
ABSTRACT
Introduction
Estrogens used in women’s healthcare have been associated with increased risks of venous thromboembolism (VTE) and breast cancer. Estetrol (E4), an estrogen produced by the human fetal liver, has recently been approved for the first time as a new estrogenic component of a novel combined oral contraceptive (E4/drospirenone [DRSP]) for over a decade. In phase 3 studies, E4/DRSP showed good contraceptive efficacy, a predictable bleeding pattern, and a favorable safety and tolerability profile.
Areas covered
This narrative review discusses E4ʹs pharmacological characteristics, mode of action, and the results of preclinical and clinical studies for contraception, as well as for menopause and oncology.
Expert opinion
Extensive studies have elucidated the properties of E4 that underlie its favorable safety profile. While classical estrogens (such as estradiol) exert their actions via both activation of nuclear and membrane estrogen receptor α (ERα), E4 presents a specific profile of ERα activation: E4 binds and activates nuclear ERα but does not induce the activation of membrane ERα signaling pathways in specific tissues. E4 has a small effect on normal breast tissue proliferation and minimally affects hepatic parameters. This distinct profile of ERα activation, uncoupling nuclear and membrane activation, is unique.
1. Introduction
The widespread physiological role of estrogen receptors (ERs) probably contributes to the pleiotropic actions of estrogens [Citation1,Citation2]. Whereas estrogens are traditionally considered as simply sex hormones responsible for the development and regulation of the female reproductive system and secondary sex characteristics, they actually induce changes in almost all vertebrate cells. Estrogens are ubiquitous, impacting on almost all physiological systems and tissues, including the urinary tract and cardiovascular system, as well as the brain, bone, breast, skin, hair, mucosa, pelvic muscles, and eating behavior [Citation3,Citation4]. Estrogens impact the liver, effecting glucose tolerance, and lipid homeostasis [Citation5]. Furthermore, estrogens are also involved in diseases, such as breast, endometrial, ovarian, colorectal, and prostate cancers, along with endometriosis, deep venous thromboembolism, and some autoimmune diseases [Citation2]. In contrast, they are protective against osteoporosis, obesity, insulin resistance, cardiovascular events, and neurodegenerative diseases [Citation2].
Four natural estrogens are found in human species over the course of life (). The names and abbreviations reflect the number of hydroxyl groups present on the 4-ring backbone, as is similar for all hormones. Estrone (E1) is present throughout life and is considered the primary estrogen during the menopausal years in women. Estradiol (E2), produced by the ovaries, is the primary estrogen during the reproductive years. Estriol (E3) is produced naturally by the placenta and is the major estrogen during pregnancy. Lastly, estetrol (E4) is the estrogen of fetal life, produced by the fetal liver, and present only during pregnancy with relatively high levels in the fetus and lower levels in the maternal circulation. Interestingly, whereas E1, E2, and E3 are found in other mammalian species, E4 is primarily only found in humans, present as early as 9 weeks of gestation. Some higher order mammals have limited levels of E4 present but only in the last few weeks of gestation. The unique role of E4 in humans, compared to lower-order mammals, is still not understood.
Figure 1. Structure of the four natural estrogens, estrogen receptors (α and β) and ligand-binding domains. A: Structure of natural estrogens. E1 and E2: adult estrogens; E3 and E4: fetal estrogens. B: Estrogen receptors α and β (ERα and ERβ). Functional domains include the DNA-binding domain (DBD), ligand-binding domain (LBD), and two transcriptional activation functions, AF-1 and AF-2. The A/B domain, at the protein amino terminus (NH2), contains AF-1. The C domain binds to DNA motifs (consensus estrogen response elements [EREs]). The D domain (the hinge region) is involved in specific DNA binding and nuclear ER localization. The E domain is the LBD: it interacts with estrogens and selective estrogen receptor modulators (SERMs) and contains the AF-2 region. The carboxy terminus (COOH) is at the F domain. C&D: Ribbon diagrams. ERα-LBD structure complexed with estrogens (blue:E2; red:E3; green:E4). C: Ball-and-stick rendering of the ligand-binding site of the ligands and their interacting residues. Dotted lines: hydrogen bonds. D: LBDs depicted in C and the glucocorticoid receptor-interacting protein 1 (GRIP1) peptide fragment coactivator protein complexed with E3 or E4 only (darker red/green). Ligand: represented as space-filled model. Helix 12 position depicted by an arrow. Panel B adapted from Arnal JF, et al (2017) [Citation3], and panels C and D from Abot A, et al (2014) [Citation48].
![Figure 1. Structure of the four natural estrogens, estrogen receptors (α and β) and ligand-binding domains. A: Structure of natural estrogens. E1 and E2: adult estrogens; E3 and E4: fetal estrogens. B: Estrogen receptors α and β (ERα and ERβ). Functional domains include the DNA-binding domain (DBD), ligand-binding domain (LBD), and two transcriptional activation functions, AF-1 and AF-2. The A/B domain, at the protein amino terminus (NH2), contains AF-1. The C domain binds to DNA motifs (consensus estrogen response elements [EREs]). The D domain (the hinge region) is involved in specific DNA binding and nuclear ER localization. The E domain is the LBD: it interacts with estrogens and selective estrogen receptor modulators (SERMs) and contains the AF-2 region. The carboxy terminus (COOH) is at the F domain. C&D: Ribbon diagrams. ERα-LBD structure complexed with estrogens (blue:E2; red:E3; green:E4). C: Ball-and-stick rendering of the ligand-binding site of the ligands and their interacting residues. Dotted lines: hydrogen bonds. D: LBDs depicted in C and the glucocorticoid receptor-interacting protein 1 (GRIP1) peptide fragment coactivator protein complexed with E3 or E4 only (darker red/green). Ligand: represented as space-filled model. Helix 12 position depicted by an arrow. Panel B adapted from Arnal JF, et al (2017) [Citation3], and panels C and D from Abot A, et al (2014) [Citation48].](/cms/asset/93cece59-4bd5-4205-a703-14f369ab4441/ierj_a_2054413_f0001_oc.jpg)
Estrogens were first synthesized in the laboratory almost 100 years ago with purification of E1 in 1929 and the discovery of E2 in 1931 and E3 in 1933. Within 5 years of their discovery, E1, E2, and E3 were introduced as potential therapeutic agents. Ethinyl estradiol (EE) was the first synthetic estrogen, produced in 1938, and marketed in 1943 for the treatment of dysmenorrhea [Citation6]. Conjugated equine estrogens (CEEs), a mixture of estrogen conjugates found in the urine of mares, were introduced in 1941 for the relief of hot flushes in postmenopausal women. Since then, no new natural estrogens have been characterized and evaluated for the benefit of women’s health for over 80 years.
The use of natural or synthetic estrogens is associated with a number of risks, especially related to liver and cardiovascular factors. These effects are related to the route of delivery, with oral methods directly impacting the liver through first-pass metabolism, and the type of estrogen, with the highly potent synthetic estrogen EE impacting the liver regardless of route of delivery.
The use of estrogens in combined oral contraceptives (COCs) has also been linked to an increased risk of thromboembolic events, including venous thromboembolic events (VTE) or pulmonary embolism [Citation7,Citation8]. An increased risk of breast cancer in women using estrogen-based hormonal contraceptives or menopausal hormone therapy (MHT) has also been reported [Citation9,Citation10]. An estrogen-based pharmaceutical that offers safer use, including a lower risk of thromboembolic events and less impact on breast tissue proliferation, has been an important unmet need in women’s health [Citation11,Citation12].
E4 has recently been approved in the United States of America (USA), Europe (EU), Canada, and Australia as a new estrogenic component of a COC and is also in clinical development for the treatment of menopausal symptoms as well as breast and prostate cancer. These clinical advancements are the culmination of decades of research elucidating the properties of E4 that underlie its favorable safety profile. Here, we review the characterization of E4 with preclinical genetically modified animal models and biochemical and molecular biology investigations that have guided the progression into clinical studies in contraception and menopause. In addition, we discuss E4ʹs unique molecular mechanism of action along with providing insight into its tissue-specific actions. Finally, we review the pivotal phase 3 studies that have supported the marketing authorization of E4.
2. Introduction to estetrol (E4)
E4 was first described by the group of Diczfalusy in 1965 [Citation13–16]. For several decades, E4 was perceived as a weak estrogen with no therapeutic potential [Citation17,Citation18]. However, in 2001 Coelingh Bennink et al. at Pantarhei Bioscience (Zeist, the Netherlands) initiated preclinical and phase 1 clinical studies that described E4ʹs unique profile of activity and its suitability for therapeutic use [Citation19]. Beginning in 2009, Pantarhei, Mithra Pharmaceuticals, and several academic groups joined efforts to examine the pharmacological characteristics of E4 and its molecular mechanisms of action with the aim of developing E4 for therapeutic use. For human use, E4 is synthesized from commercially available soy estrone. It is over 99.9% pure [Citation20] without contamination [Citation21].
The initial phase 1 and 2 clinical trials focused on using E4 in a COC. The efficacy and safety of a new COC consisting of E4 15 mg (as monohydrate, equivalent to 14.2 mg anhydrate) combined with the progestogen drospirenone (DRSP) 3 mg has been studied in two phase 3 trials in both the USA/Canada and EU/Russia [Citation22,Citation23]. Its usage has been recently approved by the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), Health Canada, and the Therapeutic Goods Administration (TGA) in Australia. Two other phase 3 trials are ongoing for the relief of menopausal symptoms: one in USA/Canada with E4 alone (15 or 20 mg) in both hysterectomized and non-hysterectomized women (NCT04090957), and one in EU/Russia/North and Latin America with E4 alone (15 or 20 mg) in hysterectomized and non-hysterectomized women or E4 (20 mg) in association with progesterone in non-hysterectomized women (NCT04209543) [Citation24–28]. Phase 2 studies examining the efficacy of E4 for the treatment of advanced breast cancer and for prostate cancer have also been conducted, demonstrating anti-tumor effects of relatively high doses of E4 (20–40 mg) in both cancers, with no safety concerns [Citation29,Citation30]. depicts the clinical applications of E4 that are currently being studied in humans.
Table 1. Clinical applications of E4
As E4 is a natural estrogen, the efficacious dose that is associated with its most optimal efficacy and safety profile is expressed in milligrams. This is similar to E2, another natural estrogen also used in milligrams for contraception, but in contrast to EE, a highly potent synthetic estrogen used in micrograms. The daily dose for the therapeutic use of other natural steroids is also in the milligram range, for example, progesterone 100–200 mg and cortisol 30–60 mg per day.
2.1. Chemistry
E4 is structurally different from the other estrogens because of the presence of an additional hydroxyl group at position 15-α of the molecule, resulting in a total of 4 hydroxyl groups (). Its concentration in fetal and maternal plasma increases throughout pregnancy to reach a maximum at the end of pregnancy (up to 1.2 ng/mL in maternal plasma). Fetal plasma levels of E4 are higher than maternal plasma levels at parturition [Citation31,Citation32].
