
ABSTRACT
Introduction: Bruton’s tyrosine kinase (BTK) inhibitors have revolutionized the treatment of B-cell lymphomas. Zanubrutinib was designed to achieve improved therapeutic concentrations and minimize off-target activities putatively accounting, in part, for the adverse effects seen with other BTK inhibitors.
Areas covered: This drug profile covers zanubrutinib clinical pharmacology and the translation of pharmacokinetics (PK) and pharmacodynamics (PD) to clinical efficacy and safety profiles, by highlighting key differences between zanubrutinib and other BTK inhibitors. We discuss PK, sustained BTK occupancy, and potential factors affecting PK of zanubrutinib, including food effects, hepatic impairment, and drug-drug interactions. These data, along with exposure-response analyses, were used to support the recommended dose of 320 mg, either once daily or as 160 mg twice daily. Translation of PK/PD attributes into clinical effects was demonstrated in a randomized, phase 3 head-to-head study comparing it with ibrutinib in patients with Waldenström macroglobulinemia.
Expert opinion: Among the approved BTK inhibitors, zanubrutinib is less prone to PK modulation by intrinsic and extrinsic factors, leading to more consistent, sustained therapeutic exposures and improved dosing convenience. Zanubrutinib PK/PD has translated into durable responses and improved safety, representing an important new treatment option for patients who benefit from BTK therapy.
1. Introduction
B-cell receptor (BCR) signaling is essential for normal B-cell development, differentiation, function, and survival [Citation1,Citation2]. Aberrant activation of protein kinases within the BCR-signaling pathway results in the development of various B-cell malignancies, including mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), Waldenström macroglobulinemia (WM), and diffuse large B-cell lymphoma (DLBCL) [Citation3]. Bruton’s tyrosine kinase (BTK), a member of the TEC family of kinases, is a key component within the BCR signaling cascade, and suppression of BCR signaling through inhibition of BTK has emerged as a promising strategy for the management of B-cell malignan-cies [Citation4]. The first-generation BTK inhibitor ibrutinib was approved by the FDA as a breakthrough therapy in 2013. Subsequently, the second-generation BTK inhibitors acalabru-tinib and zanubrutinib, which aim to reduce off-target effects, were approved in 2017 and 2019, respectively. Other irreversible or reversible non-covalent BTK inhibitors, such as orelabrutinib and pirtobrutinib (LOXO-305), are also in clinical development [Citation5,Citation6].
Zanubrutinib (BRUKINSA™; BGB-3111) was granted accelerated approval by the United States Food and Drug Administration (US FDA) in November 2019 for the treatment of MCL in patients who have received at least 1 prior therapy based on the efficacy findings from two phase 2 or phase 1/2 studies [Citation7,Citation8] as well as the safety data from an additional 3 studies. The approved dose is 160 mg twice daily (BID) or 320 mg once daily (QD). Zanubrutinib may be taken with or without food () [Citation9]. Subsequently, zanubrutinib was approved by the China National Medical Products Administration for the treatment of MCL and CLL or small lymphocytic lymphoma (SLL) in adults who have received at least 1 prior therapy [Citation10] based on two phase 2 or phase 1/2 studies [Citation11,Citation12]. The first randomized phase 3 study comparing the efficacy and safety of zanubrutinib to ibrutinib supported the authorization by Health Canada of zanubrutinib for the treatment of WM [Citation13]. More recently, interim analysis data from a second phase 3 study BGB-3111-305 (ALPINE) of zanubrutinib vs ibrutinib in patients with R/R CLL/SLL provided additional evidence to demonstrate clinical safety and efficacy superiority of zanubrutinib over ibrutinib [Citation14]. The currently approved indications and key clinical pharmacology and PK/PD data for zanubrutinib are summarized in .
Table 1. Approved dose regimens and key clinical pharmacology properties of zanubrutinib relative to those of ibrutinib and acalabrutinib
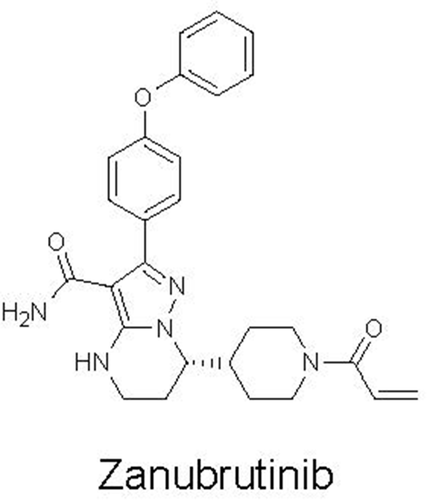
Zanubrutinib is a low-molecular-weight (471.55 g/mol) agent with a chemical structure shown in . It is differentiated from the first-generation BTK inhibitor ibrutinib [Citation17] in a number of ways, including having higher selectivity for Cys-481 in the active site of BTK compared with other structurally related kinases and better oral bioavailability. Zanubrutinib is marketed as an immediate-release capsule formulation for oral administration. Although it has pH-dependent solubility, there is only an approximately 3-fold change in solubility (ie from 0.247 mg/mL at pH 1.2 to 0.073 mg/mL at pH 4.5), and therefore, gastric acid reducing agents like proton pump inhibitors (PPIs) are not expected to affect the absorption of zanubrutinib. Because zanubrutinib exhibits low water solubility and moderate-to-high permeability, it is considered a Biopharmaceutics Classification System (BCS) Class 2 compound. The formulation has remained unchanged over the course of clinical development; therefore, relative bioavailability or bioequivalence studies have not been required to date.
Figure 1. Chemical structure of zanubrutinib
This manuscript summarizes clinical pharmacology data, including pertinent clinical pharmacokinetic (PK) and pharmacodynamic (PD) characteristics for zanubrutinib. Where appropriate, emerging data from two other BTK inhibitors in the class, ibrutinib and acalabrutinib, are also summarized to highlight attributes that distinguish zanubrutinib and support the translational understanding of clinical efficacy and safety profiles of BTK inhibitors.
1.1. Approved indications and doses
In addition to the currently approved MCL indication in the US, zanubrutinib is being developed for the treatment of additional B-cell malignancies, including CLL/SLL, WM, FL, marginal zone lymphoma (MZL), and DLBCL as a monotherapy and/or in combination with other agents. Zanubrutinib was first evaluated in Study BGB-3111-AU-003, a global phase 1/2, multicenter, open-label, dose escalation/cohort expansion study in patients with B-cell malignancies at doses ranging from 40 mg to 320 mg [Citation11]. A recommended dose of 160 mg twice daily (BID) or 320 mg once daily (QD) was selected for further study in disease-specific cohorts, including patients with WM, CLL, and MCL. Although 160 mg BID was selected as the recommended phase 2 and 3 dose, subsequently available clinical efficacy/safety data and exposure-response (E-R) analyses indicate no evidence of a difference in clinical efficacy/safety profiles between 320 mg QD and 160 mg BID (See additional summary in Section 4). The approved zanubrutinib dose for patients with relapsed or refractory (R/R) MCL and WM is 320 mg either as 160 mg BID or 320 mg QD. Zanubrutinib may be taken with or without food. In comparison, the current approved dose regimen for ibrutinib is QD, whereas acalabrutinib is administered BID. Ibrutinib is approved at doses of 420 mg or 560 mg QD and acalabrutinib at 100 mg BID. summarizes the currently approved indications and dose regimens for zanubrutinib as well as ibrutinib and acalabrutinib.
1.2. Potency and selectivity
Zanubrutinib is an oral, irreversible BTK inhibitor. Like ibrutinib, zanubrutinib forms an irreversible, covalent bond at Cys481 within the adenosine triphosphate-binding pocket of BTK. However, zanubrutinib is more selective than ibrutinib against off-target kinases, including epidermal growth factor receptor (EGFR), Janus kinase 3 (JAK3), tyrosine kinase expressed in hepatocellular Carcinoma (TEC), and IL-2 inducible tyrosine kinase (ITK), based on results from kinase inhibition and cell-based assays [Citation18]. The selectivity of BTK inhibitors is compared based on inhibitory activity of these agents on other kinases (IC50) relative to those of BTK inhibition (). Both zanubrutinib and acalabrutinib have a higher selectivity than ibrutinib [Citation19] with selectivity of >10-fold against multiple kinases (). For example, zanubrutinib demonstrated selectivity of 88-fold, 13.8-fold, and 2754-fold against TEC, HER4, JAK3, respectively. The increased BTK selectivity is thought to result in a lower incidence and severity of off-target toxicities linked to inhibition of the aforementioned kinases. Additionally, both ibrutinib and acalabrutinib have active metabolites with systemic exposures approximately 2-fold higher than those of the respective parent molecules (). The main metabolite of acalabrutinib has significant activity for kinases other than BTK, which may contribute to off-target toxicities (). Zanubrutinib is at least 10-fold weaker than ibrutinib in inhibiting rituximab-induced, antigen-dependent, cell-mediated cytotoxicity (ADCC), a potential limitation of ibrutinib-rituximab combinations [Citation20], which to date have not shown improved clinical activity over ibrutinib alone in CLL [Citation21,Citation22]. This reduced effect on rituximab-mediated ADCC may make zanubrutinib a more rational BTK inhibitor to partner with anti-CD20 therapies [Citation20]. Because BTK inhibitors are becoming a cornerstone of combination treatment with rituximab in multiple B-cell malignancies including CLL [Citation23], this attribute may maximize the benefits of BTK inhibitors in a combination setting with anti-CD20 antibodies.
