Abstract
The type and quantity of dietary fat ingested contributes to the onset and progression of chronic diseases, such as diabetes and atherosclerosis. The liver plays a central role in whole-body lipid metabolism and responds rapidly to changes in dietary fat composition. In rodents, n-3 polyunsaturated fatty acids (PUFAs) enhance hepatic fatty acid oxidation and inhibit fatty acid synthesis and very low-density lipoprotein secretion, in part, by regulating key transcription factors, including peroxisome proliferator activated receptor-α (PPAR-α), sterol regulatory element binding protein-1 (SREBP-1), carbohydrate regulatory element binding protein (ChREBP) and Max-like factor X (MLX). These transcription factors control the expression of multiple genes involved in lipid synthesis and oxidation. Changes in PPAR-α target genes correlate well with changes in intracellular non-esterified fatty acids. Insulin stimulates hepatic de novo lipogenesis by rapidly inducing SREBP-1 nuclear abundance (nSREBP-1). This mechanism is linked to insulin-induced protein kinase B (Akt) and glycogen synthase kinase (Gsk)-3β phosphorylation and inhibition of 26S proteasomal degradation of nSREBP-1. n-3 PUFAs, particularly 22:6 n-3, inhibit lipid synthesis by suppressing nSREBP-1. A major action of 22:6 n-3 is to stimulate the loss of nSREBP-1 through 26S proteasomal and extracellular regulated kinase (Erk)-dependent pathways. 22:6 n-3 is the only n-3 PUFA accumulating in livers of rodents or humans ingesting essential fatty acid-sufficient or n-3 PUFA-enriched diets. As such, 22:6 n-3 is a major feedback regulator of hepatic lipid synthesis. Finally, insulin-stimulated glucose metabolism augments de novo lipogenesis by elevating nuclear levels of ChREBP, a key regulator of glycolytic and lipogenic genes. ChREBP binding to promoters requires MLX. n-3 PUFAs repress expression of the glycolytic gene, l-pyruvate kinase and lipogenic genes by suppressing MLX nuclear abundance. In summary, n-3 PUFAs control the activity or abundance of several hepatic transcription factors that impact hepatic carbohydrate and lipid metabolism. Recent studies have identified Erk, Gsk-3β and MLX as novel targets of fatty acid-regulated gene expression.
Introduction
Dietary fat is an important macronutrient for growth and development in all animals. Excessive levels of dietary fat or an imbalance of saturated fat versus unsaturated fat or n-6 versus n-3 polyunsaturated fatty acids (PUFAs) have been implicated in the onset and progression of several chronic diseases, including atherosclerosis (coronary artery disease and stroke), diabetes and obesity, cancer, major depressive disorders and schizophrenia Citation1. Consequently, considerable clinical and basic science research has been directed towards understanding the biochemical and molecular basis of the effects of fatty acid on physiological systems impacting human health.
Complicating our understanding of fatty acid effects on chronic diseases is the fact that fatty acids have many physiological roles. Fatty acids are substrates for energy metabolism, membrane formation and signaling molecules; they also regulate gene expression. The focus of this discussion is on how fatty acids control gene transcription. Since the original description of dietary fat as a regulator of gene expression over a decade ago, many transcription factors have been identified as prospective targets for fatty acid regulation, including peroxisome proliferator activated receptor (PPAR; α, β, γ1 and γ2), sterol regulatory element binding protein-1 (SREBP-1), hepatic nuclear factor-4 (HNF-4; α and γ), retinoid X receptor-α (RXR-α), liver-X factor-α (LXR-α), carbohydrate regulatory element binding protein (ChREBP) and Max-like factor X (MLX) 1–4Citation1Citation2Citation3Citation4. Two general mechanisms characterize fatty acid control of gene transcription. Fatty acids bind directly to the transcription factor and control its activity. In this fashion, fatty acids act like hydrophobic hormones regulating the function of nuclear receptors and their control of transcriptional processes. Non-esterified fatty acids bind PPAR (α, β, γ1 and γ2) Citation5, HNF-4 (α and γ), Citation6Citation7, RXR-α Citation8 and LXR-α Citation9. All of these transcription factors are members of the nuclear receptor superfamily. Of these, PPAR subtypes are the most widely accepted fatty acid-regulated transcription factors.