2.2. Pharmacokinetics
Following oral administration, E4 undergoes fast absorption (tmax ranges between 0.25 and 0.5 hours), followed by rapid distribution [Citation33]. The elimination half-life of E4 is 24–32 hours, which is longer than other natural estrogens, such as E2 and micronized E2, which have half-lives of 1–2 hours and 10–12 hours, respectively. E4 undergoes extensive phase 2 metabolism in the liver where it is glucuronized mainly by the uridine 5’-diphospho-glucuronosyltransferase (UGT) enzyme UGT2B7 to form glucuronide and sulfate conjugates that are excreted in urine [Citation34,Citation35]. Importantly, E4 is an end-stage product of metabolism, which is not converted in vivo into active metabolites like E3, E2, or E1. Unlike E2, E4 is not converted into hydroxylated metabolites, precursors of quinone estrogens that can react and damage the DNA, which has been linked to breast cancer development [Citation36,Citation37]. Moreover, circulating levels of E1 have been linked to thrombin generation and are consequently associated with the different thrombotic risk profiles of oral and transdermal hormone therapy [Citation38]. Unlike EE and E2, cytochrome P450 (CYP450) enzymes do not play a major role in the metabolization of E4 and E4 shows minimal impact on the major CYP450 enzymes [Citation19,Citation39–41]. As a result, E4 may potentially exhibit fewer drug–drug interactions relative to other estrogens, including EE and E2.
Further, unlike other estrogens, E4 neither binds sex hormone-binding globulin (SHBG), nor increases its production by hepatic cells as much as other estrogens [Citation41]. The first is an important and unique characteristic of E4, as only non-protein bound estrogens are biologically active and able to reach target tissues [Citation42].
Enterohepatic recirculation of estrogen therapies has been linked to a 2.5-fold increased risk of biliary tract conditions, including benign or malignant gallbladder disease [Citation43]. This risk may not apply to E4 as it does not appear to enter the enterohepatic circulation. E4 is excreted in urine in the form of a ring D monoglucuronide and is otherwise metabolically unaltered [Citation44–46]. Collectively, the pharmacokinetic profile of E4 is unique, with desirable features that have formed the basis for the development of a safe once daily oral pharmaceutical compound [Citation47].
2.3. Mode of action of E4
2.3.1. Introduction to the molecular mechanisms of action of estrogens
The pleiotropic effects of estrogens are mainly mediated through the two ERs: ERα and ERβ [Citation3,Citation48]. The two ERs share a domain structure, which comprises six domains, A–F (). The central C domain binds with high affinity and specificity to specific estrogen responsive element (ERE) sequences of DNA. Ligand binding occurs at the COOH-terminal E domain via the ligand-binding domain (LBD). Two estrogen-dependent activation functions (activation function [AF]-1 and AF-2) are present within ERs.
In the classical mechanism of ER action, ERα and ERβ function as ligand-dependent factors that regulate the transcription of target genes containing the consensus ERE in their promoter regions (Supplemental Figure 1A). Estrogens bind to the ERs in the cytoplasm, and ER-ligand complexes then dimerize and translocate to the nucleus, where they interact with ERE DNA sequences in target genes. Through AF-1 and AF-2, the estrogen-ER complexes then recruit coregulators and components of the RNA polymerase II complex that will subsequently regulate the transcription of target genes and initiate nuclear actions. ERs can also indirectly initiate nuclear actions by interacting with other transcription factors, such as activator protein 1 and specificity protein 1 that mediate the transcription of genes whose promoters do not harbor an ERE (Supplemental Figure 1B). Some growth factors can also mediate nuclear effects by activating intracellular signal transduction pathways, leading to the phosphorylation and activation of ERs and the modulation of their transcriptional activity (Supplemental Figure 1C) (for a review see Arnal et al. 2017) [Citation3].
The nuclear transcriptional actions of ERα do not, however, account for all the biological functions of estrogens. A pool of ERα is also associated with plasma membrane caveolae/lipid rafts, where it can activate rapid non-nuclear signaling, termed nongenomic/membrane-initiated steroid signaling (MISS) [Citation3,Citation49–52]. Caveolae, a subset of lipid rafts, are specialized cholesterol-rich plasma membrane organelles that compartmentalize signal transduction molecules on the cell surface via, for example, myristoylation and palmitoylation. This membrane ERα activity results in the activation of intracellular signaling pathways (e.g. phosphoinositide 3-kinase [PI3K], mitogen-activated protein kinase [MAPK]), the activation of multiple kinases and the production of a variety of downstream second messengers (e.g. nitric oxide [NO], calcium flux, and cyclic adenosine monophosphate), directly influencing cell activities that contribute to the regulation of cell survival and proliferation (Supplemental Figure 1D).
Nuclear and membrane pathways interact, which is still poorly recognized. The kinases activated by the MISS pathway can phosphorylate various transcription factors, including ERs and coregulators, and therefore indirectly modulate the transcriptional activity in the nucleus [Citation53,Citation54]. Besides ERα, MISS actions can also be induced by several other receptors located at or near the membrane. ERβ is present at the membrane and induces rapid nongenomic responses [Citation55]. The more recently described G Protein-Coupled Estrogen Receptor (GPER) is present at the membrane and is involved in the rapid activation of several signaling pathways by estrogens in different cell types [Citation56].
2.3.2. The mode of action of E4: E4 has tissue specific properties
Using pharmacological tools and genetically modified animal models, E4 has been identified as a specific nuclear ERα activator in vivo, uncoupling membrane, and nuclear ERα activation. E4 activates the nuclear ERα, similarly to E2, but E4 does not activate the membrane ERα pathway in specific tissues. In a mouse model of electric injury of the carotid artery, for instance, E2 but not E4 accelerated reendothelialization; E4 even was able to inhibit the acceleration of endothelial healing induced by E2 [Citation48]. Based on several other in vivo and in vitro experiments, it was concluded that E4 is able to elicit nuclear ERα actions (for example, in the uterus) as does E2, but E4 does not elicit and even antagonizes the membrane-initiated actions of ERα at variance to E2 [Citation3,Citation48].
In contrast, several in vitro studies including our own have described the presence of MISS actions after treatment with E4, in several cell contexts. It was shown using cell cultures that E4 activates extra-nuclear signaling cascades, such as MAPK and PI3K/Protein Kinase B pathways, which are involved in many biological processes including human breast cancer cell growth and survival. However, the receptor at the origin of this MISS actions was not investigated [Citation54]. The impact of E4 on ERα MISS in a human breast cancer cell line (Michigan Cancer Foundation-7 [MCF-7]) was also investigated by highlighting the interaction at the membrane between ERα and the tyrosine kinase src using the Proximity Ligation Assay technique. E4 significantly induced this interaction but was considerably less efficient in inducing this interaction compared to E2. Importantly, when administered together, the combination of E2 and E4 totally abrogated the ERα-scr interaction [Citation48].
Finally, Giretti et al. 2014 showed that E4 weakly induced human breast cancer cell invasion and migration via a rapid phosphorylation of moesin but, again, the efficacy of E4 to induce these processes was much lower compared to E2 [Citation57]. In co-treatment with E2, E4 blocked the activation of this actin controller in a concentration-related fashion. The same group addressed the effects of E4 on the activity and expression of the endothelial nitric oxide synthase (eNOS) in cultured human umbilical vein endothelial cells (HUVEC). E4 stimulated the activation of eNOS and NO production in HUVEC but was significantly less effective compared to E2. When E2 was combined with E4, E4 antagonized NO synthesis induced by pregnancy-like E2 concentrations [Citation58].
In summary, while a complete absence of ERα MISS actions after treatment with E4 is observed in the mouse model of carotid artery reendothelialization after perivascular injury, a limited activation of ERα MISS can be observed in other experimental or cellular contexts. The efficacy of E4 to induce these ERα MISS actions always appears to be much lower compared to E2. Moreover, in all studies cited above, E4 consistently antagonized ERα MISS actions induced by E2. This unique behavior of E4 regarding the activation of ERα-MISS is believed to support, at least in part, differences between E4 and other estrogens in terms of biological and clinical effects.
In conclusion, E4 induces nuclear actions but not the MISS effects and, as a result, has the ability to act selectively as an agonist or antagonist depending on the target cell or tissue. Cellular and transgenic mouse models contributed to precisely delineating this tissue specific profile of E4, and the data indicate that E4 elicits potent estrogenic-like effects on the brain [Citation59,Citation60], bone tissue [Citation61], the cardiovascular system [Citation48], ovulation [Citation62], and the uterus [Citation48,Citation63]. These estrogenic-like effects are mediated by activation of the ERα nuclear pathway, since they are abolished in transgenic mice (ERα knock-out, ERαAF1°, ERα-AF2°) or in the presence of fulvestrant, a selective ER degrader [Citation64]. For instance, in animals, it has been demonstrated that E4 stimulates uterine gene expression, and proliferation of epithelial endometrial cells and vaginal keratinocytes. These effects are abrogated in ERαAF2° or ERαAF1° mice [Citation48,Citation65], whereas they are fully preserved using a mouse with a point mutation of the palmitoylation site of ERα (C451A-ERα), which leads to membrane-specific loss of ERα function [Citation66]. In postmenopausal women, various doses of E4 increased endometrial thickness, induced endometrial proliferation, and reversed vaginal atrophy [Citation28].
Data from the preclinical studies also suggest that E4 has anti-estrogenic-like effects in the breast and a limited impact on normal or malignant breast cell proliferation. This property of E4 is associated with its antitumor activity in the presence of E2 and is thought to be linked to differential activation of signaling pathways and not E2-induced nuclear activity blockade.
In the liver, E4 has a minimal impact on liver gene expression, which is suggested to contribute to the low impact of E4 on several hepatic functions (i.e. small changes in liver enzyme values and coagulation parameters) and which is very different from that of E2 and EE.
The selective action of E4 in tissues led to the question of whether this natural estrogen is a selective estrogen receptor modulator (SERM) like tamoxifen and raloxifene [Citation48]. Like estrogens, SERMs bind to ERs with high affinity, and regulate transcriptional events characterized by tissue-specific actions, being agonists and mimicking the effects of estrogen in some tissues, but antagonizing their effects in others [Citation67]. SERMs and estrogens differ, however, as seen in the effects that occur in the LBD of ERα [Citation68]. The LBD is composed of 12 α helices (H1-H12) and 2 antiparallel β sheets. This helical arrangement creates a scaffold that maintains a ligand-binding cavity ( & D). However, E4 is distinct from SERMs as it binds ERα differently than SERMs and in a manner more similar to estrogens.