Table 2. Kinase selectivity of zanubrutinib, ibrutinib, and acalabrutinib
In addition to improved selectivity, zanubrutinib also maintains similar or improved potency for BTK inhibition compared with other molecules in the class. The IC50 values can vary slightly depending on the type of assay and conditions. When comparing IC50 values of zanubrutinib and ibrutinib directly in the same biochemical assay, the estimated IC50 values were 0.3 nM and 0.18 nM for zanubrutinib and ibrutinib, respectively [Citation18]. In another study, IC50 values for BTK inhibition were determined in biochemical kinase assays for a comparison of three BTK inhibitors (). In this assay, the reported IC50 values were 0.5 nM, 1.5 nM, and 5.1 nM for zanubrutinib, ibrutinib, and acalabrutinib, respectively [Citation24].
1.3. Clinical PK
The PK of zanubrutinib was assessed after single- and multiple-dose, oral administrations in a phase 1, single-agent study (BGB-3111-AU-003) in patients with R/R B-cell malignancies at doses from 40 mg to 320 mg QD and 160 mg BID. The plasma concentration profiles are shown in [Citation11]. The study demonstrated that zanubrutinib was rapidly eliminated, with a mean terminal elimination half-life (t½) of approximately 2 to 4 hr. There was a dose-proportional increase in Cmax and AUC at doses from 40 mg to 320 mg. Following multiple-dose administrations of zanubrutinib, limited systemic accumulation was observed at all doses, which is consistent with its t½. Based on noncompartmental PK analysis, the geometric mean (%CV) zanubrutinib steady-state daily AUC is 2295 (37%) ng•h/mL following 160-mg BID and 2180 (41%) ng•h/mL following 320-mg QD dosing. The geometric mean (%CV) steady-state Cmax is 314 (46%) ng/mL following 160-mg BID and 543 (51%) ng/mL following 320-mg QD dosing. Consistent with the preclinical prediction, zanubrutinib achieved higher bioavailability than that of ibrutinib, with estimated oral bioavailability of 15% based on the Physiologically-based Pharmacokinetic (PBPK) model [Citation25] relative to 3.9% (fasting state) for ibrutinib [Citation26].
Figure 2. Plasma concentration-time profile of zanubrutinib after single dosing. Error bars indicate standard deviation [Citation27]
![Figure 2. Plasma concentration-time profile of zanubrutinib after single dosing. Error bars indicate standard deviation [Citation27]](/cms/asset/a3406f84-4046-476b-bd9b-490bb1ee5de5/ierj_a_1978288_f0002_oc.jpg)
As a consequence of its higher selectivity, zanubrutinib was able to achieve relatively high plasma exposures without dose-limiting toxicity (DLT). In the first-in-human dose escalation study, no DLTs were observed at any dose up to 320 mg/day and a maximally tolerated dose was not identified [Citation11].
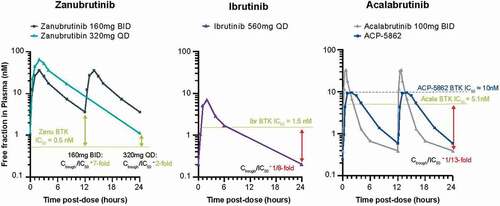
The plasma protein binding of zanubrutinib is approximately 94% compared with 97% for ibrutinib and acalabrutinib (). Thus, the unbound fraction in plasma is approximately twice that of ibrutinib and acalabrutinib [Citation9,Citation17,Citation28]. Only unbound drug is considered to elicit a pharmacological response. The unbound drug-concentration-time profiles for zanubrutinib, ibrutinib, and acalabrutinib are presented in . The daily unbound plasma exposure (AUC) for zanubrutinib at the recommended daily dose of 320 mg is estimated to be approximately 6- to 8-fold higher than that of ibrutinib at a dose of 560 mg QD [Citation11] and 2- to 3-fold higher than that of acalabrutinib at 100 mg BID [Citation29]. also shows unbound drug exposures achieved by zanubrutinib relative to the half-maximal inhibitory concentration (IC50) levels needed to inhibit BTK. Clinically, higher drug-concentration/IC50 ratios achieved at a tolerable dose would be expected to lead to more sustained BTK inhibition. Zanubrutinib has exposure coverage above its IC50 during the entire dose interval for both BID and QD dosing schedules. The ratio of Ctrough/IC50 is 7 and 2 for the 160 mg BID and 320 mg QD dosing schedules, respectively, resulting in concentrations that were significantly higher than the IC50 during the entire 24-hr dosing period for both schedules. The corresponding ratios of Ctrough/IC50 for ibrutinib and acalabrutinib are estimated to be consistently lower than 1, even with consideration of active metabolites [Citation29,Citation30]. Although the 320-mg QD regimen for zanubrutinib has a lower Ctrough than that noted for the 160 mg BID regimen, the Ctrough/IC50 (unbound) ratio following the 320-mg QD dose is consistently higher than those of ibrutinib and acalabrutinib where compelling clinical efficacy data have been reported in patients with multiple B-cell malignancies [Citation29,Citation30].
Figure 3. Free drug concentration time profiles relative to IC50 of approved BTK inhibitors
1.4. Clinical PD
The sustained plasma exposures achieved by zanubrutinib translate into deep and durable BTK inhibition in target tissues. At the 320-mg total daily dose, zanubrutinib achieves median steady-state BTK occupancy of 100% in peripheral blood mononuclear cells (PBMCs) for more than 24 hr. In addition, BTK inhibition was assessed in Study AU-003 in pre- and on-treatment lymph node biopsies, demonstrating a median steady-state (trough) BTK occupancy of 94% and 100% following the 320-mg QD and 160-mg BID regimens, respectively (). These results indicate efficient and sustained inactivation of BTK in target tissues throughout the recommended dosing interval and are consistent with high rates of objective responses reported in zanubrutinib-treated patients with B-cell malignancies, including WM, MCL, and CLL [Citation7,Citation11,Citation31]. Clinical and biological implications associated with the BTK occupancy difference (median of 94% vs 100%) in lymph nodes between QD and BID doses for zanubrutinib were unknown at the time. Thus, to maximize BTK inhibition in target tissues, the 160-mg BID dose was recommended in ongoing phase 2/3 clinical trials. Subsequently, a totality of data including clinical efficacy and safety data and exposure-response analyses showed that the observed difference in BTK occupancy between the two dose regimens is unlikely to have a meaningful clinical impact (Please see additional discussion of Dose Selection in Section 4).
Figure 4. (A) Zanubrutinib BTK occupancy in peripheral blood mononuclear cells by dose regimen. (B) Zanubrutinib BTK occupancy in lymph nodes by dose regimen. Median values are shown as lines through the individual symbols. BID twice a day, BTK Bruton’s tyrosine kinase, N number of patients, QD once a day, W1D1 Week 1 Day 1, W1D3, Week 1 Day 3, W2D1 Week 2 Day 1 [Citation11]. Predose samples were collected 24 hours after dosing for the QD dose or 12 hr after dosing for the BID dose. Samples were collected 4 hours after dose for ‘W1D1 4 hours’ samples. Republished in part with permission of Elsevier Science & Technology Journals, from Tam CSL, Trotman J, Opat S, et al. Phase 1 study of selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–859; permission conveyed through Copyright Clearance Center, Inc
![Figure 4. (A) Zanubrutinib BTK occupancy in peripheral blood mononuclear cells by dose regimen. (B) Zanubrutinib BTK occupancy in lymph nodes by dose regimen. Median values are shown as lines through the individual symbols. BID twice a day, BTK Bruton’s tyrosine kinase, N number of patients, QD once a day, W1D1 Week 1 Day 1, W1D3, Week 1 Day 3, W2D1 Week 2 Day 1 [Citation11]. Predose samples were collected 24 hours after dosing for the QD dose or 12 hr after dosing for the BID dose. Samples were collected 4 hours after dose for ‘W1D1 4 hours’ samples. Republished in part with permission of Elsevier Science & Technology Journals, from Tam CSL, Trotman J, Opat S, et al. Phase 1 study of selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–859; permission conveyed through Copyright Clearance Center, Inc](/cms/asset/84bb768a-be89-4b02-96f7-06a0891be48b/ierj_a_1978288_f0004_oc.jpg)
In comparison, median BTK occupancy in PBMCs at Ctrough was >90% at doses of 420 mg or 560 mg for ibrutinib [Citation30] and 87.6% and 95% at acalabrutinib doses of 200 mg QD and 100 mg BID, respectively [Citation32]. The BTK occupancy of ibrutinib in PBMC in some patients remains below 80% at the approved dose 420 mg [Citation33]. The BTK occupancy in lymph node biopsies at Ctrough by acalabrutinib was 90% and 95.8% following the 200-mg QD and 100-mg BID dosing schedules, respectively [Citation34]. Thus, at 160 mg BID or 320 mg QD dosing schedules, the BTK occupancy by zanubrutinib in PBMCs and lymph node biopsies is comparable to or higher than that reported in published studies of ibrutinib and acalabrutinib [Citation11].