In the second mechanism, fatty acids control the nuclear abundance of key transcription factors, such as SREBP-1, nuclear factor-κB, ChREBP and MLX Citation1Citation3Citation4. The mechanism controlling the nuclear abundance of these transcription factors is less clear, but probably does not involve direct binding of the fatty acid to the transcription factor.
This discussion will focus on n-3 PUFA control of hepatic carbohydrate and lipid metabolism. In vivo and cell-culture studies have established that dietary n-3 PUFAs regulate multiple genes involved in hepatic carbohydrate and lipid metabolism by targeting key transcription factors, i.e. PPAR-α, SREBP-1, ChREBP and MLX Citation310–15Citation10Citation11Citation12Citation13Citation14Citation15. Fatty acid control of these transcription factors provides an explanation for the diverse effects of PUFAs on hepatic and whole-body carbohydrate and lipid composition.
Fatty acid regulation of hepatic peroxisome proliferator activated receptor-α
PPARs are fatty acid-regulated nuclear receptors that form obligate heterodimer partners with RXRs and bind peroxisome proliferator regulatory elements (PPREs) in promoters of responsive genes. All PPAR subtypes (α, β/Δ, γ1 and γ2) bind fatty acids Citation5; fatty acid binding stimulates coactivator recruitment and gene transcription (). PPAR-α is the predominant PPAR subtype in rodent liver. Studies with PPAR-α null mice establish that PPAR-α is required for PUFA effects on genes encoding enzymes involved in fatty acid oxidation, desaturation and elongation, but not for PUFA suppression of enzymes involved in glycolysis or lipogenesis Citation10Citation12Citation16.
Fig. 1. Summary of polyunsaturated fatty acid (PUFA) control of hepatic peroxisome proliferator activated receptor-α (PPAR-α), sterol regulatory element binding protein-1 (SREBP-1) and Max-like factor X (MLX) nuclear abundance. PUFAs bind to and activate PPAR-α to induce the transcription of genes involved in fatty acid oxidation. PUFAs, particularly 22:6 n-3, suppress hepatic lipogenic gene expression by suppressing SREBP-1 nuclear abundance through 26S proteasome- and Erk-dependent pathways. PUFAs interfere with glucose-regulated gene expression by suppressing hepatic nuclear abundance of MLX, the obligate heterodimer partner of carbohydrate regulatory element binding protein (ChREBP). In this fashion, PUFAs inhibit the transcription of l-pyruvate kinase and other genes controlled by the ChREBP/MLX heterodimer. See text for additional explanation. ACS: acetyl coenzyme A synthetase; CoA: coenzyme A; ER: endoplasmic reticulum; FA: fatty acid; FABP: fatty acid binding protein; FAT/CD36: fatty acid transporter (CD36); FATP: fatty acid transport protein; Glut2: glucose transporter-2; NADPH: nicotinamide adenine dinucleotide phosphate (reduced); NEFA: non-esterified fatty acid; RXR: retinoid X receptor; TE: fatty acyl thioesterase.
