, and ubiquitination pathway genes. Clinically, these patients had moderate airflow obstruction and the least number of exacerbations. By GSVA, there was highest expression of signature for oxidative phosphorylation occurring in the mitochondria and for aging signature, which interestingly has a mitochondrial component. In a separate analysis of U-BIOPRED sputum proteomics, topological data analysis (TDA) identified three potential subsets or activation states of eosinophilic and neutrophilic asthma [Citation21].
3.2. Clustering of DEGs in bronchial biopsies and brushings to yield gene signatures of endotypes
In U-BIOPRED, gene expression data from bronchial biopsies and epithelial brushings were used to define molecular phenotypes of asthma by examining their gene expression profiles on the basis of important disease drivers as described by specific gene sets and derived phenotypes from the gene set clusters. Nine gene set signatures applied to genes expressed in bronchial biopsies and airway epithelial brushings identified two subtypes of patients with Type 2 eosinophilic inflammation and relative corticosteroid insensitivity. The TDA was used to provide a very clear way of visualizing the differences in gene expression and has been used elsewhere [Citation21]. Feature reduction and machine learning (shrunken centroids) analysis of these nine gene signatures identified a subgroup of genes that were associated with corticosteroid insensitivity, and Type 2 eosinophilic signatures showed an association between CD44 and the T2-associated genes, such as CCL26, IL1R2, and CST2. These were also associated with signatures of oxidative stress, and the patients exhibiting a high enrichment of these signatures were on the highest dose of daily oral corticosteroids [Citation22]. Using another machine learning tool, an inference tree framework, the inflammatory biomarkers such as sputum eosinophilia and FeNO levels were found to predict the subtypes of patients with asthma described by gene expression profiling. This approach, using a subset of gene signatures relevant to asthma pathways, defined the importance of the site of expression of these nine gene set signatures in either biopsies or brushings, or in both, in determining molecular phenotypes of asthma.
3.3. Omics analysis of clinical phenotypes
3.3.1. Type 2 pathways
We have previously used more than 100 GSVA signatures in our analysis of our clinical or molecular phenotypes. Several thousand signatures are now available in Reactome and MetaCore databases, which may help further refine subtype analysis and overcome the problem of transcriptomics in multi-omic analysis, i.e. there are far more genes (>22,000) than proteins and metabolites that can be identified with current technology (~100s to 1000s). However, a T2 signature of 34 genes that were shown to be upregulated after in vitro stimulation of airway epithelial cells with IL-13 was used to observe differences between clinical phenotypes [Citation23]. Late-onset eosinophilic asthma was analyzed by applying GSVA to a signature of IL-13-regulated genes in airway epithelium; this revealed that the T2-high phenotype was more obstructed but showed no differences in exacerbations, with worse asthma control and high blood and sputum eosinophils [Citation23]. Significant differentially enriched gene signatures were identified in nasal brushings, sputum, and endobronchial brushings in adult-onset severe asthma patients compared to childhood-onset severe asthma [Citation24]. These signatures indicated that adult-onset asthma was characterised by inflammatory pathways involving eosinophils, mast cells and Group 3 innate lymphoid cells [Citation24].
In another study of patients with chronic airflow obstruction, differentially enriched gene signatures were associated with corticosteroid sensitivity, eosinophils, interleukin-13, IFN-α, specific CD4+ T cells, and airway remodeling [Citation25]. Analysis of frequent exacerbators (two or more severe exacerbations reported in the previous year) versus infrequent exacerbations revealed higher Type 1 (T1) and T2 inflammatory pathways with steroid insensitivity pathways [Citation26]. For obesity-associated asthma, an analysis of epithelial gene expression in severe asthmatics identified three clusters, one of which was enriched for obesity and gastro-esophageal reflux disease. This was a pauci-granulocytic group as a result of reduced mechanisms of cell recruitment linked to bile acid exposure and treatment with proton pump inhibitors [Citation27]. Current smoking in severe asthma was associated with the enrichment of oxidative stress and endoplasmic reticulum stress, while CXCL5 and MMP12 expressions were upregulated in ex-smoking severe asthma patients [Citation28].
