
Abstract
Purpose
The fragmented nature of the medical device market limits our understanding of how particular sub-markets navigate the device development process. Despite the widespread use of transcutaneous neuromuscular electrical stimulation (NMES), its use for acquired dysarthria treatment has not been sufficiently explored. This study aims to provide a preliminary understanding of the stages involved in the development of NMES devices designed for neurorehabilitation. It also aims to investigate manufacturers’ perceptions concerning factors that facilitate or impede its development and determine its applicability for acquired dysarthria.
Materials and methods
In-depth semi-structured online interviews were conducted with eight NMES device manufacturers located across Europe, North America and Oceania. The interviews were video-recorded, automatically transcribed, manually reviewed, and analysed using a qualitative content analysis.
Results
NMES device development for neurorehabilitation involves six complex phases with sequential and overlapping activities. Some emerging concepts were comparable to established medical device models, while others were specific to NMES. Its adaptability to different neurological disorders, the positive academia-industry collaborations, the industry’s growth prospects and the promising global efforts for standardised regulations are all key facilitators for its development. However, financial, political, regulatory, and natural constraints emerged as barriers. Indications and challenges for the applicability of NMES for acquired dysarthria treatment were also discussed.
Conclusion
The findings provide a foundation for further investigations on the NMES market sub-sector, particularly in the context of neurorehabilitation. The study also provides insights into the potential adoption of NMES for acquired dysarthria, which can serve as a reference for future research.
IMPLICATIONS FOR REHABILITATION
The mapped neuromuscular electrical stimulation (NMES) development phases and processes can serve as a framework for new device development initiatives
Opportunities for NMES development included its adaptability to neurological disorders, its growth prospects, academia-industry relationships and regulatory standardisation initiatives
Financial, political, regulatory, and natural barriers were barriers for concern.
From an anatomical and practical standpoint, it seems feasible to use NMES to treat several features of acquired dysarthria, however, clinicians’ limited education on the use of electrical stimulation for neurorehabilitation may pose several constraints.
Introduction
Medical technology is one of the primary drivers of the healthcare industry. With more than half a million medical devices (MD) currently available in the European Union alone, covering a large and heterogenous range of technologies and health purposes, it is forecasted that the industry’s growth will continue accelerating in the years to come [Citation1, Citation2]. Nevertheless, despite its extensive development, the MD industry remains relatively “fragmented, privatised, and largely opaque” with most companies producing a limited range of devices and occupying small niche markets [Citation3, p.1]. The majority of MD companies are micro, small or medium-sized enterprises (SMEs) employing less than 50 employees [Citation4]. As a result, MD companies frequently lack the resources necessary to meet regulatory standards or obligations, making it challenging for them to withstand or survive market failures.
Neuromuscular electrical stimulation (NMES), more broadly classified as functional electrical stimulation (FES) or therapeutic electrical stimulation (TES), is a treatment modality used in neurorehabilitation [Citation5]. In line with market trends, the development and use of NMES for neurorehabilitation has been increasing exponentially since the mid-twentieth century, when Liberson et al. [Citation6] stimulated the peroneal nerve in individuals with hemiplegia. Nowadays, NMES treatment is no longer confined to the “realm” of physical therapy but has garnered interest and permeated to other health domains, including occupational therapy, prosthetics, podiatry, and speech-language therapy (the latter profession includes swallowing rehabilitation). For the latter health profession, the twenty first century saw the introduction of the first four NMES systems for the treatment of swallowing disorders (VitalStim® Therapy System, Ampcare ESP™, Guardian Way and Aspire 2, and eSWALLOW) which extended both the boundaries of the profession and the range of treatment options.
Acquired dysarthria, the most prevalent acquired communication disorder, is a speech disorder characterised by slow, weak or imprecise speech musculature that causes impaired control or coordination of movements involved in respiration, phonation, resonance, prosody and articulation [Citation7–9]. Impairment-based standard treatment methods target the affected speech subsystem with the aim of improving the overall intelligibility of speech. Therapy methods that have been investigated in the literature include but are not limited to non-speech oral motor movements, articulation, respiratory and voice exercises, and non-invasive brain stimulation [Citation10, Citation11]. Specific to non-progressive dysarthria, given the heterogeneity of treatment approaches and the limited pool of research, to date, there is limited evidence of the benefit of treatment on outcomes at an impairment level [Citation11].
Although NMES has been shown to potentially be an effective treatment for muscle weakness in progressive diseases, such as chronic respiratory disease and chronic heart failure, as well as for improving activities of daily living and swallowing disorders in non-progressive diseases, including stroke, there has been limited experimental research investigating the use of NMES in its treatment [Citation12]. Notwithstanding the ongoing debates regarding the distinctiveness of speech among motor systems, if we take the theoretical perspective of speech execution as movement production or as involving task dynamics [Citation13], then it can be proposed that similar to other motor movement systems (e.g. gross motor system), NMES may show therapeutic potential for several speech disorders. Hypothetically, NMES coupled with behavioural therapy may facilitate activation of fast and slow-twitch muscle fibres and promote transferability of motor activation to functional use during speech execution [Citation14].
The benefits and limitations of using NMES for the treatment of diverse neurological conditions have been investigated from the perspectives of practitioners [Citation15–17], as well as patients and their carergivers [Citation18, Citation19]. However, the views of manufacturers on determinants of NMES development for rehabilitation have not been widely explored. Given that NMES devices for rehabilitation frequently start off as a university research spin-off or as a start-up from professionals in different fields of health research [Citation20], with potentially limited exposure or insight in MD development and innovation, a comprehensive overview of the development processes involved can inform and guide healthcare providers, policy makers, and other relevant stakeholders of the particularities surrounding transcutaneous NMES development for neurorehabilitation. Insights into the processes and activities involved can empower clinicians interested in health technology to develop and contribute to the “evolution” of NMES technology and its applications in treating diverse groups of neurological disorders. In turn, this knowledge can also increase the evidence base for the use of NMES in neurorehabilitation to ultimately improve patient decision-making and treatment outcomes. Moreover, although the MD process, from product inception to take-off, has been covered quite comprehensively in the literature [Citation20, Citation21], this has frequently been completed in a broad manner, with little understanding of how specific MD sectors navigate the development process.
To the best of our knowledge, this is the first research investigating the views of manufacturers of NMES devices for neurorehabilitation. The aim of this paper was two-fold. Firstly, to obtain a broad understanding of the underlying processes involved in NMES device development; and secondly, to determine the potential clinical applicability of NMES technology in acquired dysarthria. The research questions included: (1) What stages and processes are implemented for the development of a NMES device for neurorehabilitation? (2) What are the barriers and facilitators faced by manufacturers during its development? (3) What are NMES manufacturers’ views of using NMES to treat acquired dysarthria?
Materials and methods
Study design and setting
This qualitative study is reported in line with the Consolidated Criteria for Reporting Qualitative research (COREQ) guidelines. A qualitative content analysis was adopted in this study to explore NMES manufacturers’ perspectives on device development and use. Data collection took place between June 2022 and December 2022 and involved online semi-structured interviews.
An interview guide was developed initially by the first author (PB), who is a certified NMES user, and refined by the research team (PB, RP, CT) (interview guide available in supplementary material). The interview guide was not pilot tested; however, its face and content validity were reviewed by a commercial intellectual property specialist (MM) working in health technology innovation and transfer. The questions of the interview guide elicited discussions around: (1) NMES device development process and use in rehabilitation; (2) facilitators and barriers for its development; and (3) the applicability of NMES for acquired dysarthria therapy.