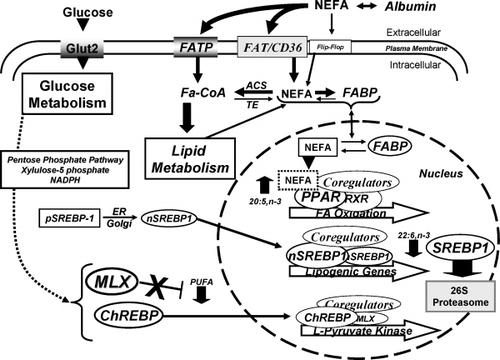
Since PPARs are considered sensors of intracellular lipid, intracellular non-esterified fatty acid (NEFA) levels were examined in primary rat hepatocytes before and after challenge with fatty acids (18:1 n-9 and 20:5 n-3) Citation17. These studies revealed several important features as to how fatty acids control PPAR-α function. First, intracellular NEFA levels are maintained at very low levels, representing <0.1% of the total lipid in the cell. Secondly, challenging cells with certain fatty acids, e.g. 20:5 n-3, induces significant changes in intracellular NEFA and promotes robust responses in PPAR-α target genes, such as cytochrome P450-4A (CYP4A) and cytosolic thioesterase (CTE-1) Citation14Citation17. Other fatty acids, e.g. 18:1 n-9, when added to cells are so rapidly assimilated as to not impact intracellular NEFA levels. As such, these fatty acids do not induce PPAR-α target genes in vivo. Thirdly, effects of fatty acids on PPAR-α signaling are rapid, but transient. Essentially, fatty acid-induced changes in PPAR-α target genes parallel the changes in intracellular NEFAs. Such studies support the concept that PPARs monitor intracellular NEFA composition and respond accordingly to induce metabolic pathways that minimize damage brought on by excessive intracellular NEFAs ().
In vivo, however, intracellular NEFA levels are determined by blood lipid composition and the turnover (lipolysis) of complex lipids in cells. Blood lipid composition is determined by lipids absorbed from the diet and mobilized from depot lipids. In vivo, cells are constantly exposed to NEFA and not the brief episodes seen under cell-culture conditions. Therefore, the response of PPARs to intracellular lipids will be controlled by diet, absorption, mobilization and intracellular lipolytic events. Factors affecting these biochemical processes will impact PPAR-regulated gene expression.
Hepatic glycolysis and lipogenesis: targets for dietary polyunsaturated fatty acid control
Glycolysis and de novo lipogenesis (DNL) are two metabolic pathways for glucose utilization for anaerobic and aerobic respiration, as well as fuel storage in the form of glycogen and triglycerides. These pathways are subject to complex hormonal and nutrient control. Insulin, thyroid hormones, glucocorticoids and glucose stimulate glycolysis and DNL, while glucagon, epinephrine (adrenaline) and PUFA suppress at least one glycolytic enzyme, i.e. l-pyruvate kinase, and several enzymes involved in DNL, i.e. ATP citrate lyase (ACL), acetyl coenzyme A carboxylase (ACC), fatty acid synthase (FAS), stearoyl coenzyme A desaturase-1 (SCD-1) and fatty acid elongase-6 (Elovl-6) Citation1Citation3Citation13Citation18Citation19.
Key transcription factors controlling expression of proteins involved in these pathways include SREBP-1 and the ChREBP/MLX heterodimer (Table 1). Insulin induces the nuclear abundance of SREBP-1 and ChREBP/MLX. In contrast to SREBP-1, however, insulin-stimulated elevation of ChREBP requires glucose metabolism Citation19. Recent advances in the control of SREBP-1 and ChREBP/MLX nuclear abundance by insulin, glucose and fatty acids are described below.