3.3.2. Non-T2 molecular phenotypes by GSVA
Similar to the use of IL-13-stimulated human bronchial epithelial cell (HBEC) signature to identify patients with T2 asthma, stimulation of HBECs with non-T2 stimuli has been used to define patients with non-T2 subtypes of asthma. Thus, exposure of HBECs to a combination of IL-6 and sIL-6R led to activation of IL-6 trans-signaling (IL-6TS), selecting a gene signature distinct from that seen with IL-13 HBEC stimulation. This signature was upregulated in asthmatic subjects who do not express high levels of the IL-13 T2 signature [Citation29]. Similarly, stimulation of HBECs with IL-17A produces a gene expression profile that is expressed inversely with the IL-13-stimulated HBEC signature in asthmatic patients, whose gene pattern resembles that reported in skin biopsies of patients with IL-17-driven psoriasis [Citation30].
Using the concept of cross-disease stimulation signatures, the transcriptomic signature of a drug used in one disease may identify individuals who may respond to the same medication in a different disease [Citation31]. Applying the transcriptomic signatures obtained by analysis of differential gene expression in the skin of atopic dermatitis patients successfully treated with the anti-IL-22 monoclonal antibody, fezakinumab, to the sputum transcriptomics of patients with severe asthma showed a significant enrichment of the signatures in those with severe neutrophilic asthma. These subjects had high levels of sputum IL-22. Nasal epithelial brushings also gave a good prediction of the potential responder population. Clinical features and biomarkers identified may be used to pick up T2-low asthma patients that might respond to Fezakinumab.
4. Gene signatures of activated mast cells
Mast cells have been implicated in asthma, with an important role both in inflammation and in innate and adaptive immunity, but their contribution in driving the pathways of severe asthma is ill-defined. Mast cells respond to the local environment of a mixture of inflammatory mediators that are liberated and are difficult to detect by standard methods. However, the effects of these mediators on the mast cells can be investigated by using gene signatures of mast cell stimulation through not only the IgE-FceR1 interaction but also IL-33 receptor, toll-like receptor 4, and IFN receptor to define severe asthma molecular phenotypes. The IL-33-stimulated mast cell signature was associated with severe neutrophilic asthma, whereas IgE-activated mast cell signature was associated with an eosinophilic phenotype [Citation32]. This was the first indication that IL-33 was associated with neutrophilic or T2-low asthma rather than T2-high asthma, which has been confirmed in subsequent clinical trials [Citation33,Citation34]. These data also indicate that mast cells can be induced to take on distinct transcriptional phenotypes associated with specific clinical phenotypes. Interestingly, in an independent cohort of severe asthma from Australia examining differentially expressed pathways in the transcriptome from endobronchial biopsies and induced sputum highlighted the role of CD4+ T cells, mast cells and pathways linked to ongoing airway remodeling as likely active mechanisms in the pathogenesis of severe asthma, a finding reproduced in U-BIOPRED cohort [Citation35].
5. Gene signatures of macrophages
Macrophages and monocytes have also been implicated in the pathogenesis of asthma in terms of innate immune response, but the pathways involved also remain unclear. We assessed the role of various types of macrophages, namely lung tissue-resident cells (TR-Mφ) and two for their polarization (classically and alternatively activated macrophages: M1 and M2, respectively), using gene signatures representative of the activation status of these macrophage subtypes. Interestingly, the expression scores for most macrophage modules were significantly reduced in severe asthma except for three associated with inflammatory responses driven by TNF and Toll-like receptors via NF-κB, eicosanoid biosynthesis via the lipoxygenase pathway, and IL-2 biosynthesis [Citation36]. The expression score for most macrophage signatures was higher in the TAC3 group compared to TAC1 and TAC2 asthmatics. However, a high enrichment was found in TAC1 for three modules showing inflammatory pathways linked to Toll-like and TNF receptor activation and arachidonic acid metabolism and in TAC2 for the inflammasome and interferon signaling pathways. TR-Mφ were enriched in TAC3 and associated with mitochondrial function. Thus, macrophage activation is attenuated in severe granulocytic asthma, highlighting defective innate immunity except for specific subsets characterized by distinct inflammatory pathways.