In addition to its PK properties to achieve and maintain desired systemic exposures, additional pharmacologic attributes should also be considered to understand the full therapeutic potential of an irreversible inhibitor such as zanubrutinib and to support an optimal dose and schedule. For example, studies have shown that the selection of an optimal-dose schedule for irreversible inhibitors may incorporate the target protein’s half-life/turnover rate in addition to the plasma half-life of the inhibitor. Multiple examples exist of less frequent dosing (i.e. weekly) for irreversible inhibitors with a plasma half-life as short as 30 min [Citation35]. To wit, the estimated BTK synthesis rate ranged from 3.6% to 31.4% per day [Citation32] and the estimated half-life of BTK was approximately 12 hours [Citation36]. Accordingly, the de novo synthesis rate of BTK is the primary driver for the loss of the PD effect and an increased de novo synthesis rate of BTK would be anticipated in patients with more rapidly proliferating B cells. In contrast to ibrutinib and acalabrutinib, zanubrutinib is able to maintain a relatively high unbound Ctrough /IC50 ratio, resulting in maintenance of consistent and complete inactivation of BTK in target tissues and subsequently accounting for the resurgence of free BTK throughout the dosing interval. This allows for sustained inhibition of BTK coincident with either dosing interval (i.e. 12 or 24 hours) and across a range of BTK turnover rates despite a short plasma half-life of 2–4 hr.
1.5. Absorption, distribution, metabolism, and excretion
Zanubrutinib is rapidly absorbed and eliminated after oral administration in humans. The median time to peak zanubrutinib plasma concentration was 2 hr. After a single, [14C]-labeled 320 mg dose was administered to healthy subjects under fasted conditions, approximately 87% of the dose was recovered in feces (38% unchanged) and 8% in urine (less than 1% unchanged) [Citation37]. Dosing with food (high- or low-fat meal) did not result in significantly meaningful effects on the AUC of zanubrutinib; therefore, zanubrutinib can be taken with or without food [Citation9]. Similarly, the mean AUC of acalabrutinib was not impacted by a high-fat meal, though Cmax was decreased by 73% compared with the fasted state. Acalabrutinib may also be administered without regard to food [Citation28]. In comparison, administration with food significantly increased ibrutinib Cmax and AUC by approximately 2- to 4-fold and 2-fold, respectively, compared with its administration after an overnight fast [Citation17]. The significant food effects may contribute to ibrutinib’s high PK variability. Despite the food impact, ibrutinib may be taken with or without food [Citation38].
Zanubrutinib undergoes extensive metabolism, primarily via a CYP3A-mediated pathway in humans. There are no major active metabolites in circulation. The most abundant metabolite in plasma is the mono-hydroxylate of the phenoxy phenyl ring (BGB-7941), which represented <10% total radioactivity in circulation and is not considered a major metabolite. In comparison, both ibrutinib and acalabrutinib have major active metabolites in circulation (). In particular, the most abundant metabolite of acalabrutinib was ACP-5862, which was about 2- to 3-fold more abundant than acalabrutinib in human plasma, accounted for 34.7% of total radioactivity and was responsible for one-half of the BTK inhibitory activity of acalabrutinib [Citation39,Citation40].
The first-pass metabolism (i.e. CYP3A expression in gut walls and in the liver) plays an important role in the clearance of zanubrutinib, as observed for ibrutinib and acalabrutinib [Citation40,Citation41]. The drug-related radioactivity for zanubrutinib was primarily excreted in feces, and to a lesser extent in urine. Renal clearance for zanubrutinib was minor, and metabolism accounts for the major clearance pathways [Citation37]. Most metabolites identified in the human ADME studies were oxidative metabolites via cytochrome P450 (CYP) 3A with a low percentage derived from glutathione/cysteine conjugation.
The brain penetration of zanubrutinib in patients relative to that of other BTK inhibitors remains to be studied (). P-glycoprotein (P-gp) is expressed at the blood–brain barrier (BBB) and limits the central nervous system (CNS) penetration of its substrates. An in vitro study indicated that zanubrutinib is a potential P-gp substrate [Citation9] that may reduce CNS penetration; however, a case describing successful treatment of Bing Neel syndrome, a CNS manifestation of WM, with zanubrutinib has been reported [Citation42]. Acalabrutinib is classified as a P-gp substrate [Citation28], but ibrutinib is not [Citation17]. The brain penetration of acalabrutinib is likely lower than that of zanubrutinib based on the reported efflux ratio (3.46 for zanubrutinib vs 42 acalabrutinib at 1 μM in MDCK-MDR1 assay) [Citation37]. The average ratio of brain to plasma AUC0−t for ibrutinib was in the range of 0.52 to 0.64 following single intraperitoneal doses at 0.6 to 6 mg/kg in healthy Swiss mice [Citation43]. Clinical data have shown promising results regarding brain penetration and the efficacy of ibrutinib in patients with central nervous system lymphoma [Citation44].
2. Impact of intrinsic and extrinsic factors
Intrinsic and extrinsic factors potentially impacting zanubrutinib PK were evaluated in dedicated clinical pharmacology studies, encompassing drug–drug interaction (DDI) and special population/hepatic impairment studies (). In addition, population PK and PBPK modeling have been conducted for zanubrutinib to evaluate other intrinsic and extrinsic covariates that may impact the PK of zanubrutinib.
Table 3. Comparison of extrinsic and intrinsic factors impacting zanubrutinib, ibrutinib, and acalabrutinib
A population PK model for zanubrutinib was developed and evaluated the impact of intrinsic and extrinsic covariates on the PK of zanubrutinib in patients and healthy volunteers [Citation45]. No statistically significant differences in the PK of zanubrutinib were observed based on age (19 to 90 years), sex, race (Asian, White, and other), body weight (36 to 144 kg), mild or moderate renal impairment (creatinine clearance ≥30 mL/min as estimated by Cockcroft-Gault), the use of acid-reducing agents, baseline aspartate aminotransferase, bilirubin, or tumor type. Baseline alanine aminotransferase (ALT) and health status (patients with B-cell malignancies vs healthy volunteers) were identified as statistically significant covariates impacting the PK of zanubrutinib. The impact of baseline ALT on zanubrutinib exposure, though statistically significant on clearance, was relatively small compared with the overall PK variability of the population. This aligns with observed clinical data in a dedicated hepatic impairment study as summarized below in which comparable exposure levels between subjects with mild or moderate hepatic impairment and those with normal hepatic function were observed [Citation46]. To assess ethnic differences in PK more robustly, a single protocol that controlled extrinsic factors was also studied using an intensive PK collection in healthy volunteers. The results show the PK of zanubrutinib was comparable between Asian and non-Asian subjects [Citation47]. Thus, various approaches all corroborated a lack of ethnic impact on PK of zanubrutinib.
Similar findings were also observed for ibrutinib and acalabrutinib, in that there were no clinically significant covariates of PK, including age, sex, race, body weight, and mild or moderate renal impairment. For ibrutinib, body weight and coadministration of antacids showed a slight impact on volume of distribution and on the duration of the absorption phase, respectively, but were not deemed clinically meaningful [Citation48]. An acalabrutinib population PK model found that age (42 to 90 years), sex, race (Caucasian, African American), body weight, mild or moderate renal impairment status, and mild hepatic impairment status did not have clinically meaningful effects on acalabrutinib PK [Citation49].