Table 1. Involvement of hepatic transcription factors in controlling glycolysis, lipid synthesis and oxidation
Sterol regulatory element binding protein-1c is a target for fatty acid control in hepatic lipid synthesis
SREBP-1c is one of three basic helix-loop-helix-Leu-Zip transcription factors (SREBP-1a, SREBP-1c and SREBP-2) that play a central role in the control of cholesterol and fatty acid synthesis. While SREBP-2 plays a major role in the regulation of cholesterol synthesis and uptake, SREBP-1 is more involved in the regulation of fatty acid synthesis and very low-density lipoprotein (VLDL) assembly Citation20Citation21. SREBP effects on gene transcription are determined by factors that control the nuclear abundance of SREBP (nSREBP). Two post-translational mechanisms are important for this process: proteolytic processing Citation21 and proteasomal degradation Citation22. All SREBPs are synthesized as precursors (pSREBP, ∼125 kDa) and tethered to the endoplasmic reticulum (ER). Precursor SREBPs are escorted from the ER to the Golgi by SREBP-cleavage activating protein (SCAP), where SREBPs are proteolytic processed to the mature nuclear form. nSREBP is transported to the nucleus, via importin-β Citation23, where it binds sterol regulatory elements as dimers in promoters of specific genes. Once bound, SREBPs recruit coactivators to the promoter and stimulate gene transcription Citation24. nSREBP is phosphorylated and ubiquitinated, a process that targets nSREBP for proteasomal degradation Citation25. Sterols regulate nSREBP levels by controlling the proteolytic processing step, not proteasomal degradation. Sterols induce the ER-resident proteins, Insig-1 or Insig-2, to bind SCAP; the Insig-SCAP-SREBP complex is retained in the ER, preventing its movement to the Golgi for cleavage and maturation Citation26. In this fashion, cholesterol functions as a feedback inhibitor of cholesterol synthesis by preventing SREBP-2 from accumulating in nuclei and inducing expression of key genes involved in cholesterol synthesis (Table 1).
While SREBP-1 and SREBP-2 are structurally similar, their regulation in the liver by nutrients and hormones is quite different. Both SREBP-1a and -1c are expressed in the liver, but SREBP-1c is the predominant subtype. In contrast to SREBP-2, SREBP-1c nuclear abundance is controlled by several mechanisms Citation2. Insulin and oxysterols (LXR agonists) induce SREBP-1c gene transcription, elevate mRNASREBP-1c and nSREBP-1c, leading to elevated de novo lipogenesis Citation21. Insulin regulates SREBP-1c gene transcription through PI3 kinase and protein kinase B (Akt) Citation27, changes in Insig-1 and -2 expression Citation28 and inhibition of 26S proteasomal degradation Citation22Citation29. Removal of insulin from primary hepatocytes leads to a prompt decline in nSREBP-1 (t1/2∼10 h). Loss of nSREBP-1 is blocked by inhibitors of 26S proteasomal degradation Citation29.
n-3 and n-6 PUFA are well-established suppressors of SREBP-1 Citation11Citation15. These fatty acids have little impact on SREBP-2 nuclear abundance. The PUFA suppression of SREBP-1 accounts for much of the PUFA suppression of DNL Citation11Citation30. The present authors and others have examined the effects of various fatty acids on nSREBP-1; fatty acids inhibit SREBP-1 gene transcription, induce mRNASREBP-1 instability and inhibit SREBP processing (for review see ref. 2). Many of these studies used high levels of fatty acids. The authors recently re-examined the PUFA control of SREBP-1 nuclear abundance and found that 22:6 n-3 was the most potent suppressor of SREBP-1 nuclear abundance (ed50<100 µM). 20:4 n-6 and 22:6 n-3 are the end-products of PUFA synthesis and the predominant n-6 and n-3 PUFAs accumulating in livers of animals fed essential fatty acid-sufficient diets Citation29. At 100 µM, 20:4 n-6, 20:5 n-3 and 22:6 n-3 had equivalent, but minor, effects on mRNASREBP-1c and SREBP-1 precursor levels. Of these three fatty acids, only 22:6 n-3 significantly (>75%) suppressed nSREBP-1 abundance. These studies indicated that 22:6 n-3 was likely to be the major regulator of nSREBP-1 abundance. Moreover, a major component of this control was directed at a post-translational level Citation29. Phenomenal post-translational control of SREBP-1 nuclear abundance was also found in vivo, particularly during postnatal development, when lipogenesis and nuclear SREBP-1 are induced at weaning Citation31.
Post-translational mechanisms controlling SREBP-1 include proteolytic conversion of the precursor to the mature (nuclear) form and 26S proteasomal degradation. 22:6 n-3 was found to have little effect on SREBP cleavage activating protein (SCAP) expression and only modestly induced Insig-1 expression. Both insulin and 22:6 n-3 strongly suppressed Insig-2 expression. When taken together, these studies provided little support for 22:6 n-3 acting at the level of SCAP or Insig to control SREBP-1 nuclear abundance Citation29.