6. Future of gene signatures in molecular phenotyping and endotyping
There are increasing number of gene signatures now available for analyzing gene expression data, and many more are being identified and developed following the treatment of cells, tissues, and patients with specific therapeutic agents and the advent of single-cell analysis. These may be used to define changes in cell composition and/or activation states in disease or after therapy using cellular deconvolution. The recent publication of the Human Lung Cell Atlas is a useful reference that will allow the wide use of this approach for lung diseases, including severe asthma [Citation11]. The combination of these new signatures along with previously identified pathway and ontology signatures will enhance our understanding of disease processes in obstructive lung diseases such as asthma and COPD. They also hold the promise of aiding in the identification of novel targets/molecular phenotypes that together with new noninvasive biomarkers will enable future therapeutic intervention in specific subsets of patients. However, we need to develop methods to generate and validate these signatures; in addition, we need to be able to pick up the best signature to use for molecular phenotyping and endotyping of patients with severe asthma.
7. Expert opinion
Despite the usefulness of gene signatures in defining the molecular phenotypes and endotypes of severe asthma patients, their application in the clinical area has hardly begun. This approach has been used to support the validity of using bedside biomarkers such as blood eosinophil counts and exhaled nitric oxide levels as markers of Type 2 inflammation [Citation23]. These biomarkers have also been used to imply a non-Type 2 inflammatory mechanism when they fall below a certain threshold level. While there is a reasonable predictability of Type 2 inflammation in the use of these biomarkers, they are not specific enough to pinpoint the type of T2 or non-T2 inflammation driving the disease process in a particular patient with severe asthma. This is an issue in terms of non-Type 2 inflammatory mechanisms, which are likely to include several disparate inflammatory and cellular pathways. The use of gene signatures would directly enhance our understanding of the potential pathways that may be driving a particular patient’s severe asthma.
It is clear that taking such an approach will necessitate the obtention of cells or tissues that can be obtained through bronchoscopic procedure or through the induction of sputum cells and running a series of gene or protein expression measurements. In addition, the data would need to be analyzed. The cost of these procedures and assays will be more expensive than a blood eosinophil count or a FeNO measurement, but they will provide more precise information as to the underlying inflammatory and immune mechanisms underlying a patient’s severe asthma and more information to establish the targets that could be aimed at for a particular patient’s asthma. Such an approach would only be available in specialized centers. Very likely, this approach may be taken in the more severe cases of severe asthma that do not show evidence of T2-inflammation with the currently used bedside biomarkers or those who have failed on the currently available biologic therapies.
Article highlights
The advent of high-throughput sequencing, enabling the simultaneous characterization of the whole gene expression of a given clinical sample, is leading to the identification of disease mechanisms.
Gene signatures can be used to examine coordinated gene expression patterns or alterations associated with a disease state or biological process or in response to drug treatment by assessing their enrichment using differentially expressed gene analysis and GSVA.
A semi-biased transcriptomic analysis together with unsupervised machine learning analysis of sputum samples identified a Type 2 high severe eosinophilic asthma endotype and two other distinct low Type 2 molecular phenotypes.
Analysis of gene expression data from bronchial biopsies and epithelial brushings identified two subtypes of patients with Type 2 eosinophilic inflammation and relative corticosteroid insensitivity.
Supervised approaches using non-T2 signatures have identified patients expressing IL-6 trans-signaling, Th17, and IL-22-associated pathways identified within the neutrophilic inflammatory phenotype.
The use of gene signatures in the clinical setting of the management of a patient with severe asthma would enhance our understanding of the pathways that may be driving the pathophysiology of severe asthma.
Declaration of interest
KF Chung has received honoraria for participating in Advisory Board meetings of GSK, AZ, Roche, Novartis, Merck, BI, and Shionogi regarding treatments for asthma, chronic obstructive pulmonary disease, and chronic cough and have also been renumerated for speaking engagements for AZ, Novartis, Sanofi, and GSK. KF Chung’s institution has received research grants that they are an Investigator on from GSK and Merck and from UKRI. CoI for Dr Ian M. Adcock: IMA has received honoraria for consulting and participating in Advisory Board meetings of Chiesi, GSK, Kinaset and Sanofi; has been renumerated for travel and speaking engagements for AZ, Eurodrug and Sanofi and has been funded by Institutional grants from EU-IMI, GSK, Sanofi and UKRI. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
The U-BIOPRED project referred to in this review was supported by the European Union’s Seventh Framework Programme (FP7/2007-2013) Innovative Medicines Initiative Joint Undertaking (grant 115010). U-BIOPRED (Unbiased Biomarkers for the Prediction of Respiratory Diseases Outcomes) was also supported by European Federation of Pharmaceutical Industries and Associations companies’ in-kind contribution (www.imi.europa.eu).
