
ABSTRACT
Introduction: The incidence of kidney dysfunction increases with age and is highly prevalent among patients with hypertension. Since many therapeutic compounds are primarily eliminated through the kidneys, impaired renal function can have negative consequences on drug disposition, efficacy and safety. Therefore, regulatory agencies such as the Food and Drug Administration (FDA) and European Medicines Agency (EMA) have issued detailed guidelines for new drug applications to determine posology requirements for patients with renal impairment.
Areas covered: The current review highlights and contrasts agency requirements for pharmacokinetic renal impairment clinical studies. While many of the guidelines are similar among the two agencies, glomerular filtration rate (GFR) determination and reporting differ. Design considerations for a reduced, full or dialysis renal impairment study, as well as modifications to the FDA’s draft guidance are discussed. Furthermore, scenarios where pharmacokinetic modelling analysis can benefit a drug development program are also reviewed. Moreover, practical solutions for patient recruitment challenges are addressed.
Expert commentary: We summarize how ‘one size does not fit all’ for GFR assessment, and recommend when to use certain modalities. Finally, we highlight the need for the pharmaceutical industry to engage therapeutic experts to assist in protocol development for renal impairment studies, as these experts understand the nuances of this special population and recommended guidelines.
1. Introduction
The kidney is involved in a number of physiological functions, such as body fluid and electrolyte balance, release of hormones that regulate blood pressure and red blood cell production, as well as the removal of waste products from the body including drugs. The kidney is a resilient organ and is able to adapt to injury, however chronic insults such as hypertension, atherosclerosis, diabetes, and smoking, as well as inherited diseases can contribute to renal dysfunction. To this end, an estimated 10% of the world population is affected by chronic kidney disease (CKD) [Citation1]. Reduced glomerular filtration rate (GFR) is the hallmark of CKD, and the degree of renal impairment based on GFR is categorized as mild, moderate, severe and end-stage renal disease (ESRD) (see ). In addition, normal aging can also impair renal function with a decline in both the upper and lower reference limit for GFR.
Table 1. Renal function stages.
As the kidneys represent a major organ for the elimination of many drugs, impairment of renal function can alter the pharmacokinetics of drugs and consequently, systemic exposure. In general, most polar compounds and those not bound to plasma proteins tend to undergo glomerular filtration or tubular secretion. Kidney damage can disturb glomerular impermeability of proteins, drugs and metabolites. Therefore, patients with impaired kidney function may be at significant risk of prolonged drug exposure or toxic effects as a result of impaired renal clearance mechanisms. Illustration for the need of dose adjustment in a renal impaired population is evidenced by elevated systemic exposure through increases in the pharmacokinetic parameter area-under-the curve (AUC) [Citation4]. A main factor influencing systemic exposure is the percent of the drug that remains unchanged (i.e. not metabolized). For instance, if a drug has high renal clearance as a percentage of total body clearance in an unchanged form, then mild or moderate renal impairment could alter AUC by twofold or more [Citation5,Citation6]. Conversely, where the percent unchanged drug (or active metabolite) excreted in urine is low, then severe renal impairment may have no effect [Citation7]. In addition, alterations in drug absorption, bioavailability, and plasma protein binding capacity can also occur in a renal impaired population (review in [Citation8]), further affecting drug exposure. Moreover, renal impairment can also influence hepatic metabolism of certain drugs by modifying CYP enzymes activity and transporter function [Citation4,Citation9,Citation10]. Therefore, it is imperative that new drug applications (NDA) include information on the impact of renal clearance in terms of total drug clearance and whether posology adjustments are required for patients with impaired renal function [Citation10].
Both the US FDA and European Medicines Agency (EMA) have issued detailed guidelines outlining when and how a pharmacokinetic renal impairment study should be included as part of a NDA. The FDA released draft guidelines in 2010 [Citation2], and an advisory committee meeting proposed several recommendations to this guidance that same year [Citation11]. Meanwhile, the EMA published full guidelines in 2016 [Citation3]. The aim of this review is to compare and contrast current FDA and EMA guidelines regarding renal impairment study design and key study considerations as well as provide practical solutions to study challenges based on prior experience.
2. Examining renal function for drug labeling requirements
The goal of a pharmacokinetic renal impairment study is to estimate the impact of varying degrees of renal impairment on the systemic availability (as typically measured by AUC and maximal plasma concentration [Cmax]) of the drug and/or relevant metabolites. Where significant alterations in AUC and Cmax are detected, a recommendation for dose adjustment may be necessary in one or more stages of renal impairment. Conversely, if a degree of impairment has not been studied then a warning that there is no information to guide a dosage adjustment recommendation in said population may be required in the drug label. For example, this type of warning was issued for TagrissoTM (osimertinib) [Citation12], a non-small cell lung cancer tyrosine kinase inhibitor.
2.1. When and when not renal impairment studies may be necessary
Both the FDA and EMA guidelines outline when a renal impairment study may be required for market registration (NDA or Marketing Authorization Application), and when these analyses are not compulsory or practical. compares guideline recommendations regarding the utility of a renal impairment study from both agencies.
Table 2. Summary of agency guidelines necessitating a renal impairment study.
If the kidney is the major organ for drug or metabolite elimination, then a renal impairment study will likely be deemed necessary. Renal elimination of a drug is defined when the compound or principal active metabolite in the urine represents ≥30% of the administered dose. In an ADME (absorptivity, distribution, metabolism, excretion) study, this is referred to as fe, the percentage of drug excreted unchanged in urine. A well-known example of a renal eliminated drug is the diabetes medication sitaglipitin. Sitagliptin is a dipeptidyl peptidase-4 (DPP-4) inhibitor which promotes incretin function and insulin sensitivity. Findings from an ADME study indicate 80% of sitagliptin is excreted in urine [Citation4], therefore warranting further investigation in a renal impairment population, and dose adjustment instructions.
Drugs intended for chronic use in CKD patients would ultimately require a pharmacokinetic renal impaired study. Since CKD is associated with hypertension, blood pressure medications are an example of a drug class necessitating pharmacokinetic evaluation with renal impaired subjects. Furthermore, compounds that may be administered acutely to renal impaired subjects may also require pharmacokinetic evaluation to determine specific dosing information. Examples of acute therapeutic agents could include antibiotics, heart-burn or motion-sickness medication. Even if there is likely little or no effect on a drug’s pharmacokinetic profile in a renal impaired population, special consideration should be given for patients on dialysis. The dialysis process may remove certain drugs, therefore dose adjustment could be required. Further discussion on the effect of dialysis in renal impairment studies is addressed in Section 3.3.