Next, 26S proteasomal degradation of SREBP-1 was examined. Insulin controls nSREBP, at least in part, by inhibiting glycogen synthase kinase-3β (Gsk-3β) phosphorylation of SREBP Citation22. Gsk-3β phosphorylation of SREBP induced binding of a ubiquitin ligase to SREBP, resulting in its ubiquitination and 26S proteasomal degradation. Gsk-3β activity is inhibited by Akt-mediated phosphorylation of Gsk-3β. Akt is a downstream target of insulin action and required for insulin control of SREBP-1 nuclear abundance. The studies established that removal of insulin from primary hepatocytes resulted in a prompt decline in nSREBP-1 (t1/2∼10 h); inhibitors of 26S proteasomal degradation blocked this decline Citation29. This observation supported the role of the 26S proteasome in insulin-mediated control of SREBP nuclear abundance Citation22.
Treatment of primary hepatocytes with 22:6 n-3 accelerates the loss of SREBP-1 (t1/2∼4 h) from nuclei. The 22:6 n-3-mediated loss of nSREBP-1 was completely blocked by 26S proteasome inhibitors. Of all the fatty acids tested, 22:6 n-3 was the most potent suppressor of nSREBP-1. Moreover, only 22:6 n-3 controlled nSREBP-1 by a 26S-proteasome-dependent pathway ().
The next objective was to identify the mechanisms that control SREBP-1 nuclear abundance. Gsk-3β-mediated phosphorylation of SREBP is clearly involved in SREBP ubiquitination and 26S proteasomal degradation SREBP Citation22. The studies, however, indicated that only SREBP-1, and not SREBP-2, was regulated by insulin and 22:6 n-3 through a 26S proteasome-dependent mechanism. Insulin suppresses Gsk-3β activity by inducing its phosphorylation through an Akt-dependent pathway. Both insulin and 22:6 n-3 induced Gsk-3β phosphorylation (unpublished). Thus, the stimulation of 26S proteasomal degradation of SREBP-1 could not be explained by inhibiting insulin-mediated phosphorylation of Gsk-3β activity. Although 22:6 n-3 transiently suppressed insulin-induced Akt-phosphorylation, i.e. its activity, overexpression of a constitutively active Akt failed to abolish the 22:6 n-3 suppression of nSREBP-1. Thus, 22:6 n-3 control of Akt or Gsk-3β could not explain the 22:6 n-3 control of SREBP-1 proteasomal degradation.
In an effort to identify other pathways that may be involved, the extracellular regulatory element (Erk) pathway, a well-established target of insulin action, was examined. Insulin rapidly, but transiently, induces Erk phosphorylation in rat primary hepatocytes. Co-treatment with 22:6 n-3 prolonged Erk-phosphorylation and elevated Erk-phosphorylation correlated with suppressed nSREBP-1. To determine whether Erk played a role in controlling nSREBP-1, primary hepatocytes were treated with MEK inhibitors (PD98059 and U0126). These inhibitors rapidly (within 30 min) inhibited 22:6 n-3-induced Erk phosphorylation. More surprisingly, mitogen-activated protein kinase (MEK) inhibitors rapidly (within 30 min) induced nSREBP-1. The studies, when taken together, indicated that 22:6 n-3 control SREBP-1 nuclear abundance through a 26S proteasomal- and Erk-dependent mechanism Citation29. Precisely how 22:6 n-3 controls Erk phosphorylation and SREBP-1 26S proteasomal degradation is under investigation.
Carbohydrate regulatory element binding protein and Max-like factor X are targets for fatty acid control in hepatic glycolysis and lipid synthesis
The effects of glucose on hepatic carbohydrate and lipid metabolism go beyond glucose control of insulin secretion Citation19. In states of excessive glucose intake, glucose is stored as glycogen or converted to fatty acids and stored as triglycerides. A key transcription factor mediating glucose effects on glycolytic and lipogenic gene transcription is ChREBP, a basic helix-loop-helix transcription factor. ChREBP, along with its obligate heterodimer partner, MLX, binds carbohydrate regulatory elements (ChoRE) in promoters of responsive genes, e.g. l-pyruvate kinase (L-PK), ACC and FAS. Glucose regulates ChREBP nuclear abundance by controlling its phosphorylation status. Glucose does not regulate MLX nuclear abundance.