Additional information
Funding
References
- Thamrin C, Frey U, Kaminsky DA, et al. Systems biology and clinical practice in Respiratory medicine. The twain shall meet. Am J Respir Crit Care Med. 2016 Nov 1;194(9):1053–1061. doi: 10.1164/rccm.201511-2288PP
- Chung KF. New treatments for severe treatment-resistant asthma: targeting the right patient. Lancet Respir Med. 2013;1(8):639–652. doi: 10.1016/S2213-2600(13)70128-0
- Chung KF, Adcock IM. Precision medicine for the discovery of treatable mechanisms in severe asthma. Allergy. 2019 Sep;74(9):1649–1659. doi: 10.1111/all.13771
- Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet. 2008 Sep 20;372(9643):1107–1119. doi: 10.1016/S0140-6736(08)61452-X
- Brusselle GG, Koppelman GH, Taichman DB. Biologic therapies for severe asthma. N Engl J Med. 2022 Jan 13;386(2):157–171. doi: 10.1056/NEJMra2032506
- Li CX, Wheelock CE, Sköld CM, et al. Integration of multi-omics datasets enables molecular classification of COPD. Eur Respir J. 2018 May;51(5):1701930. doi: 10.1183/13993003.01930-2017
- Wheelock CE, Goss VM, Balgoma D, et al. Application of ‘omics technologies to biomarker discovery in inflammatory lung diseases. Eur Respir J. 2013 Sep;42(3):802–825. doi: 10.1183/09031936.00078812
- De Meulder B, Lefaudeux D, Bansal A, et al. A computational framework for complex disease stratification from multiple large-scale datasets. BMC Syst Biol. 2018;12(1):60. doi: 10.1186/s12918-018-0556-z
- Itadani H, Mizuarai S, Kotani H. Can systems biology understand pathway activation? Gene expression signatures as surrogate markers for understanding the complexity of pathway activation. Curr Genomics. 2008;9(5):349–360. doi: 10.2174/138920208785133235
- Mallik S, Zhao Z. Identification of gene signatures from RNA-seq data using Pareto-optimal cluster algorithm. BMC Syst Biol. 2018 Dec 21;12(Suppl 8):126. doi: 10.1186/s12918-018-0650-2
- Sikkema L, Ramírez-Suástegui C, Strobl DC, et al. An integrated cell atlas of the lung in health and disease. Nat Med. 2023 Jun;29(6):1563–1577. doi: 10.1038/s41591-023-02327-2
- Vieira Braga FA, Kar G, Berg M, et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. 2019 Jul;25(7):1153–1163. doi: 10.1038/s41591-019-0468-5
- Irizarry RA, Wang C, Zhou Y, et al. Gene set enrichment analysis made simple. Stat Methods Med Res. 2009 Dec;18(6):565–575. doi: 10.1177/0962280209351908
- Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015 Apr 20;43(7):e47. doi: 10.1093/nar/gkv007
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. 2010 Jan 1;26(1):139–140. doi: 10.1093/bioinformatics/btp616
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8
- Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005 Oct 25;102(43):15545–15550. doi: 10.1073/pnas.0506580102
- Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14(1):7. doi: 10.1186/1471-2105-14-7
- Shaw DE, Sousa AR, Fowler SJ, et al. Clinical and inflammatory characteristics of the European U-BIOPRED adult severe asthma cohort. Eur Respir J. 2015 Nov;46(5):1308–1321. doi: 10.1183/13993003.00779-2015
- Kuo CS, Pavlidis S, Loza M, et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur Respir J. 2017;49(2):443–455. doi: 10.1183/13993003.02135-2016
- Schofield JPR, Burg D, Nicholas B, et al. Stratification of asthma phenotypes by airway proteomic signatures IT is tehre. J Allergy Clin Immunol. 2019;14(10):123.