2.1. Special Populations
A dedicated hepatic impairment study was conducted for zanubrutinib [Citation46]. Similar to results of the population PK analysis, no substantial difference occurred in PK parameters between patients with mild/moderate hepatic impairment (Child-Pugh class A or B) and healthy subjects. Therefore, no dose modification is recommended in patients with mild-to-moderate hepatic impairment. The total and unbound plasma exposures of zanubrutinib in subjects with severe hepatic impairment were 1.6- and 2.9-fold higher in subjects with severe hepatic impairment compared with healthy subjects; consequently, a dose modification of zanubrutinib (80 mg orally BID) is recommended in patients with severe hepatic impairment. The impact of hepatic impairment on PK appears to be lower for zanubrutinib than for ibrutinib and acalabrutinib (). Ibrutinib exposures relative to subjects with normal liver function increased on a scale of 2.7-fold to 8.2-fold in those with mild and moderate hepatic impairment, respectively, and up to 9.8-fold in those with severe hepatic impairment [Citation17]. Recommendations for ibrutinib dosing in patients with hepatic impairment include 3- to 4-fold and 6- to 8-fold dose reductions in cases of mild or moderate impairment, respectively, and use avoidance in patients with severe hepatic impairment. The AUC of acalabrutinib increased 1.9-fold in subjects with mild hepatic impairment (Child-Pugh class A), 1.5-fold in subjects with moderate hepatic impairment (Child-Pugh class B), and 5.3-fold in subjects with severe hepatic impairment (Child-Pugh class C) compared with subjects with normal liver function [Citation28]. Current label recommendations are to avoid administration of acalabrutinib in patients with severe hepatic impairment.
Zanubrutinib minimally undergoes renal elimination; mild and moderate renal impairment (estimated creatinine clearance >30 mL/min by Cockcroft-Gault equation) had no influence on the zanubrutinib exposure based on population PK analysis. Thus, no dose adjustment for zanubrutinib is proposed for patients with mild or moderate renal impairment (providing creatinine clearance ≥30 mL/min as estimated by Cockcroft-Gault). It is unlikely that zanubrutinib exposure will be affected to any clinically significant degree in patients with severe renal impairment or with end-stage renal disease; however, clinical PK and safety data in these patient populations are limited [Citation9].
Regarding renal elimination, similar to zanubrutinib, both ibrutinib and acalabrutinib undergo minimal renal elimination. No clinically significant differences in the PK of each BTK inhibitor were observed in subjects with mild or moderate renal impairment [Citation9,Citation17,Citation28]. The effects of severe renal impairment and dialysis on the PK of each compound have not been evaluated.
2.2. Clinical Drug-Drug Interaction (DDI)
2.2.1. Effects of CYP3A Modulators on the PK of Zanubrutinib
Because zanubrutinib is primarily metabolized by CYP3A, the interaction potential between zanubrutinib and a strong CYP3A inducer (rifampin) and a strong CYP3A inhibitor (itraconazole) were assessed in study BGB-3111-104 (NCT03301181) [Citation47]. Results from this dedicated DDI study [Citation47] indicated that coadministration of zanubrutinib with the strong CYP3A inhibitor itraconazole (200 mg QD) increased exposure of zanubrutinib by 3.8-fold for AUC0‑∞ and by 2.6-fold for Cmax. Coadministration of zanubrutinib with the strong CYP3A inducer rifampin (600 mg QD) decreased zanubrutinib exposure by 13.5-fold for AUC0-∞, and 12.6-fold for Cmax [Citation47]. These results are consistent with the role for CYP3A isoenzymes as the principal metabolic pathway for zanubrutinib. In addition, a PBPK model was developed and was used to predict the effect of CYP3A inhibitors and CYP3A inducers on the PK of zanubrutinib [Citation25]. PBPK simulations suggest that coadministration of multiple doses of a moderate CYP3A inhibitor (e.g. fluconazole, diltiazem, and erythromycin) may increase the Cmax and AUC of zanubrutinib by approximately 2-fold. Exposure increases were <1.5‑fold with a mild CYP3A inhibitor. PBPK simulations suggest that a moderate CYP3A inducer (e.g. efavirenz) may decrease the Cmax and AUC of zanubrutinib by approximately 2‑ to 3-fold.
Guidance for zanubrutinib currently describes dose modifications when coadministered with a strong and moderate CYP3A inhibitor and avoidance of concomitant use of strong and moderate CYP3A inducers [Citation9]. Owing to the lack of clinical experience at doses of zanubrutinib above 320 mg QD (highest administered dose) in patients with B-cell malignancies, it is recommended that patients reduce the dose to 80 mg QD (4‑fold reduction) when taking strong CYP3A inhibitors concomitantly, or 80 mg BID (2‑fold reduction) when taking moderate CYP3A inhibitors concomitantly. Current US Prescribing Information recommendations for zanubrutinib specify that coadministration of zanubrutinib with mild CYP3A inhibitors requires no dose reduction, and coadministration with strong or moderate CYP3A inhibitors (including azole antifungals, important in the management of patients with leukemia/lymphoma) is permitted at a reduced dose.
Of note, the observed magnitude of clinical DDIs with CYP3A inhibitors for zanubrutinib appears to be substantially lower than for ibrutinib [Citation50,Citation51]. This favorable attribute may be clinically relevant in patients with B-cell malignancies. In the real-world setting (n = 118), 2 of 3 CLL patients commencing ibrutinib therapy are on a concomitant medication with a potentially clinically significant DDI with ibrutinib [Citation52]. It has been shown that coadministration with ketoconazole (strong CYP3A inhibitor) increased ibrutinib Cmax by 29-fold and AUC by 24-fold in healthy volunteers under fasted conditions [Citation51]. In patients dosed under nonfasted conditions, coadministration with voriconazole (also a strong CYP3A inhibitor) increased ibrutinib steady-state Cmax by 6.7-fold and AUC by 5.7-fold. Erythromycin (a moderate CYP3A inhibitor) increased ibrutinib steady-state Cmax by 3.4-fold and AUC by 3-fold [Citation17]. PBPK simulations suggest that posaconazole (strong CYP3A inhibitor) may increase the AUC of ibrutinib 3-fold to 10-fold under fed conditions. Thus, for patients receiving ibrutinib, coadministration with strong CYP3A inhibitors other than posaconazole and voriconazole is not recommended. For short-term use (treatment for ≤7 days) of strong CYP3A inhibitors (e.g. antifungals and antibiotics), it is recommended that patients interrupt ibrutinib therapy until the CYP3A inhibitor is no longer needed. The drug interaction studies for acalabrutinib indicated a 3.9-fold increase in Cmax and a 5.1-fold increase in AUC during coadministration of 200 mg itraconazole (strong CYP3A inhibitor) in healthy subjects [Citation28]. Per US label recommendation, acalabrutinib is to be avoided in patients receiving strong CYP3A inhibitors. Zanubrutinib, however, can be coadministered with strong CYP3A inhibitors with an appropriate-dose reduction [Citation9] based on results of clinical DDI studies. This favorable clinical DDI attribute has several clinical implications. Fungal and bacterial infections are commonly observed in patients with B-cell malignancies and may require treatment with medications that are strong or moderate inhibitors of CYP3A. In these instances of comedication with CYP3A inhibitors, it would be ideal for patients to continue BTK inhibitor therapy, because it has been demonstrated that dose interruptions with BTK inhibitors can affect their efficacy. For example, patients with CLL/SLL missing ≥8 consecutive days of ibrutinib had a shorter progression-free survival compared with those missing <8 days [Citation53]. In this regard, the increased flexibility in concomitant medicine without requiring interruption of zanubrutinib is expected to improve the management of clinically relevant drug interactions.
2.2.2. Effects of concomitant medications on the PK of zanubrutinib
Zanubrutinib exhibits weak pH‑dependent solubility. Given that the use of protein pump inhibitors (PPIs) and other acid-reducing agents (ARAs) was allowed in zanubrutinib clinical studies, potential effects of ARAs on the PK of zanubrutinib were assessed using population PK [Citation45] and PBPK analyses [Citation25]. Population PK modeling for zanubrutinib indicated the lack of a significant impact for the use of PPIs and other ARAs on zanubrutinib PK. Consistently, PBPK simulations further corroborate the population PK analyses, demonstrating that zanubrutinib exposures are not expected to be affected by ARAs; thus, no dose modifications are warranted.
Similarly, the solubility of ibrutinib and acalabrutinib are both affected by changes in gastric pH. In a dedicated DDI study, omeprazole was coadministered with ibrutinib to evaluate the potential for interactions [Citation54]. Coadministration in healthy volunteers overall resulted in a decrease in ibrutinib Cmax (37.5%) with minimal impact on AUC (<10%). Similar to zanubrutinib, ibrutinib dosing would not warrant modifications when coadministered with PPIs and alternative ARAs. A clinical DDI evaluation of the impact of ARAs on acalabrutinib indicated reductions in acalabrutinib exposures with calcium carbonate (antacid) and omeprazole (PPI) by 53% and 43%, respectively [Citation28]. As such, patients receiving acalabrutinib treatment are recommended to avoid coadministration with PPIs due to the long-lasting effect on exposures [Citation28]. In addition, stagger dosing with H2-receptor antagonists and antacids are also recommended. It has been reported that the prevalence of cancer patients receiving ARAs (some available over-the-counter) may range from 20% to 55% [Citation55]. Thus, in the real-world setting, there exists a significant risk for concomitant use of PPI to decrease therapeutic exposures of acalabrutinib, resulting in sub-optimal efficacy. The flexibility of dosing around the use of ARAs for zanubrutinib is expected to be advantageous in managing clinically relevant drug interactions.