Although drugs can undergo nonrenal mechanisms of elimination such as biliary excretion of hepatically metabolized compounds, some metabolites can potentially be reabsorbed in a process termed enterohepatic cycling, and may ultimately be eliminated in urine. Therefore, both regulatory agencies require that most drugs intended for chronic or repeat use undergo evaluation in renal impaired cohort(s), whether renal or nonrenal elimination mechanisms predominate [Citation2,Citation3]. To highlight this point, an FDA survey of NDAs found that approximately 25% of compounds not primarily eliminated via the kidney demonstrated a twofold or greater increase in plasma concentration AUC in patients with renal dysfunction [Citation4]. Rosuvastatin, a cholesterol-lowering agent, is one such example. Although only 6% of rosuvastatin is eliminated via urine, plasma concentrations were reported to be increased threefold in patients with severe renal impairment, requiring dose adjustment [Citation4].
Many therapeutic proteins undergo rapid renal clearance [Citation13], necessitating investigation in a renal impaired population. As such, the FDA has suggest investigation of renal impairment for cytokines <69kDa, whereas the EMA has suggested evaluation for protein–drug conjugates, which can potentially be impacted by renal impairment [Citation2,Citation3]. Conjugating biomolecules with a polyethylene glycol (PEG) moiety, termed PEGylation, is one approach to increase the hydrodynamic radius and size (molecular weight), extending the drugs circulating half-life. A 10kDa PEG can increase the structural radius of a molecule akin to a 65.4 kDa protein [Citation14]. A prime example is filgrastrim, a recombinant granulocyte-colony stimulating factor (G-CSF), which stimulates neutrophil proliferation during myelosuppression due to chemotherapy. Filgrastrim (19 kDa) has a short half-life of approximately 4 h due to extensive renal clearance [Citation15], requiring daily administration. On the other hand, its PEGylated form, pegfilgrastrim (39 kDa, Neulasta®), is only administered once per chemotherapy cycle [Citation16]. Pegfilgrastim clearance is predominantly nonrenal and was demonstrated to not require dose adjustment in renal impaired subjects [Citation17]. Furthermore, unlike smaller biological agents, monoclonal antibodies are too large for glomerular filtration and are typically eliminated through proteolytic degradation or target-mediated process [Citation18].
Both agencies agree there are certain situations when a pharmacokinetic renal impairment study may not be required () [Citation2,Citation3]. These circumstances include drugs that are gaseous or volatile in nature and excreted by the lungs. Single-dose administration of a drug may also negate the need for a renal study if (pre)clinical safety and elimination findings support no effect on kidney function.
The EMA guidelines also include stipulations for drugs without relevant systemic absorption (i.e. topically administered) as well as those hepatically eliminated with supporting safety and clinical data indicating dose adjustments are not necessary even at a high exposure threshold [Citation3].
2.2. Role of modeling in lieu of renal impairment studies
In certain drug development scenarios, the conduct of a dedicated phase I renal impairment investigation with otherwise healthy, renal impaired subjects and healthy-matched control subjects is deemed not feasible; perhaps due to safety concerns of administering the investigational compound to healthy individuals. Moreover, for drugs classified as breakthrough therapies, the urgency in advancing the drug through clinical development may warrant by-passing or deferral of a dedicated renal impairment study. In such cases, modeling and simulation techniques may be useful to guide dosing recommendations in the target population with comorbid renal impairment. Common modeling and simulation methods can be broadly classified as top-down or bottom-up methods. Top-down methods rely on modeling of some available data, whereas bottom-up approaches take advantage of prior knowledge of anatomy and physiology of the biological system along with biochemical processes and physicochemical properties of the drug being studied. Both approaches have advantages and limitations, which to describe in detail, are outside the scope of the current review. However, the applications for each approach are not mutually exclusive in that they can be employed in a complementary manner throughout the drug development life-cycle.
Population pharmacokinetic analysis, a bottom-down approach, employing nonlinear mixed-effect modeling techniques, allows for the potential to estimate the effect of predictor variable(s) on important pharmacokinetic parameters such as total systemic clearance. In the context of renal impairment studies, the impact of renal function can be tested as a significant covariate on clearance and when sufficient data is available, the effect can be quantified at the different stages of degrees of renal function.
For axitinib (Inlyta®), an advanced kidney cancer medication, no dedicated renal impairment trial was performed to support the initial approval by the FDA. To address the objective of testing the impact of renal impairment on drug clearance, a population pharmacokinetic analysis was performed on data obtained in patients with a range of renal function. In this particular case, no significant difference in axitinib clearance was observed in patients with preexisting mild to severe renal impairment [Citation19,Citation20].
Similarly, no dedicated renal impairment study for XALKORI (crizotinib), a small-molecule kinase inhibitor for advanced non-small cell lung cancer, was performed to support the dosing information for renal impaired patients [Citation21]. Rather, as part of the clinical pharmacology assessment, the FDA reviewer(s) conducted an analysis relating trough concentrations in patients treated with crizotinib to measures of renal function (i.e. creatinine clearance [CLcr], see Section 5.2). Regression and categorical analysis were presented in the summary basis of approval with the clinical pharmacology and biopharmaceutics review demonstrating that steady state trough concentrations in patients with mild (CLcr 60–90 mL/min, N = 47) and moderate renal impairment (CLcr 30–60 mL/min, N = 27) were similar to those in patients with normal renal function (CLcr >90 mL/min, N = 33) [Citation22]. Since limited data (N = 1) were available in patients with severe renal impairment, and no data were available in patients with end-stage renal disease, no specific dosing recommendation was made for these degrees of impairment [Citation22].
As a bottom-up approach, physiologically based pharmacokinetic (PBPK) modeling tools can also be leveraged to inform the impact of varying degrees of renal impairment on systemic exposure of renal-cleared drugs. PBPK offers a mechanistic means of accounting for changes in physiologic and anatomic processes associated with normal development and pathophysiologic changes. Within the last year, the EMA has published a draft guideline on the qualification and reporting of PBPK modeling and simulation for application in drug–drug interactions and pediatric drug development [Citation23]. Similarly, reviews by Zhao et al. [Citation24], Wagner et al. [Citation25] and Jamei [Citation26] illustrate the contribution of PBPK modeling and simulation in several FDA approvals for a variety of novel compounds. Although to date, no specific waiver of a dedicated renal impairment study based on a PBPK model has been reported, the following case is presented to illustrate the value of mechanistic modeling to inform clinical development in patients with comorbid renal impairment.