- Kuo CS, Pavlidis S, Loza M, et al. A transcriptome-driven analysis of epithelial brushings and bronchial biopsies to define asthma phenotypes in U-BIOPRED. Am J Respir Crit Care Med. 2017 Feb 15;195(4):443–455. doi: 10.1164/rccm.201512-2452OC
- Pavlidis S, Takahashi K, Ng Kee Kwong F, et al. “T2-high” in severe asthma related to blood eosinophil, exhaled nitric oxide and serum periostin. Eur Respir J. 2019 Jan;53(1):1800938. doi: 10.1183/13993003.00938-2018
- Hekking PP, Loza MJ, Pavlidis S, et al. Pathway discovery using transcriptomic profiles in adult-onset severe asthma. J Allergy Clin Immunol. 2018 Apr;141(4):1280–1290. doi: 10.1016/j.jaci.2017.06.037
- Hekking PP, Loza MJ, Pavlidis S, et al. Transcriptomic gene signatures associated with persistent airflow limitation in patients with severe asthma. Eur Respir J. 2017 Sep;50(3):1602298. doi: 10.1183/13993003.02298-2016
- Hoda U, Pavlidis S, Bansal AT, et al. Clinical and transcriptomic features of persistent exacerbation-prone severe asthma in U-BIOPRED cohort. Clin Transl Med. 2022 Apr;12(4):e816. doi: 10.1002/ctm2.816
- Perotin JM, Schofield JPR, Wilson SJ, et al. Epithelial dysregulation in obese severe asthmatics with gastro-oesophageal reflux. Eur Respir J. 2019 Jun;53(6):1900453. doi: 10.1183/13993003.00453-2019
- Takahashi K, Pavlidis S, Ng Kee Kwong F, et al. Sputum proteomics and airway cell transcripts of current and ex-smokers with severe asthma in U-BIOPRED: an exploratory analysis. Eur Respir J. 2018;51(5):1280–1290. doi: 10.1183/13993003.02173-2017
- Jevnikar Z, Ostling J, Ax E, et al. Epithelial IL-6 trans-signaling defines a new asthma phenotype with increased airway inflammation. J Allergy Clin Immunol. 2019;143(2):577–590. doi: 10.1016/j.jaci.2018.05.026
- Ostling J, van Geest M, Schofield JPR, et al. IL-17-high asthma with features of a psoriasis immunophenotype. J Allergy Clin Immunol. 2019;144(5):1198–1213. doi: 10.1016/j.jaci.2019.03.027
- Badi YE, Pavel AB, Pavlidis S, et al. Mapping atopic dermatitis and anti-IL-22 response signatures to type 2-low severe neutrophilic asthma. J Allergy Clin Immunol. 2022 Jan;149(1):89–101. doi: 10.1016/j.jaci.2021.04.010
- Tiotiu A, Badi Y, Kermani NZ, et al. Association of differential mast cell activation with granulocytic inflammation in severe asthma. Am J Respir Crit Care Med. 2022 Feb 15;205(4):397–411. doi: 10.1164/rccm.202102-0355OC
- Kelsen SG, Agache IO, Soong W, et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J Allergy Clin Immunol. 2021 Apr 16;148(3):790–798. doi: 10.1016/j.jaci.2021.03.044
- Wechsler ME, Ruddy MK, Pavord ID, et al. Efficacy and safety of Itepekimab in patients with moderate-to-severe asthma. N Engl J Med. 2021 Oct 28;385(18):1656–1668. doi: 10.1056/NEJMoa2024257
- Sánchez-Ovando S, Pavlidis S, Kermani NZ, et al. Pathways linked to unresolved inflammation and airway remodelling characterize the transcriptome in two independent severe asthma cohorts. Respirology. 2022 Sep;27(9):730–738. doi: 10.1111/resp.14302
- Tiotiu A, Zounemat Kermani N, Badi Y, et al. Sputum macrophage diversity and activation in asthma: role of severity and inflammatory phenotype. Allergy. 2021 Mar;76(3):775–788. doi: 10.1111/all.14535