2.2.3. Effect of zanubrutinib on exposure of other concomitant medications
A panel of in vitro studies was performed to assess the potential for zanubrutinib to affect activities of CYP enzymes or transporters. Zanubrutinib is predicted not to cause clinically relevant inhibition of the hepatic uptake transporters, OATP1B1 and OATP1B3, or the renal uptake transporters, OAT1, OAT3, and OCT2. However, due to the estimated high concentrations in the gastrointestinal (GI) tract associated with the 160-mg BID dose, the potential for zanubrutinib as a P-gp or breast cancer resistance protein (BCRP)-mediated DDI perpetrator was further investigated. Additionally, in lieu of the in vitro prediction that zanubrutinib may also have the potential to induce and/or inhibit multiple CYP enzymes, a clinical DDI study was conducted to assess the effect of zanubrutinib on the PK of probe substrates of CYP3A (midazolam), CYP2C9 (warfarin), CYP2C19 (omeprazole), P-gp (digoxin), and BCRP (rosuvastatin) using a cocktail approach [Citation56]. The study showed that a 320-mg total daily dose of zanubrutinib had minimal or no effect on the activity of CYP2C9, BCRP, and P-gp, but was a weak inducer of CYP3A and CYP2C19 enzymes with a reduction in AUC <50% of the probe substrates midazolam and omeprazole. A PBPK model was employed to predict the effect of zanubrutinib on the exposure of CYP2C8 and CYP2B6 substrates, which were not included in the cocktail DDI study due to the lack of sensitive and validated probes for both enzymes. PBPK simulations show that concomitant use of zanubrutinib has minimal or no effect on the exposure of CYP2C8 substrate (e.g. rosiglitazone) or CYP2B6 substrate (e.g. bupropion).
Similarly, a PBPK simulation was employed to show that there were minimal effects of acalabrutinib on the PK of midazolam (CYP3A substrate) and rosiglitazone (CYP2C8 substrate) after repeated doses of acalabrutinib 100-mg BID [Citation57]. This was consistent with the in vitro data, indicating that acalabrutinib is a weak inhibitor of CYP3A4/5, CYP2C8, and CYP2C9 and is a weak inducer of CYP1A2, CYP2B6, and CYP3A4 [Citation28]. Regarding potential effects of BTK inhibitors on activities of transporters, in vitro studies suggest that ibrutinib may inhibit BCRP and P-gp transport at clinical doses. Similarly, in vitro results indicated that acalabrutinib was a weak BCRP inhibitor at the intestinal level. Acalabrutinib may increase exposure to co-administered BCRP substrates (e.g. methotrexate) by inhibition of intestinal BCRP [Citation28].
3. Clinical translation: differentiated safety and efficacy profiles of zanubrutinib
As summarized in previous sections, zanubrutinib was designed for enhanced BTK selectivity and higher therapeutic exposures relative to the first-in-class BTK inhibitor, ibrutinib. These pharmacological properties appear to translate into clinically meaningful safety and efficacy advantages for zanubrutinib in comparison with ibrutinib, as demonstrated in two randomized phase 3 clinical studies [Citation14,Citation58]. The favorable PK/PD properties of zanubrutinib appear to be consistent with the deep and durable responses demonstrated in the randomized phase 3 ASPEN study comparing the safety and efficacy of zanubrutinib and ibrutinib in patients with WM [Citation58]. Frequency of IRC-assessed VGPRs was numerically higher among zanubrutinib patients than among ibrutinib patients (28% and 19%, respectively, 2-sided P = .09). Investigator-assessed rates of VGPR were 28% and 17% in the zanubrutinib and ibrutinib arms, respectively (P = .04, descriptive only). Major differences were observed in the toxicity profiles between the two BTK inhibitors (discussed below). Clinical safety and efficacy superiority of zanubrutinib over ibrutinib, especially improvement in treatment tolerability is further substantiated by a second phase 3 study BGB-3111-305 (ALPINE) of zanubrutinib and ibrutinib in patients with R/R CLL/SLL [Citation14]. Although there are no head-to-head comparison studies of zanubrutinib and acalabrutinib, and cross-study comparison is challenging by nature, the efficacy data in patients with MCL, WM and CLL [Citation7,Citation28,Citation58] support zanubrutinib as an important treatment option in these populations.
In addition to a promising response rate demonstrated in patients treated with zanubrutinib, clinical studies are beginning to ascertain whether a more selective BTK inhibitory profile may translate into differences in tolerability and safety. BTK inhibitors as a class (including zanubrutinib) have been shown to have a low potential to cause QTc prolongation, as demonstrated in thorough QTc studies [Citation9,Citation17,Citation59,Citation60]. However, several adverse events (AEs), including diarrhea, atrial fibrillation (AF)/flutter, bleeding, and hypertension, have been associated with BTK inhibitors as a drug class. These toxicities have been observed with varying frequencies and severities across products, in part due to differences in the specificity for BTK inhibition and off-target effects [Citation61–64]. Consistent with this hypothesis, in the pivotal phase 3 ASPEN study in patients with WM, treatment with zanubrutinib was associated with improved toxicity and tolerability profiles over treatment with ibrutinib [Citation58]. This was particularly notable for cardiovascular toxicities, specifically AF/flutter and hypertension, which are known to be associated with ibrutinib treatment discontinuation. Despite a balance of risk factors for AF and hypertension across treatment arms, ibrutinib-treated patients experienced an approximately 10-fold higher incidence of AF/flutter and 2-fold increased frequency of hypertension on an exposure-adjusted basis [Citation58]. Diarrhea was also less common in patients treated in the zanubrutinib arm, which is likely attributable to its reduced EGFR inhibition and other off-target effects. Of note, an EGFR off-target effect is likely not the only mechanism underlying the frequent occurrence of diarrhea in patients treated with BTK inhibitors because acalabrutinib or tirabrutinib have also been linked to frequent occurrence of diarrhea despite minimal off-target effects toward EGFR in vitro [Citation65]. Bleeding and major bleeding occurred less frequently among patients in the zanubrutinib arm, likely due to less off-target inhibition of TEC in platelets. Conversely, all grade and Grade 3 or higher neutropenia were more frequently observed in patients treated with zanubrutinib than ibrutinib (Grade 3, 20% versus 8%). Neutropenia is likely related to the critical role of BTK in the development and maturation of myeloid progenitors. It has been reported that 13 of 50 (26%) patients with X-linked agammaglobulinemia (XLA, caused by a mutation in BTK) experienced episodes of profound neutropenia [Citation66]. Neutropenia may therefore be the result of a combination of factors including direct inhibition of BTK, immune dysregulation, and other as yet unknown etiologies. Of note, neutropenia was found to occur early during the course of zanubrutinib treatment, was manageable with growth factor support or dose reduction, and tended not to recur. Importantly, the increased incidence of neutropenia among zanubrutinib-treated patients did not correlate with an increase in Grade 3+ and serious infections. In fact, pneumonia was observed less commonly in patients treated with zanubrutinib than with ibrutinib (2% versus 12.2%). Interestingly, in the phase 1/2 study (BGB-3111-AU-003), zanubrutinib 320 mg QD dose showed a trend toward a lower rate of Grade 3 or higher neutropenia compared with 160 mg BID dose (6.3% vs 13.3%, respectively) [Citation67].
3.1. Mechanistic studies supporting translational understanding of zanubrutinib clinical profiles
Since the introduction of ibrutinib into the clinic in 2013, much effort has focused on deriving a better understanding of the mechanistic basis for BTK inhibitor-associated toxicities. The mechanism(s) behind the primary hemostatic defect associated with BTK inhibitors is arguably the best understood among all toxicities associated with the BTK inhibitor class. Platelet adhesion and aggregation are the result of a complex series of events mediated by the interaction of platelet agonists (strong agonists such as thrombin or collagen and weak agonists such as adenosine diphosphate or collagen-reactive peptide) and their cognate receptors on the platelet surface. Activation of the agonist receptors initiates signaling via specific kinase pathways (Src family kinases), ultimately resulting in the inside-out activation of the fibrinogen receptor αIIbβ3 (GPIIb/IIIa) and platelet aggregation. Glycoprotein Ib (GPIb) and glycoprotein VI (GPVI) mediate adhesion to von Willebrand factor and collagen, respectively, and that adhesion triggers signaling pathways including BTK that result in activation of other platelet receptors and further adhesion and aggregation [Citation61,Citation62,Citation68–70].