3. Types of studies
There are three general protocol designs for a renal impairment trial; a reduced, full or dialysis study. The decision to proceed with one schematic over the other can depend upon the degree of renal clearance, pharmacokinetic impact in a group of subjects with renal dysfunction as well as the safety profile of a drug. A reduced study design is intended to evaluate the initial pharmacokinetic effects in subjects at the extremes of renal function, and determine if a full study scheme is necessary. The reduced model may be considered when a drug is predominately eliminated through nonrenal processes [Citation2,Citation3], i.e. hepatic metabolism or biliary excretion. A full study design is mandatory when renal impairment is anticipated to result in clinically relevant increases in drug exposure [Citation3]; or if a reduced study demonstrated over 50% increase in systemic exposure in ESRD or severe renal impaired subjects; or if an exposure-response relationship indicate the need for further analysis [Citation2]. In addition, for safety reasons, drugs that may cause hemodynamic instability in severely impaired renal subjects may justify evaluation in a full study model in subjects with mild to moderate renal impairment. A dialysis study is intended to examine the amount of drug removed by the dialysis process, as well as the pharmacokinetic and safety profile of the treatments in ESRD patients.
3.1. Reduced study design
The reduced study is often referred to as a ‘worst case scenario’ since it compares the pharmacokinetic profile in patients with little to no renal function with a control group. Renal function is based on GFR and the modality of measurement varies between the agencies (). Operational definitions of renal function by agency are further addressed in Section 5. The reduced design includes subjects with low GFR, typically ESRD patients not yet on dialysis as well as a suitable control group. The controls should represent the intended (phase III) population with ‘normal’ renal function (see Section 6.5 for more details on control subjects). For example, a drug developed for Alzheimer treatment should be evaluated in a cohort with advanced age. However, since GFR declines by ~7.5 mL/min per decade of life [Citation27], an elderly control group would not reflect a normal renal function reference value (GFR ≥90 ml/min). Therefore, depending on the intended study population, subjects with slightly reduced renal function (GFR ≥75 ml/min) may comprise a study’s control group [Citation28].
One main challenge facing the reduced study design is the recruitment of ESRD patients not yet on dialysis. This group typically reflects a transition state before being placed on dialysis or requiring kidney transplant, and represents a very small subgroup of CKD patients. A Veteran’s Affair survey found that only 0.11% of CKD patients fit these ESRD criteria [Citation1]. To address this concern, the EMA guidelines propose the enrollment of patients with as low GFR as possible (preferably not greater than 20 mL/min) but not requiring dialysis [Citation3].
The FDA has also modified their definition of ESRD from GFR <15 to GFR <30 mL/min. This position was discussed during an advisory committee meeting for pharmaceutical science and clinical pharmacology in March 2010 [Citation11]. Recommendations to the draft guidelines included the engagement of dialysis patients on nondialysis days or patients with GFR <30 mL/min (severe renal impairment) for a reduced study design, since from a clinical practice point-of-view, a patient presenting with a GFR <30 mL/min would be recommended to begin dialysis [Citation11]. To this end, Zhang et al. suggests the recruitment of ESRD patients on nondialysis days for renal cleared drugs and patients with severe kidney impairment for drugs with a nonrenal clearance [Citation29]. If results from a reduced study show no pharmacokinetic differences among the ESRD/severely decreased GFR patients and control group, no further action is required [Citation2,Citation3].
3.2. Full study design
A full pharmacokinetic study design includes evaluating patients with various stages of renal dysfunction (). The FDA recommends the same control group as the reduced study to be used in the full trial [Citation2]. If this is not feasible, then control subjects for the full study should display similar renal function as well as intrinsic factors such as gender, age, ethnicity, body composition as the reduced study reference group. The full study should enroll approximately equal number of subjects per stage and subjects should be comparable in these intrinsic factors. While the EMA suggests 6–8 subjects per group [Citation3], the FDA draft guidance does not provide an exact number of renal impaired patients needed for such a study, however the sample size must be sufficient to determine a meaningful pharmacokinetic difference between patients and controls [Citation2].
3.3. Dialysis studies
In clinical practice, the majority of ESRD patients are on dialysis. Hemodialysis (HD) transfers uremic solutes from blood into a dialysate, a fluid solution housed outside of the body, by pumping blood through a semi-permeable membrane. Typically, this process takes 2–4 h and is performed up to three times per week. Ultimately, drugs that will be administered to ESRD patients on dialysis should undergo a dialysis study. The dialysis process may remove a drug or metabolite affecting pharmacokinetic and drug efficacy, therefore dose adjustment may be needed such as administration of a supplemental dose. In addition, a pharmacokinetic dialysis study may provide valuable information regarding dialysis treatment for drug overdose. The agency guidelines specify a dialysis pharmacokinetic study may not be required if the dialysis process is not likely to remove the drug [Citation2,Citation3]. This would be the case for drugs with a large molecular weight and those tightly bound to plasma proteins. A dialysis study may also be omitted if the process would result in <10% of drug or active metabolite elimination; this can occur with drugs not renal cleared or if the volume of distribution is >360 L [Citation2].
3.3.1. Study design and sample collection
If a dialysis study is deemed necessary, then pharmacokinetic analysis should be conducted on both nondialysis and dialysis days. One strategy is to dose patients immediately following their normal scheduled dialysis (for nondialysis assessment), and just prior to initiating the dialysis process [Citation30]. Since intermittent HD using a high-flux dialyzer is the most common method used for ESRD patients, this modality is highly recommended for pharmacokinetic studies. To examine drug clearance during dialysis; blood samples before and during the dialysis process as well as the dialysate are collected to determine drug concentration. During dialysis, both arterial and venous sides of the dialyzer should be sampled at a given interval. According to the FDA guidelines, plasma protein drug binding should also be examined in pre- and post-dialysis blood samples. The fraction of the drug recovered in the dialysate can be calculated to determine the need for supplement drug dose administration to HD patients [Citation2]. Data collection should include blood flow rates and dialysate flow rate as well as the make and model of the dialyzer. If different dialyzer models are used for a study, results can be standardized using the Renkin equation [Citation31].
As HD can result in rapid changes in fluid balance, peritoneal dialysis and continuous renal replacement therapy (CRRT) are alternative dialysis methods in patients with unstable hemodynamics. Peritoneal dialysis employs the same principles as HD yet a sterile cleansing fluid is used for diffusion and the peritoneum lining of the peritoneal cavity acts as a membrane. CRRT is typically deployed for critically ill patients with acute kidney injury, in a hospital setting. Extrapolation of HD results onto these other dialysis modalities can be difficult therefore, peritoneal dialysis and CRRT pharmacokinetic studies might be considered if the drug is likely to be influenced by these processes and administered to patients on these regimens.