Participants
Purposive sampling was used for recruitment of participants. NMES manufacturers were identified through database searches on the Devices@FDA, the Medicines and Healthcare products Regulatory Agency’s (MHRA) online registers, and electrical stimulation devices market reports. Since electrical stimulation is used in a wide range of health products, several of which were not relevant to the scope of this study (e.g. implantable electrodes in deep brain stimulation and in pacemakers), all companies identified through the database searches were reviewed based on a predetermined set of inclusion and exclusion criteria.
The inclusion criteria were as follows: (1) organisations that manufacture NMES (therapeutic and functional) devices for rehabilitation; (2) NMES devices are approved by a regulatory body (e.g. FDA or EU approved); and (3) NMES devices involve transcutaneous application. The exclusion criteria were: (1) companies that solely produce cosmetic electrotherapy products; and (2) product is a prototype and not yet available on the market. No gender, geographical location or company size restrictions were applied. For companies that met the selection criteria, an invitation email to take part in an online interview was then sent to the email address appearing on their company website.
Data collection
Ethical approval for this study was obtained from the University Research Ethics Committee of the University of Sheffield (Reference number 036240). The first author (PB) completed one semi-structured interview per participant via the Google Meet platform. All interviews were video recorded with permission and lasted between 30 and 75 min. There were no pre-existing relationships between the researcher carrying out the interviews and participants. Due to language barriers, an interpreter (a native Japanese speaker fluent in English) was present during one of the interviews.
Data analysis
Qualitative content analysis was used to analyse the interview data. All video data was initially transcribed verbatim using Descript [Citation22], an automated transcription software. All automated transcriptions were then reviewed manually by PB. This involved listening to the recordings several times to ensure the accuracy of transcriptions. Minor revisions to the transcripts were completed at this stage. Transcripts were consequently uploaded to NVivo 12 [Citation23], a qualitative research data analysis software, and coding of data was initiated by PB. A manifest analysis using an inductive coding system was applied for data analysis to the development of categories of meaning [Citation24]. To ensure methodological rigour in the coding of data and minimise risks of bias, the transcripts, initial codes and categories were then reviewed by CT, and further refined by the research team. Lastly, the findings were reported by PB and reviewed by CT, RP and MM. shows the coding tree used for the analysis of the interview data.
Figure 1. Coding tree used for analysis.
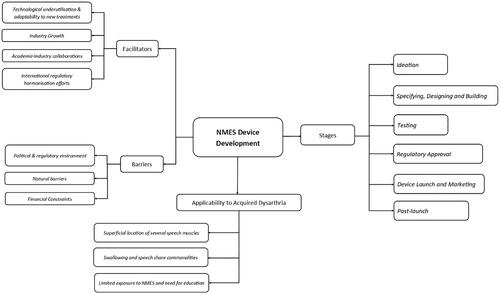
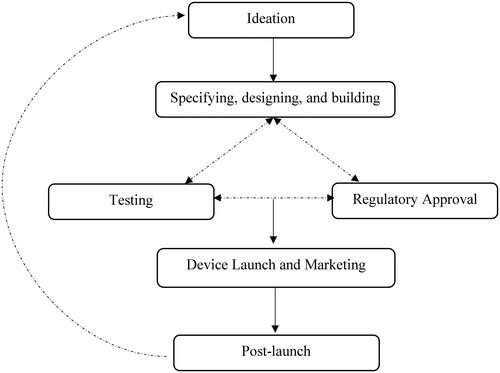
Figure 2. Simplistic schematic representation of the NMES device development process.
Results
A total of 27 NMES manufacturers were invited to participate in this research. Out of these, 17 manufacturers either declined participation or did not reply to the initial and follow-up recruitment email. The remaining 10 manufacturers agreed to take part, however, two were excluded at interview stage as they did not meet the inclusion criteria. The characteristics of the eight manufacturers (each represented by a single participant) who completed this study are displayed in .
Table 1. Characteristics of participants.
The NMES device development process
Six different common phases to NMES device development were identified from the qualitative content analysis of the interview transcripts (): (i) ideation; (ii) specifying, designing, and building; (iii) testing, (iv) regulatory approval; (v) device launch and marketing; and (vi) post-launch. The unidirectional arrows in the schematic diagram indicate a sequential flow from one development stage to another. The bi-directional arrows represent overlapping phases whereby activities in each interact with each other to direct and shape developmental processes. For instance, although the specifying, designing, and building phase precedes testing and regulatory approval, outcomes from the latter stages tend to inform and guide revisions in the former.
Ideation
For the included cases, the ideation phase was characterised by the identification of treatment gaps and opportunities, determining the intended use for the device and completing a preliminary market research. Identification of treatment gaps and opportunities primarily stemmed from clinical work with a specific clinical population or as a “spin-off project from university research activities” [M6]. This process primarily consisted of uncovering the unmet rehabilitation needs of clinical populations and identifying opportunities for innovative, clinically relevant therapy methods.
I work with speech-language pathologists, and they saw a need for…better treatment for swallowing disorders… And that’s how the idea came to fruition. It was based on patient need and the three of us being clinicians. [M5].
I see the clinical issues. I hear the patient responses and that’s valuable feedback… If you’re in a clinical setting and you see thousands of cases, these begin to help you formulate ideas [M3].
Market research revolving primarily around the analyses of patient and clinician population data was frequently completed at this stage to determine the number of potential users and revenue reach. M1 argued “that it’s very difficult to undergo a NMES development program for five or eight years unless there is a market need for the patient and for clinicians. Forecasting how many patients can the device target is essential”.
Specifying, designing and building
Once the ideation phase is complete, NMES companies go through the specifying, designing, and building stage. Fundamental activities at this stage include refining the initial idea, securing funding opportunities, ensuring appropriate technical expertise, designing the device, determining device specifications (e.g. size, shape and number of electrodes, and stimulation parameters), developing prototypes, and carrying out a detailed cost analysis. In one large company, according to M8, refining the idea included developing a launch packet.
A launch packet is a conceptual iteration of what the product should look like; what market potential it has. It includes financials and conceptual ideas for marketing. It’s kind of creating a roadmap for, from here, when we don’t have anything, to there, when we are ideally supposed to launch. It includes timelines, it includes budgets. It is a very big job. [M8].
One of the core components of this phase is creating the device design, establishing scope (the features and functions of the device) and developing prototypes. This includes activities such as: identifying and supplying materials for building the device, defining whether the NMES device is aimed to be a “workhorse that will treat all kinds of different neurological conditions or disorder-specific, for instance, specific to low extremities or swallowing” [M8], whether the intended administrator is a patient or a health professional, and whether stimulation parameters and programmes are modifiable by the user or pre-set in advance by the company. All these factors influence the degree and extent of device characteristics, such as, the number of device channels, type of waveform, pulse frequency and duration, and intensity.
There are various inputs in the design process, which are to establish how long the product would take to design? What components and hardware are needed? What will it look like? What will it require? Can we source it? How long it would take to start production? The cost of designing the product and then the unit cost of the product once it has been designed. [M7].
M2 indicated that these activities are key to ascertaining that the product scope is in line with what can be realistically achieved from a theoretical, clinical, and technological perspective.