Dephosphorylated ChREBP accumulates in nuclei and stimulates the transcription of glycolytic and lipogenic genes. Protein kinase A and adenosine monophosphate kinase (AMPK) inhibit these metabolic pathways by phosphorylating ChREBP and preventing its nuclear accumulation. When excess glucose is metabolized, glucose metabolites flow through the pentose phosphate pathway, leading to the accumulation of intracellular nicotinamide adenine dinucleotide phosphate (reduced) (NADPH) and xylulose-5-phosphate. Xylulose-5-phosphate activates protein phosphatase 2A and dephosphorylates ChREBP, leading to the accumulation of ChREBP in hepatic nuclei Citation32. The influx of ChREBP into nuclei triggers its heterodimerization with MLX and binding to promoters of responsive genes. Glucose also stimulates the accumulation of RNA polymerase II and acetylated histones on promoters of glycolytic and lipogenic genes Citation3.
PUFAs interfere with glucose activated L-PK gene transcription by targeting the ChoRE/HNF-4α region of the L-PK promoter and blocking the accumulation of RNA polymerase II and acetylated histones on the L-PK promoter Citation3Citation12Citation33Citation34. Two recent reports examined the role of ChREBP Citation4 and MLX Citation3 in this process. One study reported that PUFAs suppressed ChREBP nuclear abundance; ChREBP overexpression abrogated the effect of PUFAs on L-PK gene expression Citation4. The other report found no effect of n-3 PUFAs on ChREBP in vivo or in primary hepatocytes, and that overexpressed ChREBP failed to erase PUFA suppression of L-PK Citation3. Instead, n-3 PUFAs suppressed MLX nuclear abundance in vivo and in primary hepatocytes. Overexpressed MLX abrogated the effect of PUFAs on L-PK gene expression ().
There is no immediate explanation for the disparity between these two studies. As such, more studies are required to confirm and extend these observations. One clear agreement between the two studies, however, is that neither study advocated a role for fatty acid control of AMPK in regulating either ChREBP or MLX nuclear abundance.
Summary
Fatty acids rapidly control gene expression by regulating the activity and abundance of key hepatic transcription factors, including PPAR-α, SREBP-1, ChREBP and MLX. Identifying these transcription factors as targets of fatty acid control clarifies how fatty acids can have such diverse effects on several metabolic pathways. Hepatic pathways regulated by fatty acids include glycolysis, lipogenesis, fatty acid desaturation and elongation, as well as mitochondrial, peroxisomal and microsomal fatty acid oxidation (Table 1). The overall consequence of this action is to shift lipid metabolism away from lipid synthesis and storage, and towards fatty acid oxidation. Clearly, n-3 PUFAs play a major role in controlling intracellular lipid composition.
While considerable progress has been made in understanding the transcriptional regulatory networks controlled by fatty acids, there remain many unanswered questions. Among these is how specific fatty acids selectively control transcription factor function. While 20:5 n-3 is the preferred regulator of hepatic PPAR-α activity, 22:6 n-3 is the dominant controller of SREBP-1 nuclear abundance. PUFA control of MLX nuclear abundance is not selective (). It is suggested that selective fatty acid metabolism is important for these different control mechanisms. As such, metabolic pathways controlling intracellular NEFA composition will probably be important in controlling transcription factor function. It is also likely that the effects of PUFAs on membrane lipid composition, particularly lipid rafts, play an important role in these mechanisms. Lipid rafts are dynamic microdomains implicated in cell signaling. Fatty acid-induced changes in raft composition appear linked to changes in cell signaling Citation35.