BTK inhibitor-associated primary hemostatic defects are mediated through effects on both the von Willebrand factor-GPIb and collagen-GPVI axes in platelets, the former via BTK inhibition, the latter via BTK or TEC inhibition. In addition, the activity of C-type lectin-like receptor 2 (CLEC-2), which mediates thrombus stability after platelet adhesion, is impaired by BTK and TEC inhibition. Patients with XLA are not prone to bleedings, but, when tested, show mildly diminished collagen mediated platelet activation, suggesting that the activity of TEC can compensate for the absence of BTK in signaling axes where both tyrosine kinases are active [Citation71]. Moreover, patients with congenital or acquired GPVI deficiency have a similar bleeding diathesis to that noted for ibrutinib [Citation71]. These data suggest that the bleeding risk is a consequence of inhibition of other platelet kinases, such as TEC [Citation68] and is consistent with the observation that the second-generation, more selective BTK inhibitors, such as zanubrutinib and acalabrutinib, have reported lower rates of major bleeding compared with ibrutinib in clinical studies. This suggests that off-target effects of ibrutinib may contribute to bleeding [Citation29,Citation72,Citation73]. Previous studies have demonstrated that ibrutinib affects collagen- and von Willebrand factor-dependent platelet functions but not G-protein coupling-dependent functions [Citation62]. In addition, ibrutinib inhibits platelet integrin αIIbβ3 outside-in signaling and thrombus stability but not adhesion to collagen [Citation74]. Consistent with clinical findings, various mechanistic studies (in vitro, mouse model, and ex vivo) have demonstrated that compared with ibrutinib, zanubrutinib appears to be more selective in its BTK inhibition without the observed effects on platelet function and receptor shedding [Citation75].
The unintended and differential AE profiles among BTK inhibitors including AF may be putatively related to reduced inhibition of TEC or HER2 by zanubrutinib relative to ibrutinib. In particular, zanubrutinib demonstrated selectivity of 88-fold and 176-fold against TEC and HER2, much higher than for ibrutinib, (6.7- and 4.3-fold; ), respectively. It has been hypothesized that ibrutinib-induced AF may be due to on-target inhibition of BTK and related kinases such as TEC, which hitherto were known to have functions in the human heart [Citation76]. McMullen et al. have shown that both BTK and TEC transcripts are expressed in human heart tissue and that the expression of BTK and TEC transcripts is higher in atrial tissue under conditions of AF than sinus rhythm (P < .05), suggesting that BTK and TEC may have functional roles under conditions of cardiac stress [Citation76]. Furthermore, one of the pathways regulated by BTK and TEC is the PI3K-Akt pathway, which is a critical regulator of cardiac protection under stress conditions. Pretorius et al. reported that mice with reduced cardiac PI3K-Akt activity were highly susceptible to the development of AF [Citation77]. C-terminal src kinase (Csk) inhibition as the mechanism by which ibrutinib leads to AF has also been reported [Citation78], and zanubrutinib has been shown to have much lower activity than ibrutinib to inhibit CSK kinase [Citation18]. In another report, it was shown that inhibition of ERBB2/HER2 results in cardiomyocyte dysfunction and reduced heart contractile efficiency [Citation79]. It was also shown that conditional ERBB2/HER2 mutation in ventricular cardiomyocytes leads to impaired cardiac conduction, and the protein is required for atrial electrical activity during development [Citation79].
Similar to ibrutinib, which could improve immunity of CLL/SLL patients, zanubrutinib can regulate immunity primarily by improving T cell exhaustion, inhibiting suppressor cells and disrupting CLL cell migration through downregulation of adhesion/homing receptors [Citation80]. In addition, several BTK inhibitors including evobrutinib, tolebrutinib and fenebrutinib are in clinical development addressing autoimmune disorders (e.g. multiple sclerosis).
The association between kinase inhibition and infection risk in patients treated with BTK inhibitors is unclear. Ibrutinib has been shown to inhibit TEC family kinases TEC, ITK, and BMX. Of these, zanubrutinib has significant less inhibition of JAK3, TEC, and ITK (); it has been postulated that combined inhibition of BTK and TEC may cause the augmented susceptibility to infection and impairment of the ITK kinase could contribute to the increased incidence of infection [Citation65]. Other non-clinical studies on potential mechanisms underlying kinase-associated toxicities for immune functions have also been reported [Citation81]. Off-target inhibition of JAK 3 results in immune dysfunction which may result in greater risk of serious infection [Citation81].
4. Dose selection
The totality of data, including PK, PD, safety, and efficacy data, summarized here was used to support the recommendation of a 320-mg total daily dose (either as 160 mg twice daily or 320 mg once daily) in patients with B-cell malignancies. Given the limited clinical data in patients treated in the QD dose regimen, population pharmacokinetics and exposure–response analyses were employed to bridge the two regimens [Citation67]. To maximize BTK inhibition in target tissues, the 160-mg BID zanubrutinib dose has been extensively studied in completed and ongoing phase 2/3 clinical trials. The clinical efficacy and safety results demonstrated in the pivotal phase 2/3 studies [Citation7,Citation58] provided the primary evidence of effectiveness to support the proposed 160-mg BID dose in patients with B-cell malignancies. However, accumulated and emerging data called for consideration of the 320-mg QD regimen as an option in addition to the 160-mg BID regimen. Although clinical efficacy data are limited at the 320-mg QD dose compared with those at the 160-mg BID dose, both regimens demonstrated comparable daily plasma exposures (AUC) based on population PK analysis [Citation45] and nearly complete BTK occupancy in both PBMC and lymph node tissues. Furthermore, numerically comparable objective responses were observed using QD or BID doses in patients with MCL and other B-cell malignancies including WM and CLL (). Comparable safety profiles were observed with both dosing schedules in patients with various B-cell malignancies [Citation67]. The exposure–response (E-R) analysis for patients with MCL showed that efficacy (ORR) does not appear to be impacted by Cmax or Cmin. In addition, there were no evident E–R relationships between PK exposure (AUC0-24, Cmax, or Cmin) and the safety endpoints. The analyses showed that differences in trough concentration and maximum plasma concentration between the two regimens are unlikely to have a meaningful impact on efficacy and safety endpoints. Taken together, available clinical efficacy and safety data, as well as the E–R analysis, indicate no evidence of differences in clinical efficacy and/or safety profiles as a function of dosing schedule, thus supporting the recommendation for a 320-mg dose administered either once daily or 160-mg BID.
Table 4. Comparison of response rates between two dose regimens of zanubrutinib in patients with B-cell malignancies in study BGB-3111-AU-003
5. Conclusion
Zanubrutinib is an oral, irreversible BTK inhibitor designed to have enhanced selectivity for BTK and improved bioavailability relative to the first-in-class BTK inhibitor, ibrutinib. Consistent with preclinical design principles and hypothesis, data from the randomized phase 3 study that compared zanubrutinib to ibrutinib demonstrate comparable-to-improved efficacy, superior safety, and advantages in quality of life for zanubrutinib compared with ibrutinib in patients with WM. Zanubrutinib treatment was associated with a trend toward better response quality and a lower incidence in AF, hypertension, diarrhea, and bleeding/major bleeding. These clinical observations are consistent with less off-target inhibition of zanubrutinib, including HER2, CSK (AF), EGFR (diarrhea) and TEC (AF, bleeding).
A review of the literature also indicated that among the approved BTK inhibitors, zanubrutinib is less prone to modulations of its PK by intrinsic and extrinsic factors, including food, and hepatic impairment as well as DDI with strong or moderate CYP3A inhibitors and PPIs. These attributes can lead to more consistent and sustained therapeutic exposures and improved dosing convenience. Of note, prophylactic or therapeutic use of azole antifungals (many of which are CYP3A inhibitors) is frequent in the treatment of patients with B-cell malignancies. The magnitude of zanubrutinib exposure impact by concomitant CYP3A inhibitors and hepatic impairment is significantly lower than that for ibrutinib, alleviating potential safety concerns due to a large increase in drug exposure from DDI or organ impairment. In addition, unlike acalabrutinib, zanubrutinib exposure is minimally impacted by concomitant PPI usage. Given a wide prevalence of cancer patients receiving PPI (20% to 55%), zanubrutinib can avoid potential for sub-optimal efficacy due to decreased therapeutic exposures from concomitant use of PPI. Therefore, the increased flexibility in concomitant medication for CYP3A inhibitors and PPI with zanubrutinib is expected to significantly improve the management of clinically relevant drug interactions.
A totality of data and additional analyses were employed to bridge the clinical data of 2 dose regimens (160 mg BID and 320 mg QD) and provided the basis for the recommended 320-mg total daily dose for approved indications. An oral dose of 320 mg QD in addition to the 160 mg BID dose may provide patients and caregivers with additional dosing flexibility and the potential for increased drug adherence.