Almost putting a list around what we ideally want with this device… And then there’s a bit of a backwards and forwards of what is possible and what isn’t possible from a technological and clinical standpoint. It’s having an optimal wishlist in terms of functions and parameters, and then realising what can actually be done. [M2].
Device design and scope processes in NMES device development are visibly tied to access to technical expertise. Since the development is complex, several professionals from different job sectors, such as, health, business, engineering, software development, regulatory, and legal work are often required to guide and support specific activities during device development.
When that packet is ready, we then have a meeting with various stakeholders from several departments. So, it’s engineers, experts in NMES, marketing executives, finance, and the person that’s in charge of creating the package. Engineering is intimately involved. Marketing will start working as well – will work on colours, form of the product, what it should look like, how heavy or light, etcetera. [M8].
Given the high costs associated with NMES designing and building, testing and regulatory approval, small companies included in this study argued that funding opportunities, mainly through governmental grants and private investors, are often necessary. Despite their benefits, funding grants are often “hard and competitive, include a long review cycle and involve lengthy decision timelines. So, it may take several years to get funding” [M2]. In comparison to small companies, medium-sized and large enterprises indicated that although most of their products are self-funded, research and development is still rigidly regulated, and any new device developments “are closely planned and monitored from a financial point of view” [M7].
Another important process at this stage is carrying out a thorough cost analysis. Cost analysis “ensures that the expected price and upkeep of the product, the expected demand from our customers, and the expected cost of developing the product means that the company would be profitable by developing that new product” [M4]. M2 suggested that cost analysis also includes whether new spin-off ideas, such as new intended purposes for use and functionalities, can be generated from one specific device development.
Testing
Once the initial design and development phases are completed and prototypes are developed, companies start the testing process. For the enterprises in our study, primarily this phase involved feasibility testing with users, generating clinical evidence, and measuring device safety. Feasibility testing involves “focus groups with the future users of the product [patients and clinicians] and allowing them to test NMES prototypes” [M8]. Activities often include measuring usability and acceptability through qualitative data collection procedures to evaluate concepts around device design and functionality, and to inform device modifications.
We’ll put the prototype in the hands of the end-users. And then they’ll tell you what they like and don’t like about it. Let them use it, let them try to break it. Ideally, they will break it, and then we can then go back and look at it, figure out why it broke, change the design to make it better, give 'em a new, better one, and re-test. [M6].
Safety testing primarily revolves around ensuring that the NMES device meets electrical safety standards and requirements. It also includes determining possible adverse effects and contraindications for use. NMES developers in this study argued that, although generating clinical evidence is a feature of the testing stage, the degree and extent of clinical data required depends primarily on the type of regulatory application being submitted and the country where you plan to sell the device. For instance, M1 indicated that for devices that were not like others already available on the market in terms of site of stimulation and function, clinical data from human participants had to be collected to determine the clinical effectiveness of the device. This was a prerequisite for regulatory approval (e.g. U.S. Food and Drug [FDA] Premarket Approval). M8 debated that “prototype testing on patients is possible, but only as part of a clinical trial. The more onerous part of the testing is to ensure electrical safety and electrical compliance; that (a) the machine is safe enough to use on patients and (b) the machine does not emit electrical interference with other equipment” [M8]. On the contrary, for other devices, clinical effectiveness was based on establishing substantial equivalence with a predicate, a marketed device already approved for use by a regulatory body (e.g. FDA 510(k)).
Ideally, a device is launched under a 510(k) approval as costs related to research are considerably less. If it cannot be done, it now becomes a different category altogether where the FDA requires studies where it is used on patients, and they are wanting to see efficacy, as well as safety data. [M8].
When we plan to implement new treatment that we never tested before, then we need clinical evidence. First pilot studies or basic research to show that the NMES device shows effects in specific patient groups. And later we need a randomised control trial. Only when these studies show effectiveness, then we can get regulatory approval and go on the market. [M1].
Regulatory approval
The regulatory approval phase includes determining the type of application and documentation required for a specific regulatory submission. M1 indicated that since regulatory approval involves a large volume of intricate documentation, it is often initiated during the specifying, designing and building phase and elaborated further in the testing phase.
The type of regulatory approval required is directly related to the geographic target market, that is, the country or countries in which developers aim to market the device. There are two main global standards for regulatory approval of new MDs: the CE mark certificate for commercialisation of devices in the European Union (EU); and the FDA’s approval for the US market. M7 contended that although endorsements from these two governing bodies may speed up or facilitate device approvals in other foreign regulatory authorities, they neither offer nor ensure direct market access in other countries. M6 indicated that there is a medical device single audit programme (MDSAP) that allows you to obtain one regulatory approval that covers all regulatory bodies participating in the programme reducing the number of audits a manufacturer must undergo and facilitating access to multiple international markets.
These two standards can be then applied in most other countries because most other countries recognise both standards as being a globally acceptable standard. And will give you shortcut entry into regulatory approval. For instance, if you’re trying to sell a product into Singapore, if the product is CE marked or FDA approved, it’s a much quicker, easier process to get it approved. [M7].
The amount of scrutiny and control posed by regulatory bodies for the approval of new MDs is also related directly to their risk classification system. Manufacturers reported that since NMES devices are classified as class II (posing medium risks) the process for approval is often complex and time-consuming. Besides, since most developers in this study were working in small-sized companies and were health professionals by background, they did not possess sufficient knowledge nor internal human resources to go through the regulatory approval process on their own without the backing and support of a regulatory firm.
We tried to take it to the FDA ourselves as clinicians. We got letters from the FDA for additional information, and we needed some help. So that’s when we went and hired a regulatory firm to help us understand what the FDA needs and help us develop an appropriate submission. [M5].
Device launch and marketing
Once the testing phase is completed, the hardware and software components of the device are modified and integrated, and regulatory clearance is obtained, the device is then ready to be launched and marketed. NMES manufacturers suggested that activities at this stage include identifying and partnering with medical equipment distributors and suppliers and promoting the new device to medical and health professionals working in the field of rehabilitation.
We gradually brought in and started talking to a Japanese company. That was one of the largest powered muscle stimulation companies, oldest companies, in Japan. And we eventually struck up a deal with them that they would market our product. [M5].
Although the benefits of using standard marketing strategies, such as digital marketing approaches, were recognised by most companies, greater emphasis was placed on educating and increasing awareness of medical and health professionals. The main reasons for focusing on education included: (1) NMES can treat various neurological conditions and related disorders with different stimulation parameters for adjustment; (2) training on the physiological foundations and use of electrical stimulation for rehabilitation is not always covered during university education; (3) in certain countries, physician-initiated referrals or treatment authorisation is necessary for the provision of NMES therapy.
We are probably targeting a lot of therapists who have not studied electrical stimulation in their education or have never used it before, rather than always targeting the therapists who are very familiar with electrical stimulation. And that’s a big problem because there’s a big gap. [M1].
Post-launch
The sixth and last phase of the process is post-launch. It includes user support and the planning of new iterations of the device. M1 indicated that user support primarily involves generic video e-support and user-focused one-to-one support. Another key component of this stage is collecting additional feedback from the user base to identify and generate additional or novel device value propositions or design concepts. M8 suggested that NMES device development is a dynamic process that involves integrating technological advances to generate or expand additional device specifications and functions. As a result, this last phase should ideally loop the manufacturer back to the first phase, the ideation process.