Finally, the pleiotropic nature of the effects of PUFAs on cell function guarantees many years of new discoveries of PUFA action. Clarification of these actions will aid in our understanding of how changes in dietary lipid intake can affect the onset and progression of chronic diseases.
| Acronyms | ||
| ACC | = | acetyl coenzyme A carboxylase |
| ACL | = | ATP citrate lyase |
| Akt/PKB | = | protein kinase B |
| AMPK | = | adenosine monophosphate kinase |
| ChoRE | = | carbohydrate regulatory element |
| ChREBP | = | carbohydrate regulatory element binding protein |
| CTE-1 | = | cytosolic thioesterase 1 |
| CYP4A | = | cytochrome P450-4A |
| DNL | = | de novo lipogenesis |
| Elovl | = | fatty acid elongase |
| ER | = | endoplasmic reticulum |
| Erk | = | extracellular regulated kinase |
| FAS | = | fatty acid synthase |
| Gsk | = | glycogen synthase kinase |
| HNF | = | hepatic nuclear factor |
| L-PK | = | l-type pyruvate kinase |
| LXR | = | liver X receptor |
| MEK | = | mitogen-activated protein kinase |
| MLX | = | Max-like factor X |
| NADPH | = | nicotinamide adenine dinucleotide phosphate (reduced) |
| NEFA | = | non-esterified fatty acid |
| PPAR | = | peroxisome proliferator activated receptor |
| PPRE | = | peroxisome proliferator regulatory element |
| PUFA | = | polyunsaturated fatty acid |
| RXR | = | retinoid X receptor |
| SCAP | = | SREBP cleavage activating protein |
| SCD | = | stearoyl coenzyme A desaturase |
| SREBP | = | sterol regulatory element binding protein |
| VLDL | = | very low-density lipoprotein. |
This project was supported by the National Institutes of Health (DK43220), the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service (2003-5200-3400) and the Michigan Agriculture Experiment Station.
References
- JumpDB. Fatty acid regulation of gene transcriptionCrit Rev Clin Lab Sci2004414178
- JumpDBBotolinDWangYXuJChristianBDemeureO. Fatty acid regulation of gene transcriptionJ Nutr200513525036
- Xu J, Christian B, Jump DB. Regulation of rat hepatic lpyruvate kinase promoter composition and activity by glucose, n-3 PUFA and peroxisome proliferator activated receptor-alpha agonist. J Biol Chem 2006;281: 18351–62.
- DentinRBenhamedFPegorierJPFoufelleFViolletB.VaulontS Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocationJ Clin Invest2005115284354
- XuHELambertMHMontanaVGParksDJBlanchardSGBrownPJ Molecular recognition of fatty acids by peroxisome proliferator-activated receptorsMol Cell19993397403
- WiselyGBMillerABDavisRGThornquestADJrJohnsonRSpitzerT Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acidsStructure (Camb)200210122534
- Dhe-PaganonSDudaKIwamotoMChiYIShoelsonSE. Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligandJ Biol Chem2002277379736
- Mata de UrquizaALiuSSjobergMZetterstromRHCriffithWSjovallJPerlmannT. Docosahexeanoic acid, a ligand for retinoic X receptor in mouse brainScience200029021404
- OuJTuHShanBLukADeBose-BoydRABashmakovY Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXRProc Natl Acad Sci U S A200198602732
- RenBAThelenPPetersJMGonzalezFJJumpDB. Polyunsaturated fatty acid suppression of hepatic fatty acid synthase and S14 gene expression does not require peroxisome proliferator-activated receptor alphaJ Biol Chem19972722682732
- MaterMKThelenAPPanDAJumpDB. Sterol regulatory element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcriptionJ Biol Chem19992743272532
- PanDAMaterMKThelenAPPetersJMGonzalezFJJumpDB. Evidence against the peroxisome proliferator-activated receptor alpha (PPARalpha) as the mediator for polyunsaturated fatty acid suppression of hepatic l-pyruvate kinase gene transcriptionJ Lipid Res20004174251
- WangYBotolinDChristianBBusikCXuJJumpDB. Tissue-specific, nutritional and developmental regulation of rat fatty acid elongasesJ Lipid Res20054670615
- PawarABotolinDMangelsdorfDJJumpDB. The role of liver X receptor-alpha (LXR-alpha) in the fatty acid regulation of hepatic gene expressionJ Biol Chem20032784073643
- WorgallTSSturleySLSeoTOsborneTFDeckelbaumRJ. Polyunsaturated fatty acids decrease expression of promoters with sterol regulatory elements by decreasing levels of mature sterol regulatory element-binding proteinJ Biol Chem19982732553740
- Wang Y, Botolin D, Xu Jinghua, Christian B, Mitchell E, Jayaprakasam B, Regulation of hepatic fatty acid elongase and desaturase expression in diabetes and obesity. J Lipid Res 2006;47: 2028–41.