Estrogen binding induces a major structural reorganization of the LBD, which converts the inactive ER to the functionally active form. When E2 binds to its LBD, H12 packs against helices 3, 5/6, and 11, forming the coactivator-binding groove recognized by the leucine (L) amino acid (X)XLL motifs of coactivators. This positioning of H12 is a prerequisite for transcriptional activation as it induces the activation of the receptor’s AF2 transactivation function that binds numerous co‐regulatory proteins. In contrast, SERMs alter the conformation of the LBD of ERα. The position of H12 in relation to the other helices of the LBD differs when SERMs occupy the ligand-binding pocket. This characteristic orientation of H12 in the presence of tamoxifen occludes the coactivator-binding groove, whereas it allows the release of the A domain and possible recruitment of corepressors [Citation69,Citation70]. Crystallographic analysis shows that E4 interacts with ERα in a similar way to other estrogens and induces similar conformational changes (), unlike SERMs. Ligands are perfectly superimposable and interact equally with identical residues within the ligand-binding pocket. E4 binds ERα, in a similar way to E2 and E3, forming a complex that binds a key coactivator protein, steroid receptor coactivator (SRC3) [Citation48].
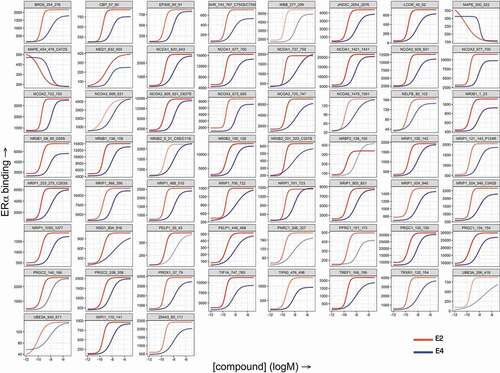
Functional comparison of E2- and E4-binding to ERα has also been performed using MARCoNI (Microarray Assay for Real-time Coregulator-Nuclear receptor Interaction). This platform measures the binding of a nuclear receptor to a set of peptides with coregulator-derived LXXLL motifs, thus providing in vitro coregulator recruitment data and a functional readout of receptor conformation [Citation71]. E2 and E4 modulate coregulator recruitment in a very similar manner (). At receptor saturating concentrations, the binding pattern and affinities of all coregulators in the presence of E4 were identical to that elicited by E2 but with a 50-fold lower potency. A SERM-induced ERα conformation would result in coregulator displacement and virtually opposite behavior to E4 on the MARCoNI platform [Citation72,Citation73]. These data together with analyses of the crystal structures of E2, E3, and E4 complexes within the ERα binding domain confirm that E4 interacts with nuclear ERα in a manner identical to that of other estrogens [Citation74–76].
Figure 2. Comparison of E2- and E4-induced in vitro coregulator recruitment by ERα. Full-length estrogen receptor α (ERα) was stimulated with estradiol (E2) or estetrol (E4) and incubated with a set of 154 immobilized peptides representing coregulator-derived binding motifs; binding was detected using a fluorescently labeled ERα-antibody. The subset with ERα-interacting peptides is shown here. Each panel displays concentration-dependent (log(M), x-axis) modulation of ERα binding (arbitrary units’ fluorescence, y-axis) to a particular coregulator motif by E2 (red) or E4 (blue).
In summary, estrogen gene regulation is a complex multi-receptor mediated mechanism, which controls cellular functions by cross-talk between MISS and nuclear actions [Citation77]. The properties of E4 show that it has specific action in tissue activity due to the activation and antagonization of nuclear and membrane-bound ERα, respectively [Citation3]. Studies of molecular interactions of E4 with nuclear ERα point to an estrogenic mode of action, that is distinct from SERMs.
3. Phase 2 clinical trial results
3.1. Contraception
Phase 2 clinical trials were performed using different E4 doses in combination with different doses of DRSP and levonorgestrel (LNG) to develop a formulation with suitable contraceptive efficacy and cycle control. An early phase 2 clinical trial showed that E4 5 to 20 mg in combination with LNG 150 µg or DRSP 3 mg inhibited ovarian activity adequately to prevent pregnancy [Citation78]. Additionally, bleeding pattern, cycle control, tolerability, body weight control, and user satisfaction were better with the E4/DRSP combinations than with the E4/LNG combinations, with E4 15 mg/DRSP 3 mg being most favorable [Citation79,Citation80]. Based on the cumulative results of these different test products, E4 15 mg/DRSP 3 mg (E4/DRSP) was selected for late phase 2 and phase 3 clinical studies. The results of subsequent phase 2 studies showed that the effect on ovulation inhibition and suppression of ovarian function for E4/DRSP was comparable to EE 20 μg/DRSP 3 mg, a marketed COC [Citation81]. E4/DRSP also demonstrated limited effects on endocrine and metabolic parameters; the effects on gonadotropins, cortisol, corticosteroid-binding globulin (CBG), angiotensinogen, SHBG, and triglycerides were less pronounced compared to EE 30 μg/LNG 150 μg or EE 20 μg/DRSP 3 mg [Citation82,Citation83]. In addition, changes in hemostasis parameters, such as endogenous thrombin potential based activated protein C (APC) resistance (expressed as normalized APC sensitivity ratio, nAPCsr), and prothrombin fragment 1 + 2 were all significantly less with E4/DRSP than with EE 30 μg/LNG 150 μg and EE 20 μg/DRSP 3 mg. SHBG was significantly less with E4/DRSP than with EE 20 μg/DRSP 3 mg [Citation84]. These results highlight the key clinical advantages that E4-containing COCs may have in relation to other estrogen-containing COCs.
3.2 Enhanced safety: tissue-specific effects of E4 in the liver (hemostasis, lipids, renin-angiotensin-aldosterone system), breast, and cardiovascular system
3.2.1. Impact of E4 on hemostasis
Liver function is closely linked to the hemostatic system as liver parenchymal cells produce the majority of factors involved in the clotting and fibrinolytic systems. Interestingly, E4 displays a specific transcriptional program in the liver, which differs from those induced by E2 and tamoxifen. This underlines the fact that each ERα ligand causes a highly specific profile of gene regulation in the liver, consistent with the differential action of E4, E2, and tamoxifen on hepatic coagulation factors in humans [Citation82,Citation84,Citation85]. Although additional molecular studies are necessary to precisely understand the complex interactions between the extranuclear/membrane and nuclear sub-populations of ERα, current knowledge suggests that the absence of ERα MISS activity could contribute to the limited impact of E4 on the liver that has been observed in several studies and which is very different from that of E2 and EE [Citation82,Citation85,Citation86].
The above presented phase 2 clinical hemostasis data for E4 demonstrate that the impact of COCs on hemostasis parameters is modulated by the total liver estrogenicity of the COC [Citation84]. The progestin can modulate the overall effect, with more liver ‘neutral’ progestins having less impact and allowing the full estrogen effect to be realized. The lower the estrogenicity of the combination of the estrogen and progestin in a COC equates to lower thrombogenic potential and thereby reduced VTE risk. Indeed, in a meta-analysis it was reported that EE/DRSP use is associated with a greater VTE risk than the combination of EE/LNG [Citation87]. Among many factors that could explain this difference (e.g. bias, such as preferential prescribing), it is likely that the anti-estrogenic properties of LNG counteract the estrogenic properties of EE on the liver more than DRSP, and as a result EE/LNG has a less thromboembolic risk than EE/DRSP. Since E4 is a less potent estrogen, the combination of DRSP with E4 results in a total estrogenicity that has a similar or even lower hemostatic impact as the EE/LNG combination [Citation84]. Therefore, choosing DRSP instead of LNG in a COC as the progestin with E4 was justified, also because of the better bleeding pattern observed (the combination with LNG resulted in a COC with anti-estrogenic properties that were too high), and better tolerability and user preference [Citation79,Citation80]. In this context, it is also good to note that studies examining usage of progestin-only COCs, including DRSP only, have shown no increased VTE risk [Citation88].
Changes to hemostasis parameters elicited by the E4/DRSP combination in a phase 2 trial were equivalent to or less than that of an EE/LNG COC and were more favorable than an EE/DRSP COC () [Citation84]. The impact of E4 alone (2.5, 5, 10, or 15 mg) on hemostasis parameters in postmenopausal women was also found to be limited compared to placebo when used once daily over 12 weeks [Citation89]. Hemostasis markers including fibrinogen, prothrombin, prothrombin fragment 1 + 2, D-dimer, factor VIII activity, protein C, protein S, antithrombin, and tissue factor pathway inhibitor, did not show significant changes from baseline compared to placebo in postmenopausal women treated with E4. Moreover, E4 had a limited effect on nAPCsr, a parameter associated with the relative risk of developing thrombosis () [Citation90]. The effect on nAPCsr and other relevant hemostatic parameters was also lower with E4 use relative to those obtained with contraceptive combinations containing EE, including the combination considered as the safest option by the EMA (i.e. EE/LNG) () [Citation84,Citation91].
Figure 3. Effect of estetrol (E4) on hemostasis markers and sex hormone binding globulin (SHBG). A: Changes of hemostasis markers are significantly lower in the E4 15 mg/ drospirenone (DRSP) 3 mg group than in the ethinyl estradiol (EE)/DRSP group and always equivalent or lower than in the EE/levonorgestrel group. Columns represent median of percentage change from baseline to cycle 6. Parameters examined: PAI-1, plasminogen activator inhibitor type 1; SHBG, sex hormone-binding protein; t-PA, tissue plasminogen activator; TFPI, tissue factor pathway inhibitor; VWF, von Willebrand factor. B: nAPCsr is significantly lower in the E4/DRSP group than in the EE/LNG or EE/DRSP groups. C: Relative risk of venous thromboembolism (VTE RR) as a function of the change in normalized activated protein C sensitivity ratio (nAPCsr) during combined oral contraceptive (COC) use. * p < 0.05 vs baseline; # p < 0.05 vs E4/DRSP. Panel A and B: Published in Douxfils et al. 2020 [Citation84]. Permission to use granted. Panel C: In silico-modeling based on the Cochrane network meta-analysis of de Bastos M. et al. Cochrane Database Syst Rev. 2014 published in Morimont, L., Dogné J-M., and Douxfils J. Permission to use granted [Citation90].
![Figure 3. Effect of estetrol (E4) on hemostasis markers and sex hormone binding globulin (SHBG). A: Changes of hemostasis markers are significantly lower in the E4 15 mg/ drospirenone (DRSP) 3 mg group than in the ethinyl estradiol (EE)/DRSP group and always equivalent or lower than in the EE/levonorgestrel group. Columns represent median of percentage change from baseline to cycle 6. Parameters examined: PAI-1, plasminogen activator inhibitor type 1; SHBG, sex hormone-binding protein; t-PA, tissue plasminogen activator; TFPI, tissue factor pathway inhibitor; VWF, von Willebrand factor. B: nAPCsr is significantly lower in the E4/DRSP group than in the EE/LNG or EE/DRSP groups. C: Relative risk of venous thromboembolism (VTE RR) as a function of the change in normalized activated protein C sensitivity ratio (nAPCsr) during combined oral contraceptive (COC) use. * p < 0.05 vs baseline; # p < 0.05 vs E4/DRSP. Panel A and B: Published in Douxfils et al. 2020 [Citation84]. Permission to use granted. Panel C: In silico-modeling based on the Cochrane network meta-analysis of de Bastos M. et al. Cochrane Database Syst Rev. 2014 published in Morimont, L., Dogné J-M., and Douxfils J. Permission to use granted [Citation90].](/cms/asset/84ec3f9f-1c0c-41e8-a78b-23027cebe154/ierj_a_2054413_f0003_oc.jpg)
The hemostasis results realized in the phase 2 clinical studies are in line with those obtained with thrombotic mouse model studies. However, it is important to remember that in mice, estrogens do not alter the circulating coagulation factors toward a procoagulant profile, as they do in humans [Citation85,Citation92–94]. In one study, a chronic high physiologic dose of E2 (reminiscent of pregnancy) decreased platelet responsiveness, increased tail-bleeding times, and protected animals against collagen/epinephrine-induced thromboembolism, through hematopoietic ERα, and independently of hematopoietic ERβ [Citation93]. Following chronic E4 treatment, mice exhibited a prolonged tail-bleeding time and were protected from arterial and also venous thrombosis in vivo [Citation95]. In addition, E4 treatment decreased ex vivo thrombus growth on collagen under arterial flow conditions [Citation93,Citation95].