We get feedback from doctors and therapists. And of course, we also get recommendations from them - what device or how the device could be improved in further generations. So that’s very important. [M1].
Thinking about iterations of the original device needs to be an ongoing process. The portfolio needs to expand, even if you think that your product is a crown jewel. It cannot stay static. This is necessary in medical innovation because our understanding is continuously evolving. New research comes out, so products need to be updated from time to time. [M8].
Facilitators and barriers to NMES development
Several facilitators and barriers for NMES device development were identified.
Facilitators
Insights from manufacturers converged into four distinct facilitators: (1) underutilisation and adaptability to new treatments; (2) industry growth; (3) academia-industry collaborations; and (4) international regulatory harmonisation efforts.
Technological underutilisation and adaptability to new treatments
Manufacturers indicated that the fact that NMES technologies in general are still underemployed in the treatment of neurological diseases and related disorders is a key facilitator for embarking on the development of new devices. M6 stated that NMES technology can be adapted to meet satisfactorily the physiological and functional demands of specific anatomical regions and muscle groups. Thus, it has the potential to contribute considerably more to the rehabilitation of neurological diseases and related disorders.
There’s a lot of product opportunities, all of them based on the idea that NMES, in my opinion, is grossly underutilised. Even though it’s been a commercial success, I still think people don’t use it enough. I think there is much more potential. [M8].
Industry growth
It was commonly identified between manufacturers that global growth of NMES is expected to continue to increase in the years to come. Indicators for expanding growth included both internal and external factors. These included results from company data driven insights, increased product adoption to new neurorehabilitation treatments, improved acceptance and use of NMES devices by clinicians, and the fact that the MD industry is still an unsaturated market.
We will see more growth as it takes off in different countries, and it is used by different clinicians for different purposes. There should be growth from the market. [M7].
We project long term increases in the use of NMES devices. It’s not just predicting. We’ve got eight years of data. [M5].
Academia-industry collaborations
One common facilitator across several small-sized manufacturers was early collaboration with academic institutions, particularly for the testing phase of development. Manufacturers reported that collaborations with academia allowed them to obtain access to additional resources, such as, the ancillary use of additional medical equipment, for instance, radiographic instruments, and access to clinical populations. For most, the latter factor was crucial for the successful identification and recruitment of clinical populations for testing and for determining the feasibility and clinical effectiveness of the novel NMES device.
And we’ve also got cooperation with universities for the clinical studies field. Research, such as, case-studies, pre and post studies in the US. One specific teaching hospital was enormous for us. They did case studies for us and a few feasibility trials early on that really helped us with the clinical data. [M5].
International regulatory harmonisation efforts
Although manufacturers felt that obtaining regulatory approval is an inherently complex and time-consuming process, it was indicated that recent reforms in regulation, primarily, the introduction of international regulatory harmonisation processes offer new opportunities to established manufacturers and new ones alike. Two main international efforts were mentioned by participants. These include the Medical Device Single Audit Program (MDSAP) and the International Medical Device Regulators Forum (IMDRF). Interviewees indicated that aligning regulatory requirements for the development and marketing of MDs across countries through the adoption of international guidance documentation and scientific standards, will reduce current challenges around regulations governing access to international jurisdictions and markets.
The Medical Device Single Audit Program is an example of harmonisation that’s going on. And the EU and some others are participating in that program as observers right now. But we expect that they will participate at some point. And then there is the IMDRF who are specifically trying to harmonise processes. Despite limitations such as, most stringent rules, if and when they can pull that off, I think that it’ll be very helpful for manufacturers as it will reduce the barriers to international market access. [M5].
Barriers
Three categories of barriers around the NMES device development process were identified from patterns in the transcribed data. These include barriers in the: (1) political and regulatory; (2) natural; and (3) financial environments.
Political and regulatory environment
Political hurdles in NMES device development included regulatory changes, bureaucracy, and limited treatment reimbursements. Regarding bureaucracy, interviewees indicated that regulatory requests and approvals have become more rigid, inflexible, and costly due to more stringent criteria. Requests for documentation have also increased exponentially over the years, increasing the overall duration and complexity of the process. M7 argued that the regulatory approval for class II devices is often uncertain as it involves “special controls” which are specific to the device being submitted for approval.
The main barrier nowadays is the medical regulation. It’s not so easy anymore. 20 to 30 years ago, someone with a good idea, would just design a new medical device to bring on the market. It’s much more regulated in the last few years. So, that makes it extremely hard and makes it nearly impossible for small companies to achieve. [M4].
Regulatory changes secondary to legislative amendments also impact NMES development opportunities. Manufacturers discussed how changes in the political landscape, such as, the withdrawal process of the UK from the EU (Brexit) and the shift from the EU Medical Device Directive to the EU Medical Device Regulation (MDR) [Citation25] will impact commercially available products, hinder new development efforts, and increase regulatory compliance and human resource costs. For instance, M1 suggested that “since the MDR will lead the market towards individualisation of NMES devices, so that you have a device for each specific condition or therapy, the future existence of workhorse NMES devices that are developed and sold for multiple neurological indications is in jeopardy”. Similarly, M7 stated that Brexit is likely to give rise to regulatory changes that will impact the developmental and manufacturing processes.
Brexit will have an impact. If you’re selling a device into the UK market, you’ll need to design it to comply with UK standards. Currently, because it’s so complicated, the UK government has said, as long as your product is CE marked, you can still sell it in the UK, but there will come a time when your device has to comply with UK specific requirements and not the CE marking. If you are just restricting yourself to the UK, those regulatory compliance costs must be lower because they’ve still gotta produce a profitable return, but from a smaller market. So, this regulation might influence market access and the choice of where you decide to regulate the device. [M7].
Another obstacle mentioned by participants was the fact that health insurers are either not covering NMES therapy costs or are restricting monetary reimbursement limits. As a result, clinical groups that may benefit from such treatment may not have the economic capability to access these services and therapists are reluctant to offer NMES treatment as part of their services. In turn, these factors reduce revenue generation and foresight for NMES manufacturers.
Reimbursement from insurance is very difficult, so therapists are reluctant to get involved. They are trying to implement a law where they want to reduce the fees by 50%. You have all the overhead, especially related to the FDA regulations, but suddenly the reimbursement people have decided you’re going to be cut by 50%. You know, there’s a huge disconnect between the regulatory people, insurers and the lawmakers which directly influences, and to a certain extent hinders our operational strategies and financial growth. [M3].
Natural barriers
The Covid-19 pandemic was mentioned as a major impact to NMES device development and manufacturing. M5 argued that natural barriers, which are often unforeseen, can give rise to company bankruptcy, especially for small-sized companies. Besides, M3 and M8 mentioned that the unprecedented difficulties with purchasing and delivering of device components, testing processes, supply chain and human resources, and reduced marketing, educational and training opportunities arose from the pandemic. M5 debated that adequate risk assessment plans need to be in place to minimise financial failure risks that may arise from these situations.