4. Dose selection and regimen
For safety purposes, it is recommended that the lowest effective dose be administered for renal impairment studies. Since the drug to be evaluated in the renal impaired patient population is not intended to treat the renal patient, other factors to consider include comorbidities, drug-drug interaction and drug safety limits as well as bioanalytical method limitations (e.g. limits of detection).
The majority of reduced or full studies will administer a single dose of the investigational product. Prior findings must support that a single dose accurately reflects the drug’s pharmacokinetic profile. This is often observed when a parent drug or metabolite displays a linear and time-independent profile. For compounds with a nonlinear or time-dependent pharmacokinetics, evaluating following multiple doses may be required. Multiple-dose studies should be of sufficient duration to achieve steady state. A loading dose strategy might be considered to expedite reaching steady state if the elimination half-life is relatively long, necessitating repeat dosing for several weeks. However, if a drug with a long half-life can achieve clinically relevant concentrations following single dose administration, administering a loading doses or assessing the pharmacokinetic impact at steady-state may not be necessary [Citation32]. In addition, a loading dose could be considered for drugs intended to be administered as an IV infusion [Citation33]. Moreover, lower doses or less frequent dosing may be an important consideration for the renal impaired patient to avoid drug or metabolite accumulation and potential toxic side-effects during a reduced or full study.
5. Operational definition of renal impairment
GFR is an ideal index to assess kidney function as it calculates nephron plasma volume filtration per unit time during urine formation (milliliters per minute). Reference GFR ranges are based on BSA-normalization and are approximately 105 mL/min per 1.73 m2 [Citation34]. A key difference between the two agency guidelines is their requirements for how GFR is determined. The EMA guidelines recommend that for the classification of renal function in clinical studies, an exogenous marker (ex. inulin) be used to assess GFR. Conversely, the FDA acknowledge that exogenous methods for GFR determination are not usually employed or available in a clinical research setting, and therefore allows for estimates of GFR (eGFR) based on endogenous markers.
5.1. Inulin clearance
Inulin clearance is often considered the gold standard for assessment of renal excretion. Inulin is an inert polysaccharide exclusively eliminated by glomerular filtration. For GFR assays, inulin or sinistrin (a more water soluble analog) is continuously infused intravenously while timed urine samples are collected. Although heralded as the reference standard, this assessment is not particularly practical for patients, therefore variations in the procedure were developed to remove the need for multiple urine samples to assess systemic inulin clearance in plasma. Unfortunately, these results showed poor correlation to the reference method [Citation35]. Moreover, the inulin methodology is associated with other drawbacks including the feasibility of acquiring inulin or sinistrin, a lengthy infusion period, and although generally considered safe, one case of anaphylaxis was observed upon sinistrin infusion [Citation36].
Other exogenous GFR markers include radiolabeled 51Cr-EDTA, 99mTc-DTPA or 125I-iothalamate, as well as nonradiolabeled contrast agents. The strengths and limitations of these measured GFR approaches are summarized elsewhere (reviewed in [Citation37–Citation39]). The chief criticism to these direct methodologies is that they are expensive and time-consuming. Another disadvantage is the discrepancy in results between exogenous measurements and estimations of GFR based on endogenous metabolites at baseline and end of study respectively.
5.2. Serum creatinine
Phosphocreatine is metabolized in the muscle to generate creatinine at a fairly constant rate. Creatinine is freely filtered by the glomerulus and is completely cleared by renal excretion. Approximately 10–20% of excreted creatinine is secreted by the proximal tubules, which can overestimate GFR when measured by creatinine clearance (in urine). Before the used of isotope dilution mass spectrophotometry (IDMS) calibration, the creatinine measurement was commonly afflicted by up to 20% assay error [Citation34]. In addition, assay interferences such as bilirubin, glucose and uric acid are also known. Furthermore, serum creatinine is influenced by gender, age, ethnicity, muscle mass and dietary protein intake, factors which can affect assay accuracy. Therefore, serum creatinine alone is not recommended for diagnosis or to evaluate kidney dysfunction. However, mathematical formulas have been developed to overcome these compounding factors and improve eGFR accuracy.
The Cockcroft-Gault (CG) equation was first described in 1976 [Citation40] and was historically used in drug labeling to provide guidance on dose adjustment for the renal impaired patient. CG adjusts for age, weight and gender according to the following formula:
Since minor increases in serum creatinine concentrations can result in an underestimation of GFR, an alternative formula was proposed. The Modification of Diet in Renal Disease (MDRD) eGFR formula was developed to adjust for body surface area and ethnicity, as well as considerations such as age and gender [Citation41].
In many ways the MDRD is superior to the CG calculation as it is more precise and reliable in predicting GFR (reviewed in [Citation42]). However, the MDRD estimation holds limited accuracy for CKD patients with a normal serum creatinine level as well as those with advanced renal failure [Citation42].
Despite these limitations, the majority of CKD patients maintain the same recommended drug dose adjustments regardless of the equation used to estimate kidney function. Parks et al. compared the performance of MDRD to CG for classification of renal function [Citation43]. Their analysis revealed that only 12% of subjects requiring dose adjustment differed by a higher or lower dosing category with MDRD compared to the CG estimation. Variables such as low body weight, advanced age and mild serum creatinine elevation were all factors for the discordant dose category [Citation43]. Although kidney disease research initiatives such as KDIGO (Kidney Disease: Improving Global Outcomes) are promoting eGFR calculation using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation [Citation44], at this time neither agency provides guidance regarding the use of this formula.
5.3. Urine creatinine clearance
Twenty-four hour urine creatinine clearance rates are typically used in medical settings and not clinical research since this method tends to overestimate GFR due to creatinine secretion in the proximal renal tubules. This eGFR assessment is subject to urine collection alone errors, diurnal variation in creatinine concentration and day-to-day changes in creatinine excretion. Therefore, only under certain circumstances can 24-hour creatinine urine collection be assessed for clinical research studies. The FDA grants limited use of this measurement only for conditions that can influence creatinine levels in diet such as a vegetarian diet or creatinine supplementation, or factors affecting muscle mass (muscle wasting, amputation) [Citation2].