In summary, the presented data show that E4 might have less residual thromboembolic risk than other estrogens, including those used in COCs or MHT, due to its limited impact on the liver [Citation91].
3.2.2. Impact on lipids
Phase 2 studies demonstrated also that the E4/DRSP combination or E4 alone (15 mg) had a minimal impact on plasma triglycerides levels, which were more increased with the EE/LNG or EE/DRSP combination. E4 alone or in combination with DRSP significantly increased high-density lipoprotein cholesterol, and non-significantly decreased low-density lipoprotein (LDL) cholesterol [Citation28,Citation75,Citation82,Citation83].
3.2.3. Impact on the renin-angiotensin aldosterone system
Interestingly, in the phase 2 studies, E4/DRSP demonstrated limited Renin-angiotensin Aldosterone System (RAAS) stimulation [Citation83]. Other estrogens stimulate RAAS, a key regulator of fluid and electrolyte balance, arteriolar resistances and thereby long-term blood pressure [Citation96]. Enhanced stimulation of RAAS can lead to the development of salt retention and of arterial hypertension. EE-containing COCs result in the stimulation of angiotensinogen production, a rate limiting substrate of renin enzymatic activity of the generation of angiotensin I. This subsequently leads to the activation of RAAS, and thereby results in a rise of exchangeable sodium levels [Citation97–102]. In healthy normotensive women, however, renin and aldosterone increments are limited due to the RAAS negative feedback loop. Prolonged exposure to EE can lead to persistent increases in angiotensinogen levels, but the negative feedback loop leads to a reduction in plasma renin concentration, and to only small elevations in plasma renin activity (PRA) and thereby angiotensin I, II and aldosterone, with no substantial impact on arterial blood pressure. In women that become hypertensive due to EE-based pharmaceutical usage, the compensatory inhibition of renin concentration is not sufficient, and thereby leads to larger increases in PRA, angiotensin II, aldosterone, and blood pressure. In contrast, E4 seems to have a limited effect on angiotensinogen, which would explain the limited changes in weight and blood pressure observed in the clinical trial program. E4 treatment is also able to prevent angiotensin II–induced hypertension by selective activation of nuclear ERα in transgenic mice [Citation63]. In addition, the anti-mineralocorticoid activity of DRSP attenuates salt and water retention, thereby preventing elevation in blood pressure [Citation102]. Beside these mild diuretic characteristics, DRSP also displays anti-androgenic activities, thereby reducing side effects associated with many COCs [Citation103].
3.2.4. Impact of E4 on normal and malignant breast tissue
In vitro studies involving normal and malignant breast epithelial cell cultures demonstrated that E4 acts as a weak estrogen in stimulating the growth of normal human breast epithelial cells and of hormone-dependent breast cancer (BC). E4 displayed a stimulatory impact only at concentrations exceeding menopausal therapeutic needs [Citation54]. Concentrations of E4 were 1,000-fold higher (1x10−8M) than that of E2 in order to promote growth. In addition, E4 presented antitumor activity in the presence of E2, by reducing the strong proliferative stimulation induced by E2. The dual weak-estrogenic and anti-estrogenic-like properties of E4 result from differential signaling pathway activation and not from a capacity to antagonize E2-induced nuclear activity.
Estrogens paradoxically induce apoptosis in long-term estrogen-deprived BC cells. Long-term treatment with SERMs and anti-estrogens leads to cell populations in which multiple stress- and inflammation-associated transcription factors and pathways are activated and participate in the promotion of acquired endocrine resistance [Citation104]. Simultaneously, these stress and inflammatory responses create a microenvironment facilitating E2- and E4-induced apoptosis in the acquired resistant BC cells [Citation105]. In a recent study, the pharmacology of E4 was examined in eight biologically different human BC cell lines with the full agonists E1, E2, and E3, a partial agonist triphenylethylene bisphenol, and the antagonists tamoxifen and endoxifen. E4 was classified as a less-potent full-estrogen agonist, causing tumor regression in long-term estrogen-deprived BC cells by triggering a rapid unfolded protein response (UPR) and apoptosis. Based on the results, the authors concluded that the use of E4 as a full agonist to treat advanced BCs is potentially superior to a partial agonist, given the partial agonist delayed induction of UPR and apoptosis, with a higher probability of tumor clonal evolution and resistance [Citation106].
In animal studies, E4 is weakly estrogenic in the normal breast and mammary gland, but is an E2-antagonist when administered in combination with E2 [Citation54,Citation107]. E4, dose-dependently, prevented the growth of chemically induced (7,12-dimethylbenz(a)anthracene, DMBA) mammary tumors in female rats [Citation108]. While E4 alone (3–10 mg/kg/day) moderately enhanced the tumor growth of human MCF7 breast carcinoma cells inoculated to ovariectomized nude mice, it reduced tumor growth in the presence of E2 [Citation54]. In a recent study, E4 (0.3 mg/kg/day) was administered continuously (either alone or with progesterone or DRSP) to PymT-transgenic mice or to patient-derived xenograft mice to mimic the steady-state plasma concentrations observed in women receiving a therapeutic dose of 15–20 mg E4 [Citation109]. This dose increased the proliferation and gene expression of uterine epithelial cells but did not affect BC growth nor metastatic dissemination to the lung, nor breast gene expression. Nevertheless, when E4 was used at 10-fold the therapeutic dose (3 mg/kg/day), it exerted pro-tumoral activity similar to that observed with E2.
In women with recently diagnosed BC, short-term exposure to 20 mg E4 induced apoptosis, and not proliferation, of breast carcinoma cells [Citation110]. E4 used in patients with end-stage endocrine resistant BC at a dose of 20–60 mg was safe and well tolerated during 12 weeks of treatment, with anti-tumor effects in five of nine heavily pre-treated patients with progressive, anti-estrogen resistant, advanced BC [Citation29].
In summary, both preclinical and initial clinical study results indicate that E4 is a weak estrogen in the breast and almost neutral at concentrations used for contraception and menopause. Limited results suggest that the combined estrogenic and anti-estrogenic properties of E4 on BC may provide a safe therapeutic window for the treatment of menopausal symptoms in healthy women. Also, as described, the beneficial effect of E4 in a phase 2 study for the treatment of women with late stage, endocrine resistant, advanced BC is promising [Citation29]. Extensive clinical studies are needed to further delineate the effect of E4 on the normal and malignant breast. It is noteworthy to mention that the use of E4/DRSP as a contraceptive is contraindicated in women with BC or with a past history of BC.
3.2.5. Nuclear ERα mediated cardiovascular protection
Several cardiovascular protective effects (including reendothelization, atheroprotection after arterial injury, and those mediated by nitric oxide), which were studied using mouse models, have until now been shown to rely on membrane ERα actions. However, as already depicted above, E4 appears as a specific ERα nuclear agonist and membrane antagonist. These actions of E4 have been extensively evaluated in various animal models of vascular protection, which demonstrated that E4 prevents atherosclerosis (Supplemental Figure 2A) [Citation48], inhibits postinjury arterial vascular smooth muscle cell proliferation, and neointimal hyperplasia (Supplemental Figure 2C) [Citation111]. Guivarc’h et al., for instance, demonstrated that selective activation of nuclear ERα with E4 elicited vasculoprotective effects by preventing angiotensin II–induced hypertension along with restoring flow-mediated arterial dilation and remodeling (Supplemental Figure 2B) [Citation63]. These results reveal an unexpected and prominent role of nuclear ERα in the vasculoprotective action of estrogens, in contrast to the wide belief that the vasculoprotection conveyed by estrogens is the consequence of their selective activation of the membrane ER. In line with this, membrane ERα participates in flow-mediated dilation in a ligand-independent manner [Citation112]. Interestingly and in agreement, the presence or absence of E4 does not alter the flow-mediated dilation [Citation112].
Acceleration of endothelial healing and increased NO production are considered as two important protective mechanisms of arteries. This, however, does not necessarily mean that the before mentioned less effective activation of eNOS and NO production in HUVEC with E4 in comparison to E2, results in a deficit of vasodilation and in a poorer endothelium-mediated vascular protection in comparison to E2. The main physiological driver of NO production is not estrogen but shear stress [Citation113–115] and estrogens are considered to play a limited role in regulating endothelial-derived NO production and vasodilation. Also, several studies confirmed that NO production is controlled by multiple mechanisms besides estrogen and shear stress, including reduction in temperature as well as by a large number of neurohumoral mediators through the activation of specific endothelial cell membrane receptors and/or posttranslational modifications [Citation74–76]. In addition, E4 induces vasodilation in uterine arteries [Citation116] and relaxing responses in rat uterine, thoracic, aortic, carotid, mesenteric, pulmonary, renal, middle cerebral, and septal coronary arteries, by a specific mechanism distinct from NO production [Citation117]. It was shown that this vasodilation was ER dependent since it was abrogated by ICI 182780, an ERα antagonist. Blockade of eNOS by N(ω)-nitro-l-arginine methyl ester (L-NAME) blunted E2- but not E4-mediated relaxing responses, demonstrating that E2 but not E4 induces vasodilation by stimulating eNOS activity. Only, the soluble guanylate cyclase inhibitor, ODQ, blocked E4 relaxation. These studies also showed that E4 inhibited smooth muscle cell Ca++ entry and contraction, and it was concluded that E4 caused relaxation of precontracted rat uterine arteries via both an endothelium-dependent mechanism involving ER and a guanylate cyclase mechanism. Finally, tamoxifen-like E4 but unlike E2, does not accelerate endothelial healing after electric injury of the carotid artery that destroys the endothelial and smooth muscle cell layers. However, in other models of injury of carotid artery where only the endothelial cells are destroyed, tamoxifen accelerates endothelial healing through activation of the nuclear ERα in smooth muscle cells. It is therefore tempting to speculate that such a mechanism could be operational for E4 [Citation118].