Small companies may have a way to make it, but then COVID-19 or some other pandemic or natural disaster occur that disrupt your operations. You better have risk assessment plans or you’re going to run into some serious headaches. [M5]
Financial constraints
The financial burden associated with developing a new NMES device was another commonly identified barrier. Manufacturers argued that the costs for developing and finalising the device prior to commercialisation is one of the biggest threats to start-ups and established companies alike. Capital funding sources were closely linked to the time it takes to develop a new NMES device. Indeed, companies with more financial resources were likely to develop and market NMES devices in a shorter time span. It was also debated that financial restrictions can “influence the quality of the product, from design aspects, to methodological rigour, to what can be carried out during the launch and post-launch phases” [M7]. Specific to product quality, M6 argued that manufacturers often must mitigate and strike a balance between NMES system parameters and functions with the total cost of producing the device. Even though software features and ancillary technologies can be added to improve the overall application and benefit of NMES systems, these are frequently too cost prohibitive to develop and market and may not render any return on investment.
So, for example, like I've got this device, this is a two-channel stimulator, a NMES device. And, you know, that device is $200, and it’s got two channels of stimulation, and that’s it. This device is a has limited function versus our system, which is an entire cycling system with 10-channels of electrical stimulation. There’s just a lot more complexity, but that makes it a much more useful and effective tool. So, in my mind, a large trouble with NMES in general is striking a balance between cost and complexity. [M6].
Applicability of NMES for acquired dysarthria treatment
Most manufacturers had heard of dysarthria and could identify it as a speech disorder that causes impaired intelligibility. This awareness was primarily related to the fact that most of the interviewees were health professionals who previously worked in rehabilitation settings. Whilst the majority of manufacturers were not interested in, nor received requests for developing or adopting a NMES device for dysarthria, M1 did not feel comfortable answering and commenting about this. M8 elaborated further on this and reported that even though they had received requests from researchers to use their NMES device, manufactured for swallowing disorders, to assess its effectiveness for treating dysarthria, this was never completed due to methodological weaknesses in the proposed research protocols.
M5 and M8 noted that the speech-language profession employs NMES stimulation for clinical dysphagia treatment. Research endeavours have also explored its applications for dysphagia and voice disorders. It was indicated that NMES can be applied to several aspects of dysarthria asspeaking and swallowing share several small muscles that are reciprocally active during both activities. Several of the primary muscles of speech production, which are superficially located close to the epidermis at the facial, oral, and submental regions, can easily be reached and functionally activated with electrical stimulation. M8 also indicated that anecdotal evidence from speech-language therapists suggests that in individuals with co-occurring dysphagia and dysarthria, often show a co-current improvement in both physiological functions when treated using NMES.
It depends on what causing the dysarthria, but in principle, the input of electricity can produce effects that might be beneficial for dysarthria patients. In fact, dysarthria is one of those symptoms that improves quicker than the dysphagia improves in patients who have both. And, you know, it is one of the most frequent impairments in stroke. And reported effects of using NMES for swallowing is that their ability to articulate and voice is improving as well, which makes sense. I mean, it’s relatively the same muscles. [M8].
M5 and M8 debated that the biggest challenge for the use of NMES for dysarthria rehabilitation is the fact that the speech and language therapists have limited knowledge, exposure, and experience with using this treatment modality. Both manufacturers referred to their experience with developing and marketing NMES devices for dysphagia to argue that training on the foundations and fundamentals of electrical stimulation, and the use of NMES as a rehabilitation technique is thus inevitably necessary to promote device awareness and use, improve understanding in the field and ascertain safe and effective administration.
I just think the biggest challenge is that the speech language discipline still doesn’t know what this modality can do. I believe, they think electrical stimulation is all the same. I believe that training gives them the opportunity to understand it a little bit better, and once they grasp it, then you can build from it and use it accordingly. [M8].
Discussion
NMES device development
Despite differences in the number of device cycle phases and terminology, the six phases of the NMES device development identified from the qualitative content analysis of the eight in-depth interviews are reasonably in line with well-established generic product [Citation26] and MD development models [Citation20, Citation21, Citation27]. Similar to the Pietzsch et al.’s [Citation27] MD development model, NMES development starts off from ideation, which involves the identification of a clinical unmet need based on structured or unstructured activities, such as: observations and personal experiences, literature reviews, and determining whether there is scope for MD development by completing a preliminary market analysis.
The specifying, designing, and building phase of NMES development consists of several distinct but connected processes that influence the who, how, why and when of the product. A principal activity of this phase, refining the initial idea, involves creating a list of criteria, requirements, and time-limits that will serve as a roadmap for successful device development. Identifying and securing funding opportunities is also central to this phase. Manufacturers, particularly, small stand-alone enterprises, argued that even though the process for applying for a grant is complex and lengthy, funding sources are normally essential for kick-starting and developing new MDs. Research by Moses et al. [Citation28] indicates that when compared to other high-tech industries, biomedical innovations rely substantially on governmental funding opportunities. This could be related to the fact that the MD sector is a high-risk industry in which most enterprises are not able to provide sufficient profit and return on investment [Citation29]. Without the necessary sustained support from funding sources, NMES research and innovation is also poised to rapidly decline.
Central to the designing and building aspects of this phase is the recruitment of external professionals with specific clinical and technical expertise Kaplan et al. [Citation30] argue that this activity is frequently necessary to bring the concept through the design-building-test-redesign process. Akin to the broad MD development processes [Citation29], once the development team is formed, device features and parameters are selected, hardware components are sourced, software is developed, initial device designs are created, and prototypes are built for testing purposes.
The testing of prototypes and regulatory approval are distinct but closely tied phases. In line with EU Medical Device Regulation (EU 745/2017) and the Food and Drug Administration’s (FDA) regulatory approval processes, the type of regulatory compliance required will influence the amount of time and financial resources needed for testing. Manufacturers argued that NMES devices are classified as class II devices and hence, regulatory approval involves device-specific “special controls”. Whilst several U.S. developers were able to prove “substantial equivalence” to existing approved NMES devices on the market and received 510(k) clearance, others had to go through a Premarket Approval (PMA) which is substantially more complex, lengthy, and costly. A significant association between the likelihood of a MD being recalled and its acquisition of 510(k) clearance indicates that the concept of substantial equivalence may be deficient in specific contexts. This is particularly true when safety risks of the predicate device are not well established or when there exist significant variances between the “novel” device and its predicate [Citation31, Citation32].
For NMES manufacturers, regardless of whether regulatory approval was through the FDA, the EU, or another regulatory body, evidence of safety standards was taken to be more important than clinical efficacy data. This result corroborates findings from other MD sectors confirming that obtaining approval to commercialise a MD in the EU is mainly dependent on being able to demonstrate that it is safe and functions as per its intended use [Citation30]. Similarly, Feldman et al. [Citation33] argue that the FDA may grant commercial approval of MDs without the requirement of high-quality studies focused on efficacy and outcomes.
Approving novel MD technologies without the need of extensive effectiveness data decreases costs and time to market for new MD technologies. However, one major drawback associated with the limited generation of robust efficacy and outcomes evidence during the development process is that clinical practice guidelines and standards are unable to support the use of these new technologies when these are launched on the market. For instance, even though the first NMES device for dysphagia was approved by the FDA in 2002, to date, the National Institute for Health and Care Excellence [Citation34] and consequently, the UK National Health System (NHS), have still not advocated its use in post-stroke dysphagia rehabilitation. This is primarily related to the fact that despite an indication of potential benefit and the absence of any significant safety issue related to its administration, the quality and quantity of evidence supporting its efficacy and effectiveness is still scarce [Citation34]. Undeniably, limited evidence complicates patient informed treatment decisions with potentially minimal therapeutic benefit [Citation35].