5.4. Agency guidelines for reporting GFR
Renal impairment must be defined by an accurate (exogenous) method for consideration by the EMA. Estimated GRF should also be presented from results obtained during a pharmacokinetic study from serum creatinine or creatinine clearance measurements. The EMA guidelines indicate that all GFR values should be in absolute units, which are not adjusted for body surface area (1.73 m2). While either equation can be used to categorize the stages of renal impairment, current FDA guidelines suggest that both CLcr and MDRD eGFR be calculated [Citation2]. In addition, the FDA draft guidelines also recommend that urine and plasma estimates of creatinine clearance results be reported.
6. Subject inclusion/exclusion criteria
Complications due to diabetes, hypertension, renal inflammation (glomerulnephritis) or inherited diseases such as polycystic kidney disease or Alport syndrome are all associated with renal dysfunction. Hypertension and diabetes are highly prevalent in a CKD population. A 2012 survey found that approximately 61% and 36% of CKD patients >60 years of age reported these comorbidities, respectively [Citation1]. Therefore, study inclusion criteria should reflect these conditions with allowances for high blood pressure limits and elevated HbA1c levels, as well as concomitant medications. In contrast, other complications associated with CKD such as anemia, malnutrition, bone disease, and neuropathy are typically exclusionary for pharmacokinetic studies. CKD is medically defined as renal damage or reduced GFR over 3 months or more [Citation34]. For most clinical studies, CKD patients should display stable renal function with no clinically significant change in status at least 1 month prior to study initiation. GFR inclusion criterion is typically based on the mean of two measurements at baseline usually 14 days to 2 months apart. In addition, inclusion/exclusion criteria related to age, weight, cardiodynamics and smoking status can substantially differ from a typical health participant study.
6.1. Age
The average age range for CKD patient enrollment in a pharmacokinetic renal impairment study is typically between 18 and 80 years old. With age, the number of viable nephrons gradually declines and is paralleled by an increased incidence of kidney damage such as nephrosclerosis and glomerulosclerosis [Citation45]. Therefore, it is estimated that half of adults >70 years displayed age-related loss in renal function, associated with eGFR <60 mL/min resulting in CKD diagnosis [Citation46]. In subjects with advanced age, overestimation of eGFR as a result of declining serum creatinine, due to age-related loss of muscle mass, can be overcome by standardizing GFR to metabolic rate rather than body surface area [Citation47]. However, this approach is not addressed in either FDA or EMA guidelines.
6.2. Weight
Body mass index (BMI) is a measure of weight relative to height, and is usually preferred over weight alone as an inclusion/exclusion criterion for many clinical studies. Obesity (>30 kg/m2) and a higher BMI are considered independent risk factors for CKD and progression to ESRD requiring dialysis [Citation48–Citation51]. Therefore, a BMI range between 18 and 40 kg/m2 is typical for CKD patient enrollment in a renal impairment study. Since weight is a component of CG eGFR, for subjects with elevated BMI the use of lean body weight is preferred over actual body weight to improve accuracy and reduce bias [Citation43], or an alternative approach is to determine eGFR with the MDRD model.
6.3. Cardiac function
Abnormal measures of cardiac function in a CKD cohort are thought to be related to the increased age of this population and the negative impact of years of hypertension. It is not uncommon to detect prolonged QT, QRS, and PR intervals during electrocardiogram readings in patients with renal dysfunction. Furthermore, corrected QT (QTc) prolongation significantly increases across disease stage severity [Citation52]. In addition, the hemodialysis process can also contribute to increased QTc intervals in CKD patients associated with changes in serum potassium and calcium levels [Citation53]. Therefore, accommodations for baseline QTc >450 ms may be warranted for a renal impairment clinical study, if deemed safe by the principle investigator and medical monitor.
6.4. Smoking status
Tobacco smoke elevates blood pressure [Citation54], increases urine albumin excretion [Citation55] and negatively impacts renal hemodynamics [Citation56]. Approximately 12% of Stage 4 CKD patients in the US are current smokers, and this value almost triples for Stage 1 CKD patients [Citation1]. Furthermore, within distinct ethnic groups, smoking tobacco has been implicated as an independent risk factor for the development and progression of CKD [Citation49,Citation57,Citation58]. From a pharmacology perspective, tobacco smoke induces several hepatic drug metabolizing enzyme isoforms such as CYP1A1, CYP1A2, and CYP2E1 [Citation59,Citation60], and therefore may have an effect on pharmacokinetic profiles. CYP modulation is not just limited to tobacco use, as marijuana consumption is also implicated in altering CYP activity. For instance, cannabidiol, a component of marijuana, is a potent inhibitor of CYP2D6 [Citation61]. Although CYP2D6 represents approximately 2% of total hepatic P450 content [Citation62], it is responsible for the metabolism of a number of drug classes [Citation63]. As the legalization of marijuana continues to expand, this will become a relevant issue for clinical research. Therefore, inclusion/exclusion criterion for smoking (tobacco and/or marijuana) status is an important consideration for clinical studies.
6.5. Healthy control matching strategies
Regardless of the design employed (i.e. reduced or full), both EMA and FDA guidelines require pharmacokinetic exposure comparison between the varying degrees of renal impairment to control subjects. The matched control group should exhibit renal function consistent with the typical patient population for whom the drug in question is likely to be prescribed (e.g. elderly cohort for Alzheimer medication), and are similar in terms of demographic and anthropometric characteristics. Two commonly applied matching schemes are the 1:1 pairing [Citation30] and matching to the reference group mean [Citation64,Citation65] ().
Figure 1. Methods for assignment of matching healthy controls in renal impaired pharmacokinetic studies.
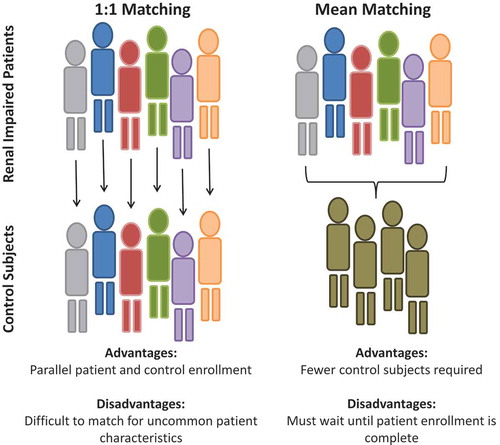
6.5.1. One-to-one matching
Matching 1:1 implies each renal impaired subject recruited into the trial is paired with a corresponding control subject with the same gender. With the impaired subject treated as the reference subject, the control is matched for demographic and anthropometric measures such as race, age, body weight and/or BMI to be within some reasonable range of the reference individual. Neither the EMA nor FDA guidelines codify a particular range for age and weight; however, our experience matching controls to the reference impaired age ±10 years and BMI ±5–20% is reasonable to recruit and have supported labeling [Citation32,Citation66] Use of tobacco products, when permitted by protocol, should also be considered when matching control subjects to impaired, as smoking in particular, has been demonstrated to influence GFR in both healthy and renal impaired individuals, as address in Section 6.4.