3.2.6. Effects of E4 on metabolism
The decreased estrogen production by ovaries at menopause causes a redistribution of body fat, with visceral adiposity that is associated with an increased risk of cardiovascular diseases, type 2 diabetes, and steatosis [Citation119–121]. Chronic E4 exposure prevented aortic atherosclerosis, body weight gain, glucose intolerance, and steatosis in ovariectomized C57Bl/6 mice and in the atheroprone LDL receptor-/- mouse fed with a Western diet [Citation122]. The beneficial effects of E4 on body weight gain, glucose tolerance, and atheroma is dependent of nuclear ERα but persisted in mice harboring a hepatocyte-specific ERα deletion. However, hepatic ERα was necessary to prevent steatosis in mice.
In summary, the phase 2 trials demonstrated that the combined properties of E4 and DRSP could make this a promising COC due to its reliable contraceptive properties, good bleeding profile, cycle control, user satisfaction, and potentially less residual thromboembolic risk than its comparators [Citation79,Citation80].
4. Phase 3 clinical trial results
Two open-label, non-comparative, phase 3 trials were conducted in EU/Russia (EU/RUS) and the USA/Canada (USA/CAN) to confirm the contraceptive efficacy and safety of E4/DRSP given for 1 year, in healthy reproductive age women () [Citation22,Citation23]. With over 1,500 women enrolled in each study, the primary contraceptive efficacy endpoint was determined in women 35 years and younger. In the EU/RUS trial, the Pearl Index (PI) was 0.44 (95% confidence interval [CI] 0.14–1.03) [Citation22], while in the USA/CAN trial the PI was 2.65 (95% CI 1.73–3.88) [Citation23]. These PI values are consistent with other contemporary COCs and show that E4/DRSP indeed provides a high level of contraceptive protection. The higher PI values in the USA/CAN relative to the EU/RUS trial are also typical as the PI has increased in US-based trials over the last few decades [Citation123]. The two most likely important contributors to this increase in PIs in the USA are more frequent pregnancy testing with more sensitive tests and less adherent study populations [Citation124]. In women aged 18–35 years, compliance based on expected pill intake averaged 99.4% across all cycles in the EU/RUS study and 98.7% in the USA/CAN study. Overall, respectively, 90.1% or 82.0% of participants missed no pills, with 6.1% or 9.1%, 2.0% or 3.6% and 1.8% or 5.3% missing an average of one, two, or more than two pills across 13 consecutive cycles, respectively, in the EU/RUS study and in the USA/CAN study, indicating a lower adherence to treatment in the USA and Canada [Citation22,Citation23].
Table 2. Phase 3 studies evaluating the E4 15 mg/DRSP 3 mg COC (24 active/4 inactive regimen)
In these phase 3 trials, E4/DRSP also showed excellent cycle control, which is an important factor influencing contraceptive selection, adherence, and treatment continuation [Citation22,Citation23]. These findings confirmed previous phase 2 findings [Citation80]. In the EU/RUS trial, more than 92% of women had regular cycles, while in the USA/CAN trial more than 83% of women had regular cycles. Only a few women did not have scheduled bleeding/spotting (EU/RUS: 7.9%; USA/CAN: 16.0%). In addition, the percentage of women that reported at least one unscheduled bleeding/spotting during treatment was 16.4% in the EU/RUS study and 20.3% in the USA/CAN study, the majority of which were qualified by participants as spotting (not needing sanitary protection). The percentage of women that discontinued treatment due to bleeding problems was 3.5% in EU/RUS and 2.7% in USA/CAN. In addition, E4/DRSP showed excellent tolerability with no unexpected safety concerns [Citation22,Citation23]. A single case of venous thrombosis was observed in the 1,553 participants in the EU/RU trial and none in the 1,864 participants in the USA/CAN trial of whom 23% had a body mass index (BMI) ≥30.0 kg/m2, a risk factor for VTE [Citation125]. In comparison, the number of VTE cases in other contraceptive US trials was 3 for EE 10 µg/norethindrone acetate 1 mg (N = 1,683, 18% obese) [Citation123], 4 for a vaginal ring delivering EE 13 µg and segesterone acetate 150 µg per day (N = 1,188) [Citation126], and 4 for a patch with dosing equivalent to an EE 30 µg/levonorgestrel 120 µg oral contraceptive (all in obese participants [N = 2,031, 35% obese]) [Citation127]. Thus, even with low-dose EE short-acting combination contraceptives, thrombosis risk remains.
In conclusion, phase 3 clinical trials examining E4 15 mg in combination with DRSP 3 mg (E4/DRSP) in a 24/4-day regimen showed high efficacy, excellent cycle control, high user ssatisfaction,and a good tolerability and safety profile. In addition, minimal impact was noted on hemostasis and metabolic parameters in women aged 16–50 years inclusive with a BMI ≤35.0 kg/m2.
The classical contraindications and warnings for the use of COCs remain valid with the E4/DRSP combination. The decision to prescribe E4/DRSP should take into consideration the individual woman’s current risk factors, particularly those for VTE. Finally, the beneficial impact of E4/DRSP on hemostasis as indicated by their limited impact on coagulation markers has to be confirmed in a larger post-authorization safety study investigating the incidence of VTE events, as agreed with regulatory authorities.
5. Other therapeutic applications for E4
5.1. Hormone replacement for menopause
In the MHT development program, E4 is used as a stand-alone compound. E4 efficacy for menopausal vasomotor symptoms (VMS) relief was initially tested in rat models, whereby E4 effectively prevented temperature increases (representative of hot flushes/VMS) in a dose-dependent manner and a potency 10-fold less than EE [Citation19,Citation128]. An early clinical trial in postmenopausal women showed that oral E4 in multiple rising doses (from 2 to 20 mg) resulted in a decrease in the mean number of hot flushes and sweating, and in an expected increase in endometrial thickness. A dose-dependent decrease in anti-gonadotropic activity was observed in both serum follicle stimulating hormone (FSH) and luteinizing hormone levels [Citation86].
Based on these early findings, a phase 2 multicenter, randomized, placebo-controlled, double-blinded, dose-finding study was conducted to determine the minimum effective dose of E4 in relieving VMS in postmenopausal women [Citation27,Citation28,Citation129]. The study included 257 postmenopausal women from 40 to 65 years old. Women received E4 2.5, 5, 10, or 15 mg or placebo once-daily for 12 weeks. All E4 doses demonstrated a reduction in the frequency of moderate to severe hot flushes. However, the E4 15 mg dosage group demonstrated the most pronounced improvement with a significant difference in the percentage change of weekly hot-flush frequency relative to placebo and a statistically significant absolute decrease in severity of VMS. Menopause-related complaints, as assessed by the menopause rating scale [Citation130], showed a reduction in hot-flush severity and sweating with E4 15 mg compared to placebo. The incidence and severity of symptoms of genitourinary syndrome of menopause (GSM) and vaginal cytology parameters also improved from baseline to week 12 in all E4 dosage groups. However, the greatest reduction in the incidence and severity of GSM symptoms compared to placebo was in the E4 15 mg group [Citation27,Citation28,Citation129]. In addition, health-related quality-of-life was improved, most prominently in the E4 15 mg group [Citation27]. Based on these results, two phase 3 studies examining E4 15 mg and 20 mg in menopausal women were initiated in 2019 and are currently ongoing [Citation24,Citation25].
5.2. Perimenopause, central nervous system disorders, vascular protection, and oncology
The therapeutic potential of E4 as a pharmaceutical component for contraception, as well as its utility in menopause, suggests the potential of E4 to relieve symptoms while providing both contraception and cycle control during the perimenopause. E4 may thus offer women an alternative estrogen with an improved risk/benefit ratio to cater for these needs during this transitional period of life.
While the impact of E4 on the reproductive system is better understood than its impact on the central nervous system, initial studies have also shown that E4 has neuroprotective, promyelinating (i.e. promotion of myelin deposits around neuronal processes and fibers, important in effective electrical transmission), neurogenic, and cerebro-angiogenic effects. E4 can effectively reduce blood markers of brain damage (S100 Calcium-Binding Protein B and glial fibrillary acidic protein) and its antioxidative actions are navigated through E4ʹs impact on estrogen receptors ERα and ERβ [Citation131]. Antioxidant properties of estrogens potentially correlate with their phenolic moiety, whereby the presence of free phenolic hydroxy groups is linked to their capacity to protect against oxidative stress. E4 has the greatest number of free phenolic hydroxy groups among natural estrogens, and thereby may have strong antioxidant effects [Citation132]. E4 may therefore have therapeutic potential in newborns, especially premature neonates (hypoxic-ischemic encephalopathy), and adults with neurological disorders [Citation133,Citation134].
Preclinical studies have also shown that E4 performs as an estrogen antagonist in breast tumor tissue due to its antagonistic activity on the membrane ERα. Clinical studies of E4 in postmenopausal women with estrogen-receptor positive early breast cancer demonstrated a significant pro-apoptotic effect on tumor tissue, and that Ki67 expression, which is a marker of cell proliferation, was not changed [Citation110]. A dose-escalation study with E4 (20, 40, and 60 mg) in postmenopausal patients with ER+/HER2- breast cancer resistant to anti-estrogens has been completed with favorable anti-tumor effects [Citation29]. Furthermore, high-dose E4 has been investigated in advanced prostate cancer as co-treatment with a luteinizing hormone-releasing hormone agonist in androgen-deprivation therapy (ADT) [Citation30]. Phase 2 results show that co-treatment in ADT with high-dose E4 is well tolerated and safe. Moreover, E4 provides highly effective estrogen substitution, and further suppresses free testosterone, prostate-specific antigen, and FSH, suggesting enhanced disease control [Citation30].
6. Conclusions
Natural and synthetic estrogens have been used for decades in contraception and MHT, with known but rare potential adverse events, such as increasing thrombosis and breast tissue proliferation. New estrogens with a better safety profile are an unmet medical need. The unique pharmacologic properties and molecular mechanisms of action of E4 suggest that it would provide a better benefit/risk ratio compared to other estrogens.
E4 is a native estrogen with differential action in tissues. The molecular nuclear mode of action of E4 is very similar to the actions of classic estrogens and is not like SERMs. Unlike other estrogens, the tissue selectivity of E4 is potentially a consequence of the differential activation of the nuclear and membrane ERα pathways; E4 does not stimulate the membrane ERα and antagonizes the membrane effects induced by E2. Clinical studies demonstrate that E4, in contrast to EE or E2, has little or no effect on circulating SHBG, angiotensinogen, triglycerides, or coagulation factors. This limited impact of E4 on the liver represents an important facet of this innate molecule, which may have a safer profile regarding the risk of thromboembolic events.