The device launch and post-launch stages also follow similar patterns to other MD development models. However, one major difference between this and other models was the education paradigms associated with product commercialisation. Whilst Pietzsch et al. [Citation27] indicate that a peer-to-peer education model is frequently employed with physicians to stimulate marketing and promote the use of a new MD, in this study, NMES device education and marketing frequently involved formal training via face-to-face and online learning certifications. In addition to promoting the NMES device, a primary purpose of these training opportunities is to equip health professionals, particularly those with limited experience in the field, with foundational knowledge of electrical stimulation and its effective and safe application.
Facilitators and barriers
Although technological underutilisation is often considered as a barrier, NMES manufacturers in this study contended that the lack of NMES usage and its adaptability to new treatments, along with forecasted industry growth, offer substantial technological and commercial opportunities for the future. Based on Grandjean and Mortimer’s [Citation36] propositions that the biophysical properties of electrical stimulation are well defined, acquiring anatomical data is relatively simple, and quantifying the absolute and relative effects of parameters from several electrode setups is feasible, Loeb [Citation37, p.380] argues that “one might never guess this from the clinical literature, whose methods are rarely presented in a useful way and are often irrational and even self-defeating …[yet] the science and the technology are ready. All we need is the discipline to focus collectively on real problems and real opportunities”.
Academia-industry collaborations were also reported to be important contributors to facilitating NMES device development. This finding is in line with results from the broader biomedical industry confirming that collaborations between academia and industry, which often entail academic institutions offering access to trial participants as well as clinical and methodological knowledge, and industries offering financing and expertise for carrying out clinical trials, are mutually advantageous [Citation38]. The significance of this relationship is amplified when the need for additional high-quality research evidence for supporting the clinical implementation and use of NMES is considered.
NMES manufacturers regarded the implementation of international harmonisation initiatives by the IMDRF and the MDSAP as facilitators for the development, growth, and distribution of MDs in the future. Even though the number of participating countries is limited (i.e. the U.S., Japan, Australia, Canada, and Brazil) and the process may involve additional audit criteria and costs, it was asserted that the harmonisation of regulatory procedures enhances manufacturers’ ability to access foreign markets and expedite the release of new NMES devices. A crucial step towards enhancing the scale of harmonisation efforts would be the successful integration of the EU and the UK from their current status as MDSAP observers to participating members, while also increasing the engagement of other external but prominent markets. The importance of future efforts to harmonize regulatory disparities between the EU and the US cannot be emphasized enough, given that these two markets, along with China, account for the largest MD markets worldwide [Citation39].
In terms of barriers, this study indicates that the political and regulatory domains pose significant obstacles to the development and manufacturing of NMES devices. At EU level, manufacturers argued that whilst increased patient safety is a positive development, the transition from the Medical Device Directives (MDD) to the Medical Devices Regulation 2017/745 (MDR) imposes more rigorous and supplementary data prerequisites that could impact the viability and survival of NMES companies and their NMES devices. For instance, the MDR’s increased requirements for clinical evidence [Citation40], heightens the possibility of non-compliance of workhorse NMES devices. Furthermore, the MDR will result in an exponential increase in regulatory costs for companies [Citation41]. As a result, small organisations, including NMES manufacturers, are at risk of not being able to absorb the additional costs associated with the regulatory process and post-market surveillance.
NMES manufacturers also discussed the impact of the UK Medical Device Reform, a consequence of Brexit, on device development. Whilst other studies that involved stakeholders from the MD sector contended that regulatory divergence from international standards is likely to increase delays and costs in placing the devices on the UK market to ultimately decrease the availability of devices for UK patients [Citation42], NMES manufactures in our study asserted that decreased development and access of NMES devices in the UK will be the result of restricted market access, diminished market sizes, and reduced return on investment.
Limited health insurance coverage for the costs of NMES therapy or restrictions on monetary reimbursement were also identified as barriers to the manufacturing of NMES devices. The lack of high-quality evidence for the use of NMES in therapy limits its reimbursement coverage and often excludes this treatment modality from standard treatment practices [Citation43]. For instance, in the U.S., reimbursement of NMES therapy is restricted to the treatment of disuse atrophy where neural innervation to the muscle is intact, including brain, spinal cord, and peripheral nerves (e.g. prolonged immobilisation following total knee arthroplasty), and other disuse atrophy causes that are not related to neurological damage [Citation44]. At present, NMES therapy for neurorehabilitation is still largely considered an experimental or investigational modality.
Financial constraints also pose a major obstacle to NMES device development. In line with the broader MD market, NMES manufacturers are primarily small enterprises that offer a limited range of devices [Citation4, Citation30]. As a result, their focus is on targeting smaller markets and often must rely on public or private funding opportunities to achieve product launch [Citation45]. Development of devices is too often constrained by the high cost of additional features and ancillary technologies, which frequently renders them cost-prohibitive for integration into a new device. This limitation could affect one of the key determinants of MD effectiveness, that is, the intrinsic net efficacy of the device resulting from its characteristic features and parameters [Citation46]. Since financial resources are a critical factor that affect the time required for the production and commercialisation of NMES devices, inadequate funding and financing can lead to a longer time-to-market, elevating the risks associated with a short product life cycle.
Unanticipated natural calamities were also mentioned barriers to NMES development. Similar to other MD sectors, the COVID-19 pandemic brought about substantial disruptions to the NMES sector across the diverse stages of development, from testing to device launch and marketing. However, the primary cause of NMES device scarcity was the dearth of essential components necessary for device manufacturing, rather than a demand-supply predicament, as seen in the case of emergency and critical medical products such as COVID-19 vaccines and personal protective equipment [Citation47, Citation48].
Applicability of NMES to acquired dysarthria
Two NMES developers in the study, well-versed in the application of NMES for swallowing disorders, argued that NMES for acquired dysarthria is an option from a theoretical and practical perspective. From a practical standpoint, it was argued that several muscles of speech production are located superficially and can thus be easily activated with this modality. For instance, the orbicularis oris, the masseter, the zygomaticus major, and the anterior belly of digastric are some examples of superficial muscles that can be easily stimulated with NMES [Citation49].
From a theoretical perspective, the use of NMES to treat facial and submental muscles is still in its infancy when compared to other rehabilitation fields such as upper and lower limb movements [Citation50, Citation51]. However, initial indication for use has been accrued for several neurological disorders, including facial palsy and dysphagia [Citation50, Citation52, Citation53]. Drawing from this preliminary evidence and the understanding that acquired dysarthria shares several commonalities with neurological disorders resulting from damage to cranial nerves V, VII, IX, X, and XII, it is possible to hypothesise that superficial NMES application may be a feasible therapeutic option for acquired dysarthria.
Regarding commercialisation and post-launch, manufacturers reported that since the speech-language therapy profession is still new to NMES and is relatively “unfamiliar” with the technology, more output efforts, such as structured training opportunities, will be required for a successful roll-out of a newly launched product. This position concurs with pharmaceutical research that suggests that inadequate familiarity and education regarding a novel medical product could lower its prescription and utilisation rates, thereby slowing down its adoption in clinical practice [Citation54–55].