One-to-one matching also provides an operational benefit in that, renal impaired and paired control subjects can be recruited in parallel as some sites employ a priori matching of control and impaired subjects within the recruiting database. This method allows for timelier enrollment and completion of the trial. A limitation of this approach is when an impaired subject presents with intrinsic characteristics which are inherently difficult to match (e.g. an uncommon race or ethnicity for the study center’s typical population), potentially leading to a delay in enrollment and study completion.
6.5.2. Mean matching
Matching to the reference group mean, entails awaiting completion of one or more impaired cohorts and enrolling matched control subjects which fall within a prespecified range relative to the group for one or more demographic and/or anthropometric characteristics.
Ultimately this approach is intended to spare the number of control match subjects required and in the full-study design (including dialysis cohorts). This approach could result in a net reduction of up to 24 subjects, and thus have a significant impact on the total cost of the study. However, enrollment of matched controls can only occur after the completion of the impaired cohorts to calculate the mean and corresponding range. In a reduced design, mean matching poses a challenge in that the control subjects can only be matched to the completed impaired cohorts. As described earlier, full designs are typically conducted in a sequential manner and conducting a subsequent impaired cohort could result in control subjects falling outside the prespecified range when the two impaired cohorts are pooled. Enrolling further control subjects with demographic and anthropometric factors closer to the mean of the pooled impaired group is one way to overcome this limitation.
7. Conclusion
The current draft FDA [Citation2] and full EMA [Citation3] guidelines describe how pharmaceutical companies should develop posology instructions for renal impaired subjects. While the guidelines are quite similar, agency-specific requirements such as methodology for GFR assessment differ. Furthermore, the FDA has modified their definition of ESRD to meet the growing challenges of ‘worst case scenario’ patient identification and recruitment [Citation11]. It is anticipated that the FDA full guidelines will include this new position and will address the use of updated GFR estimates (e.g. CKD-EPI), currently recommended by numerous kidney research organizations and initiatives.
8. Expert commentary
The methods of assessing renal function for renal pharmacokinetic studies have substantially changed over the last 20 years as seen by the evolution from 24-h urine collection for creatinine clearance to Cockroft-Gault formula, and now MDRD (with a small number of physicians applying the CKD-EPI equation, which yields similar results to MDRD). A weakness in CG is that it can overestimate GFR and hence misclassify ‘true’ severe renal impaired subjects as moderate subjects. While MDRD is most accurate to assess ‘true’ moderate and severe subjects, it is not as sensitive in identifying healthy subjects, especially >80 or 90 mL/min. Normal renal function was typically considered >80 mL/min; however, since KDOQI staging renal function into five categories [Citation67], normal has been pushed up to >90 mL/min making it considerably more difficult to find matching controls to the older, heavier renal impaired subjects. Since the goal of these studies is to address posology in moderate or severe renal impaired subjects, our opinion is the preferential use of MDRD. Since most physicians in practice and labs commonly report renal function as eGFR (from MDRD), use of this formula is easy to implement to facilitate proper dosage adjustment for their patients.
For the reduced renal study design, the FDA considers both ESRD on HD as well as severe and ESRD not on HD as acceptable representatives of a worst case scenario [Citation11]. From our experience conducting and reviewing the data from renal pharmacokinetic studies, it seems that both groups show similar results; sometimes the ESRD subjects on HD are slightly worse, and other times the severe or ESRD patients not on HD are slightly worse. In practice, we prefer the reduced study design to enroll ESRD patients on HD, as a larger pool of subjects are available and the subjects tend to be younger and overall healthier with minimal concomitant diseases. In addition, vital HD data can be obtained to help support whether HD removes a drug as an overdose treatment option. Furthermore, there is less risk of the investigational product worsening renal function in this population, as this is a concern with severe and ESRD patients not yet on HD. The scarcity of severe and ESRD subjects not on HD, often leads to recruitment of older and more fragile subjects, thereby also contributing to an increase in risk.
Another issue in the current state of practice is the lack of industry awareness regarding appropriate study design and subject criteria. As a result, the inclusion and exclusion criteria initially written in a protocol may not be realistic for this special population. This can result in prolonged participant enrollment times as well as protocol amendments. The most successful renal pharmacokinetic studies are those where the pharmaceutical sponsor and/or clinical research organization (CRO) involve the nephrology expert, often the Principal Investigator, in the design of the protocol, especially with respect to inclusion and exclusion criteria for both realistic criteria and safety of the subjects in this special population group.
Finally, there seems to be some confusion about what the regulatory agencies want. The last published FDA guidance was in 1998 and a draft guidance was posted in 2010. The FDA advisory committee met immediately after the release of the draft guidance and voted down some of the recommendations in the document [Citation11]. While, the subcommittee minutes are not as easy to find, the FDA has also expressed its view through various national meetings such as the DIA [Citation68,Citation69] and publications [Citation29], which are summarized in this article. But some sponsors feel they need to mimic the recommendations in the 2010 published draft guidance and are reluctant to vary from that document. The EMA has also published guidance and seems to favor Cockcroft-Gault assessment and asks for a measurement by another external marker, but in practice this is only happening in a minority of studies. Regulatory guidance documents are published infrequently and it is important to seek current and expert advice when designing and implementing renal impaired pharmacokinetic studies.
9. Five-year view
The current methods to categorize renal function (CG and MDRD) suffer from limitations and lack of accuracy over the entire range of renal function. The future of renal research will be to develop new formulas to calculate eGFR over the range from normal to ESRD as well as to identify alternative biomarkers of renal function. Over the next 5 years, our opinion is that for renal impaired pharmacokinetic studies, the utility of eGFR formulas will switch from CG to MDRD and we could possibly see an increase in the use of CKD-EPI. Moreover, we anticipate more research nephrologists will opt to use 24-h urine collection for the healthy match subjects, if CG or MDRD does not truly reflect GFR.
A recent advancement in this field is the attempt of pharmaceutical sponsors and CROs to develop templates for protocol design or engage people with expertise to provide uniformity in procedural development. However, even with a template or standardized study design, investigational drugs may have different toxicities and pharmacokinetic properties so expert input is needed to customize these protocols for this special patient population.
Key issues
Renal impaired pharmacokinetic studies are necessary to determine if a therapeutic compound will require dose adjustment in patients with renal dysfunction or are contraindicated for patients with certain stages of chronic kidney disease.