6. Expert opinion
The market for covalent BTK inhibitors is competitive and includes three licensed compounds, all of which have shown activity in patients with a diverse range of B-cell malignancies. It is likely that the major differentiating factors in the choice of available BTK inhibitor will relate to tolerability of the compound, and ease of prescribing. Major issues with the first-generation drug ibrutinib include cardiovascular and bleeding side-effects, including cardiac arrhythmias, hypertension, and hemorrhages; many of these side-effects are partly ‘on-target’ due to BTK inhibition, exacerbated by off-target inhibition of related enzymes, including TEC, EGFR, HER2, CSK and JAK3. In this respect, the selective profile of zanubrutinib has proven in a head-to-head comparison with ibrutinib to result in reduced side-effects, and improved tolerance. Although it is likely that acalabrutinib will be similarly well tolerated, head-to-head studies against ibrutinib have not been published to date. Additionally, important considerations, such as drug-drug interactions, ability to be prescribed in the presence of PPI and CYP3A4 modulators, and safety in patients with mild-moderate renal and hepatic impairments, all favor zanubrutinib as a safe and effective drug when prescribed in accordance with guidelines. Finally, the recommended total dose of 320 mg, either once daily or as 160 mg twice daily may provide patients and caregivers with additional dosing flexibility and the potential for increased drug adherence to maximize therapeutic benefits.
Article highlights
Zanubrutinib is an oral inhibitor of Bruton’s tyrosine kinase (BTK) designed for greater target selectivity and higher therapeutic exposures than the first-in-class BTK inhibitor ibrutinib.
In clinical studies of B-cell malignancies, zanubrutinib PK properties were unaffected by factors including renal (estimated glomerular filtration rate ≥30 mL/min) and mild/moderate hepatic impairment (Child-Pugh class A or B), and with appropriate dose reductions, it could be administered with moderate or strong CYP3A inhibitors; zanubrutinib can be administered concurrently with proton pump inhibitors (PPI)/acid-reducing agents without restriction.
The totality of evidence—including PK/PD properties, safety, efficacy, and exposure-response analyses—provided the basis for the recommended 320-mg dose either once daily or as 160 mg BID.
The greater selectivity of zanubrutinib as well as its PK/PD profiles translates into clinically impactful benefits, including improved dosing flexibility, safety, and efficacy.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author Contributions
All authors have substantially contributed to the conception and design of the review article and interpreting the relevant literature, and been involved in writing the review article or revising for intellectual content.
Acknowledgments
We thank study patients, their supporters, and investigators and clinical research staff at study centers. Editorial assistance was provided by Bio Connections, LLC, (Chicago, IL), and funded by BeiGene USA, Inc.
Disclosure Statement
CS Tam receives research funding from Janssen and AbbVie; honoraria from Janssen, AbbVie, BeiGene, Novartis, and Roche. YC Ou is an employee of BeiGene, Inc. and may own shares/stock options in BeiGene. J Trotman receives research funding from BeiGene, Roche, Pharmacyclics, Celgene, and Takeda. S Opat has a consulting/advisory role with AbbVie, Janssen, Gilead, Roche, Mundipharma, Merck, BMS, and Celgene; receives research funding from AbbVie, BeiGene, Janssen, Gilead, Roche, Celgene, and Epizyme; and honoraria from AbbVie, Janssen, Gilead, Roche, Mundipharma, Merck, BMS, and Celgene. Authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Additional information
Funding
References
- Dal Porto JM, Gauld SB, Merrell KT, et al. B cell antigen receptor signaling 101. Mol Immunol. 2004;41(6–7):599–613.
- Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2(12):945–956.
- Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013;13(8):578–591.
- Niemann CU, Wiestner A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin Cancer Biol. 2013;23(6):410–421.
- Xu W, Song Y, Li Z, et al. Safety, tolerability and efficacy of orelabrutinib, once a day, to treat Chinese patients with relapsed or refractory chronic lymphocytic leukemia/small cell leukemia. Blood. 2019;134(Suppl_1):4319.
- Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892–901.
- Song Y, Zhou K, Zou D, et al. Treatment of patients with relapsed or refractory mantle-cell lymphoma with zanubrutinib, a selective inhibitor of Bruton’s tyrosine kinase. Clin Cancer Res. 2020;26(16): 4216–4224.
- Tam CS, Opat S, Simpson D, et al. Zanubrutiib for the treatment of relapsed or refractory mantle cell lymphoma. Blood Adv. 2021;5(12):2577–2585.
- Brukinsa® [package insert]. Beijing China: BeiGene Co. Ltd; 2019 November.
- BeiGene Press Release. BeiGene announces the approval of BRUKINSATM (Zanubrutinib) in China for patients with relapsed/refractory chronic lymphocytic leukemia or small lymphocytic lymphoma and relapsed/refractory mantle cell lymphoma 2020. [cited 2021 Aug 1]. Available from: http://ir.beigene.com/news-releases/news-release-details/beigene-announces-approval-brukinsatm-zanubrutinib-china]
- Tam CSL, Trotman J, Opat S, et al. Phase 1 study of selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851–859.
- Xu W, Yang S, Zhou K, et al. Treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma with the BTK inhibitor zanubrutinib: phase 2, single-arm, multicenter study. J Hematol Oncol. 2020;13(1):48.
- BeiGene Press release. Health Canada approves brukinsa® (zanubrutinib) for the treatment of waldenstrom’s macroglobulinemia. 2021. [cited 2021 Aug 1]. Available from https://ir.beigene.com/news-releases/news-release-details/health-canada-approves-brukinsar-zanubrutinib-treatment].
- Hillmen P, Eichhorst B, Brown JR, et al. First interim analysis of ALPINE study: results of a phase 3 randomized study of zanubrutinib vs ibrutinib in patients with relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma. EHA 2021. LB 1900.
- US Food and Drug Administration Center for Drug Evaluation and Research (Ibrutinib ImbruvicaTM). Application number 20552Orig2s000. [cited 2021 Aug 1]. Available at https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205552Orig2s000ClinPharmR.pdf]
- Herman SE, Mustafa RZ, Gyamfi JA, et al. Ibrutinib inhibits BCR and NF-kappa B signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123(21):3286–3295.
- Imbruvica® (ibrutinib) [package insert]. Sunnyvale Ca: Pharmacyclics, LLC; 2015.
- Guo Y, Liu Y, Hu NY, et al., Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J Med Chem. 62(17): 7923–7940. 2019.
- Barf T, Covey T, Izumi R, et al. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J Pharmacol Exp Ther. 2017;363(2):240–252.
- Kohrt HE, Sagiv-Barfi I, Rafiq S, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123(12):1957–1960.
- Burger JA, Sivina M, Jain N, et al. Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood. 2018;133(10):1011–1019.
- Woyach J, Ruppert AS, Perez G, et al. Alliance A041702: a randomized phase III study of ibrutinib plus obinutuzumab versus ibrutinib plus venetoclax and obinutuzumab in untreated older patients (≥ 70 years of age) with chronic lymphocytic leukemia (CLL). Blood. 2019;134(Supplement_1):1751.
- Shanafelt TD, Wang XV, Kay NE, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–443.
- Kaptein A, De Bruin G, Emmelot-van Hoek M, et al. Potency and selectivity of BTK inhibitors in clinical development for B-Cell malignancies. Blood. 2018;132(Supplement 1):1871.
- Wang K, Yao X, Zhang M, et al. Comprehensive PBPK model to predict drug interaction potential of zanubrutinib as a victim or perpetrator. CPT Pharmacometrics Syst Pharmacol. 2021;10(5):441–454. epub ahead of print.
- De Vries R, Smit JW, Hellemans P, et al. Stable isotope-labelled intravenous microdose for absolute bioavailability and effect of grapefruit juice on ibrutinib in healthy adults. Br J Clin Pharmacol. 2016;81(2):235–245.
- Canada H. Brukinsa®Product Monograph. Canada; BeiGene, 2021.
- Calquence® [package insert]. AstraZeneca Gaithersburg MD. November 2019.
- Byrd JC, Harrington B, O’Brien S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–332.
- Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94.
- DImopoulos M, Opat S, Lee H-P, et al. Major responses in MYD88 wildtype (MYD88WT) Waldenstrom macroglobulinemia (WM) patients treated with Bruton tyrosine kinase (BTK) inhibitor zanubrutinib (BGB-3111) 2019 [cited 2021 Aug 1]. Available from: https://library.ehaweb.org/eha/2019/24th/266287/meletios.a.dimopoulos.major.responses.in.myd88.wildtype.%28myd88wt%29.waldenstrm.html].
- Alsadhan A, Cheung J, Gulrajani M, et al. Pharmacodynamic analysis of BTK inhibition in patients with chronic lymphocytic leukemia treated with acalabrutinib. Clin Cancer Res. 2020;26(12):2800–2809.
- Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. 69.
- Sun C, Nierman P, Kendall EK, et al. Clinical and biological implications of target occupancy in CLL treated with the BTK inhibitor acalabrutinib. Blood. 2020;136(1):93–105.
- Moreau P, Mateos MV, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018;19(7):953–964.
- Saffran DC, Parolini O, Fitch-Hilgenberg ME, et al. A point mutation in the SH2 domain of Bruton’s tyrosine kinase in atypical X-linked agammaglobulinemia. N Engl J Med. 1994;330(21):1488–1491.