Limitations
This qualitative research is subject to several limitations. Firstly, despite efforts to recruit a larger number of NMES developers, the sample size was limited to eight participants. Although the pool of participants was homogenous on several demographic characteristics and several data redundancy signals were identified, risks of not reaching data saturation cannot be excluded. Secondly, this research was solely based on semi-structured interviews. Methodological triangulation, involving focus groups, direct process observations and document reviews would have improved the validity and comprehensiveness of the findings. Thirdly, this study primarily provides a synoptic macro-level understanding of the phases involving in NMES device development for neurorehabilitation. Specific focus on partial aspects of the development process would have yielded richer insights about the micro-level aspects of NMES device development.
Conclusion
The development of a transcutaneous NMES device involves six phases with several activities at each level. It includes the identification of treatment opportunities, conceptually grounding the device’s scope and purpose, exploration of funding sources, specification of NMES parameters and features, collaboration with internal and external partners to design and build prototypes, and generation of evidence on the device’s safety and effectiveness for regulatory approval. Post-regulatory approval activities include commercialisation, education, increasing product awareness, and brainstorming for new iterations of the product.
Device development is facilitated by the underutilisation of NMES technology, the capacity to adapt to new neurorehabilitation treatments, industry growth, partnerships between academic and industry sectors, and international efforts towards harmonised international regulatory standards. However, financial, political, regulatory, and natural constraints may pose significant barriers to successful device development.
Manufacturers anticipate that utilising NMES for treating acquired dysarthria is plausible from an anatomical and practical standpoint, however, clinicians’ limited education on the use of electrical stimulation for neurorehabilitation may present difficulties in testing and translating research finding to clinical practice for this speech impairment. Future studies exploring the views of speech-language therapists and persons with dysarthria on using NMES for acquired dysarthria treatment are required to identify patient needs for this population and determine their interest in trialling this treatment modality.
Supplemental Material
Download MS Word (24.2 KB)Acknowledgments
We thank the research participants for their valuable time and expertise in the field of transcutaneous NMES.
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Funding
Notes on contributors
Pasquale Balzan
Pasquale Balzan is a PhD researcher at the Division of Human Communication Sciences of the University of Sheffield. His research focuses on the potential use of transcutaneous neuromuscular electrical stimulation for dysarthria treatment.
Catherine Tattersall
Catherine Tattersall is a senior university teacher at the School of Allied Health Professions, Nursing, and Midwifery of the University of Sheffield and leads the specialist motor speech disorders clinic at the university. She earned her PhD in object naming at the University of York and completed a research fellowship on aphasia in Philadelphia, USA.
Rebecca Palmer
Rebecca Palmer is a professor in communication and stroke rehabilitation at the University of Sheffield. She holds a PhD in dysarthria treatment using speech recognition technology, was the chief investigator of the Big CACT US trial investigating the clinical and cost-effectiveness of aphasia computer treatment, and currently leads the NIHR-funded Aphasia Partnership Training (AP T) project, focused on communication partner training for individuals with aphasia and their families.
Michael Murray
Michael Murray is an Entrepreneur in Residence at the University of Sheffield. He is a dealmaker and commercial intellectual property (IP) specialist working in technology transfer in the biopharmaceutical industry.
References
- European Commission. Questions and Answers: commission proposes an extension of the transitional periods for the application of the Medical Devices Regulation. [Internet]. European Commission. Available from: https://ec.europa.eu/commission/presscorner/detail/en/qanda_23_24
- Ramakrishna S, Tian L, Wang C, et al. Medical devices: regulations, standards and practices. Cambridge: Woodhead Publishing, an imprint of Elsevier; 2015.
- McCulloch P. The EU’s system for regulating medical devices. BMJ. 2012;345(1):e7126–6–e7126. doi: 10.1136/bmj.e7126.
- MedTech Europe. The European medical technology industry in figures 2020 [Internet]. MedTech Europe; 2020. Available from: https://www.medtecheurope.org/wp-content/uploads/2020/05/The-European-Medical-Technology-Industry-in-figures-2020.pdf
- Takeda K, Tanino G, Miyasaka H. Review of devices used in neuromuscular electrical stimulation for stroke rehabilitation. Med Devices (Auckl). 2017;10:207–213. doi: 10.2147/MDER.S123464.
- Liberson WT. Functional electrotherapy. Trans Am Soc Artif Intern Organs. 1962;8(1):373–377. doi: 10.1097/00002480-196204000-00075.
- Duffy JR. Motor speech disorders: substrates, differential diagnosis, and management. St. Louis (MO): Elsevier Mosby; 2013.
- Emerson J, Enderby P. Does speech and language therapy work. London: Whurr; 1995.
- Yorkston KM. Treatment efficacy. J Speech Lang Hear Res. 1996;39(5):S46–S57. doi: 10.1044/jshr.3905.s46.
- Chiaramonte R, Pavone P, Vecchio M. Speech rehabilitation in dysarthria after stroke: a systematic review of the studies. Eur J Phys Rehabil Med. 2020;56(5):547–562. doi: 10.23736/S1973-9087.20.06185-7.
- Mitchell C, Bowen A, Tyson S, et al. Interventions for dysarthria due to stroke and other adult-acquired, non-progressive brain injury. Cochrane Database Syst Rev. 2017;1(1):CD002088. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6464736/ doi: 10.1002/14651858.CD002088.pub3.
- Balzan P, Tattersall C, Palmer R. Non-invasive brain stimulation for treating neurogenic dysarthria: a systematic review. Ann Phys Rehabil Med. 2022;65(5):101580. doi: 10.1016/j.rehab.2021.101580.
- Smith A. The control of orofacial movements in speech. Crit Rev Oral Biol Med. 1992;3(3):233–267. doi: 10.1177/10454411920030030401.
- Manske RC, Reiman MP. Muscle weakness. In: Cameron MH, Monroe LG, editors. Physical rehabilitation: evidence-Based examination, evaluation, and intervention. St. Louis, (MI): saunders, Elsevier; 2007. p. 64–86.
- Auchstaetter N, Luc J, Lukye S, et al. Physical therapists’ use of functional electrical stimulation for clients with stroke: frequency, barriers, and facilitators. Phys Ther. 2016;96(7):995–1005. doi: 10.2522/ptj.20150464.
- Abouzakhm N, Choy S, Feld R, et al. Evaluating the validity of a functional electrical stimulation clinical decision making tool: a qualitative study. Front Neurol. 2022;13:1001123. doi: 10.3389/fneur.2022.1001123.
- O'Connor D, Lennon O, Minogue C, et al. Design considerations for the development of neuromuscular electrical stimulation (NMES) exercise in cancer rehabilitation. Disabil Rehabil. 2021;43(21):3117–3126. doi: 10.1080/09638288.2020.1726510.
- O'Connor D, Lennon O, Wright S, et al. Self-directed home-based neuromuscular electrical stimulation (NMES) in patients with advanced cancer and poor performance status: a feasibility study. Support Care Cancer. 2020;28(11):5529–5536. doi: 10.1007/s00520-020-05394-0.
- Walker DM, Fletcher-Smith J, Sprigg N, et al. Designing a trial of early electrical stimulation to the stroke-affected arm: qualitative findings on the barriers and facilitators. Br J Occup Therapy. 2022;85(3):181–186. doi: 10.1177/03080226211008706.
- Santos IC, Gazelle GS, Rocha LA, et al. Medical device specificities: opportunities for a dedicated product development methodology. Expert Rev Med Devices. 2012;9(3):299–311. doi: 10.1586/erd.12.3.