Both the FDA and EMA have issued detailed guidelines for renal impaired pharmacokinetic studies, while many of the requirements are similar between the two agency instructions, GFR methodology and reporting differ.
An updated position on the FDA draft guidelines redefines end-stage renal disease as <30 mL/min, and states this patient population is suitable for a ‘worst case scenario’ reduced study.
Under certain circumstances, population pharmacokinetic and PBPK modeling analysis may provide sufficient information to instruct renal impairment labeling
Declaration of interest
S Paglialunga, E Offman, N Ichhpurani, and BH Morimoto are employees of Celerion. TC Marbury is an employee and equity owner of Orlando Clinical Research Center. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Acknowledgments
We would like to thank all of the patients and families who have participated in these renal impaired pharmacokinetic studies as well as the pharmaceutical and biotech sponsors we have worked with.
Additional information
Funding
References
- Centers for Disease Control and Prevention. Chronic Kidney Disease Surveillance System—United States. 2016 [ cited 2016 Oct 10]. Available from: http://www.cdc.gov/ckd
- Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry: pharmacokinetics in patients with impaired renal function - study design, data analysis, and impact on dosing and labeling. 2010. [cited 2016 Dec 26]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP). Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. 2016. [cited 2016 Dec 26]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/02/WC500200841.pdf
- Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85:305–311.
- Rabinovich-Guilatt L, Siegler KE, Schultz A, et al. The effect of mild and moderate renal impairment on the pharmacokinetics of pridopidine, a new drug for Huntington’s disease. Br J Clin Pharmacol. 2016;81:246–255.
- Boulton DW. Clinical pharmacokinetics and pharmacodynamics of saxagliptin, a dipeptidyl peptidase-4 inhibitor. Clin Pharmacokinet. 2016. doi:10.1007/s40262-016-0421-4. [Epub ahead of print]
- Fradette C, Pichette V, Sicard E, et al. Effects of renal impairment on the pharmacokinetics of orally administered deferiprone. Br J Clin Pharmacol. 2016;82:994–1001.
- Ibrahim S, Honig P, Huang SM, et al. Clinical pharmacology studies in patients with renal impairment: past experience and regulatory perspectives. J Clin Pharmacol. 2000;40:31–38.
- Yeung CK, Shen DD, Thummel KE, et al. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2014;85:522–528.
- Huang SM, Temple R, Xiao S, et al. When to conduct a renal impairment study during drug development: US Food and Drug Administration perspective. Clin Pharmacol Ther. 2009;86:475–479.
- Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Summary minutes of the advisory committee for pharmaceutical science and clinical pharmacology. 2010 Mar 17. [cited 2016 Dec 26]. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM248402.pdf
- Food and Drug Administration. TagrissoTM (osimertinib) highlights for prescribing information. 2015 [ cited 2016 Oct 26]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208065s000lbl.pdf
- Di L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015;17:134–143.
- Pasut G, Veronese FM. State of the art in PEGylation: the great versatility achieved after forty years of research. J Control Release. 2012;161:461–472.
- Layton JE, Hockman H, Sheridan WP, et al. Evidence for a novel in vivo control mechanism of granulopoiesis: mature cell-related control of a regulatory growth factor. Blood. 1989;74:1303–1307.
- Food and Drug Administration. Neulasta® (pefilgrastim) highlights for prescribing information. 2010 [ cited 2016 Oct 26]. Available from: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/PediatricAdvisoryCommittee/UCM235408.pdf2015
- Yang BB, Kido A, Salfi M, et al. Pharmacokinetics and pharmacodynamics of pegfilgrastim in subjects with various degrees of renal function. J Clin Pharmacol. 2008;48:1025–1031.
- Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–558.
- Food and Drug Administration. Inlyta® (axitinib) Highlights for prescribing information. 2012 [ cited 2016 Oct 26]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202324lbl.pdf
- Food and Drug Administration. Inlyta® (axitinib) Clinical pharmacology and biopharmaceutics review(s). 2012 [ cited 2016 Oct 26]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202324Orig1s000ClinPharmR.pdf
- Food and Drug Administration. Xalkori® (crizotinib) Highlights for prescribing information. 2011 [ cited 2016 Oct 26]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202570s000lbl.pdf
- Food and Drug Administration. Xalkori® (crizotinib) Clinical pharmacology and biopharmaceutics review(s). 2011 [ cited 2016 Oct 26]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000ClinPharmR.pdf
- European Medicines Agency, Committee for medicinal products for human use (CHMP). Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling (draft). 2016 Jul 21. [cited 2016 Dec 26]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf
- Zhao P, Zhang L, Grillo JA, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89:259–267.
- Wagner C, Pan Y, Hsu V, et al. Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin Pharmacokinet. 2015;54:117–127.
- Jamei M. Recent advances in development and application of physiologically-based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161–169.
- Lindeman RD, Tobin J, Shock NW. Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc. 1985;33:278–285.
- Nagy CF, Kumar D, Cullen EI, et al. Steady-state pharmacokinetics and safety of donepezil HCl in subjects with moderately impaired renal function. Br J Clin Pharmacol. 2004;58(Suppl 1):18–24.
- Zhang L, Xu N, Xiao S, et al. Regulatory perspectives on designing pharmacokinetic studies and optimizing labeling recommendations for patients with chronic kidney disease. J Clin Pharmacol. 2012;52:79S–90S.
- Tatosian DGS, Caceres M, Grenier J, et al. Pharmacokinetics of omarigliptin (Mk-3102), a once-weekly dipeptidyl peptidase-IV (DPP-4) inhibitor, in patients with renal impairment. Poster presentation: ASCPT; 2014; Atlanta, GA. Available from: http://www.celerion.com/wordpress/wp-content/uploads/2014/04/Celerion_ASCPT-2014_Pharmacokinetics-of-Omarigliptin-Mk-3102-in-Patients-With-Renal-Impairment.pdf
- Em R. The relation between dialysance, membrane area, permeability and blood flow in the artificial kidney. Tr Am Soc Artific Organs. 1956;2:3.
- Nguyen L, Holland J, Ramies D, et al. Effect of renal and hepatic impairment on the pharmacokinetics of cabozantinib. J Clin Pharmacol. 2016;56:1130–1140.
- Roy MJ, Erdman KA, Abeyratne AT, et al. Pharmacokinetics of intravenous conivaptan in subjects with hepatic or renal impairment. Clin Pharmacokinet. 2013;52:385–395.
- Levey AS, Coresh J, Balk E, et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137–147.