- US Food and Drug Administration Center for Drug Evaluation and Research. Application number 213217Orig1s00 multi-discipline review package. 2019 [ Bruinksa] [cited 2021 Aug 1]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213217Orig1s000MultidisciplineR.pdf].
- de Jong J, Sukbuntherng J, Skee D, et al. The effect of food on the pharmacokinetics of oral ibrutinib in healthy participants and patients with chronic lymphocytic leukemia. Cancer Chemother Pharmacol. 2015;75(5):907–916.
- US Food and Drug Administration Center for Drug Evaluation and Research. Approval package for application number 210259Orig1S006, 210259Orig1s007 (CALQUENCE capsules). [cited 2021 Aug 1]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/210259Orig1s006,%20s007.pdf].
- Podoll T, Pearson PG, Evarts J, et al. Bioavailability, biotransformation, and excretion of the covalent Bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab Dispos. 2019;47(2):145–154.
- Scheers E, Leclercq L, de Jong J, et al. Absorption, metabolism, and excretion of oral (1)(4)C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos. 2015;43(2):289–297.
- Wong J, Cher L, Griffiths J, et al. Efficacy of zanubrutinib in the treatment of bing-neel syndrome. Hemasphere. 2018;2(6):e155.
- Goldwirt L, Beccaria K, Ple A, et al. Ibrutinib brain distribution: a preclinical study. Cancer Chemother Pharmacol. 2018;81(4):783–789.
- Bernard S, Goldwirt L, Amorim S, et al. Activity of ibrutinib in mantle cell lymphoma patients with central nervous system relapse. Blood. 2015;126(14):1695–1698.
- Ou YC, Liu L, Tariq B, et al. Population pharmacokinetic analysis of the BTK inhibitor zanubrutinib in healthy volunteers and patients with B-Cell malignancies. Clin Transl Sci. 2021;14(20):764–772.
- Ou YC, Preston RA, Marbury TC, et al. A phase 1, open-label, single-dose study of the pharmacokinetics of zanubrutinib in subjects with varying degrees of hepatic impairment. Leuk Lymphoma. 2020;61(6):1355–1363.
- Mu S O, Tang Z, Novotny W, et al. Effect of rifampin and itraconazole on the pharmacokinetics of zanubrutinib (a Bruton’s tyrosine kinase inhibitor) in Asian and non-Asian healthy subjects. Cancer Chemother Pharmacol. 2020;85(2):391–399.
- Marostica E, Sukbuntherng J, Loury D, et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol. 2015;75(1):111–121.
- Edlund H, Lee SK, Andrew MA, et al. Population pharmacokinetics of the BTK inhibitor acalabrutinib and its active metabolite in healthy volunteers and patients with B-cell malignancies. Clin Pharmacokinet. 2019;58(5):659–672.
- Bruggemann RJ, Alffenaar JW, Blijlevens NM, et al. Clinical relevance of the pharmacokinetic interactions of azole antifungal drugs with other coadministered agents. Clin Infect Dis. 2009;48(10):1441–1458.
- de Jong J, Skee D, Murphy J, et al. Effect of CYP3A perpetrators on ibrutinib exposure in healthy participants. Pharmacol Res Perspect. 2015;3(4):e00156.
- Finnes HD, Chaffee KG, Call TG, et al. Pharmacovigilance during ibrutinib therapy for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) in routine clinical practice. Leuk Lymphoma. 2017;58(6):1376–1383.
- Barr PM, Brown JR, Hillmen P, et al. Impact of ibrutinib dose adherence on therapeutic efficacy in patients with previously treated CLL/SLL. Blood. 2017;129(19):2612–2615.
- de Jong J, Hellemans P, Jiao J, et al. An open-label, sequential-design drug interaction study of the effects of omeprazole on the pharmacokinetics of ibrutinib in healthy adults. Blood. 2016;128(22):1588.
- Budha NR, Frymoyer A, Smelick GS, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92(2):203–213.
- Ou YC, Tang Z, Novotny W, et al. Evaluation of drug interaction potential of zanubrutinib with cocktail probes representative of CYP3A4, CYP2C9, CYP2C19, P-gp and BCRP. Br J Clin Pharmacol. 2021;87(7):2926–2936.
- Zhou D, Podoll T, Xu Y, et al. Evaluation of the drug-drug interaction potential of acalabrutinib and Its active metabolite, ACP-5862, using a physiologically-based pharmacokinetic modeling approach. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):489–499.
- Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib versus ibrutinib in symptomatic Waldenstrom macroglobulinemia: the Aspen study. Blood. 2020136(18):2038–2050.
- de Jong J, Hellemans P, Jiao JJ, et al. Ibrutinib does not prolong the corrected QT interval in healthy subjects: results from a thorough QT study. Cancer Chemother Pharmacol. 2017;80(6):1227–1237.
- Mu S, Darpo B, Tang Z, et al. No QTc prolongation with zanubrutinib: results of concentration-QTc analysis from a thorough QT study in healthy subjects. Clin Transl Sci. 2020;13(5):923–931.
- Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–787.
- Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124(26):3991–3995.
- Tam CS, Opat S, Zhu J, et al. Pooled analysis of safety data from monotherapy studies of the Bruton tyrosine kinase (BTK) inhibitor, zanubrutinib (BGB-3111) in B-cell malignancies. 24th European Hematology Association Congress; June 13–16, 2019; Amsterdam, The Netherlands 2019.
- Tang CPS, McMullen J, Tam C. Cardiac side effects of bruton tyrosine kinase (BTK) inhibitors. Leuk Lymphoma. 2018;59(7):1554–1564.
- Estupiñán YH, Berglöf A, Zain R, et al. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front Cell Dev Biol. 2021;9:630942.
- Farrar JE, Rohrer J, Conley ME. Neutropenia in X-linked agammaglobulinemia. Clin Immunol Immunopathol. 1996;81(3):271–276.
- Ou YC, Tang Z, Novotny W, et al. Rationale for once-daily or twice-daily dosing of zanubrutinib in patients with mantle cell lymphoma. Leuk Lymphoma. 2021;1–13. epub ahead of print. https://doi.org/10.1080/10428194.2021.1929961.
- Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592–3599.
- Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–1140.
- Rigg RA, Aslan JE, Healy LD, et al. Oral administration of Bruton’s tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am J Physiol Cell Physiol. 2016;310(5):C373–80.
- Shatzel JJ, Olson SR, Tao DL, et al. Ibrutinib-associated bleeding: pathogenesis, management and risk reduction strategies. J Thromb Haemost. 2017;15(5):835–847.
- Bye AP, Unsworth AJ, Desborough MJ, et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1(26):2610–2623.
- Tam CS, LeBlond V, Novotny W, et al. A head-to-head Phase III study comparing zanubrutinib versus ibrutinib in patients with Waldenstrom macroglobulinemia. Future Oncol. 2018;14(22):2229–2237.
- Bye AP, Unsworth AJ, Vaiyapuri S, et al. Ibrutinib inhibits platelet integrin αIIbβ3 outside-in signaling and thrombus stability but not adhesion to collagen. Arterioscler Thromb Vasc Biol. 2015;35(11):2326–2335.
- Dobie G, Kuriri FA, Omar MMA, et al., Ibrutinib, but not zanubrutinib, induces platelet receptor shedding of GPIb-IX-V complex and integrin αIIbβ3 in mice and humans. Blood Adv. 2019;3(24):4298–4311.
- McMullen JR, Boey EJ, Ooi JY, et al. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124(25):3829–3830.
- Pretorius L, Du XJ, Woodcock EA, et al. Reduced phosphoinositide 3-kinase (p110alpha) activation increases the susceptibility to atrial fibrillation. Am J Pathol. 2009;175(3):998–1009.
- Xiao L, Salem JE, Clauss S, et al. Ibrutinib-mediated atrial fibrillation attributable to inhibition of C-terminal Src kinase. Circulation. 2020;142(25):2443–2455.
- Berglof A, Hamasy A, Meinke S, et al. Targets for ibrutinib beyond B cell malignancies. Scand J Immunol. 2015;82(3):208–217.
- Zou YX, Zhu HY, Li XT, et al. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemica/small lymphocytic lymphoma. Hematol Oncol. 2019;37(4):392–400.
- Choy EH. Clinical significance of Janus kinase inhibitor selectivity. Rheumatology (Oxford). 2019;58(6):953–962.
- Tam CS, Wang M, SImpson D, et al. Updated safety and efficacy data in the phase 1 trial of patients with mantle cell lymphoma (MCL) treated with Bruton tyrosine kinase (BTK) inhibitor zanubrutinib (BGB-3111). Hematol Oncol. 2019;37:245–247.
- Trotman J, Opat S, Gottlieb D, et al. Zanubrutinib for the treatment of patients with waldenstrom macroglobulinemia: 3 years of follow-up. Blood. 2020;136(18):2027–2037.