The development of E4 in contraception, menopause, and prevention/treatment of hypoxic-ischemic encephalopathy is an example of how basic research and preclinical studies have guided the clinical characterization of E4 activities in humans and enabled precise delineation of its differences from the three other natural estrogens. These results also highlight the importance of the estrogenic component of COCs; the specific estrogen/progestin combination has an impact on the total estrogenicity of the formulation, its clinical use, and linked benefit/risk balance. Both preclinical and clinical data have shown the cardiovascular safety of E4, acting as a classic estrogen to induce a series of cardiovascular/arterial benefits through activation of the ERα nuclear pathway. At the same time, E4 may pose less of a risk on thromboembolic events, such as VTE due to its neutral profile on the synthesis of hemostatic proteins. Finally, E4 has been shown to have a lower impact on breast tissue compared to other estrogens. Nevertheless, the effects of the tissue selectivity of E4, in particular in the liver and breast, will require further molecular studies for validation.
7. Expert opinion
With its unique profile of activity, E4 is a new chemical entity, distinct from all other estrogens as recognized by the EMA, FDA, and TGA. This estrogen, first described over 80 years, could fill the gap for a safer hormone therapy to address the needs of women during the reproductive (contraception), perimenopause, and menopause years. With its minimal impact on liver metabolism, hemostasis, lipids, RAAS, and breast, a low risk of drug–drug interactions, and its favorable VTE risk profile, E4 has the potential to be a safer estrogen. Overall, E4 in combination with DRSP offers excellent cycle control with additional safety in a contraceptive indication.
Previous attempts to reduce the increased risk of VTE by decreasing the EE content resulted in poor cycle control and miserable tolerance. Replacement of EE by E2 also resulted in an increase in high rates of transient amenorrhea, spotting, and bleeding. This is the consequence of the transformation of E2 in the endometrium into estrone by the 17-β hydroxysteroid dehydrogenase type 2 induced by all progestins present in all COCs [Citation135]. E4 escapes this inactivation, stabilizes the endometrium and provides a bleeding/spotting profile equivalent to that of the best EE containing COCs, together with comparable efficacy and tolerance, but with the potential for a considerably improved safety profile.
Many women will spend ≥40% of their lives in post-menopause. Current guidelines still highlight the need to inform women that there could be an increased risk of breast cancer, stroke, and VTE associated with MHT. They recommend limiting the MHT to the shortest possible period. E4 alone or in combination could become an ideal chronic treatment for aging women beyond a short-term treatment to alleviate hot flushes only. Other menopausal symptoms including changes in mood, sleep patterns, memory, body shape, as well as the onset urogenital symptoms can also be very distressing and affect a woman’s personal and social life.
Early-stage clinical oncologic studies point also to the safety of this new estrogen. We therefore consider that the introduction of E4 in women’s healthcare is a breakthrough with multiple therapeutic opportunities throughout women’s lives.
Article highlights
Estetrol (E4) is an estrogen that activates the nuclear estrogen receptor (ER) α signaling pathway but does not activate membrane ERα signaling pathway in specific tissues. This unique molecular mode of action of E4 is distinct from other estrogens.
E4 has been referred to as the first native estrogen with selective tissue activity due to its tissue-specific actions mainly supported by a differential activity on nuclear and membrane ERα activation.
E4 has recently been approved in the United States of America, Europe, Canada, and Australia as a new estrogenic component of a combined oral contraceptive (E4 monohydrate 15 mg/drospirenone 3 mg [DRSP]).
E4/DRSP displays good contraceptive efficacy, safety, and user tolerability.
E4 has a low impact in the liver and breast and a more potent effect in the endometrium, vaginal keratinocytes, and the cardiovascular system.
Relative to other contraceptives, E4/DRSP showed less impact on several hemostasis parameters, including markers of prothrombotic states. This suggests that E4/DRSP may have less residual risk of thromboembolic events than other contraceptives.
Clinical trials examining the therapeutic potential of E4 treatment for menopausal symptoms, as well as for breast and prostate cancer, are underway.
Declaration of interest
C Gérard and M Jost are employees of Estetra SRL (an affiliate company of Mithra Pharmaceuticals). The laboratory of J-F Arnal, F Lenfant, and C Fontaine received financial support from Mithra Pharmaceuticals for preclinical studies. MD Creinin has received speaking honorarium from Gedeon Richter, serves on an advisory board for Fuji Pharma, and is a consultant for Danco, Estetra SRL (an affiliate company of Mithra Pharmaceuticals), Mayne, Medicines360, and Merck. The University of California, Davis receives contraceptive research funding for MD Creinin from Chemo Research SL, Evofem, HRA Pharma, Medicines360, Merck, and Sebela. J Douxfils is the director and founder of Qualiblood, a contract research organization that received funding from Mithra Pharmaceuticals. He also reports personal fees from Mithra Pharmaceuticals and Gedeon Richter as relevant financial activities outside the submitted work. R Houtman was the Nuclear Receptor Group Leader at PamGene, a contract research organization that received funding from Mithra Pharmaceuticals. He is currently employed as VP Research at Precision Medicine Lab that receives funding from Mithra Pharmaceuticals. DF Archer owns stock or options in Agile Therapeutics and InnovaGyn, Inc., and is a consultant for AbbVie, Agile Therapeutics, Exeltis, Mayne Pharma, Mithra Pharmaceuticals, and TherapeuticsMD. Eastern Virginia Medical School receives research funding from AbbVie, Bayer Healthcare, Estetra SRL (an affiliate company of Mithra Pharmaceuticals), Myovant, ObsEva, and TherapeuticsMD. RL Reid is an advisory board member and speaker for Pfizer, Biosyent, and Duschenay, and serves on an advisory board for Searchlight. He received personal fees from Merck Canada, Searchlight Pharma, and Mithra Pharmaceuticals, outside the submitted work. RA Lobo is a member of the scientific advisory board of Mithra Pharmaceuticals. U Gaspard is a senior consultant at Mithra Pharmaceuticals. HJT Coelingh Bennink is president and a shareholder of Pantarhei Oncology, an affiliate of Pantarhei Bioscience BV. He has commercial interests in the development of Estetrol for oncological applications. J-M Foidart is co-founder of Mithra Pharmaceuticals, shareholder, and member of the Board. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
A reviewer on this manuscript has disclosed speaker’s honoraria from Gedeon Richter, who owns commercial rights on the association of estetrol and drospirenone for contraceptive use. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Supplemental Material
Download JPEG Image (1.4 MB){kind=link}
Supplemental Material
Download JPEG Image (811.2 KB){kind=link}
Supplementry material
Supplemental data for this article can be accessed publisher’s website.
Additional information
Funding
References
- Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11(8):597–608.
- Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116(3):242–246.
- Arnal JF, Lenfant F, Metivier R, et al. Membrane and nuclear estrogen receptor alpha actions: from tissue specificity to medical implications. Physiol Rev. 2017;97(3):1045–1087.
- Valera MC, Fontaine C, Dupuis M, et al. Towards optimization of estrogen receptor modulation in medicine. Pharmacol Ther. 2018;189:123–129.
- Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34(3):309–338.
- Frobenius W. The rabbits are prepared … ” - the development of ethinylestradiol and ethinyltestosterone. J Reproduktionsmed Endokrinol. 2011;8(Suppl.1):32–57.
- Lidegaard O, Lokkegaard E, Svendsen AL, et al. Hormonal contraception and risk of venous thromboembolism: national follow-up study. BMJ. 2009;339:b2890.
- Lidegaard O, Nielsen LH, Skovlund CW, et al. Risk of venous thromboembolism from use of oral contraceptives containing different progestogens and oestrogen doses: Danish cohort study, 2001-9. BMJ. 2011;343:d6423.
- Morch LS, Skovlund CW, Hannaford PC, et al. Contemporary hormonal contraception and the risk of breast cancer. N Engl J Med. 2017;377(23):2228–2239.
- Chlebowski RT, Hendrix SL, Langer RD, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the women’s health initiative randomized trial. JAMA. 2003;289(24):3243–3253.
- Manson JE, Kaunitz AM. Menopause management–getting clinical care back on track. N Engl J Med. 2016;374(9):803–806.
- Vinogradova Y, Coupland C, Hippisley-Cox J. Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the qResearch and CPRD databases. BMJ. 2019;364:k4810.
- Hagen AA, Barr M, Diczfalusy E. Metabolism of 17-beta-oestradiol-4-14-C in early infancy. Acta Endocrinol (Copenh). 1965;49:15–21.
- Gurpide E, Schwers J, Welch MT, et al. Fetal and maternal metabolism of estradiol during pregnancy. J Clin Endocrinol Metab. 1966;26(12):1355–1365.
- Schwers J, Eriksson G, Diczfalusy E. 15 alpha-hydroxylation: a new pathway of estrogen metabolism in the human fetus and newborn. Biochim Biophys Acta. 1965;100:313–316.
- Schwers J, Govaerts-Videtsky M, Wiqvist N, et al. Metabolism of oestrone sulphate by the previable human foetus. Acta Endocrinol (Copenh). 1965;50(4):597–610.
- Holinka CF, Gurpide E. In vivo effects of estetrol on the immature rat uterus. Biol Reprod. 1979;20(2):313–316.
- Holinka CF, Bressler RS, Zehr DR, et al. Comparison of effects of estetrol and tamoxifen with those of estriol and estradiol on the immature rat uterus. Biol Reprod. 1980;22(4):913–926.
- Coelingh Bennink HJ, Holinka CF, Diczfalusy E. Estetrol review: profile and potential clinical applications. Climacteric. 2008;Suppl 11:47–58.
- Warmerdam EG, Visser M, Coelingh Bennink HJ, et al. A new route of synthesis of estetrol. Climacteric. 2008;Suppl 11:59–63.
- Estetra SA. Process for the production of estetrol intermediates. International patent application WO 2012/164096 A1; 2012 Jun 1.
- Gemzell Danielsson K, Apter D, Zatik J, et al. Estetrol-drospirenone combination oral contraceptive: a clinical study of contraceptive efficacy, bleeding pattern, and safety in Europe and Russia. Br J Obstet Gynaecol. 2022;129(1):63–71.
- Creinin MD, Westhoff CL, Bouchard C, et al. Estetrol-drospirenone combination oral contraceptive: north American phase 3 efficacy and safety results. Contraception. 2021;104(3):222. .
- Estetra SPRL. A randomized double-blind placebo controlled phase 3 trial to evaluate the efficacy and safety of estetrol for the treatment of moderate to severe vasomotor symptoms in postmenopausal women (E4Comfort Study I). EUDRACT 2019-001289-14. Clinicaltrials.gov NCT04209543.
- Estetra SPRL. A randomized double-blind placebo controlled phase 3 trial to evaluate the efficacy and safety of estetrol for the treatment of moderate to severe vasomotor symptoms in postmenopausal women (E4Comfort Study II). Clinicaltrials.Gov NCT04090957.
- Utian WH, Lobo R, Mawet M, et al. A phase 3 protocol to assess the efficacy and safety of estetrol (E4), a promising new treatment for menopausal vasomotor symptoms. Menopause. 2019;26(12):1480.
- Gaspard U, Taziaux M, Jost M, et al. Estetrol (E4), the next generation hormone therapy (HT) for menopausal symptoms: phase 2b clinical trial results. 12th European Congress on Menopause and Andropause. Berlin, Germany. Maturitas. 2019 May 15-17;124(153): Abstract P09.