- Orlando R, Floreani M, Padrini R, et al. Determination of inulin clearance by bolus intravenous injection in healthy subjects and ascitic patients: equivalence of systemic and renal clearances as glomerular filtration markers. Br J Clin Pharmacol. 1998;46:605–609.
- Chandra R, Barron JL. Anaphylactic reaction to intravenous sinistrin (Inutest). Ann Clin Biochem. 2002;39:76.
- Rahn KH, Heidenreich S, Bruckner D. How to assess glomerular function and damage in humans. J Hypertens. 1999;17:309–317.
- Stevens LA, Levey AS. Measured GFR as a confirmatory test for estimated GFR. J Am Soc Nephrol. 2009;20:2305–2313.
- Thomas C, Thomas L. Renal failure–measuring the glomerular filtration rate. Dtsch Arztebl Int. 2009;106:849–854.
- Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41.
- Levey AS, Bosch JP, Lewis JB, et al. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med. 1999;130:461–470.
- Helou R. Should we continue to use the Cockcroft-Gault formula? Nephron Clin Pract. 2010;116:c172–185; discussion c186.
- Park EJ, Wu K, Mi Z, et al. A systematic comparison of cockcroft-gault and modification of diet in renal disease equations for classification of kidney dysfunction and dosage adjustment. Ann Pharmacother. 2012;46:1174–1187.
- Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612.
- Denic A, Glassock RJ, Rule AD. Structural and functional changes with the aging kidney. Adv Chronic Kidney Dis. 2016;23:19–28.
- Schaeffner ES, Ebert N, Delanaye P, et al. Two novel equations to estimate kidney function in persons aged 70 years or older. Ann Intern Med. 2012;157:471–481.
- Daugirdas JT, Meyer K, Greene T, et al. Scaling of measured glomerular filtration rate in kidney donor candidates by anthropometric estimates of body surface area, body water, metabolic rate, or liver size. Clin J Am Soc Nephrol. 2009;4:1575–1583.
- Chen J, Muntner P, Hamm LL, et al. The metabolic syndrome and chronic kidney disease in U.S adults. Ann Intern Med. 2004;140:167–174.
- Fox CS, Larson MG, Leip EP, et al. Predictors of new-onset kidney disease in a community-based population. JAMA. 2004;291:844–850.
- Kramer H, Luke A, Bidani A, et al. Obesity and prevalent and incident CKD: the hypertension detection and follow-up program. Am J Kidney Dis. 2005;46:587–594.
- Hsu CY, McCulloch CE, Iribarren C, et al. Body mass index and risk for end-stage renal disease. Ann Intern Med. 2006;144:21–28.
- Sherif KA, Abo-Salem E, Panikkath R, et al. Cardiac repolarization abnormalities among patients with various stages of chronic kidney disease. Clin Cardiol. 2014;37:417–421.
- Khosoosi Niaki MR, Saravi M, Oliaee F, et al. Changes in QT interval before and after hemodialysis. Caspian J Intern Med. 2013;4:590–594.
- Cellina GU, Honour AJ, Littler WA. Direct arterial pressure, heart rate, and electrocardiogram during cigarette smoking in unrestricted patients. Am Heart J. 1975;89:18–25.
- Gupta RK, Gupta R, Maheshwari VD, et al. Impact of smoking on microalbuminuria and urinary albumin creatinine ratio in non-diabetic normotensive smokers. Indian J Nephrol. 2014;24:92–96.
- Ritz E, Benck U, Franek E, et al. Effects of smoking on renal hemodynamics in healthy volunteers and in patients with glomerular disease. J Am Soc Nephrol. 1998;9:1798–1804.
- Lash JP, Go AS, Appel LJ, et al. Chronic Renal Insufficiency Cohort (CRIC) Study: baseline characteristics and associations with kidney function. Clin J Am Soc Nephrol. 2009;4:1302–1311.
- Hallan SI, Orth SR. Smoking is a risk factor in the progression to kidney failure. Kidney Int. 2011;80:516–523.
- Anderson GD, Chan LN. Pharmacokinetic drug interactions with tobacco, cannabinoids and smoking cessation products. Clin Pharmacokinet. 2016;55:1353–1368.
- Czekaj P, Wiaderkiewicz A, Florek E, et al. Tobacco smoke-dependent changes in cytochrome P450 1A1, 1A2, and 2E1 protein expressions in fetuses, newborns, pregnant rats, and human placenta. Arch Toxicol. 2005;79:13–24.
- Yamaori S, Okamoto Y, Yamamoto I, et al. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab Dispos. 2011;39:2049–2056.
- Shimada T, Yamazaki H, Mimura M, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423.
- Zhou SF, Liu JP, Lai XS. Substrate specificity, inhibitors and regulation of human cytochrome P450 2D6 and implications in drug development. Curr Med Chem. 2009;16:2661–2805.
- Yeh W, Caro L, Guo Z, et al. Pharmacokinetics of co-Administered HCV protease inhibitor grazoprevir (MK-5172) and NS5A inhibitor elbasvir (MK-8742) in volunteers with end-stage renal disease on hemodialysis or severe renal impairment not on hemodialysis. Poster presentation: AASLD; 2014; Boston, MA. Available from: http://celerion.com/wordpress/wp-content/uploads/2015/01/Celerion_2014-AASLD_Pharmacokinetics-of-Co-Administered-HCV-Protease-Inhibitor-Grazoprevir-MK-5172-and-NS5A-Inhibitor-Elbasvir-MK-8742.pdf
- Grant TM, Obaidi M, Chai P, et al. Effect of renal insufficiency on the pharmacokinetics of avanafil, a new, potent, selective PDE-5 inhibitor, in male subjects. Poster presentation: ASCPT; 2012; National Harbour, MD. Available from: http://www.celerion.com/wordpress/wp-content/uploads/2012/03/ASCPT-2012-Effect-of-Renal-Insufficiency-on-the-Pharmacokinetics-of-Avanafil-a-New-Potent-Selective-PDE-5-Inhibitor-in-Male-Subjects.pdf
- Lauring B, Li XS, Liu Y, et al. Influence of renal and hepatic impairment on the pharmacokinetics of anacetrapib. J Clin Pharmacol. 2014;54:1247–1255.
- National Kidney F. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis. 2002;39:S1–266.
- Xu N. Phase 1 Studies in subjects with renal impairment: an FDA perspective. Conference proceeding: DIA; 2014; San Diego, CA.
- Marbury TC. Optimizing design and conduct of pharmacokinetic studies in patients with impaired kidney function. Conference Proceeding: DIA; 2014; San Diego, CA.