- Medina LA, Kremer GEO, Wysk RA. Supporting medical device development: a standard product design process model. J Eng Des. 2013;24(2):83–119. doi: 10.1080/09544828.2012.676635.
- Descript. [Internet]. 2021. Available from: https://www.descript.com/tour
- QSR International Pty Ltd. NVivo (Version 12). [Internet]. 2018. Available from: https://www.qsrinternational.com/nvivo-qualitative-data-analysis-software/home
- Bengtsson M. How to plan and perform a qualitative study using content analysis. NursingPlus Open [Internet]. 2016;2(2):8–14. doi: 10.1016/j.npls.2016.01.001.
- European Union. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (Text with EEA relevance.) [Internet]. 2017. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
- Ogot MM, Okudan-Kremer G. Engineering design. Bloomington, IN: Trafford Publishing; 2004.
- Pietzsch JB, Shluzas LA, Paté-Cornell ME, et al. Stage-Gate process for the development of medical devices. J Med Dev. 2009;3(2):021004-1–021004-15. doi: 10.1115/1.3148836.
- Moses H, Dorsey ER, Matheson DHM, et al. Financial anatomy of biomedical research. JAMA. 2005;294(11):1333–1342. doi: 10.1001/jama.294.11.1333.
- Marešová P, Klímová B, Honegr J, et al. Medical device development process, and associated risks and legislative Aspects-Systematic review. Front Public Health. 2020;8:308. doi: 10.3389/fpubh.2020.00308.
- Kaplan AV, Baim DS, Smith JJ, et al. Medical device development. Circulation. 2004;109(25):3068–3072. doi: 10.1161/01.CIR.0000134695.65733.64.
- Everhart AO, Sen S, Stern AD, et al. Association between regulatory submission characteristics and recalls of medical devices receiving 510(k) clearance. JAMA. 2023;[329(2):144–156.
- Kadakia KT, Beckman AL, Ross JS, et al. Renewing the call for reforms to medical device safety—the case of penumbra. JAMA Intern Med. 2022;182(1):59–65.
- Feldman MD, Petersen AJ, Karliner LS, et al. Who is responsible for evaluating the safety and effectiveness of medical devices? The role of independent technology assessment. J Gen Intern Med. 2008;23 Suppl 1(Suppl 1):57–63. doi: 10.1007/s11606-007-0275-4.
- National Institute for Health and Care Excellence. Transcutaneous neuromuscular electrical stimulation for oropharyngeal dysphagia in adults Interventional procedures guidance Your responsibility Your responsibility [Internet]. 2018. Available from: https://www.nice.org.uk/guidance/ipg634/resources/transcutaneous-neuromuscular-electrical-stimulation-for-oropharyngeal-dysphagia-in-adults-pdf-1899874043109061
- Hwang TJ, Sokolov E, Franklin JM, et al. Comparison of rates of safety issues and reporting of trial outcomes for medical devices approved in the European union and United States: cohort study. BMJ. 2016;353:i3323. doi: 10.1136/bmj.i3323.
- Grandjean PA, Mortimer JT. Recruitment properties of monopolar and bipolar epimysial electrodes. Ann Biomed Eng. 1986;14(1):53–66. doi: 10.1007/BF02364648.
- Loeb GE. Galvani’s delayed legacy: neuromuscular electrical stimulation. Expert Rev Med Devices. 2005;2(4):379–381. doi: 10.1586/17434440.2.4.379.
- Rasmussen K, Bero L, Redberg R, et al. Collaboration between academics and industry in clinical trials: cross sectional study of publications and survey of lead academic authors. BMJ. 2018;363:k3654. doi: 10.1136/bmj.k3654.
- Euromonitor International. Global Overview of the Pharmaceuticals and Medical Equipment Industry. 2022.
- Niemiec E. Will the EU medical device regulation help to improve the safety and performance of medical AI devices? Digit Health. 2022;8:20552076221089079. doi: 10.1177/20552076221089079.
- Garzotto F, Comoretto RI, Dorigo L, et al. Preparing healthcare, academic institutions, and notified bodies for their involvement in the innovation of medical devices under the new european regulation. Expert Rev Med Devices. 2022;19(8):613–621. doi: 10.1080/17434440.2022.2118046.
- Han JED, Ibrahim H, Aiyegbusi OL, et al. Opportunities and risks of UK medical device reform. Ther Innov Regul Sci. 2022;56(4):596–606. doi: 10.1007/s43441-022-00394-0.
- Yan D, Vassar R. Neuromuscular electrical stimulation for motor recovery in pediatric neurological conditions: a scoping review. Dev Med Child Neurol. 2021;63(12):1394–1401. doi: 10.1111/dmcn.14974.
- Centers for Medicare & Medicaid Services. NCD - Neuromuscular Electrical Stimulation (NMES) (160.12) [Internet]. 2006 [cited 2023 Feb 18]. Available from: https://www.cms.gov/medicare-coverage-database/view/ncd.aspx?ncdid=175&ncdver=2
- Kirisits A, Redekop WK. The economic evaluation of medical devices. Appl Health Econ Health Policy. 2013;11(1):15–26. doi: 10.1007/s40258-012-0006-9.
- Ramsey SD, Luce BR, Deyo R, et al. The limited state of technology assessment for medical devices: facing the issues. Am J Manag Care. 1998;4: SP188–99.
- Bown CP, Bollyky TJ. How COVID‐19 vaccine supply chains emerged in the midst of a pandemic. World Econ. 2022;45(2):468–522. doi: 10.1111/twec.13183.
- World Health Organization. Shortage of personal protective equipment endangering health workers worldwide [Internet]. 2020. Available from: https://www.who.int/news/item/03-03-2020-shortage-of-personal-protective-equipment-endangering-health-workers-worldwide
- Rubin JP, Neligan P. Plastic surgery. In Aesthetic surgery. Vol. 2. Amsterdam: Elsevier; 2018.
- Alamer A, Melese H, Nigussie F. Effectiveness of neuromuscular electrical stimulation on Post-Stroke dysphagia: a systematic review of randomized controlled trials. Clin Interv Aging. 2020;15:1521–1531. doi: 10.2147/CIA.S262596.
- Kristensen MGH, Busk H, Wienecke T. Neuromuscular electrical stimulation improves activities of daily living Post-Stroke: a systematic review and meta-analysis. Arch Rehabil Res Clin Transl. 2022;4(1):100167. doi: 10.1016/j.arrct.2021.100167.
- Mäkelä E, Venesvirta H, Ilves M, et al. Facial muscle reanimation by transcutaneous electrical stimulation for peripheral facial nerve palsy. J Med Eng Technol. 2019;43(3):155–164. doi: 10.1080/03091902.2019.1637470.
- Marotta N, Demeco A, Inzitari MT, et al. Neuromuscular electrical stimulation and shortwave diathermy in unrecovered bell palsy. Medicine (Baltimore). 2020;99(8):e19152. doi: 10.1097/MD.0000000000019152.
- McGettigan P, Golden J, Fryer J, et al. Prescribers prefer people: the sources of information used by doctors for prescribing suggest that the medium is more important than the message. Br J Clin Pharmacol. 2001;51(2):184–189. doi: 10.1111/j.1365-2125.2001.01332.x.
- Lublóy Á. Factors affecting the uptake of new medicines: a systematic literature review. BMC Health Serv Res. 2014;14:469. doi: 10.1186/1472-6963-14-469.