- Gaspard U, Taziaux M, Mawet M, et al. A multicenter, randomized study to select the minimum effective dose of estetrol (E4) in postmenopausal women (E4Relief): part 1. Vasomotor symptoms and overall safety. Menopause. 2020;27(8): 848–857
- Schmidt M, Lenhard H, Hoenig A, et al. Tumor suppression, dose-limiting toxicity and wellbeing with the fetal estrogen estetrol in patients with advanced breast cancer. J Cancer Res Clin Oncol. 2021;147(6):1833–1842.
- Coelingh Bennink HJT, Van Moorselaar JA, Crawford ED, et al. Estetrol cotreatment of androgen deprivation therapy in infiltrating or metastatic, castration-sensitive prostate cancer: a randomized, double-blind, phase II trial (PCombi). Eur Urol Open Sci. 2021; 28: 52–61.
- Coelingh Bennink F, Holinka CF, Visser M, et al. Maternal and fetal estetrol levels during pregnancy. Climacteric. 2008;Suppl 11:69–72.
- Hickey M, Hart R, Keelan JA. The relationship between umbilical cord estrogens and perinatal characteristics. Cancer Epidemiol Biomarkers Prev. 2014;23(6):946–952.
- Visser M, Holinka CF, Coelingh Bennink HJ. First human exposure to exogenous single-dose oral estetrol in early postmenopausal women. Climacteric. 2008;Suppl 11:31–40.
- Nextstellis. Prescibing information. [cited Jul 2021]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214154s000lbl.pdf
- Drovelis (estetrol/drospirenone). Summary of product characteristics [cited Jul 2021]. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/drovelis
- Santen RJ, Yue W, Wang JP. Estrogen metabolites and breast cancer. Steroids. 2015;99(Pt A):61–66.
- Yager JD. Mechanisms of estrogen carcinogenesis: the role of E2/E1-quinone metabolites suggests new approaches to preventive intervention–a review. Steroids. 2015;99(Pt A):56–60.
- Bagot CN, Marsh MS, Whitehead M, et al. The effect of estrone on thrombin generation may explain the different thrombotic risk between oral and transdermal hormone replacement therapy. J Thromb Haemost. 2010;8(8):1736–1744.
- Pugeat MM, Dunn JF, Nisula BC. Transport of steroid hormones: interaction of 70 drugs with testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53(1):69–75.
- Hodgert Jury H, Zacharewski TR, Hammond GL. Interactions between human plasma sex hormone-binding globulin and xenobiotic ligands. J Steroid Biochem Mol Biol. 2000;75(2–3):167–176.
- Hammond GL, Hogeveen KN, Visser M, et al. Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells. Climacteric. 2008;Suppl 11:41–46.
- Siiteri PK, Murai JT, Hammond GL, et al. The serum transport of steroid hormones. Recent Prog Horm Res. 1982;38:457–510.
- Cirillo DJ, Wallace RB, Rodabough RJ, et al. Effect of estrogen therapy on gallbladder disease. JAMA. 2005;293(3):330–339.
- Jirku H, Kadner S, Levitz M. Pattern of estetrol conjugation in the human. Steroids. 1972;19(4):519–534.
- Nagatomi K, Osawa Y, Kirdani RY, et al. Studies on phenolic steroids in human subjects. XVII. The kidney and fate of 1 5 alpha-hydroxyestrogens. J Clin Endocrinol Metab. 1973;37(6):887–900.
- Schut HA, Bowman JM, Solomon S. Precursor role of 15 alpha-hydroxyestradiol and 15alpha-hydroxyandrostenedione in the formation of estetrol. Can J Biochem. 1978;56(2):887–900.
- Coelingh Bennink HJT, Foidart JM. Estetrol, a fetal steroid for the treatment of adults. J Reprod Med Endokrinol Online. 2015;12(4):399–403.
- Abot A, Fontaine C, Buscato M, et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor alpha modulation, uncoupling nuclear and membrane activation. EMBO Mol Med. 2014;6(10):1328–1346. •Describes the characterisation of E4 as an selective ER modulator.
- Levin ER, Pietras RJ. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat. 2008;108(3):351–361.
- Mendelsohn ME, Karas RH. Rapid progress for non-nuclear estrogen receptor signaling. J Clin Invest. 2010;120(7):2277–2279.
- Wu Q, Chambliss K, Umetani M, et al. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286(17):14737–14743.
- Darblade B, Caillaud D, Poirot M, et al. Alteration of plasmalemmal caveolae mimics endothelial dysfunction observed in atheromatous rabbit aorta. Cardiovasc Res. 2001;50(3):566–576.
- Zhang D, Trudeau VL. Integration of membrane and nuclear estrogen receptor signaling. Comp Biochem Physiol A Mol Integr Physiol. 2006;144(3):306–315.
- Gerard C, Mestdagt M, Tskitishvili E, et al., Combined estrogenic and anti-estrogenic properties of estetrol on breast cancer may provide a safe therapeutic window for the treatment of menopausal symptoms. Oncotarget. 6(19): 17621–17636. 2015.
- Marino M, Ascenzi P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids. 2008;73(9–10):853–858.
- Zimmerman MA, Budish RA, Kashyap S, et al. GPER-novel membrane oestrogen receptor. Clin Sci (Lond). 2016;130(12):1005–1016.
- Giretti MS, Montt Guevara MM, Cecchi E, et al. Effects of estetrol on migration and invasion in T47D breast cancer cells through the actin cytoskeleton. Front Endocrinol (Lausanne). 2014;5:80.
- Montt-Guevara MM, Giretti MS, Russo E, et al. Estetrol modulates endothelial nitric oxide synthesis in human endothelial cells. Front Endocrinol (Lausanne). 2015;6:111.
- Pluchino N, Santoro AN, Casarosa E, et al. Effect of estetrol administration on brain and serum allopregnanolone in intact and ovariectomized rats. J Steroid Biochem Mol Biol. 2014;143:285–290.
- Tskitishvili E, Nisolle M, Munaut C, et al. Estetrol attenuates neonatal hypoxic-ischemic brain injury. Exp Neurol. 2014;261:298–307.
- Coelingh Bennink HJ, Heegaard AM, Visser M, et al. Oral bioavailability and bone-sparing effects of estetrol in an osteoporosis model. Climacteric. 2008;Suppl 11:2–14.
- Coelingh Bennink HJ, Skouby S, Bouchard P, et al. Ovulation inhibition by estetrol in an in vivo model. Contraception. 2008;77((3)):186–190.
- Guivarc’h E, Buscato M, Guihot A-L, et al. Predominant Role of Nuclear Versus Membrane Estrogen Receptor α in Arterial Protection: implications for Estrogen Receptor α Modulation in Cardiovascular Prevention/Safety. J Am Heart Assoc. 2018;7(13):e008950.
- Wardell SE, Marks JR, McDonnell DP. The turnover of estrogen receptor alpha by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem Pharmacol. 2011;82(2):122–130.
- Benoit T, Valera MC, Fontaine C, et al. Estetrol, a fetal selective estrogen receptor modulator, acts on the vagina of mice through nuclear estrogen receptor alpha activation. Am J Pathol. 2017;187(11):2499–2507.
- Adlanmerini M, Solinhac R, Abot A, et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. 2014;111(2):E283–90.
- Komm BS, Mirkin S. An overview of current and emerging SERMS. J Steroid Biochem Mol Biol. 2014;143:207–222.
- Heery DM, Kalkhoven E, Hoare S, et al. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387(6634):733–736.
- Metivier R, Stark A, Flouriot G, et al. A dynamic structural model for estrogen receptor-alpha activation by ligands, emphasizing the role of interactions between distant A and E domains. Mol Cell. 2002;10(5):1019–1032.
- Katzenellenbogen BS, Sun J, Harrington WR, et al. Structure-function relationships in estrogen receptors and the characterization of novel selective estrogen receptor modulators with unique pharmacological profiles. Ann N Y Acad Sci. 2001;949:6–15.
- Broekema MF, Hollman DAA, Koppen A, et al. Profiling of 3696 nuclear receptor-coregulator interactions: a resource for biological and clinical discovery. Endocrinology. 2018;159(6):2397–2407.
- Guan J, Zhou W, Hafner M, et al. Therapeutic ligands antagonize estrogen receptor function by impairing its mobility. Cell. 2019;178(4):949–963 e18.
- Nwachukwu JC, Srinivasan S, Zheng Y, et al. Predictive features of ligand-specific signaling through the estrogen receptor. Mol Syst Biol. 2016;12(4):864.
- Foidart JM, Arnal J, Lenfant F, et al. Estetrol (E4) is a unique estrogen with selective actions in tissues which are distinctly different from the actions of SERMs. Menopause. 2019;26:1464–1465.
- Foidart JM, Arnal JF, Lenfant F, et al. Estetrol (E4) is a unique estrogen with selective actions in tissues which are distinctly different from the actions of SERMs. J Endocr Soc. 2019;3(Suppl.1):SUN–LB001.
- Foidart JM, Gaspard U, Pequeux C, et al. Unique vascular benefits of estetrol, a native fetal estrogen with specific actions in tissues (NEST). In: Diaz Brinton RD, Genazzani AR, Simoncini T, et al.editors. Sex steroids’ effects on brain, heart and vessels: volume 6: frontiers in gynecological endocrinology. Cham: Springer International Publishing; 2019. p. 169–195.
- Yasar P, Ayaz G, User SD, et al. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. 2017;16(1):4–20.
- Duijkers IJM, Klipping C, Zimmerman Y, et al. Inhibition of ovulation by administration of estetrol in combination with drospirenone or levonorgestrel: results of a phase II dose-finding pilot study. Eur J Contracept Reprod Health Care. 2015;20(6):476–489.
- Apter D, Zimmerman Y, Beekman L, et al. Estetrol combined with drospirenone: an oral contraceptive with high acceptability, user satisfaction, well-being and favourable body weight control. Eur J Contracept Reprod Health Care. 2017;22(4):260–267.
- Apter D, Zimmerman Y, Beekman L, et al. Bleeding pattern and cycle control with estetrol-containing combined oral contraceptives: results from a phase II, randomised, dose-finding study (FIESTA). Contraception. 2016;94:366–373.
- Duijkers I, Klipping C, Kinet V, et al., Effects of an oral contraceptive containing estetrol and drospirenone on ovarian function. Contraception. 103(6): 386–393. 2021.
- Mawet M, Maillard C, Klipping C, et al. Unique effects on hepatic function, lipid metabolism, bone and growth endocrine parameters of estetrol in combined oral contraceptives. Eur J Contracept Reprod Health Care. 2015;20(6):463–475.
- Klipping C, Duijkers I, Mawet M, et al., Endocrine and metabolic effects of an oral contraceptive containing estetrol and drospirenone. Contraception. 103(4): 213–221. 2021.
- Douxfils J, Klipping C, Duijkers I, et al., Evaluation of the effect of a new oral contraceptive containing estetrol and drospirenone on hemostasis parameters. Contraception. 102(6): 396–402. 2020.