- PawarAJumpDB. Unsaturated fatty acid regulation of peroxisome proliferator activated receptor-alpha activity in primary rat hepatocytesJ Biol Chem2003278359319
- IizukaKBruickRKLiangGHortonJDUyedaK. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysisProc Natl Acad Sci U S A200410172816
- TowleHC. Glucose as a regulator of eukaryotic gene transcriptionTrends Endocrinol Metab20051648994
- HortonJDShahNAWarringtonJAAndersonNNParkSWBrownMSGoldsteinJL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genesProc Natl Acad Sci U S A20031001202732
- HortonJDGoldsteinJLBrownMS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liverJ Clin Invest2002109112531
- SundqvistABengoechea-AlonsoMTYeXLukiyanchuk JinJHarperJWEricssonJ. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCGFBW7Cell Metabolism2005137991
- NagoshiEImamotoNSatoRYonedaY. Nuclear import of sterol regulatory element-binding protein-2, a basic helix-loop-helix-leucine zipper (bHLH-Zip)-containing transcription factor, occurs through the direct interaction of importin beta with HLH-ZipMol Biol Cell199910222133
- BennettMTothJIOsborneTF. Selective association of sterol regulatory element binding protein isoforms with target promoters in vivoJ Biol Chem2004279373607
- HiranoYYoshidaMShimizuMSatoR. Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathwayJ Biol Chem2001276364317
- AdamsCMReitzJDe BrabanderJKFeramiscoJDLiLBrownMSGoldsteinJL. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and InsigsJ Biol Chem20042795277280
- RibauxPGIynedjianPB. Analysis of the role of protein kinase B (cAKT) in insulin-dependent induction of glucokinase and sterol regulatory element-binding protein 1 (SREBP1) mRNAs in hepatocytesBiochem J2003376697705
- YabeDKomuroRLiangGGoldsteinJLBrownMS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesisProc Natl Acad Sci U S A2003100315560
- BotolinDWangYChristianBJumpDB. Docosahexaneoic acid [22:6, n-3] stimulates rat hepatic sterol regulatory element binding protein-1c (SREBP-1c) degradation by an Erk- and 26S proteasome-dependent pathwayJ Lipid Res20064718192
- YahagiNShimanoHHastyAHAmemiya-KudoMOkazakiHTamuraY A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acidsJ Biol Chem1999274358404
- BotolinDJumpDB. Selective proteolytic processing of rat hepatic sterol regulatory element binding protein-1 (SREBP-1) and SREBP-2 during postnatal developmentJ Biol Chem2003278695962
- KabashimaTKawaguchiTWadzinskiBEUyedaK. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liverProc Natl Acad Sci U S A2003100510712
- JumpDBClarkeSDThelenALiimattaM. Coordinate regulation of glycolytic and lipogenic gene expression by polyunsaturated fatty acidsJ Lipid Res199435107684
- LiimattaMTowleHCClarkeSJumpDB. Dietary polyunsaturated fatty acids interfere with the insulin/glucose activation of l-type pyruvate kinase gene transcriptionMol Endocrinol19948114753
- JumpDB. The biochemistry of n-3 polyunsaturated fatty acidsJ Biol Chem200227787558