- Kluft C, Zimmerman Y, Mawet M, et al. Reduced haemostatic effects with drospirenone-based oral contraceptives containing estetrol versus ethinyl estradiol. Contraception. 2017;95(2):140–147.
- Coelingh Bennink HJT, Zimmerman Y, Verhoeven C, et al. Clinical effects of the fetal estrogen estetrol in a multiple-rising-dose study in postmenopausal women. Maturitas. 2016;91:93–100.
- Dragoman MV, Tepper NK, Fu R, et al. A systematic review and meta-analysis of venous thrombosis risk among users of combined oral contraception. Int J Gynaecol Obstet. 2018;141(3):287–294.
- Mantha S, Karp R, Raghavan V, et al. Assessing the risk of venous thromboembolic events in women taking progestin-only contraception: a meta-analysis. BMJ. 2012;345(aug07 2):e4944.
- Foidart JM, Lobo RA, Rosing J, et al. Estetrol (E4) is a unique native estrogen that does not modify coagulation markers in postmenopausal women and maintains sensitivity to activated protein C (APC). Menopause. 2019;26(12):1464–1465.
- Morimont L, Dogne JM, Douxfils J. Letter to the editors-in-chief in response to the article of Abou-Ismail, et al. Entitled “estrogen and thrombosis: a bench to bedside review. Thromb Res. 2020;193(193):221–223. (Thrombosis Research 192 (2020) 40-51).
- Morimont L, Haguet H, Dogné J-M, et al. Combined oral contraceptives and venous thromboembolism: review and perspective to mitigate the risk. Front Endocrinol (Lausanne). 2021; 12. 1592 https://doi.org/10.3389/fendo.2021.769187
- Shapiro S. Oral contraceptives, hormone therapy and cardiovascular risk. Climacteric. 2008;11(5):355–363.
- Valera MC, Gratacap MP, Gourdy P, et al. Chronic estradiol treatment reduces platelet responses and protects mice from thromboembolism through the hematopoietic estrogen receptor alpha. Blood. 2012;120(8):1703–1712.
- Valera MC, Fontaine C, Lenfant F, et al. Protective hematopoietic effect of estrogens in a mouse model of thrombosis: respective roles of nuclear versus membrane estrogen receptor alpha. Endocrinology. 2015;156(11):4293–4301.
- Valera MC, Noirrit-Esclassan E, Dupuis M, et al. Effect of estetrol, a selective nuclear estrogen receptor modulator, in mouse models of arterial and venous thrombosis. Mol Cell Endocrinol. 2018;477:132–139.
- Oelkers WK. Effects of estrogens and progestogens on the renin-aldosterone system and blood pressure. Steroids. 1996;61(4):166–171.
- Oelkers W, Blumel A, Schoneshofer M, et al. Effects of ethinylestradiol on the renin-angiotensin-aldosterone-system and on plasma transcortin in women and men. J Clin Endocrinol Metab. 1976;43(5):1036–1040.
- Crane MG, Harris JJ. Effect of estrogens and gestagens on the renin–aldosterone system in oral contraceptives and high blood pressure. In: Fregly MJ, Fregly MS, editors. Oral contraceptives and high blood pressure. Gainesville: Dolphin press; 1974. p. 100–119.
- Crane MG, Harris JJ. Effect of estrogens and gestagens on exchangeable sodium. In: Fregly MJ, Fregly MS, editors. Oral contraceptives and high blood pressure. Gainesville: Dolphin press; 1974. p. 159–168.
- Oelkers W, Schoneshofer M, Blumel A. Effects of progesterone and four synthetic progestagens on sodium balance and the renin-aldosterone system in man. J Clin Endocrinol Metab. 1974;39(5):882–890.
- Oelkers W, Berger V, Bolik A, et al. Dihydrospirorenone, a new progestogen with antimineralocorticoid activity: effects on ovulation, electrolyte excretion, and the renin-aldosterone system in normal women. J Clin Endocrinol Metab. 1991;73(4):837–842.
- Oelkers W, Foidart JM, Dombrovicz N, et al. Effects of a new oral contraceptive containing an antimineralocorticoid progestogen, drospirenone, on the renin-aldosterone system, body weight, blood pressure, glucose tolerance, and lipid metabolism. J Clin Endocrinol Metab. 1995;80(6):1816–1821.
- Foidart JM. Added benefits of drospirenone for compliance. Climacteric. 2005;8(3):28–34.
- Fan P, Jordan VC. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist. 2019;2:198–209.
- Yue W, Verhoeven C, Bernnink HC, et al. Pro-Apoptotic Effects of Estetrol on Long-Term Estrogen-Deprived Breast Cancer Cells and at Low Doses on Hormone-Sensitive Cells. Breast Cancer : Basic and Clinical Research. 2019;13:1178223419844198.
- Abderrahman B, Maximov PY, Curpan RF, et al. Rapid induction of the unfolded protein response and apoptosis by estrogen mimic TTC-352 for the treatment of endocrine-resistant breast cancer. Mol Cancer Ther. 2021;20(1):11–25.
- Gerard C, Blacher S, Communal L, et al. Estetrol is a weak estrogen antagonizing estradiol-dependent mammary gland proliferation. J Endocrinol. 2015;224(1):85–95.
- Coelingh Bennink HJT, Singer C, Simoncini T, et al. Estetrol, a pregnancy-specific human steroid, prevents and suppresses mammary tumor growth in a rat model. Climacteric. 2008;11(Suppl.1):29.
- Gallez A, Blacher S, Maquoi E, et al. Estetrol combined to progesterone for menopause or contraception indication is neutral on breast cancer. Cancers (Basel). 2021;13:2486.
- Singer CF, Bennink HJ, Natter C, et al. Antiestrogenic effects of the fetal estrogen estetrol in women with estrogen-receptor positive early breast cancer. Carcinogenesis. 2014;35(11):2447–2451.
- Smirnova NF, Fontaine C, Buscato M, et al. The activation function-1 of estrogen receptor alpha prevents arterial neointima development through a direct effect on smooth muscle cells. Circ Res. 2015;117(9):770–778.
- Favre J, Vessieres E, Guihot AL, et al. Membrane estrogen receptor alpha (ERα) participates in flow-mediated dilation in a ligand-independent manner. ELife. 2021;10. https://doi.org/10.7554/eLife.68695.
- Vanhoutte PM. Nitric oxide: from good to bad. Ann Vasc Dis. 2018;11(1):41–51.
- Vanhoutte PM, Zhao Y, Xu A, et al. Thirty years of saying no: sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circ Res. 2016;119(2):375–396.
- Yamazaki Y, Kondo Y, Kamiyama Y. Estimation of shear-stress-induced endothelial nitric oxide production from flow-mediated dilation. Annu Int Conf IEEE Eng Med Biol Soc. 2013;2013:41–51.
- Levine MG, Miodovnik M, Clark KE. Uterine vascular effects of estetrol in nonpregnant ewes. Am J Obstet Gynecol. 1984;148(6):735–738.
- Hilgers RH, Oparil S, Wouters W, et al. Vasorelaxing effects of estetrol in rat arteries. J Endocrinol. 2012;215(1):97–106.
- Zahreddine R, Davezac M, Smirnova N, et al. Tamoxifen accelerates endothelial healing by targeting ERα in smooth muscle cells. Circ Res. 2020;127(12):1473–1487.
- Carr MC. The emergence of the metabolic syndrome with menopause. J Clin Endocrinol Metab. 2003;88(6):29.
- Mauvais-Jarvis F, Manson JE, Stevenson JC, et al. Menopausal hormone therapy and type 2 diabetes prevention: evidence, mechanisms, and clinical implications. Endocr Rev. 2017;38(3):173–188.
- Thong EP, Codner E, Laven JSE, et al. Diabetes: a metabolic and reproductive disorder in women. Lancet Diabetes Endocrinol. 2020;8(2):134–149.
- Buscato M, Davezac M, Zahreddine R, et al. Estetrol prevents western diet-induced obesity and atheroma independently of hepatic estrogen receptor alpha. Am J Physiol Endocrinol Metab. 2021;320(1):E19–E29.
- Archer DF, Nakajima ST, Sawyer AT, et al. Norethindrone acetate 1.0 milligram and ethinyl estradiol 10 micrograms as an ultra low-dose oral contraceptive. Obstet Gynecol. 2013;122(3):E19–E29.
- Trussell J, Portman D. The creeping pearl: why has the rate of contraceptive failure increased in clinical trials of combined hormonal contraceptive pills? Contraception. 2013;88(5):604–610.
- Abdollahi M, Cushman M, Rosendaal FR. Obesity: risk of venous thrombosis and the interaction with coagulation factor levels and oral contraceptive use. Thromb Haemost. 2003;89(3):493–498.
- Gemzell-Danielsson K, Sitruk-Ware R, Creinin MD, et al. Segesterone acetate/ethinyl estradiol 12-month contraceptive vaginal system safety evaluation. Contraception. 2019;99(6):323–328.
- Nelson AL, Kaunitz AM, Kroll R, et al. Efficacy, safety, and tolerability of a levonorgestrel/ethinyl estradiol transdermal delivery system: phase 3 clinical trial results. Contraception. 2021;103(3):137–143.
- Holinka CF, Brincat M, Coelingh Bennink HJ. Preventive effect of oral estetrol in a menopausal hot flush model. Climacteric. 2008;Suppl 11:15–21.
- Utian WH, Gaspard U, Jost M, et al. Estetrol, the next generation of hormone therapy: results of a phase 2b dose-finding study in postmenopausal women (E4 Relief). Menopause. 2018;25:1489–1490.
- Schneider HP, Heinemann LA, Rosemeier HP, et al. The menopause rating scale (MRS): reliability of scores of menopausal complaints. Climacteric. 2000;3(1):59–64.
- Tskitishvili E, Pequeux C, Munaut C, et al. Estrogen receptors and estetrol-dependent neuroprotective actions: a pilot study. J Endocrinol. 2017;232(1):85–95.
- Prokai-Tatrai K, Perjesi P, Rivera-Portalatin NM, et al. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids. 2008;73(3):280–288.
- Tskitishvili E, Foidart JM. Estetrol and its effects on the damaged brain. In: Diaz Brinton R, Genazzani AR, Simoncini T, et al., editors. Sex steroids’ effects on brain, heart and vessels: volume 6: frontiers in gynecological endocrinology. Cham: Springer International Publishing; 2019. p. 43–92.
- Tskitishvili E, Pequeux C, Munaut C, et al. Use of estetrol with other steroids for attenuation of neonatal hypoxic-ischemic brain injury: to combine or not to combine? Oncotarget. 2016;7(23):33722–33743.
- Yang S, Fang Z, Gurates B, et al. Stromal PRs mediate induction of 17beta-hydroxysteroid dehydrogenase type 2 expression in human endometrial epithelium: a paracrine mechanism for inactivation of E2. Mol Endocrinol. 2001;15(12):2093–2105.